

A Cysteine Residue within the Kinase Domain of Zap70 Regulates Lck Activity and Proximal TCR Signaling
 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Antibodies
2.2. Cell Culture
2.3. DNA Constructs and Site-Directed Mutagenesis
- Zap70 C564A, forward: tacagttcgggtggagcctctggtgggcactc;
- reverse: gagtgcccaccagaggctccacccgaactgta.
- Zap70 C564R, forward: gtgcccaccagagcgtccacccgaact;
- reverse: agttcgggtggacgctctggtgggcac.
- Successful mutations were confirmed by sequencing.
2.4. Cell Transfections
2.5. Stimulation and Lysis of Cells
2.6. Immunoprecipitation
2.7. In Vitro Kinase Assay
2.8. Calcium-Flux
2.9. PLA
2.10. Statistics and Graphics
3. Results
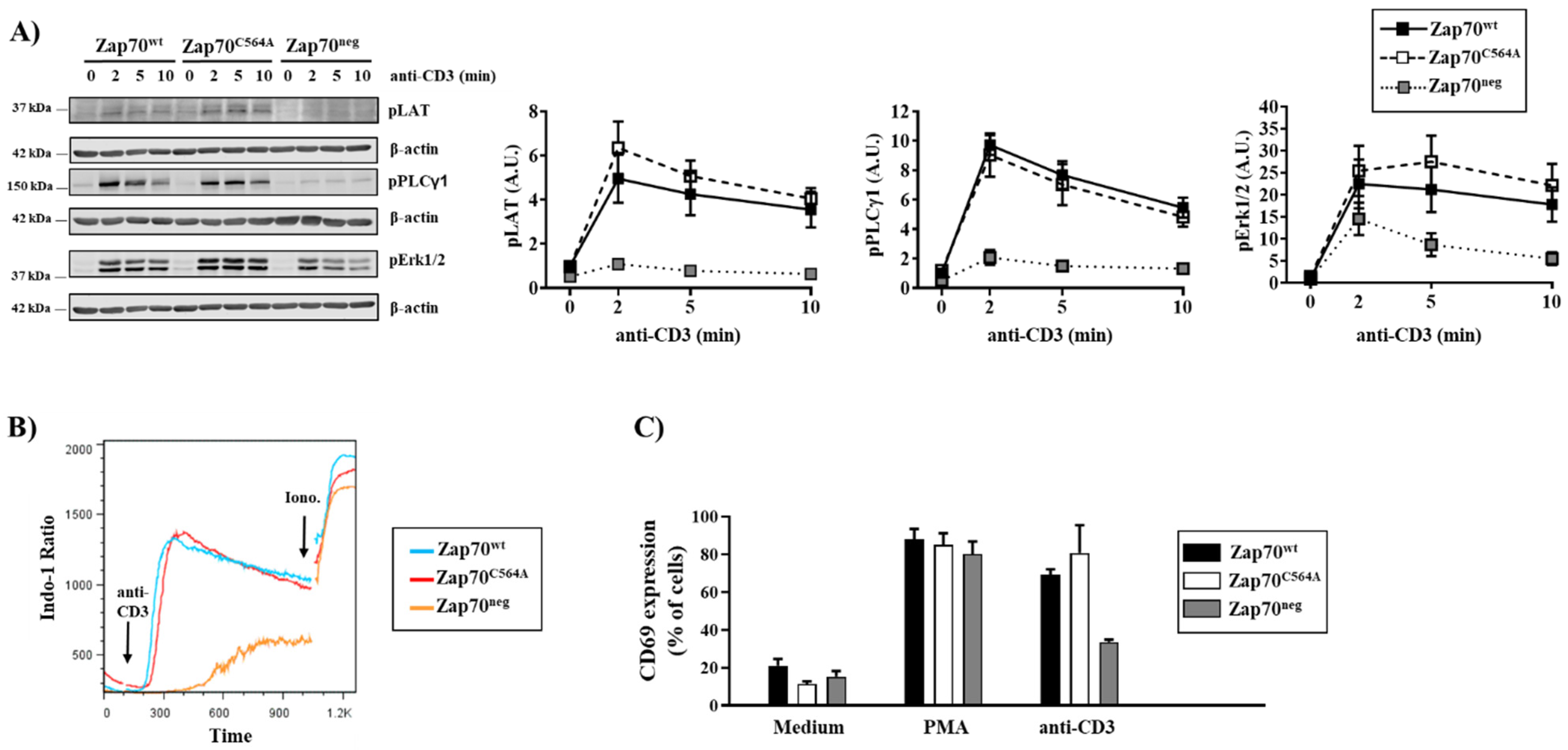
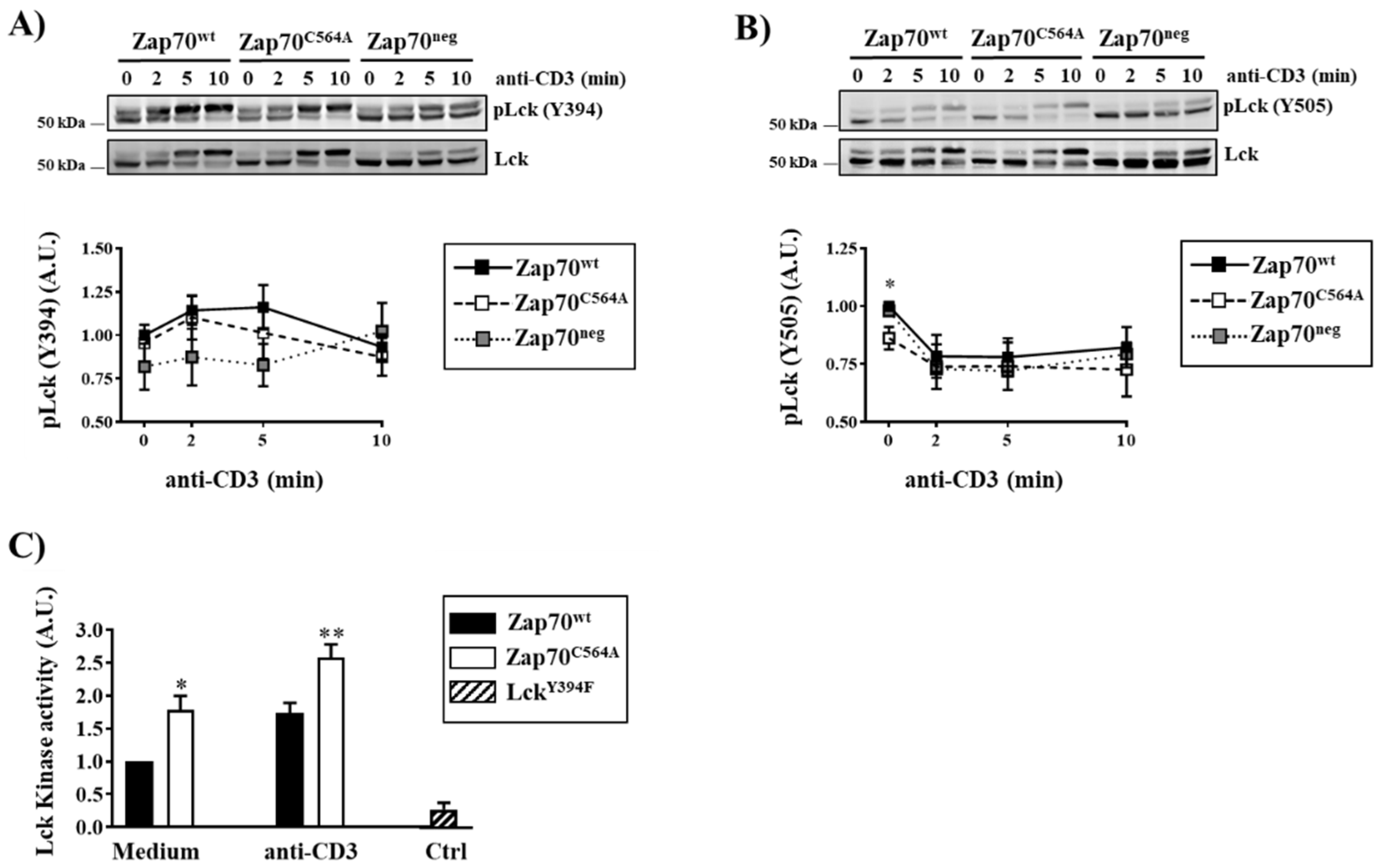
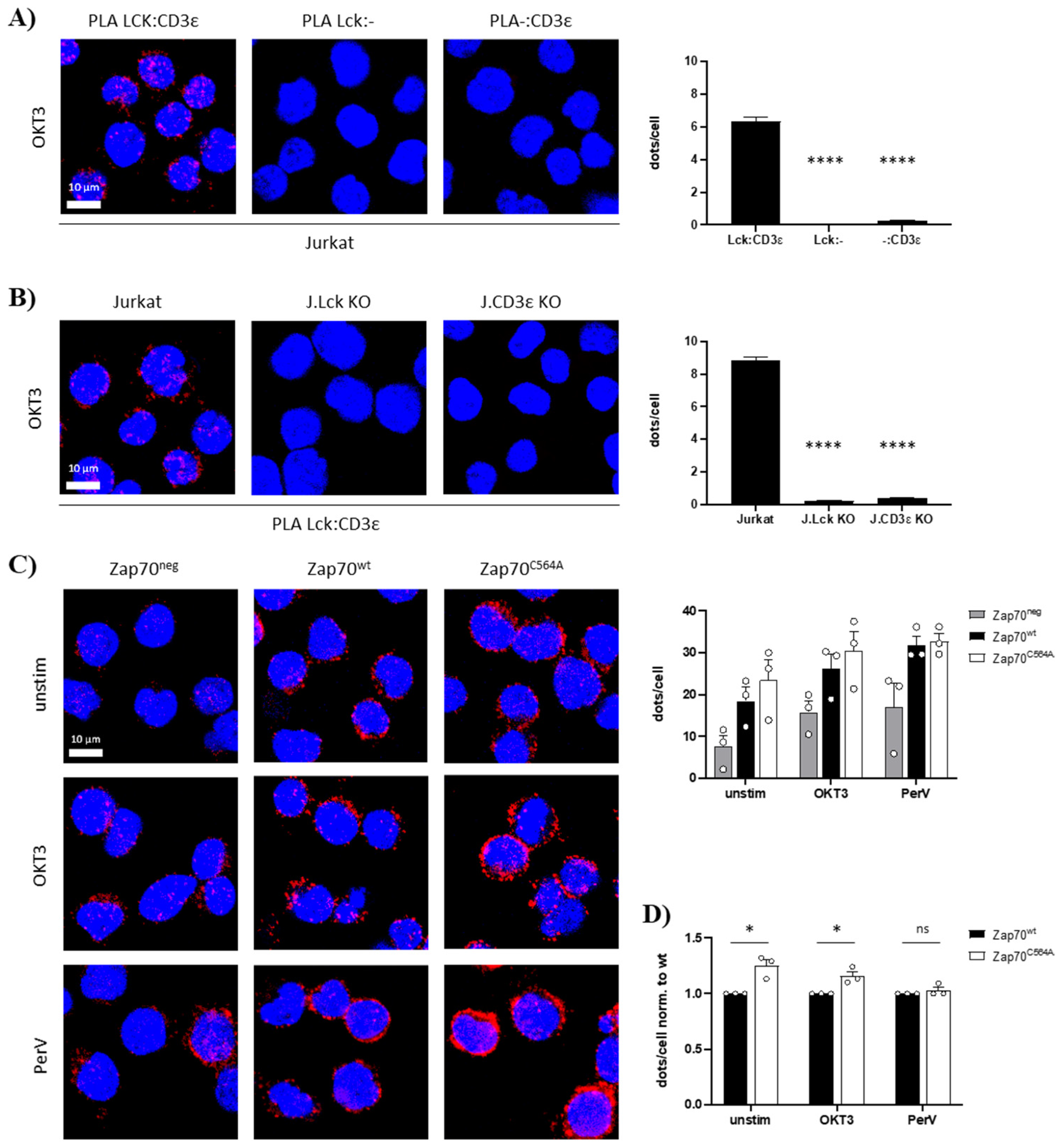
3.1. S-Acylation of C564 Is Dispensable for Signal Propagation and T-Cell Activation
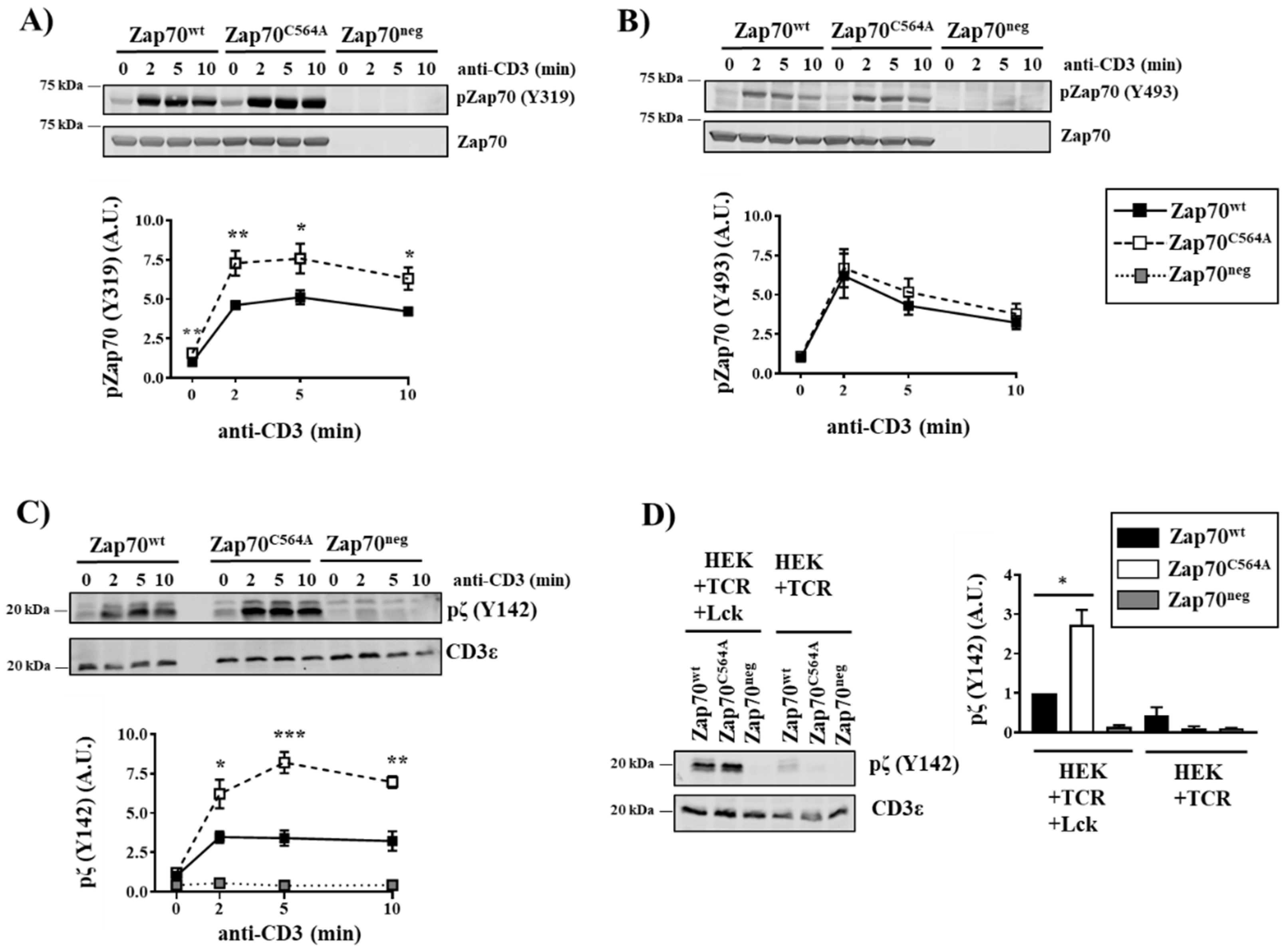
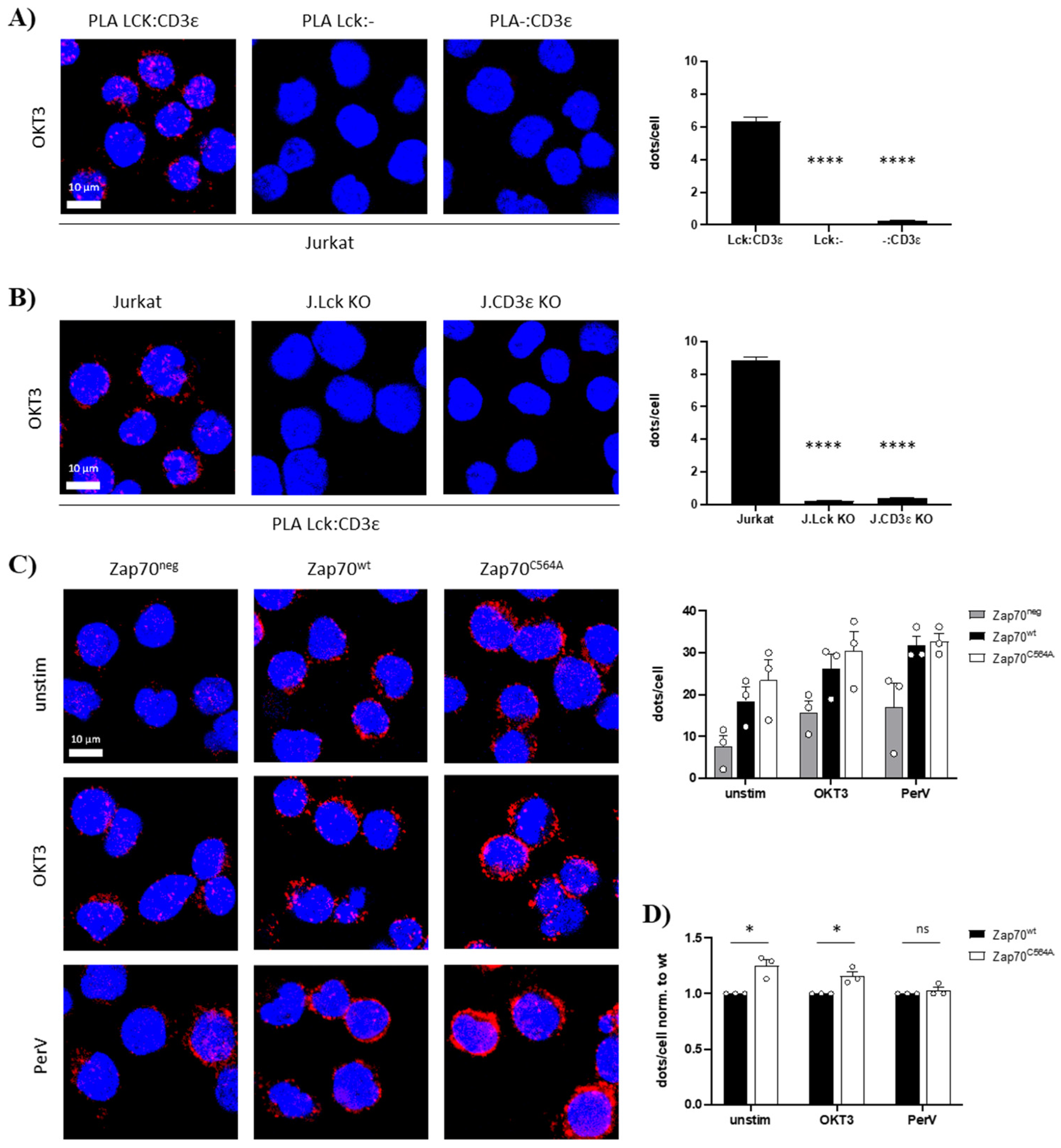
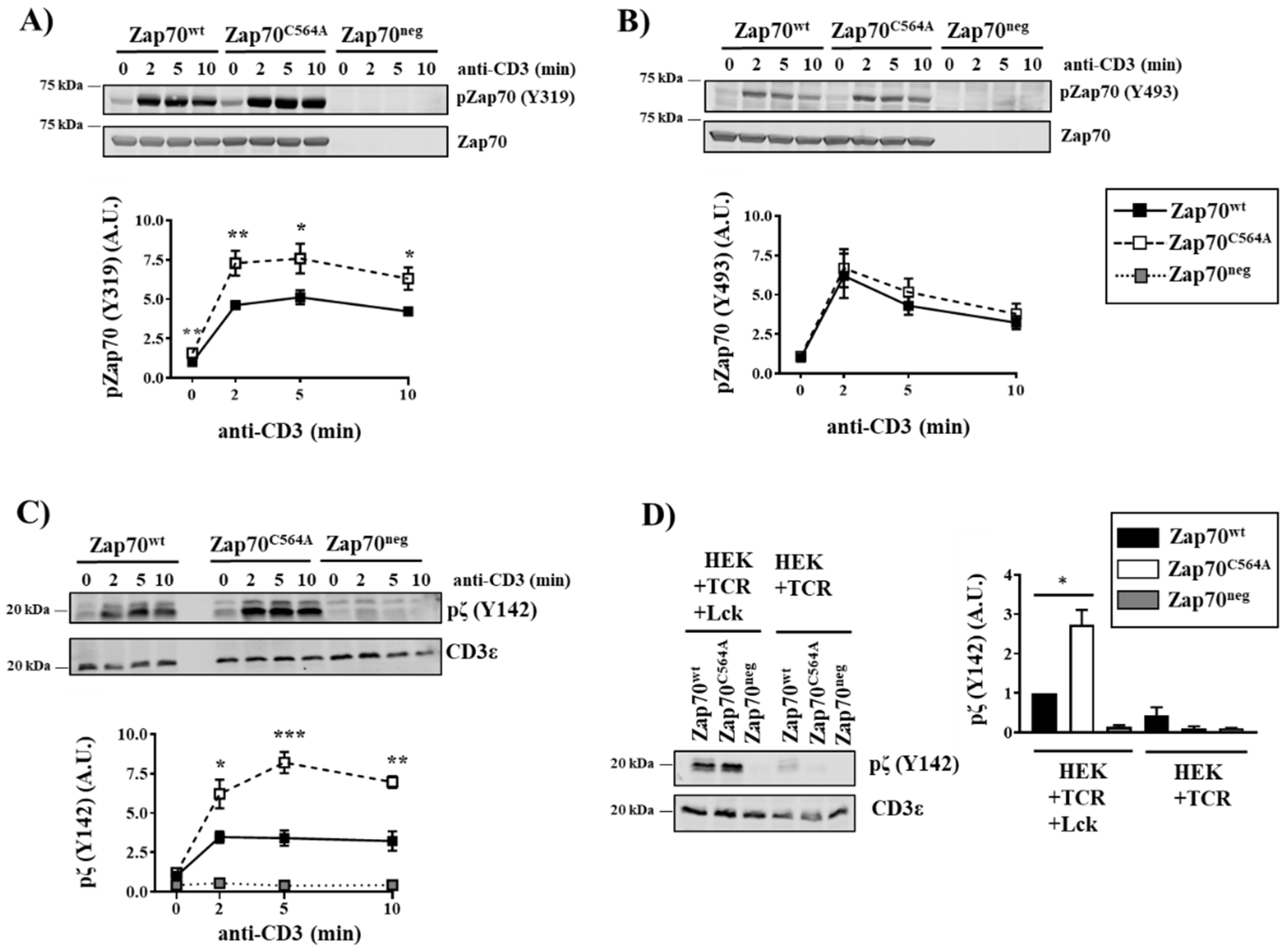
3.2. Zap70C564A Regulates Lck Activity and Proximal TCR Signaling
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Courtney, A.H.; Lo, W.L.; Weiss, A. TCR Signaling: Mechanisms of Initiation and Propagation. Trends Biochem. Sci. 2018, 43, 108–123. [Google Scholar] [CrossRef]
- Au-Yeung, B.B.; Shah, N.H.; Shen, L.; Weiss, A. ZAP-70 in Signaling, Biology, and Disease. Annu. Rev. Immunol. 2018, 36, 127–156. [Google Scholar] [CrossRef]
