
Apigetrin Promotes TNFα-Induced Apoptosis, Necroptosis, G2/M Phase Cell Cycle Arrest, and ROS Generation through Inhibition of NF-κB Pathway in Hep3B Liver Cancer Cells
 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Line and Chemicals
2.2. Apigetrin Treatment and Cell Viability Assay
2.3. Colony Formation Assay
2.4. DAPI Staining of Apoptotic Cells
2.5. Hematoxylin Staining for Observing Cell Death Morphology
2.6. Cell Cycle Analysis
2.7. Annexin V-Staining for Cell Death Analysis
2.8. Determination of Reactive Oxygen Species
2.9. Western Blot Analysis
2.10. Statistical Analysis
3. Results
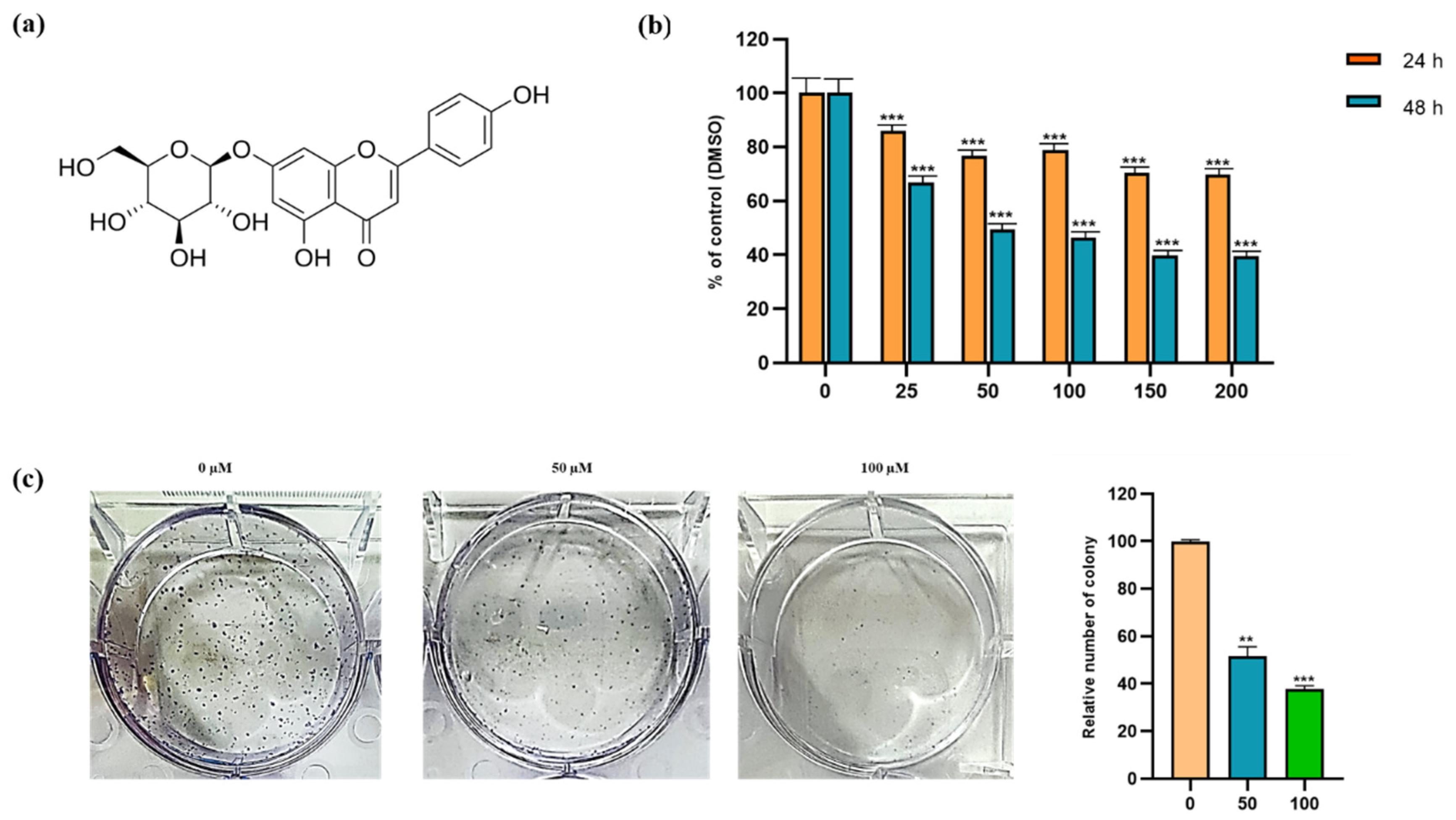
3.1. Cytotoxic and Inhibitory Effect of Apigetrin on Hep3B Cells
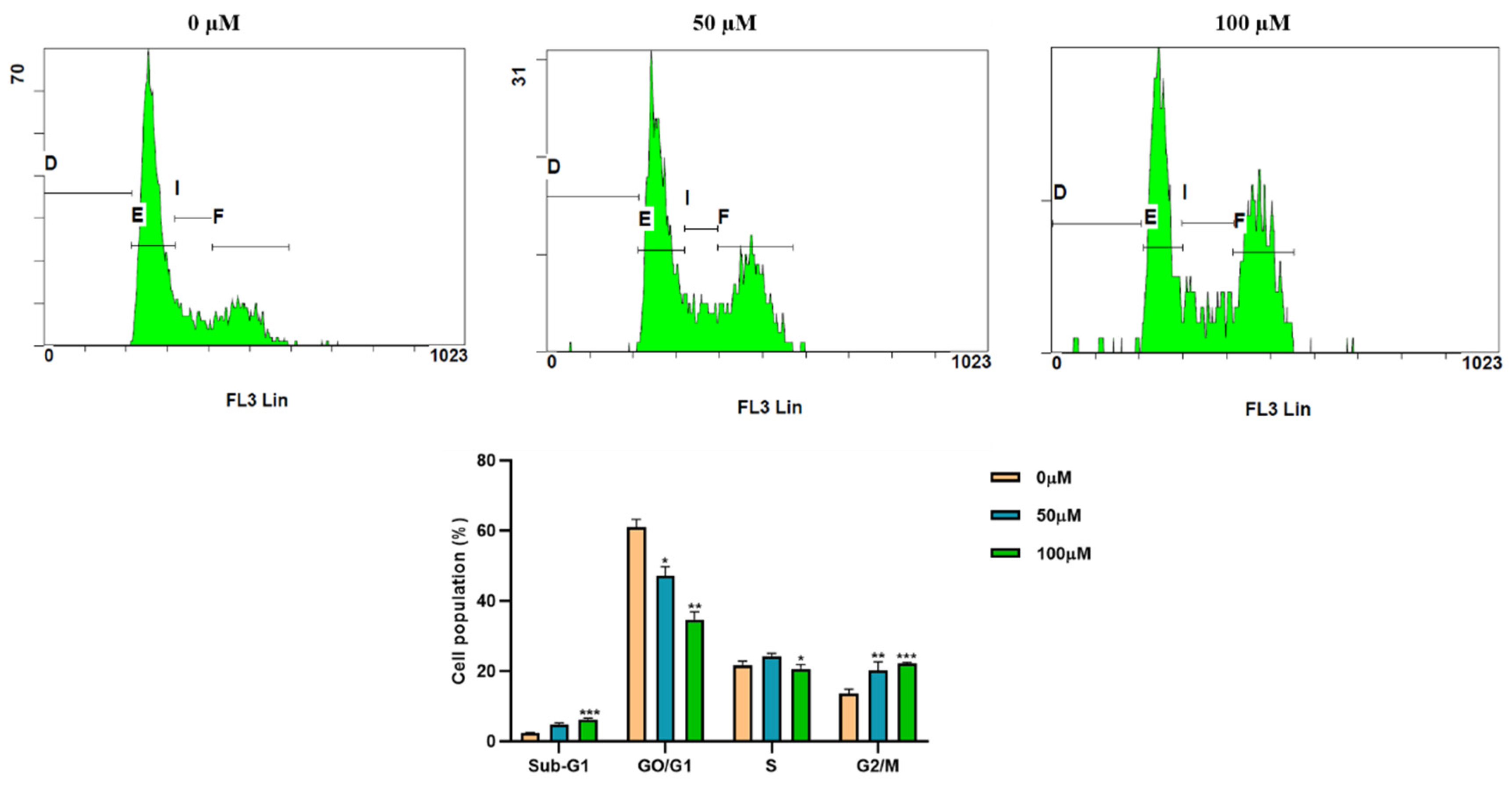
3.2. Apigetrin Induces G2/M Phase Cell Cycle Arrest in Hep3B Cells
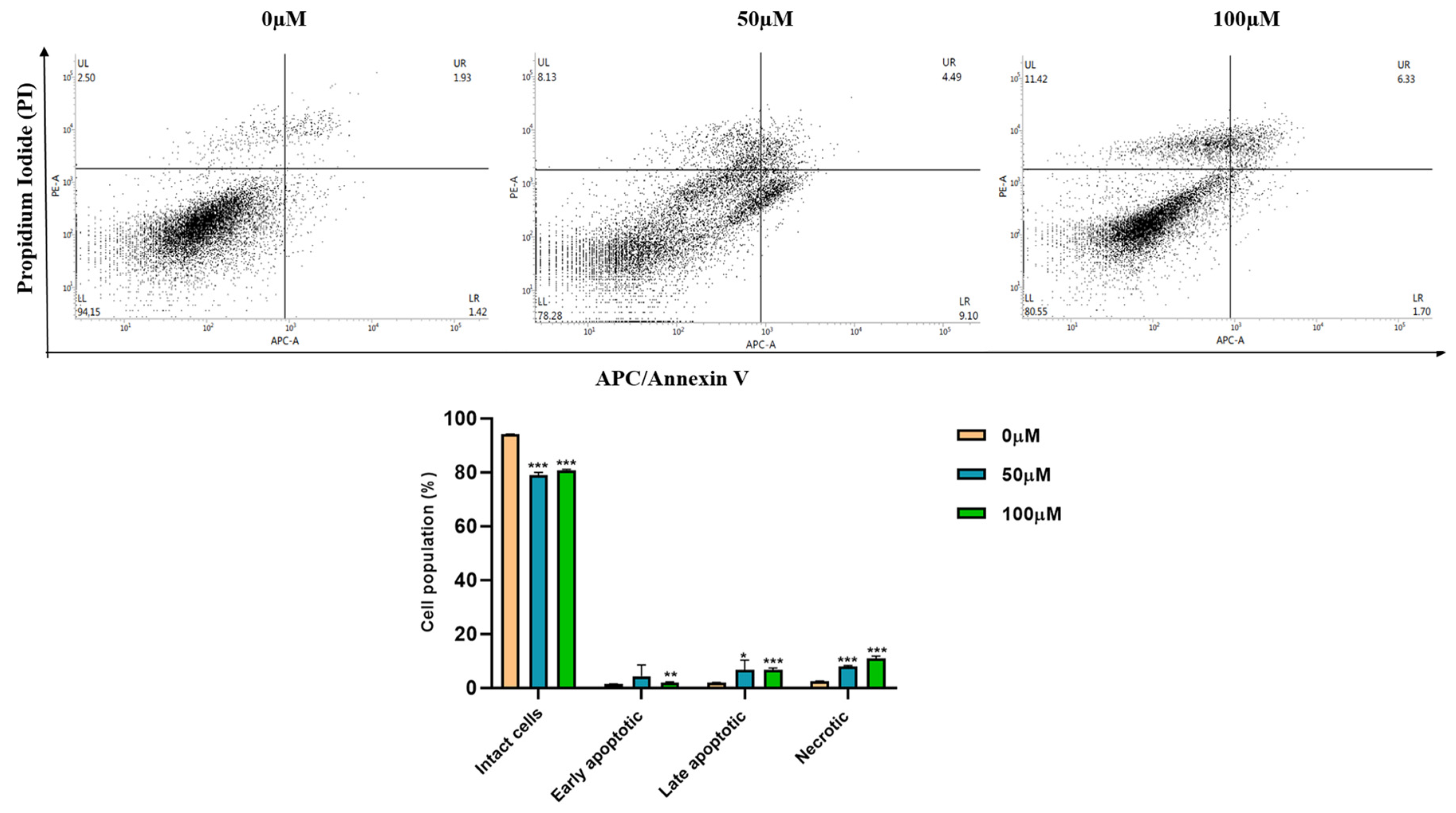
3.3. Apigetrin Induces Apoptotic and Necroptotic Cell Death in Hep3B Cells
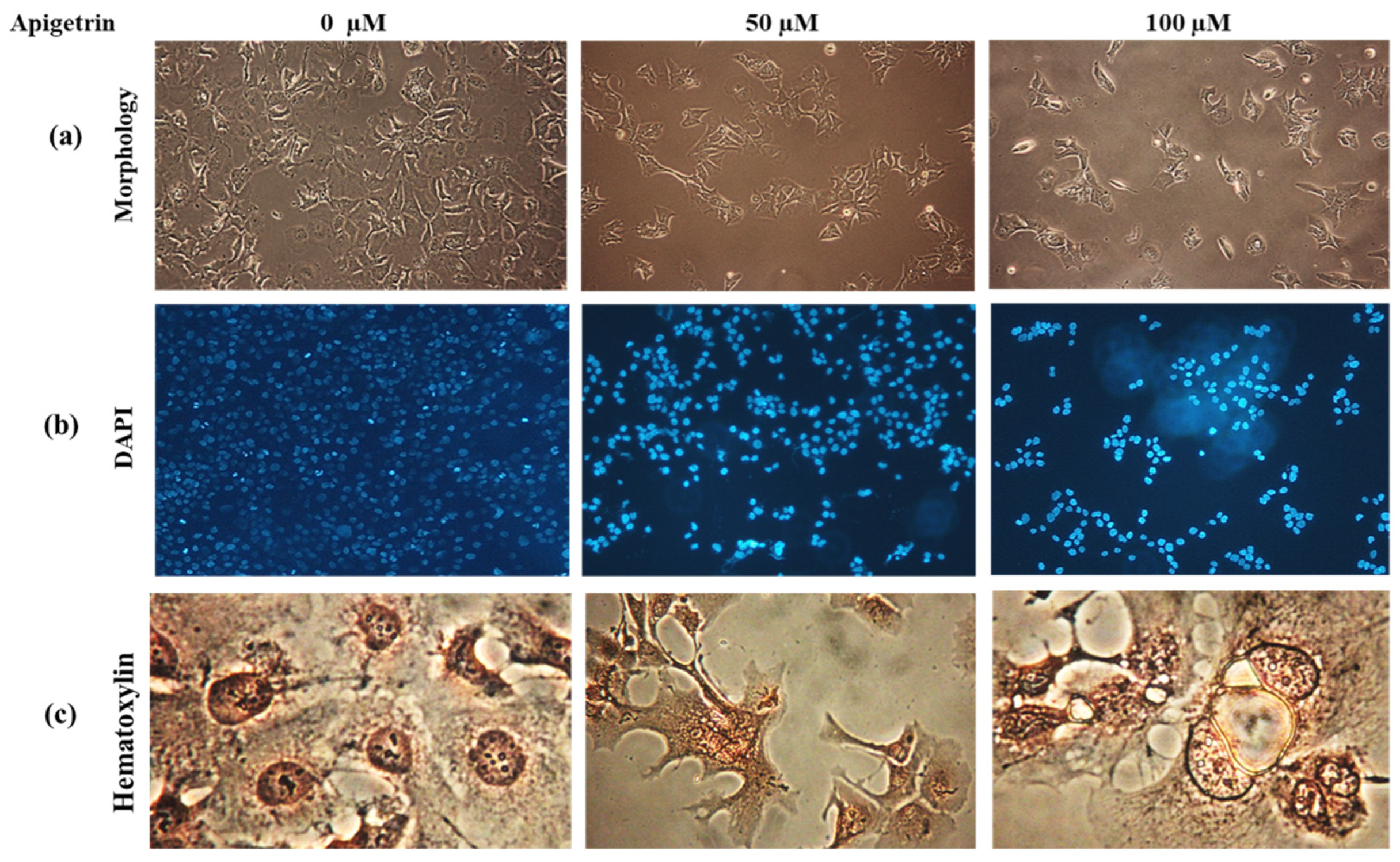
3.4. Apigetrin Causes Morphological and Nuclear Alterations of Hep3B Cells
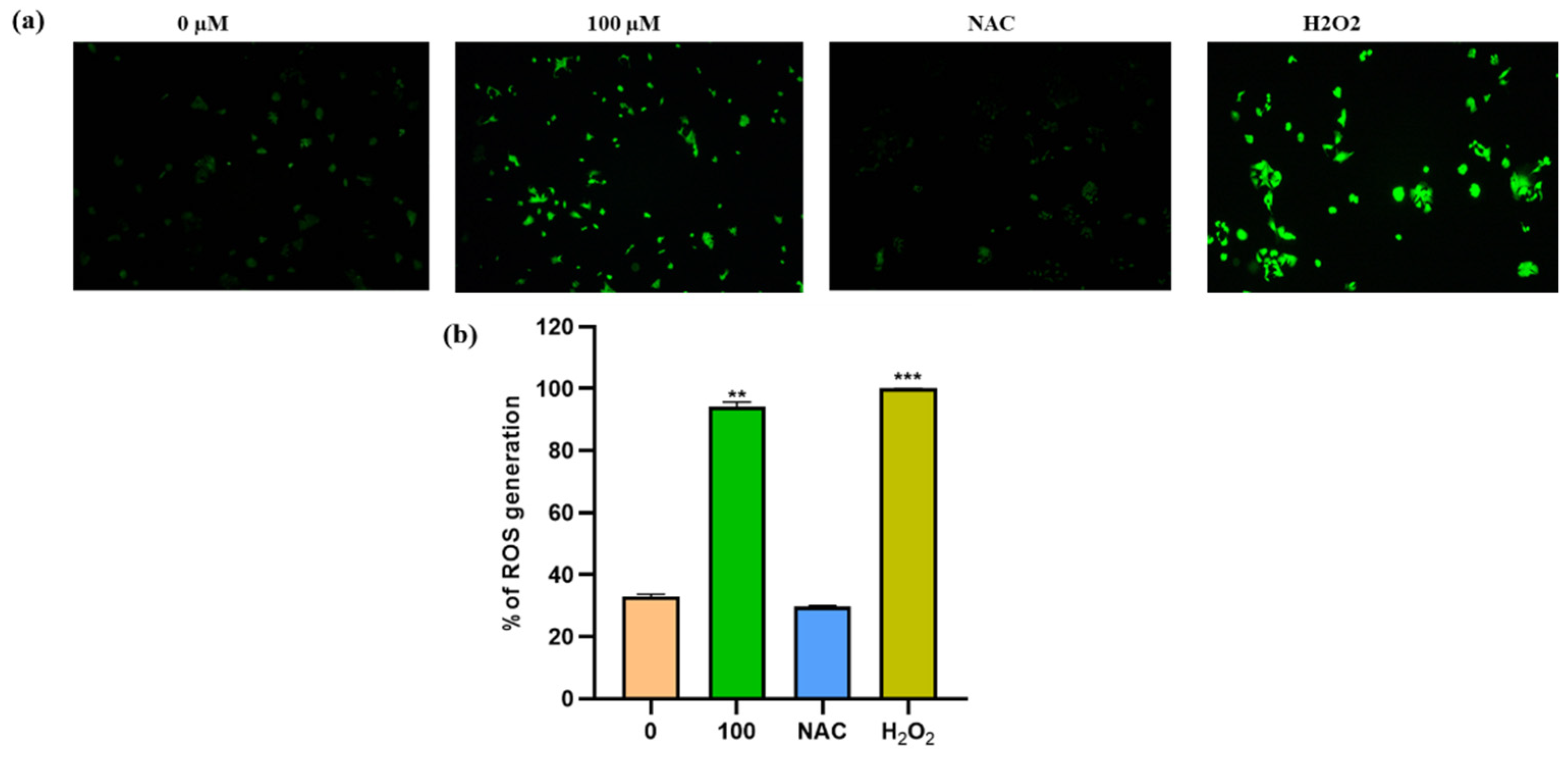
3.5. Apigetrin Increases the Generation of ROS in Hep3B Cells
3.6. Effect of Apigetrin on Inflammatory Pathway in Hep3B Cells
3.7. Effects of Apigetrin on Apoptotic Proteins in Treated on Hep3B Cells
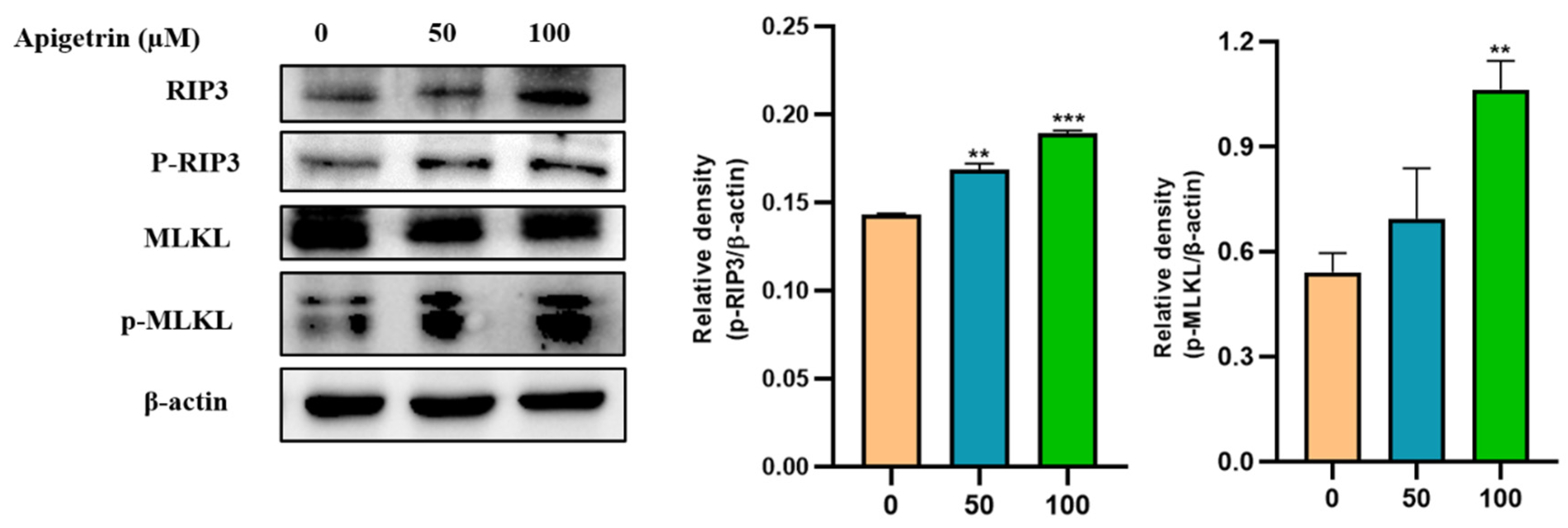
3.8. Effects of Apigetrin on Necroptotic Markers in Hep3B Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Llovet, J.M.; Kelley, R.K.; Augusto, V.; Singal, A.G.; Eli, P.; Sasan, R.; Riccardo, L.; Kazuhiko, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma (Primer). Nat. Rev. Dis. Primers 2021, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Roskilly, A.; Rowe, I.A. Surveillance for hepatocellular cancer. Clin. Med. 2018, 18, s66–s69. [Google Scholar] [CrossRef] [PubMed]
