

IgG+ Extracellular Vesicles Measure Therapeutic Response in Advanced Pancreatic Cancer

 , , , ,
, , , , 
 and
and
Abstract
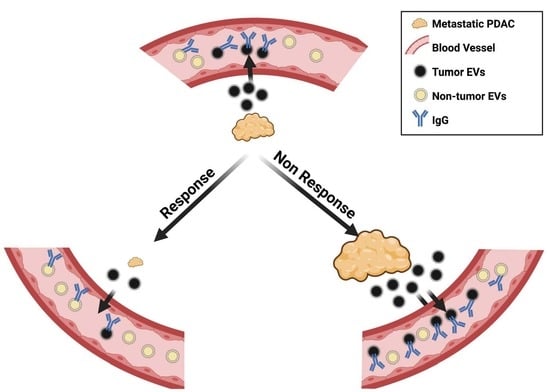
:
1. Introduction
2. Materials and Methods
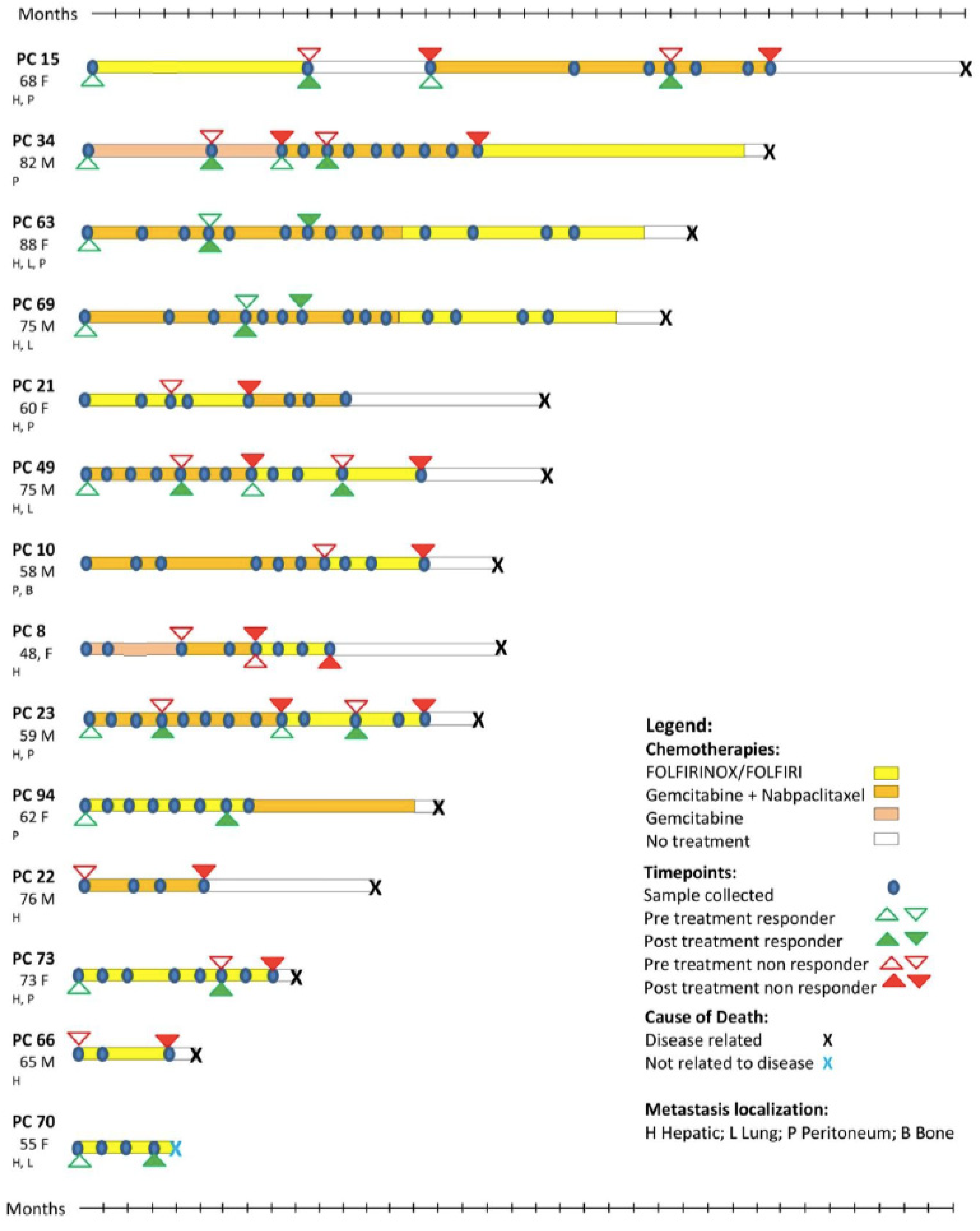
2.1. Patients and Controls
2.2. Purification and Characterization of EVs from Plasma
2.3. Characterization of EV Protein Composition by Mass Spectrometry (MS)
2.4. Western Blotting
2.5. Analysis of IgG+ EV Population by Vesicle Flow Cytometry
2.6. Total Plasma Immunoglobulin G Quantification
2.7. Identification of EV Surface Proteins Associated with IgG
2.8. Statistical Analysis
3. Results
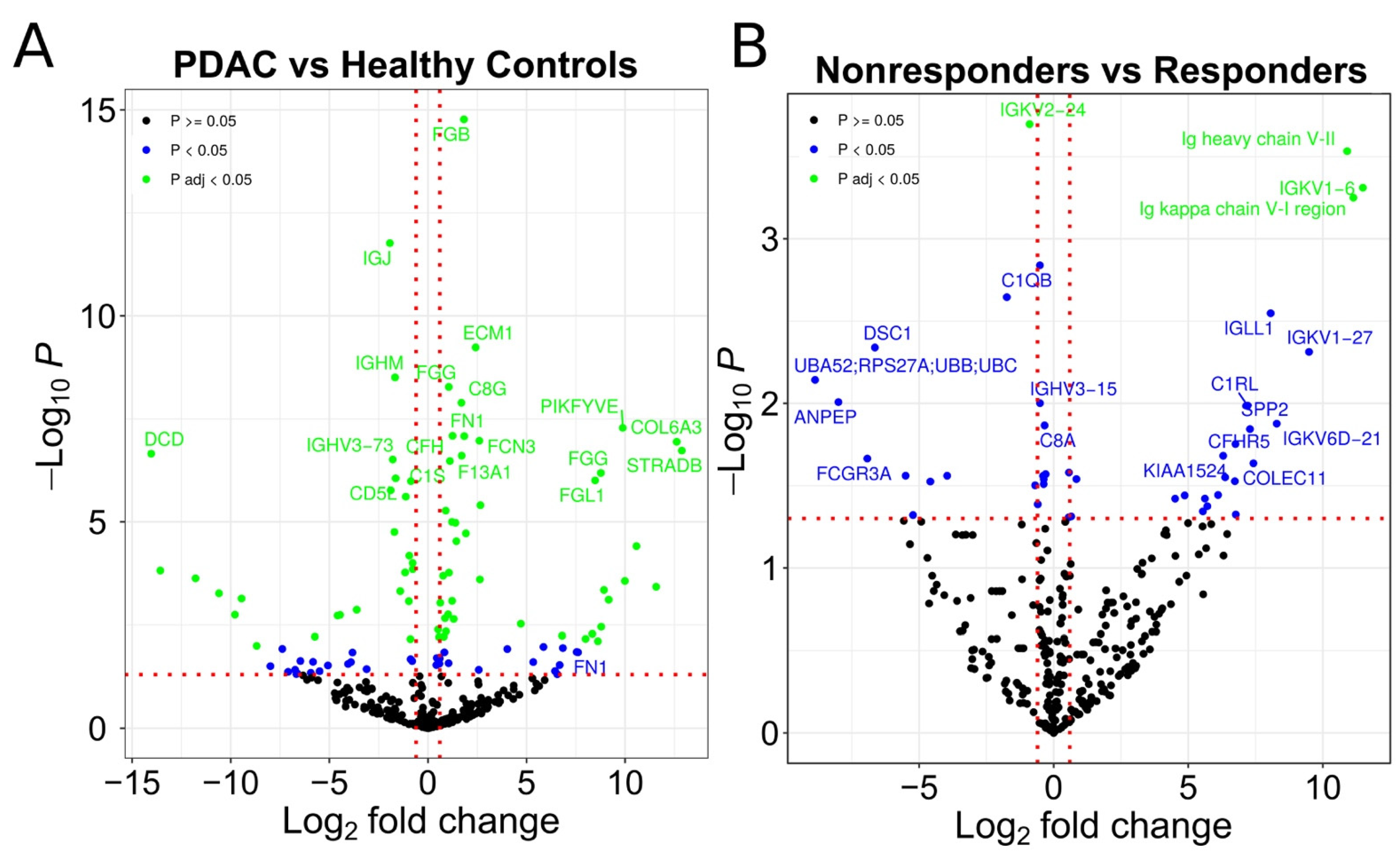
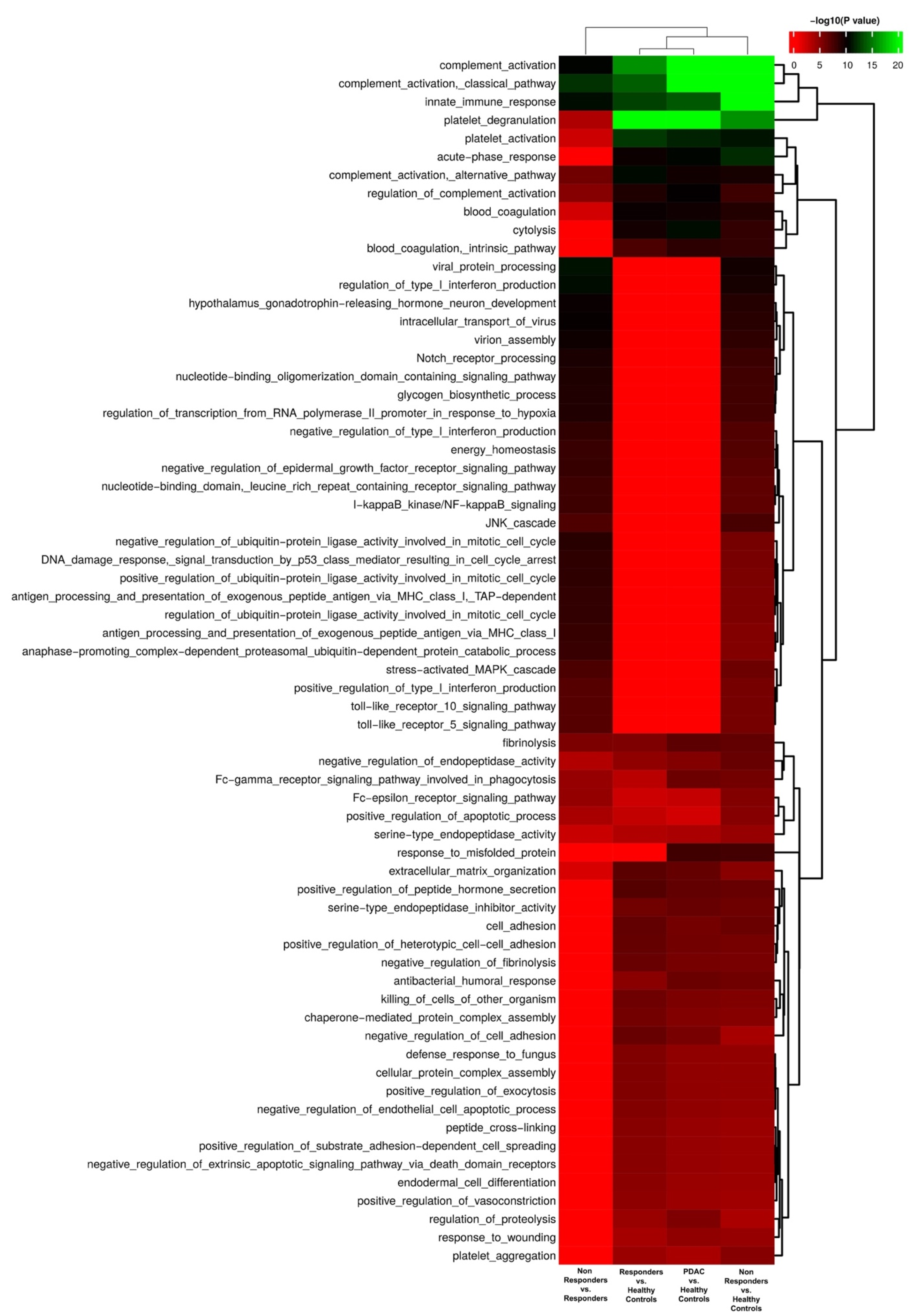
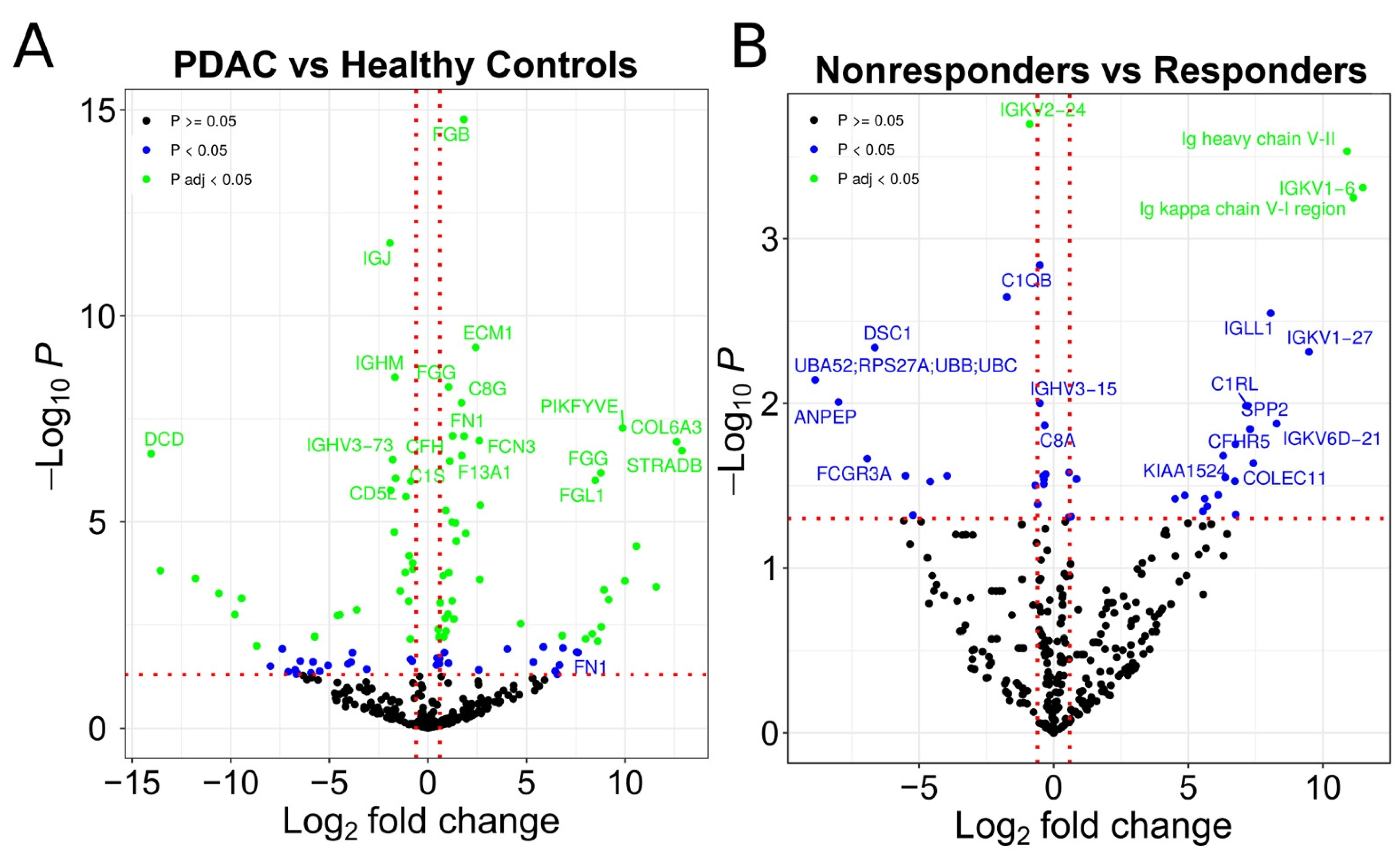
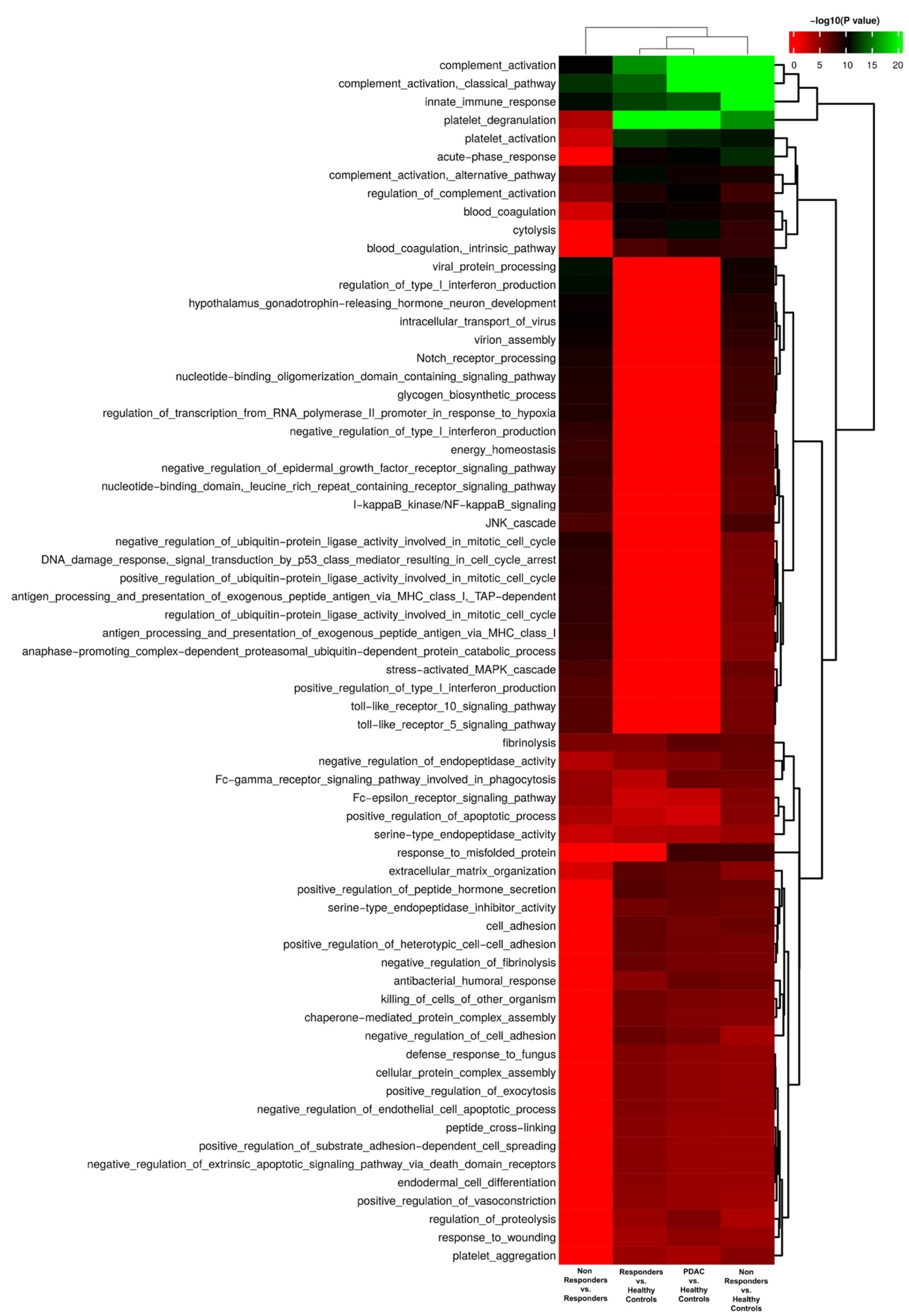
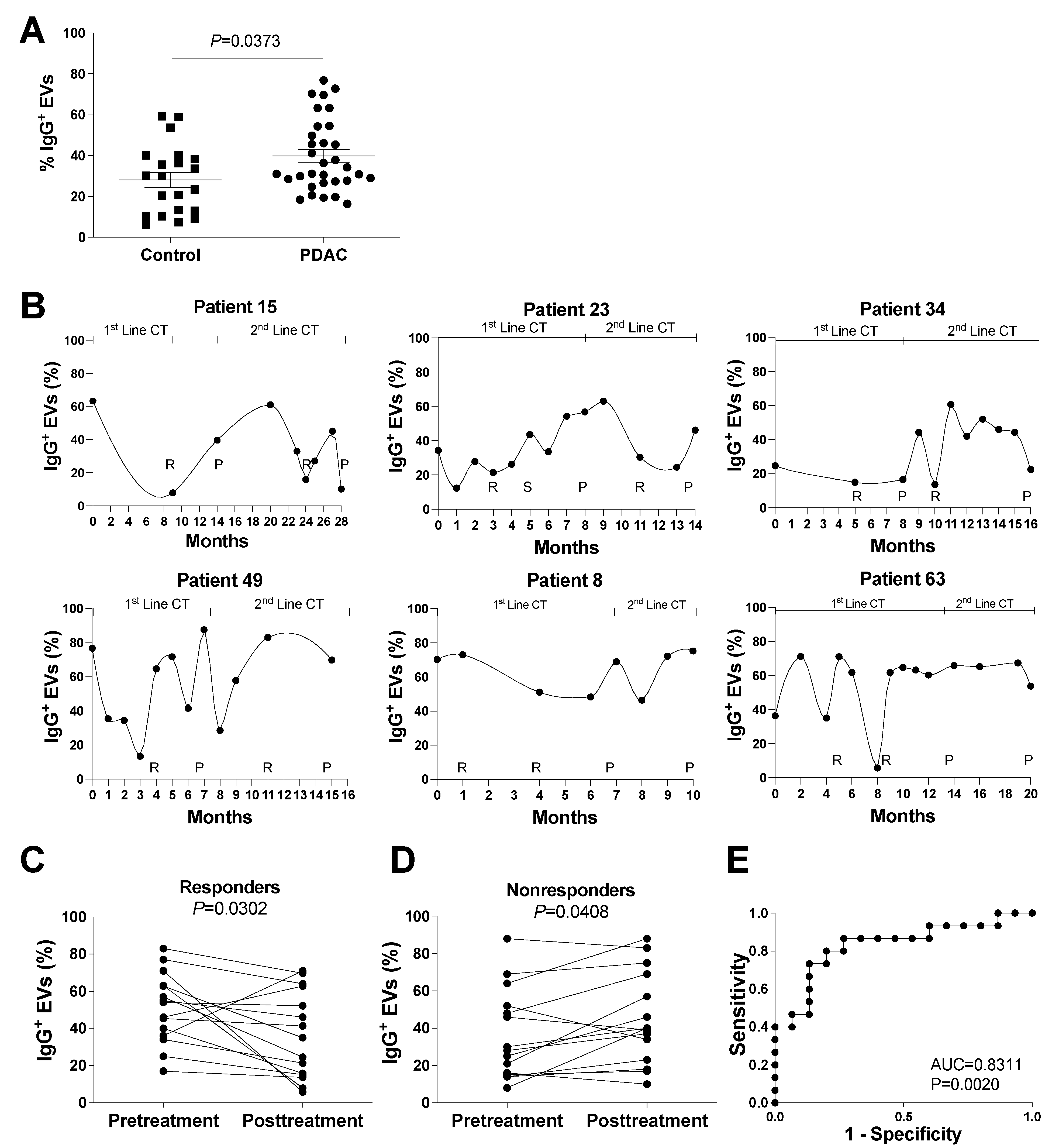
3.1. Identification of Possible EV Markers for PDAC Diagnosis and Therapeutic Response
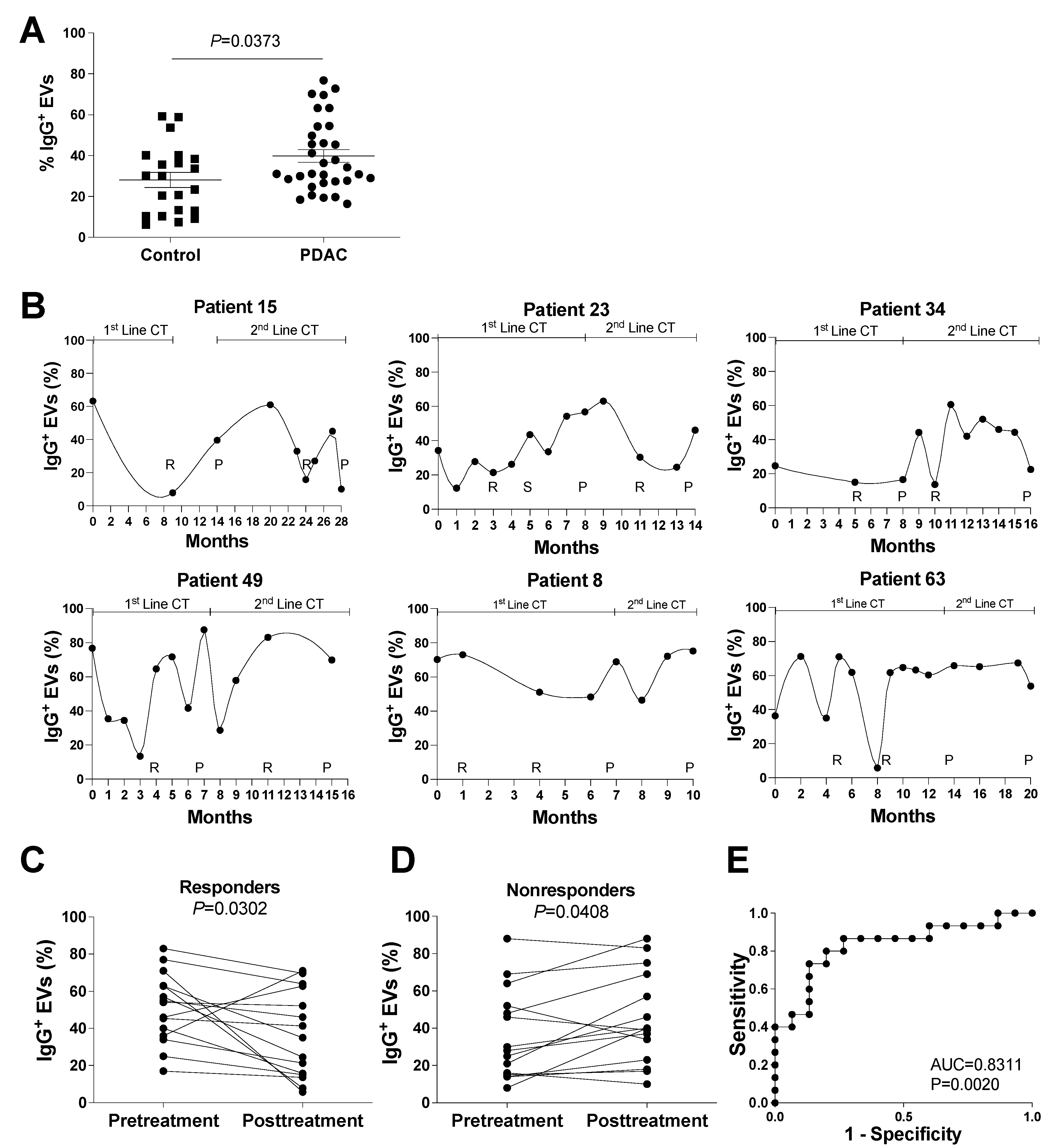
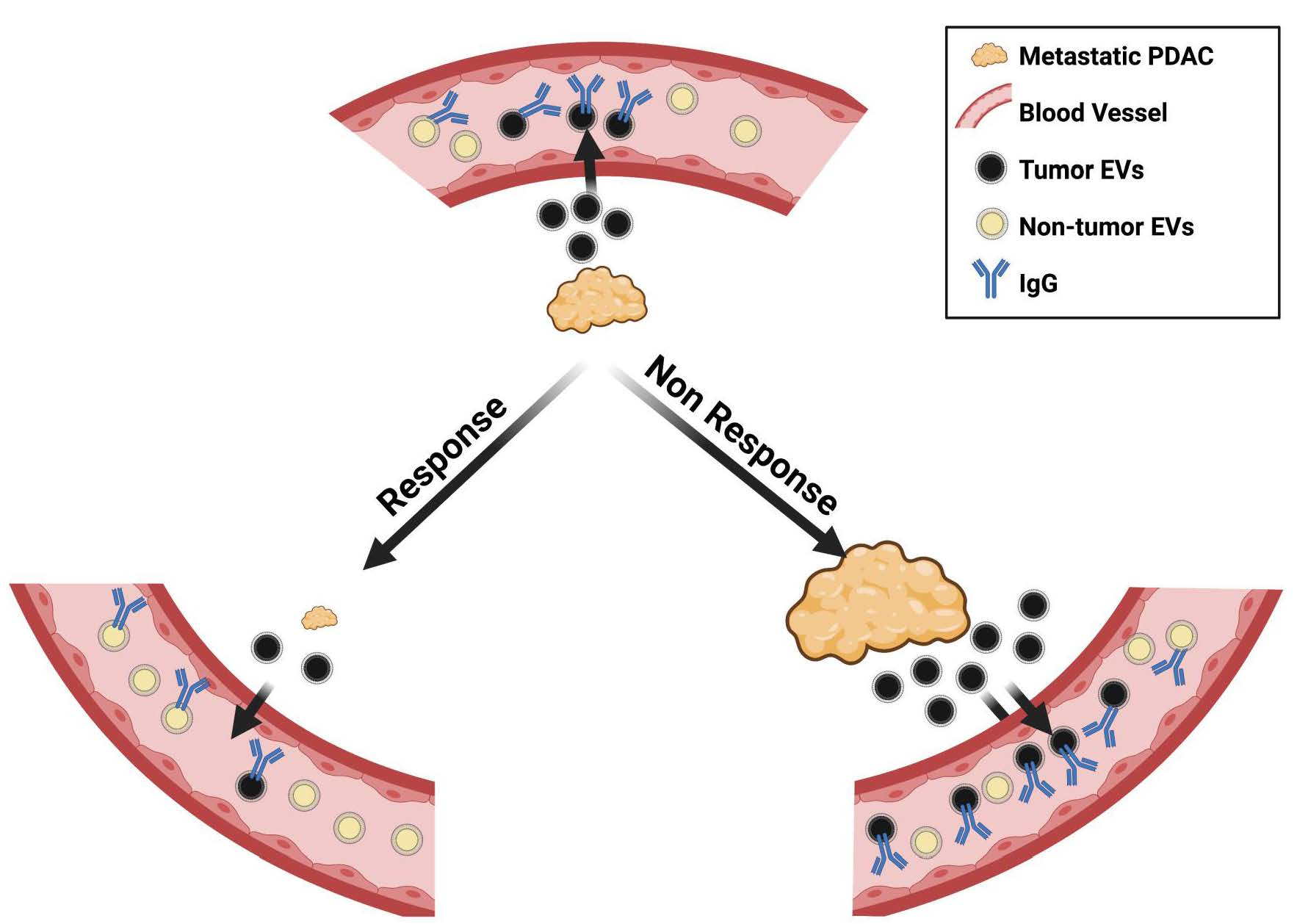
3.2. Evaluation of IgG+ EVs as Possible Markers of PDAC Treatment Response
3.3. Evaluation of the potential role of Plasmatic IgG Levels and Inflammation Status in IgG+ EV Proportion
3.4. Identification of IgG-Associated Proteins on the Surface of PDAC Patient EVs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA A Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Carioli, G.; Malvezzi, M.; Bertuccio, P.; Boffetta, P.; Levi, F.; La Vecchia, C.; Negri, E. European cancer mortality predictions for the year 2021 with focus on pancreatic and female lung cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2021, 32, 478–487. [Google Scholar] [CrossRef]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting Cancer Incidence and Deaths to 2030: The Unexpected Burden of Thyroid, Liver, and Pancreas Cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardière, C.; et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef]
- Gränsmark, E.; Bågenholm Bylin, N.; Blomstrand, H.; Fredrikson, M.; Åvall-Lundqvist, E.; Elander, N.O. Real World Evidence on Second-Line Palliative Chemotherapy in Advanced Pancreatic Cancer. Front. Oncol. 2020, 10, 1176. [Google Scholar] [CrossRef]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer (Oxford Engl. 1990) 2009, 45, 228–247. [Google Scholar] [CrossRef]
- Marom, E.M.; Sarvis, S.; Herndon, J.E.; Patz, E.F., Jr. T1 lung cancers: Sensitivity of diagnosis with fluorodeoxyglucose PET. Radiology 2002, 223, 453–459. [Google Scholar] [CrossRef]