- Liaunardy-Jopeace, A.; Murton, B.L.; Mahesh, M.; Chin, J.W.; James, J.R. Encoding optical control in LCK kinase to quantitatively investigate its activity in live cells. Nat. Struct. Mol. Biol. 2017, 24, 1155–1163. [Google Scholar] [CrossRef] [PubMed]
- Martelli, M.P.; Lin, H.; Zhang, W.; Samelson, L.E.; Bierer, B.E. Signaling via LAT (linker for T-cell activation) and Syk/ZAP70 is required for ERK activation and NFAT transcriptional activation following CD2 stimulation. Blood 2000, 96, 2181–2190. [Google Scholar] [CrossRef] [PubMed]
- Griffith, C.E.; Zhang, W.; Wange, R.L. ZAP-70-dependent and -independent activation of Erk in Jurkat T cells. Differences in signaling induced by H2O2 and CD3 cross-linking. J. Biol. Chem. 1998, 273, 10771–10776. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.L.; Schreiber, K.L.; Zhang, W.; Wange, R.L.; Samelson, L.E.; Leibson, P.J.; Abraham, R.T. Genetic evidence for differential coupling of Syk family kinases to the T-cell receptor: Reconstitution studies in a ZAP-70-deficient Jurkat T-cell line. Mol. Cell Biol. 1998, 18, 1388–1399. [Google Scholar] [CrossRef] [PubMed]
- Kadlecek, T.A.; van Oers, N.S.; Lefrancois, L.; Olson, S.; Finlay, D.; Chu, D.H.; Connolly, K.; Killeen, N.; Weiss, A. Differential requirements for ZAP-70 in TCR signaling and T cell development. J. Immunol. 1998, 161, 4688–4694. [Google Scholar] [PubMed]