- Hartke, J.; Johnson, M.; Ghabril, M. The diagnosis and treatment of hepatocellular carcinoma. Semin. Diagn. Pathol. 2017, 34, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Connell, L.C.; Harding, J.J.; Abou-Alfa, G.K. Advanced Hepatocellular Cancer: The Current State of Future Research. Curr. Treat. Options Oncol. 2016, 17, 43. [Google Scholar] [CrossRef] [PubMed]
- Anwanwan, D.; Singh, S.K.; Singh, S.; Saikam, V.; Singh, R. Challenges in liver cancer and possible treatment approaches. Biochim. Biophys. Acta 2019, 1873, 188314. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- D’Arcy, M.S. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Bhosale, P.B.; Ha, S.E.; Vetrivel, P.; Kim, H.H.; Kim, J.-A.; Park, K.-I.; Kim, S.M.; Kim, G.S. Flavonoid-induced apoptotic cell death in human cancer cells and its mechanisms. J. Biomed. Transl. Res. 2020, 21, 50–58. [Google Scholar] [CrossRef]
- Pistritto, G.; Trisciuoglio, D.; Ceci, C.; Garufi, A.; D’Orazi, G. Apoptosis as anticancer mechanism: Function and dysfunction of its modulators and targeted therapeutic strategies. Aging 2016, 8, 603–619. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.-S.; Kim, J.-Y.; Lee, J.-H.; Lee, B.-W.; Park, K.-H.; Shim, K.-H.; Lee, M.-K.; Seo, K.-I. Celastrol isolated from Tripterygium regelii induces apoptosis through both caspase-dependent and -independent pathways in human breast cancer cells. Food Chem. Toxicol. 2011, 49, 527–532. [Google Scholar] [CrossRef]