- Tirkes, T.; Hollar, M.A.; Tann, M.; Kohli, M.D.; Akisik, F.; Sandrasegaran, K. Response criteria in oncologic imaging: Review of traditional and new criteria. Radiographics 2013, 33, 1323–1341. [Google Scholar] [CrossRef]
- Azizian, A.; Ruhlmann, F.; Krause, T.; Bernhardt, M.; Jo, P.; Konig, A.; Kleiss, M.; Leha, A.; Ghadimi, M.; Gaedcke, J. CA19-9 for detecting recurrence of pancreatic cancer. Sci. Rep. 2020, 10, 1332. [Google Scholar] [CrossRef] [PubMed]
- Scarà, S.; Bottoni, P.; Scatena, R. CA 19-9: Biochemical and Clinical Aspects. Adv. Exp. Med. Biol. 2015, 867, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Deng, S.; Jin, K.; Gong, Y.; Cheng, H.; Fan, Z.; Qian, Y.; Huang, Q.; Ni, Q.; Luo, G.; et al. Lewis antigen-negative pancreatic cancer: An aggressive subgroup. Int. J. Oncol. 2020, 56, 900–908. [Google Scholar] [CrossRef]
- Boulay, B.R.; Parepally, M. Managing malignant biliary obstruction in pancreas cancer: Choosing the appropriate strategy. World J. Gastroenterol. 2014, 20, 9345–9353. [Google Scholar] [CrossRef] [PubMed]
- Kahlert, C.; Melo, S.A.; Protopopov, A.; Tang, J.; Seth, S.; Koch, M.; Zhang, J.; Weitz, J.; Chin, L.; Futreal, A.; et al. Identification of double-stranded genomic DNA spanning all chromosomes with mutated KRAS and p53 DNA in the serum exosomes of patients with pancreatic cancer. J. Biol. Chem. 2014, 289, 3869–3875. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, A.; Kim, H.S.; Bojmar, L.; Gyan, K.E.; Cioffi, M.; Hernandez, J.; Zambirinis, C.P.; Rodrigues, G.; Molina, H.; Heissel, S.; et al. Extracellular Vesicle and Particle Biomarkers Define Multiple Human Cancers. Cell 2020, 182, 1044–1061.e1018. [Google Scholar] [CrossRef]
- Costa-Silva, B.; Aiello, N.M.; Ocean, A.J.; Singh, S.; Zhang, H.; Thakur, B.K.; Becker, A.; Hoshino, A.; Mark, M.T.; Molina, H.; et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat. Cell Biol. 2015, 17, 816–826. [Google Scholar] [CrossRef]
- Hoshino, A.; Costa-Silva, B.; Shen, T.-L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef]
- Thakur, B.K.; Zhang, H.; Becker, A.; Matei, I.; Huang, Y.; Costa-Silva, B.; Zheng, Y.; Hoshino, A.; Brazier, H.; Xiang, J.; et al. Double-stranded DNA in exosomes: A novel biomarker in cancer detection. Cell Res. 2014, 24, 766–769. [Google Scholar] [CrossRef]