- Negishi, I.; Motoyama, N.; Nakayama, K.; Nakayama, K.; Senju, S.; Hatakeyama, S.; Zhang, Q.; Chan, A.C.; Loh, D.Y. Essential role for ZAP-70 in both positive and negative selection of thymocytes. Nature 1995, 376, 435–438. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Moore, A.; Ringshausen, I. ZAP-70 Shapes the Immune Microenvironment in B Cell Malignancies. Front. Oncol. 2020, 10, 595832. [Google Scholar] [CrossRef]
- Chan, A.Y.; Punwani, D.; Kadlecek, T.A.; Cowan, M.J.; Olson, J.L.; Mathes, E.F.; Sunderam, U.; Fu, S.M.; Srinivasan, R.; Kuriyan, J.; et al. A novel human autoimmune syndrome caused by combined hypomorphic and activating mutations in ZAP-70. J. Exp. Med. 2016, 213, 155–165. [Google Scholar] [CrossRef]
- Sharifinejad, N.; Jamee, M.; Zaki-Dizaji, M.; Lo, B.; Shaghaghi, M.; Mohammadi, H.; Jadidi-Niaragh, F.; Shaghaghi, S.; Yazdani, R.; Abolhassani, H.; et al. Clinical, Immunological, and Genetic Features in 49 Patients With ZAP-70 Deficiency: A Systematic Review. Front. Immunol. 2020, 11, 831. [Google Scholar] [CrossRef]
- Turul, T.; Tezcan, I.; Artac, H.; de Bruin-Versteeg, S.; Barendregt, B.H.; Reisli, I.; Sanal, O.; van Dongen, J.J.; van der Burg, M. Clinical heterogeneity can hamper the diagnosis of patients with ZAP70 deficiency. Eur. J. Pediatrics 2009, 168, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Tewari, R.; Shayahati, B.; Fan, Y.; Akimzhanov, A.M. T cell receptor-dependent S-acylation of ZAP-70 controls activation of T cells. J. Biol. Chem. 2021, 296, 100311. [Google Scholar] [CrossRef] [PubMed]