- Idriss, H.T.; Naismith, J.H. TNFα and the TNF receptor superfamily: Structure-function relationship (s). Microsc. Res. Tech. 2000, 50, 184–195. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. Regulation of NF-κB by TNF family cytokines. Semin. Immunol. 2014, 26, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Grootjans, S.; Vanden Berghe, T.; Vandenabeele, P. Initiation and execution mechanisms of necroptosis: An overview. Cell Death Differ. 2017, 24, 1184–1195. [Google Scholar] [CrossRef] [PubMed]
- Auten, R.L.; Davis, J.M. Oxygen Toxicity and Reactive Oxygen Species: The Devil Is in the Details. Pediatr. Res. 2009, 66, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Kopustinskiene, D.M.; Jakstas, V.; Savickas, A.; Bernatoniene, J. Flavonoids as anticancer agents. Nutrients 2020, 12, 457. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.-Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef]
- Sauter, E.R. Cancer prevention and treatment using combination therapy with natural compounds. Expert Rev. Clin. Pharmacol. 2020, 13, 265–285. [Google Scholar] [CrossRef]
- Ponte, L.G.S.; Pavan, I.C.B.; Mancini, M.C.S.; Da Silva, L.G.S.; Morelli, A.P.; Severino, M.B.; Bezerra, R.M.N.; Simabuco, F.M. The Hallmarks of Flavonoids in Cancer. Molecules 2021, 26, 2029. [Google Scholar] [CrossRef] [PubMed]
- Hazafa, A.; Rehman, K.-U.; Jahan, N.; Jabeen, Z. The Role of Polyphenol (Flavonoids) Compounds in the Treatment of Cancer Cells. Nutr. Cancer 2019, 72, 386–397. [Google Scholar] [CrossRef]
- Zheng, Y.; Zhang, W.; Xu, L.; Zhou, H.; Yuan, M.; Xu, H. Recent Progress in Understanding the Action of Natural Compounds at Novel Therapeutic Drug Targets for the Treatment of Liver Cancer. Front. Oncol. 2022, 11, 795548. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.U.; Dagur, H.S.; Khan, M.; Malik, N.; Alam, M.; Mushtaque, M. Therapeutic role of flavonoids and flavones in cancer prevention: Current trends and future perspectives. Eur. J. Med. Chem. Rep. 2021, 3, 100010. [Google Scholar] [CrossRef]