- Skog, J.; Würdinger, T.; van Rijn, S.; Meijer, D.H.; Gainche, L.; Curry, W.T.; Carter, B.S.; Krichevsky, A.M.; Breakefield, X.O. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 2008, 10, 1470–1476. [Google Scholar] [CrossRef]
- Cui, M.; Huang, J.; Zhang, S.; Liu, Q.; Liao, Q.; Qiu, X. Immunoglobulin Expression in Cancer Cells and Its Critical Roles in Tumorigenesis. Front. Immunol. 2021, 12, 613530. [Google Scholar] [CrossRef] [PubMed]
- Edge, S.B. AJCC Cancer Staging Manual; Springer: New York, NY, USA, 2017. [Google Scholar]
- Maia, J.; Otake, A.H.; Pocas, J.; Carvalho, A.S.; Beck, H.C.; Magalhaes, A.; Matthiesen, R.; Strano Moraes, M.C.; Costa-Silva, B. Transcriptome Reprogramming of CD11b (+) Bone Marrow Cells by Pancreatic Cancer Extracellular Vesicles. Front Cell Dev. Biol. 2020, 8, 592518. [Google Scholar] [CrossRef] [PubMed]
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, A.S.; Baeta, H.; Henriques, A.F.A.; Ejtehadifar, M.; Tranfield, E.M.; Sousa, A.L.; Farinho, A.; Silva, B.C.; Cabecadas, J.; Gameiro, P.; et al. Proteomic Landscape of Extracellular Vesicles for Diffuse Large B-Cell Lymphoma Subtyping. Int. J. Mol. Sci. 2021, 22, 11004. [Google Scholar] [CrossRef]
- Carvalho, A.S.; Ribeiro, H.; Voabil, P.; Penque, D.; Jensen, O.N.; Molina, H.; Matthiesen, R. Global mass spectrometry and transcriptomics array based drug profiling provides novel insight into glucosamine induced endoplasmic reticulum stress. Mol. Cell. Proteom. MCP 2014, 13, 3294–3307. [Google Scholar] [CrossRef]
- Matthiesen, R.; Prieto, G.; Amorim, A.; Aloria, K.; Fullaondo, A.; Carvalho, A.S.; Arizmendi, J.M. SIR: Deterministic protein inference from peptides assigned to MS data. J. Proteom. 2012, 75, 4176–4183. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Maia, J.; Batista, S.; Couto, N.; Gregório, A.C.; Bodo, C.; Elzanowska, J.; Strano Moraes, M.C.; Costa-Silva, B. Employing Flow Cytometry to Extracellular Vesicles Sample Microvolume Analysis and Quality Control. Front. Cell Dev. Biol. 2020, 8, 593750. [Google Scholar] [CrossRef]
- Cvjetkovic, A.; Jang, S.C.; Konečná, B.; Höög, J.L.; Sihlbom, C.; Lässer, C.; Lötvall, J. Detailed Analysis of Protein Topology of Extracellular Vesicles-Evidence of Unconventional Membrane Protein Orientation. Sci. Rep. 2016, 6, 36338. [Google Scholar] [CrossRef]
- Holmberg, A.; Blomstergren, A.; Nord, O.; Lukacs, M.; Lundeberg, J.; Uhlén, M. The biotin-streptavidin interaction can be reversibly broken using water at elevated temperatures. Electrophoresis 2005, 26, 501–510. [Google Scholar] [CrossRef]