- Courtney, A.H.; Amacher, J.F.; Kadlecek, T.A.; Mollenauer, M.N.; Au-Yeung, B.B.; Kuriyan, J.; Weiss, A. A Phosphosite within the SH2 Domain of Lck Regulates Its Activation by CD45. Mol. Cell 2017, 67, 498–511. [Google Scholar] [CrossRef] [PubMed]
- Kastle, M.; Merten, C.; Hartig, R.; Kaehne, T.; Liaunardy-Jopeace, A.; Woessner, N.M.; Schamel, W.W.; James, J.; Minguet, S.; Simeoni, L.; et al. Tyrosine 192 within the SH2 domain of the Src-protein tyrosine kinase p56(Lck) regulates T-cell activation independently of Lck/CD45 interactions. Cell Commun. Signal. 2020, 18, 183. [Google Scholar] [CrossRef] [PubMed]
- Thurm, C.; Poltorak, M.P.; Reimer, E.; Brinkmann, M.M.; Leichert, L.; Schraven, B.; Simeoni, L. A highly conserved redox-active Mx(2)CWx(6)R motif regulates Zap70 stability and activity. Oncotarget 2017, 8, 30805–30816. [Google Scholar] [CrossRef] [PubMed]
- Stirnweiss, A.; Hartig, R.; Gieseler, S.; Lindquist, J.A.; Reichardt, P.; Philipsen, L.; Simeoni, L.; Poltorak, M.; Merten, C.; Zuschratter, W.; et al. T cell activation results in conformational changes in the Src family kinase Lck to induce its activation. Sci. Signal. 2013, 6, ra13. [Google Scholar] [CrossRef] [PubMed]