- Piasek, A.; Bartoszek, A.; Namieśnik, J. Phytochemicals that counteract the cardiotoxic side effects of cancer chemotherapy. Postepy Hig. I Med. Dosw. 2009, 63, 142–158. [Google Scholar]
- Pathak, N.; Khan, S.; Bhargava, A.; Raghuram, G.V.; Jain, D.; Panwar, H.; Samarth, R.M.; Jain, S.K.; Maudar, K.K.; Mishra, D.K.; et al. Cancer Chemopreventive Effects of the Flavonoid-Rich Fraction Isolated from Papaya Seeds. Nutr. Cancer 2014, 66, 857–871. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.-S.; Kim, O.-S.; Kim, B.-Y.; Jeong, S.-J. Apigetrin from Scutellaria baicalensis Georgi Inhibits Neuroinflammation in BV-2 Microglia and Exerts Neuroprotective Effect in HT22 Hippocampal Cells. J. Med. Food 2016, 19, 1032–1040. [Google Scholar] [CrossRef]
- Smiljkovic, M.; Stanisavljevic, D.; Stojkovic, D.; Petrovic, I.; Vicentic, J.M.; Popovic, J.; Grdadolnik, S.G.; Markovic, D.; Sankovic-Babice, S.; Glamoclija, J.; et al. Apigenin-7-O-glucoside versus apigenin: Insight into the modes of anticandidal and cytotoxic actions. EXCLI J. 2017, 16, 795–807. [Google Scholar] [CrossRef]
- Sun, Q.; Lu, N.-N.; Feng, L. Apigetrin inhibits gastric cancer progression through inducing apoptosis and regulating ROS-modulated STAT3/JAK2 pathway. Biochem. Biophys. Res. Commun. 2018, 498, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.-M.; Ma, R.-H.; Ni, Z.-J.; Thakur, K.; Cespedes-Acuña, C.L.; Jiang, L.; Wei, Z.-J. Apigenin 7-O-glucoside promotes cell apoptosis through the PTEN/PI3K/AKT pathway and inhibits cell migration in cervical cancer HeLa cells. Food Chem. Toxicol. 2020, 146, 111843. [Google Scholar] [CrossRef]