- Dawson, S.J.; Tsui, D.W.; Murtaza, M.; Biggs, H.; Rueda, O.M.; Chin, S.F.; Dunning, M.J.; Gale, D.; Forshew, T.; Mahler-Araujo, B.; et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N. Engl. J. Med. 2013, 368, 1199–1209. [Google Scholar] [CrossRef] [PubMed]
- Daamen, L.A.; Groot, V.P.; Heerkens, H.D.; Intven, M.P.W.; van Santvoort, H.C.; Molenaar, I.Q. Systematic review on the role of serum tumor markers in the detection of recurrent pancreatic cancer. HPB 2018, 20, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Li, X.; Xie, K.-P. Coupled liquid biopsy and bioinformatics for pancreatic cancer early detection and precision prognostication. Mol. Cancer 2021, 20, 34. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.; You, L.; Zheng, B.; Huang, X.; Liu, Q.; Huang, J.; Pan, B.; Qiu, X.; Liao, Q.; Zhao, Y. High Expression of Cancer-Derived Glycosylated Immunoglobulin G Predicts Poor Prognosis in Pancreatic Ductal Adenocarcinoma. J. Cancer 2020, 11, 2213–2221. [Google Scholar] [CrossRef]
- Mei, Z.; Shi, L.; Wang, B.; Yang, J.; Xiao, Z.; Du, P.; Wang, Q.; Yang, W. Prognostic role of pretreatment blood neutrophil-to-lymphocyte ratio in advanced cancer survivors: A systematic review and meta-analysis of 66 cohort studies. Cancer Treat. Rev. 2017, 58, 1–13. [Google Scholar] [CrossRef]
- Allin, K.H.; Nordestgaard, B.G. Elevated C-reactive protein in the diagnosis, prognosis, and cause of cancer. Crit. Rev. Clin. Lab. Sci. 2011, 48, 155–170. [Google Scholar] [CrossRef]
- Schmitz-Winnenthal, F.H.; Galindo-Escobedo, L.V.; Rimoldi, D.; Geng, W.; Romero, P.; Koch, M.; Weitz, J.; Krempien, R.; Niethammer, A.G.; Beckhove, P.; et al. Potential target antigens for immunotherapy in human pancreatic cancer. Cancer Lett. 2007, 252, 290–298. [Google Scholar] [CrossRef]
- Record, M.; Carayon, K.; Poirot, M.; Silvente-Poirot, S. Exosomes as new vesicular lipid transporters involved in cell-cell communication and various pathophysiologies. Biochim. Biophys Acta 2014, 1841, 108–120. [Google Scholar] [CrossRef]
- Taylor, D.D.; Gercel-Taylor, C. MicroRNA signatures of tumor-derived exosomes as diagnostic biomarkers of ovarian cancer. Gynecol. Oncol. 2008, 110, 13–21. [Google Scholar] [CrossRef]