- Philipsen, L.; Reddycherla, A.V.; Hartig, R.; Gumz, J.; Kastle, M.; Kritikos, A.; Poltorak, M.P.; Prokazov, Y.; Turbin, E.; Weber, A.; et al. De novo phosphorylation and conformational opening of the tyrosine kinase Lck act in concert to initiate T cell receptor signaling. Sci. Signal. 2017, 10, eaaf4736. [Google Scholar] [CrossRef]
- Hartl, F.A.; Beck-Garcia, E.; Woessner, N.M.; Flachsmann, L.J.; Cardenas, R.M.V.; Brandl, S.M.; Taromi, S.; Fiala, G.J.; Morath, A.; Mishra, P.; et al. Noncanonical binding of Lck to CD3epsilon promotes TCR signaling and CAR function. Nat. Immunol. 2020, 21, 902–913. [Google Scholar] [CrossRef]
- Lo, W.L.; Shah, N.H.; Ahsan, N.; Horkova, V.; Stepanek, O.; Salomon, A.R.; Kuriyan, J.; Weiss, A. Lck promotes Zap70-dependent LAT phosphorylation by bridging Zap70 to LAT. Nat. Immunol. 2018, 19, 733–741. [Google Scholar] [CrossRef]
- Goodfellow, H.S.; Frushicheva, M.P.; Ji, Q.; Cheng, D.A.; Kadlecek, T.A.; Cantor, A.J.; Kuriyan, J.; Chakraborty, A.K.; Salomon, A.; Weiss, A. The catalytic activity of the kinase ZAP-70 mediates basal signaling and negative feedback of the T cell receptor pathway. Sci. Signal. 2015, 8, ra49. [Google Scholar] [CrossRef]
- Kastle, M.; Merten, C.; Hartig, R.; Plaza-Sirvent, C.; Schmitz, I.; Bommhardt, U.; Schraven, B.; Simeoni, L. Type of PaperY192 within the SH2 Domain of Lck Regulates TCR Signaling Downstream of PLC-gamma1 and Thymic Selection. Int. J. Mol. Sci. 2022, 23, 7271. [Google Scholar] [CrossRef]