- Abdulridha, M.K.; Al-Marzoqi, A.H.; Al-Awsi, G.R.L.; Mubarak, S.M.H.; Heidarifard, M.; Ghasemian, A. Anticancer Effects of Herbal Medicine Compounds and Novel Formulations: A Literature Review. J. Gastrointest. Cancer 2020, 51, 765–773. [Google Scholar] [CrossRef]
- Ijaz, S.; Akhtar, N.; Khan, M.S.; Hameed, A.; Irfan, M.; Arshad, M.A.; Ali, S.; Asrar, M. Plant derived anticancer agents: A green approach towards skin cancers. Biomed. Pharmacother. 2018, 103, 1643–1651. [Google Scholar] [CrossRef]
- Barberán, F.A.T.; Gil, M.I.; Tomás, F.; Ferreres, F.; Arques, A. Flavonoid Aglycones and Glycosides from Teucrium gnaphalodes. J. Nat. Prod. 1985, 48, 859–860. [Google Scholar] [CrossRef]
- Bhosale, P.-B.; Vetrivel, P.; Ha, S.-E.; Kim, H.-H.; Heo, J.-D.; Won, C.-K.; Kim, S.-M.; Kim, G.-S. Iridin Induces G2/M Phase Cell Cycle Arrest and Extrinsic Apoptotic Cell Death through PI3K/AKT Signaling Pathway in AGS Gastric Cancer Cells. Molecules 2021, 26, 2802. [Google Scholar] [CrossRef] [PubMed]
- Vetrivel, P.; Kim, S.M.; Ha, S.E.; Kim, H.H.; Bhosale, P.B.; Senthil, K.; Kim, G.S. Compound Prunetin Induces Cell Death in Gastric Cancer Cell with Potent Anti-Proliferative Properties: In Vitro Assay, Molecular Docking, Dynamics, and ADMET Studies. Biomolecules 2020, 10, 1086. [Google Scholar] [CrossRef] [PubMed]
- Uzbekov, R.; Prigent, C. A Journey through Time on the Discovery of Cell Cycle Regulation. Cells 2022, 11, 704. [Google Scholar] [CrossRef]
- Kim, S.M.; Vetrivel, P.; Ha, S.E.; Kim, H.H.; Kim, J.-A.; Kim, G.S. Apigetrin induces extrinsic apoptosis, autophagy and G2/M phase cell cycle arrest through PI3K/AKT/mTOR pathway in AGS human gastric cancer cell. J. Nutr. Biochem. 2020, 83, 108427. [Google Scholar] [CrossRef] [PubMed]
- Saraste, A.; Pulkki, K. Morphologic and biochemical hallmarks of apoptosis. Cardiovasc. Res. 2000, 45, 528–537. [Google Scholar] [CrossRef]