- Nuzhat, Z.; Kinhal, V.; Sharma, S.; Rice, G.E.; Joshi, V.; Salomon, C. Tumour-derived exosomes as a signature of pancreatic cancer-liquid biopsies as indicators of tumour progression. Oncotarget 2017, 8, 17279–17291. [Google Scholar] [CrossRef] [Green Version]
- Couto, N.; Caja, S.; Maia, J.; Strano Moraes, M.C.; Costa-Silva, B. Exosomes as emerging players in cancer biology. Biochimie 2018, 155, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Bernard, V.; Kim, D.U.; San Lucas, F.A.; Castillo, J.; Allenson, K.; Mulu, F.C.; Stephens, B.M.; Huang, J.; Semaan, A.; Guerrero, P.A.; et al. Circulating Nucleic Acids Are Associated With Outcomes of Patients With Pancreatic Cancer. Gastroenterology 2019, 156, 108–118.e14. [Google Scholar] [CrossRef] [PubMed]
- Liang, K.; Liu, F.; Fan, J.; Sun, D.; Liu, C.; Lyon, C.J.; Bernard, D.W.; Li, Y.; Yokoi, K.; Katz, M.H.; et al. Nanoplasmonic quantification of tumour-derived extracellular vesicles in plasma microsamples for diagnosis and treatment monitoring. Nat. Biomed. Eng. 2017, 1, 0021. [Google Scholar] [CrossRef] [PubMed]
- Capello, M.; Vykoukal, J.V.; Katayama, H.; Bantis, L.E.; Wang, H.; Kundnani, D.L.; Aguilar-Bonavides, C.; Aguilar, M.; Tripathi, S.C.; Dhillon, D.S.; et al. Exosomes harbor B cell targets in pancreatic adenocarcinoma and exert decoy function against complement-mediated cytotoxicity. Nat. Commun. 2019, 10, 254. [Google Scholar] [CrossRef]
- Hofmann, L.; Ludwig, S.; Schuler, P.J.; Hoffmann, T.K.; Brunner, C.; Theodoraki, M.N. The Potential of CD16 on Plasma-Derived Exosomes as a Liquid Biomarker in Head and Neck Cancer. Int. J. Mol. Sci. 2020, 21, 3739. [Google Scholar] [CrossRef]
- Cloutier, N.; Tan, S.; Boudreau, L.H.; Cramb, C.; Subbaiah, R.; Lahey, L.; Albert, A.; Shnayder, R.; Gobezie, R.; Nigrovic, P.A.; et al. The exposure of autoantigens by microparticles underlies the formation of potent inflammatory components: The microparticle-associated immune complexes. EMBO Mol. Med. 2013, 5, 235–249. [Google Scholar] [CrossRef]
- Buzas, E.I.; Gyorgy, B.; Nagy, G.; Falus, A.; Gay, S. Emerging role of extracellular vesicles in inflammatory diseases. Nat. Rev. Rheumatol. 2014, 10, 356–364. [Google Scholar] [CrossRef]
- Fortin, P.R.; Cloutier, N.; Bissonnette, V.; Aghdassi, E.; Eder, L.; Simonyan, D.; Laflamme, N.; Boilard, E. Distinct Subtypes of Microparticle-containing Immune Complexes Are Associated with Disease Activity, Damage, and Carotid Intima-media Thickness in Systemic Lupus Erythematosus. J. Rheumatol. 2016, 43, 2019–2025. [Google Scholar] [CrossRef]