- Swamy, M.; Beck-Garcia, K.; Beck-Garcia, E.; Hartl, F.; Morath, A.; Yousefi, O.S.; Pashupati Dopfer, E.; Molnár, E.; Schulze, A.K.; Blanco, R.; et al. A Cholesterol-Based Allostery Model of T Cell Receptor Phosphorylation. Immunity 2016, 44, 1091–1101. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhu, Y.; Li, X.; Gao, W.; Zhen, Z.; Dong, D.; Huang, B.; Ma, Z.; Zhang, A.; Song, X.; et al. Cholesterol inhibits TCR signaling by directly restricting TCR-CD3 core tunnel motility. Mol. Cell 2022, 82, 1278–1287. [Google Scholar] [CrossRef] [PubMed]
- Katz, Z.B.; Novotna, L.; Blount, A.; Lillemeier, B.F. A cycle of Zap70 kinase activation and release from the TCR amplifies and disperses antigenic stimuli. Nat. Immunol. 2017, 18, 86–95. [Google Scholar] [CrossRef]
- Corcoran, A.; Cotter, T.G. Redox regulation of protein kinases. FEBS J. 2013, 280, 1944–1965. [Google Scholar] [CrossRef]
- Giannoni, E.; Buricchi, F.; Raugei, G.; Ramponi, G.; Chiarugi, P. Intracellular reactive oxygen species activate Src tyrosine kinase during cell adhesion and anchorage-dependent cell growth. Mol. Cell Biol. 2005, 25, 6391–6403. [Google Scholar] [CrossRef]
- Leonberg, A.K.; Chai, Y.C. The functional role of cysteine residues for c-Abl kinase activity. Mol. Cell Biochem. 2007, 304, 207–212. [Google Scholar] [CrossRef]
- Yoo, S.K.; Starnes, T.W.; Deng, Q.; Huttenlocher, A. Lyn is a redox sensor that mediates leukocyte wound attraction in vivo. Nature 2011, 480, 109–112. [Google Scholar] [CrossRef]