- Atale, N.; Gupta, S.; Yadav, U.; Rani, V. Cell-death assessment by fluorescent and nonfluorescent cytosolic and nuclear staining techniques. J. Microsc. 2014, 255, 7–19. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, K.; Shen, H.; Yao, X.; Sun, Q.; Chen, G. Necroptosis: A novel manner of cell death, associated with stroke. Int. J. Mol. Med. 2018, 41, 624–630. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Fan, J.; Ai, G.; Liu, J.; Luo, N.; Li, C.; Cheng, Z. Berberine in combination with cisplatin induces necroptosis and apoptosis in ovarian cancer cells. Biol. Res. 2019, 52, 37. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Yang, Y.; Wang, X.; Wei, M.; Guo, Q.; Zhao, L. Oroxyloside ameliorates acetaminophen-induced hepatotoxicity by inhibiting JNK related apoptosis and necroptosis. J. Ethnopharmacol. 2020, 258, 112917. [Google Scholar] [CrossRef] [PubMed]
- Schumacker, P.T. Reactive oxygen species in cancer cells: Live by the sword, die by the sword. Cancer Cell 2006, 10, 175–176. [Google Scholar] [CrossRef]
- Slika, H.; Mansour, H.; Wehbe, N.; Nasser, S.A.; Iratni, R.; Nasrallah, G.; Shaito, A.; Ghaddar, T.; Kobeissy, F.; Eid, A.H. Therapeutic potential of flavonoids in cancer: ROS-mediated mechanisms. Biomed. Pharmacother. 2022, 146, 112442. [Google Scholar] [CrossRef]
- Kastl, L.; Sauer, S.; Ruppert, T.; Beissbarth, T.; Becker, M.; Süss, D.; Krammer, P.; Gülow, K. TNF-α mediates mitochondrial uncoupling and enhances ROS-dependent cell migration via NF-κB activation in liver cells. FEBS Lett. 2014, 588, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Xu, X.; Sho, T.; Zhang, J.; Xu, W.; Yao, J.; Xu, J. ROS-induced autophagy regulates porcine trophectoderm cell apoptosis, proliferation, and differentiation. Am. J. Physiol.-Cell Physiol. 2019, 316, C198–C209. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F. TNF-α in promotion and progression of cancer. Cancer Metastasis Rev. 2006, 25, 409–416. [Google Scholar] [CrossRef]