- Perez-Hernandez, J.; Redon, J.; Cortes, R. Extracellular Vesicles as Therapeutic Agents in Systemic Lupus Erythematosus. Int. J. Mol. Sci. 2017, 18, 717. [Google Scholar] [CrossRef]
- Buzas, E.I.; Toth, E.A.; Sodar, B.W.; Szabo-Taylor, K.E. Molecular interactions at the surface of extracellular vesicles. Semin Immunopathol. 2018, 40, 453–464. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.L.; Feizi, A.; Uhlen, M.; Nielsen, J. A Systematic Investigation of the Malignant Functions and Diagnostic Potential of the Cancer Secretome. Cell Rep. 2019, 26, 2622–2635.e5. [Google Scholar] [CrossRef] [PubMed]
- Walker, C.; Mojares, E.; Del Rio Hernandez, A. Role of Extracellular Matrix in Development and Cancer Progression. Int. J. Mol. Sci. 2018, 19, 3038. [Google Scholar] [CrossRef] [PubMed]
- Donati, M.B.; Lorenzet, R. Coagulation factors and tumor cell biology: The role of tissue factor. Pathophysiol. Haemost. Thromb. 2003, 33, 22–25. [Google Scholar] [CrossRef] [PubMed]
- Kochanek, D.M.; Ghouse, S.M.; Karbowniczek, M.M.; Markiewski, M.M. Complementing Cancer Metastasis. Front. Immunol. 2018, 9, 1629. [Google Scholar] [CrossRef]
- Lee, Y.T. Quantitative change of serum protein and immunoglobulin in patients with solid cancers. J. Surg. Oncol. 1977, 9, 179–187. [Google Scholar] [CrossRef]
- Seve, P.; Ray-Coquard, I.; Trillet-Lenoir, V.; Sawyer, M.; Hanson, J.; Broussolle, C.; Negrier, S.; Dumontet, C.; Mackey, J.R. Low serum albumin levels and liver metastasis are powerful prognostic markers for survival in patients with carcinomas of unknown primary site. Cancer 2006, 107, 2698–2705. [Google Scholar] [CrossRef]
- Pleiman, C.M.; D’Ambrosio, D.; Cambier, J.C. The B-cell antigen receptor complex: Structure and signal transduction. Immunol. Today 1994, 15, 393–399. [Google Scholar] [CrossRef]
- DeFranco, A.L. Structure and function of the B cell antigen receptor. Annu. Rev. Cell Biol. 1993, 9, 377–410. [Google Scholar] [CrossRef]
- Li, X.; Ni, R.; Chen, J.; Liu, Z.; Xiao, M.; Jiang, F.; Lu, C. The presence of IGHG1 in human pancreatic carcinomas is associated with immune evasion mechanisms. Pancreas 2011, 40, 753–761. [Google Scholar] [CrossRef]
- Wan, X.; Lei, Y.; Li, Z.; Wang, J.; Chen, Z.; McNutt, M.; Lin, D.; Zhao, C.; Jiang, C.; Li, J.; et al. Pancreatic Expression of Immunoglobulin G in Human Pancreatic Cancer and Associated Diabetes. Pancreas 2015, 44, 1304–1313. [Google Scholar] [CrossRef]