- Paulsen, C.E.; Truong, T.H.; Garcia, F.J.; Homann, A.; Gupta, V.; Leonard, S.E.; Carroll, K.S. Peroxide-dependent sulfenylation of the EGFR catalytic site enhances kinase activity. Nat. Chem. Biol. 2011, 8, 57–64. [Google Scholar] [CrossRef] [Green Version]
- Kato, M.; Iwashita, T.; Akhand, A.A.; Liu, W.; Takeda, K.; Takeuchi, K.; Yoshihara, M.; Hossain, K.; Wu, J.; Du, J.; et al. Molecular mechanism of activation and superactivation of Ret tyrosine kinases by ultraviolet light irradiation. Antioxid. Redox Signal. 2000, 2, 841–849. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Clone | Company |
|---|---|---|
| APC anti-human CD3 | HIT3a | BD Pharmingen™ |
| APC anti-human CD69 | FN 50 | BD Pharmingen™ |
| Goat IgG anti-Mouse IgG + IgM (H + L) | polyclonal | Dianova |
| Antibody | Clone/Lot | Company |
|---|---|---|
| β-actin | AC-15 | Sigma-Aldrich |
| CD3ε | EB12592 | Everest Biotech Ltd. |
| Lat pY191 | 3584S | Cell Signaling Technology |
| Lck | 06-583 | Merck Millipore, upstate |
| Lck | 3A5 | Santa Cruz Biotechnology |
| Lck pY505 | 2751S | Cell Signaling Technology |
| PLCγ pY783 | 2821S | Cell Signaling Technology |
| p44/42 MAPK (Erk1/2) (pT202/pY204) | D13.14.4E | Cell Signaling Technology |
| Src pY416 | 2101S | Cell Signaling Technology |
| Zap70 pY319 | 2701S | Cell Signaling Technology |
| Zap70 pY493 | 2704S | Cell Signaling Technology |
| Zap70 | 1E7.2 | Santa Cruz Biotechnology |