- Khandelwal, N.; Simpson, J.; Taylor, G.; Rafique, S.; Whitehouse, A.; Hiscox, J.; Stark, L. Nucleolar NF-κB/RelA mediates apoptosis by causing cytoplasmic relocalization of nucleophosmin. Cell Death Differ. 2011, 18, 1889–1903. [Google Scholar] [CrossRef] [PubMed]
- Suh, Y.; Afaq, F.; Johnson, J.J.; Mukhtar, H. A plant flavonoid fisetin induces apoptosis in colon cancer cells by inhibition of COX2 and Wnt/EGFR/NF-κB-signaling pathways. Carcinogenesis 2009, 30, 300–307. [Google Scholar] [CrossRef]
- Fas, S.C.; Baumann, S.; Zhu, J.Y.; Giaisi, M.; Treiber, M.K.; Mahlknecht, U.; Krammer, P.H.; Li-Weber, M. Wogonin sensitizes resistant malignant cells to TNFα- and TRAIL-induced apoptosis. Blood 2006, 108, 3700–3706. [Google Scholar] [CrossRef]
- Li, J.; Cheng, Y.; Qu, W.; Sun, Y.; Wang, Z.; Wang, H.; Tian, B. Fisetin, a Dietary Flavonoid, Induces Cell Cycle Arrest and Apoptosis through Activation of p53 and Inhibition of NF-Kappa B Pathways in Bladder Cancer Cells. Basic Clin. Pharmacol. Toxicol. 2010, 108, 84–93. [Google Scholar] [CrossRef]
- Strasser, A.; Vaux, D.L. Cell Death in the Origin and Treatment of Cancer. Mol. Cell 2020, 78, 1045–1054. [Google Scholar] [CrossRef]
- Yan, J.; Wan, P.; Choksi, S.; Liu, Z.-G. Necroptosis and tumor progression. Trends Cancer 2022, 8, 21–27. [Google Scholar] [CrossRef]
- Sundarraj, K.; Raghunath, A.; Panneerselvam, L.; Perumal, E. Fisetin, a phytopolyphenol, targets apoptotic and necroptotic cell death in HepG2 cells. BioFactors 2020, 46, 118–135. [Google Scholar] [CrossRef] [PubMed]