- Fujie, T.; Mori, M.; Ueo, H.; Sugimachi, K.; Akiyoshi, T. Expression of MAGE and BAGE genes in Japanese breast cancers. Ann. Oncol. 1997, 8, 369–372. [Google Scholar] [CrossRef] [PubMed]
- Kubuschok, B.; Xie, X.; Jesnowski, R.; Preuss, K.-D.; Romeike, B.F.M.; Neumann, F.; Regitz, E.; Pistorius, G.; Schilling, M.; Scheunemann, P.; et al. Expression of cancer testis antigens in pancreatic carcinoma cell lines, pancreatic adenocarcinoma and chronic pancreatitis. Int. J. Cancer 2004, 109, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Rogner, U.C.; Wilke, K.; Steck, E.; Korn, B.; Poustka, A. The melanoma antigen gene (MAGE) family is clustered in the chromosomal band Xq28. Genomics 1995, 29, 725–731. [Google Scholar] [CrossRef]
- Shantha Kumara, H.M.C.; Grieco, M.J.; Caballero, O.L.; Su, T.; Ahmed, A.; Ritter, E.; Gnjatic, S.; Cekic, V.; Old, L.J.; Simpson, A.J.; et al. MAGE-A3 is highly expressed in a subset of colorectal cancer patients. Cancer Immun. 2012, 12, 16. [Google Scholar] [PubMed]
- Sienel, W.; Varwerk, C.; Linder, A.; Kaiser, D.; Teschner, M.; Delire, M.; Stamatis, G.; Passlick, B. Melanoma associated antigen (MAGE)-A3 expression in Stages I and II non-small cell lung cancer: Results of a multi-center study. Eur. J. Cardio-Thorac. Surg. 2004, 25, 131–134. [Google Scholar] [CrossRef]
- Kim, J.; Reber, H.A.; Hines, O.J.; Kazanjian, K.K.; Tran, A.; Ye, X.; Amersi, F.F.; Martinez, S.R.; Dry, S.M.; Bilchik, A.J.; et al. The clinical significance of MAGEA3 expression in pancreatic cancer. Int. J. Cancer 2006, 118, 2269–2275. [Google Scholar] [CrossRef]
- Das, B.; Senapati, S. Functional and mechanistic studies reveal MAGEA3 as a pro-survival factor in pancreatic cancer cells. J. Exp. Clin. Cancer Res. 2019, 38, 294. [Google Scholar] [CrossRef] [PubMed]
- Poojary, M.; Jishnu, P.V.; Kabekkodu, S.P. Prognostic Value of Melanoma-Associated Antigen-A (MAGE-A) Gene Expression in Various Human Cancers: A Systematic Review and Meta-analysis of 7428 Patients and 44 Studies. Mol. Diagn. Ther. 2020, 24, 537–555. [Google Scholar] [CrossRef]
- Padoan, A.; Plebani, M.; Basso, D. Inflammation and Pancreatic Cancer: Focus on Metabolism, Cytokines, and Immunity. Int. J. Mol. Sci. 2019, 20, 676. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Patients | |
| Immunoglobulin lambda variable 3–21 | |
| Melanoma-associated antigen B1 | |
| Albumin | |