| CD3 ζ pY142 | SAB4301233 | Sigma-Aldrich |
| CD3 ζ | 6B10.2 | Santa Cruz Biotechnology |
| Antibody | Clone | Company |
|---|---|---|
| CD3ε | UCHT1/OKT3 | BioLegend |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schultz, A.; Schnurra, M.; El-Bizri, A.; Woessner, N.M.; Hartmann, S.; Hartig, R.; Minguet, S.; Schraven, B.; Simeoni, L. A Cysteine Residue within the Kinase Domain of Zap70 Regulates Lck Activity and Proximal TCR Signaling. Cells 2022, 11, 2723. https://doi.org/10.3390/cells11172723
Schultz A, Schnurra M, El-Bizri A, Woessner NM, Hartmann S, Hartig R, Minguet S, Schraven B, Simeoni L. A Cysteine Residue within the Kinase Domain of Zap70 Regulates Lck Activity and Proximal TCR Signaling. Cells. 2022; 11(17):2723. https://doi.org/10.3390/cells11172723
Chicago/Turabian StyleSchultz, Annika, Marvin Schnurra, Ali El-Bizri, Nadine M. Woessner, Sara Hartmann, Roland Hartig, Susana Minguet, Burkhart Schraven, and Luca Simeoni. 2022. "A Cysteine Residue within the Kinase Domain of Zap70 Regulates Lck Activity and Proximal TCR Signaling" Cells 11, no. 17: 2723. https://doi.org/10.3390/cells11172723
APA StyleSchultz, A., Schnurra, M., El-Bizri, A., Woessner, N. M., Hartmann, S., Hartig, R., Minguet, S., Schraven, B., & Simeoni, L. (2022). A Cysteine Residue within the Kinase Domain of Zap70 Regulates Lck Activity and Proximal TCR Signaling. Cells, 11(17), 2723. https://doi.org/10.3390/cells11172723