- Khorsandi, L.; Orazizadeh, M.; Niazvand, F.; Abbaspour, M.R.; Mansouri, E.; Khodadadi, A. Quercetin induces apoptosis and necroptosis in MCF-7 breast cancer cells. Bratisl. Med. J. 2017, 118, 123–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhosale, P.B.; Abusaliya, A.; Kim, H.H.; Ha, S.E.; Park, M.Y.; Jeong, S.H.; Vetrivel, P.; Heo, J.D.; Kim, J.-A.; Won, C.k.; et al. Apigetrin Promotes TNFα-Induced Apoptosis, Necroptosis, G2/M Phase Cell Cycle Arrest, and ROS Generation through Inhibition of NF-κB Pathway in Hep3B Liver Cancer Cells. Cells 2022, 11, 2734. https://doi.org/10.3390/cells11172734
Bhosale PB, Abusaliya A, Kim HH, Ha SE, Park MY, Jeong SH, Vetrivel P, Heo JD, Kim J-A, Won Ck, et al. Apigetrin Promotes TNFα-Induced Apoptosis, Necroptosis, G2/M Phase Cell Cycle Arrest, and ROS Generation through Inhibition of NF-κB Pathway in Hep3B Liver Cancer Cells. Cells. 2022; 11(17):2734. https://doi.org/10.3390/cells11172734
Chicago/Turabian StyleBhosale, Pritam Bhagwan, Abuyaseer Abusaliya, Hun Hwan Kim, Sang Eun Ha, Min Yeong Park, Se Hyo Jeong, Preethi Vetrivel, Jeong Doo Heo, Jin-A Kim, Chung kil Won, and et al. 2022. "Apigetrin Promotes TNFα-Induced Apoptosis, Necroptosis, G2/M Phase Cell Cycle Arrest, and ROS Generation through Inhibition of NF-κB Pathway in Hep3B Liver Cancer Cells" Cells 11, no. 17: 2734. https://doi.org/10.3390/cells11172734
APA StyleBhosale, P. B., Abusaliya, A., Kim, H. H., Ha, S. E., Park, M. Y., Jeong, S. H., Vetrivel, P., Heo, J. D., Kim, J.-A., Won, C. k., Kim, H.-W., & Kim, G. S. (2022). Apigetrin Promotes TNFα-Induced Apoptosis, Necroptosis, G2/M Phase Cell Cycle Arrest, and ROS Generation through Inhibition of NF-κB Pathway in Hep3B Liver Cancer Cells. Cells, 11(17), 2734. https://doi.org/10.3390/cells11172734