| Probable non-functional immunoglobulin heavy variable 3–16 | |
| Healthy Donors + Cancer Patients | |
| Keratin, type II cytoskeletal 1 | |
| Chloride intracellular channel protein 4 | |
| Alpha-2-macroglobulin | |
| Glutamate receptor ionotropic, kainate 3 | |
| Keratin, type II cytoskeletal 6B | |
| HUMAN Ig kappa chain V-I region Lay | |
| HUMAN Ig kappa chain V-III region NG9 (Fragment) | |
| HUMAN Ig kappa chain V-III region POM | |
| HUMAN Ig kappa chain V-III region CLL | |
| HUMAN Ig kappa chain V-III region VH (Fragment) | |
| Immunoglobulin kappa variable 3D-7 | |
| Immunoglobulin kappa variable 3/OR2-268 (non-functional) | |
| Immunoglobulin kappa constant | |
| Immunoglobulin kappa variable 3–15 | |
| Probable non-functional immunoglobulin kappa variable 3–7 | |
| Ig lambda chain V-II region NIG-84 | |
| Chondroitin sulfate synthase 3 | |
| Ig lambda chain V-II region BUR | |
| Keratin, type I cytoskeletal 10 | |
| Protein argonaute-2 | |
| Golgi integral membrane protein 4 | |
| Healthy Donors | |
| S100A9 | |
| Ig kappa chain V-III region Ti | |
| Uncharacterized protein (Fragment) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Couto, N.; Elzanowska, J.; Maia, J.; Batista, S.; Pereira, C.E.; Beck, H.C.; Carvalho, A.S.; Strano Moraes, M.C.; Carvalho, C.; Oliveira, M.; et al. IgG+ Extracellular Vesicles Measure Therapeutic Response in Advanced Pancreatic Cancer. Cells 2022, 11, 2800. https://doi.org/10.3390/cells11182800
Couto N, Elzanowska J, Maia J, Batista S, Pereira CE, Beck HC, Carvalho AS, Strano Moraes MC, Carvalho C, Oliveira M, et al. IgG+ Extracellular Vesicles Measure Therapeutic Response in Advanced Pancreatic Cancer. Cells. 2022; 11(18):2800. https://doi.org/10.3390/cells11182800
Chicago/Turabian StyleCouto, Nuno, Julia Elzanowska, Joana Maia, Silvia Batista, Catarina Esteves Pereira, Hans Christian Beck, Ana Sofia Carvalho, Maria Carolina Strano Moraes, Carlos Carvalho, Manuela Oliveira, and et al. 2022. "IgG+ Extracellular Vesicles Measure Therapeutic Response in Advanced Pancreatic Cancer" Cells 11, no. 18: 2800. https://doi.org/10.3390/cells11182800
APA StyleCouto, N., Elzanowska, J., Maia, J., Batista, S., Pereira, C. E., Beck, H. C., Carvalho, A. S., Strano Moraes, M. C., Carvalho, C., Oliveira, M., Matthiesen, R., & Costa-Silva, B. (2022). IgG+ Extracellular Vesicles Measure Therapeutic Response in Advanced Pancreatic Cancer. Cells, 11(18), 2800. https://doi.org/10.3390/cells11182800