
The Central Domain of MCPH1 Controls Development of the Cerebral Cortex and Gonads in Mice



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice and Mating Scheme
2.2. Cell Culture
2.3. RNA Extraction and RT-PCR Analysis
2.4. Histological Analysis
2.5. Immunofluorescence (IF) Staining on Cells and Brain Sections
2.6. Western Blotting
2.7. Statistical Analysis
3. Results
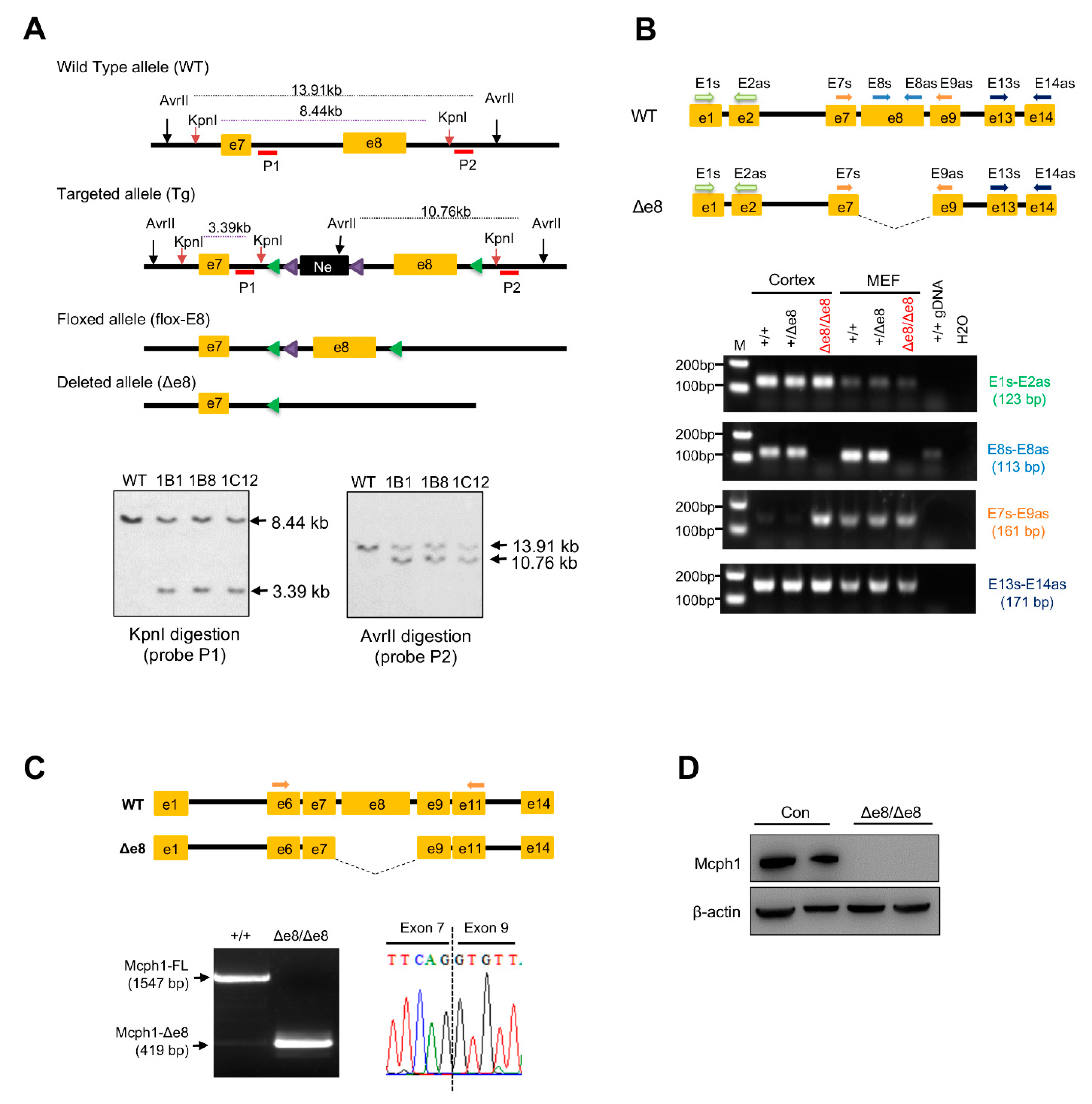
3.1. Generation of Mcph1-Δe8 Mice
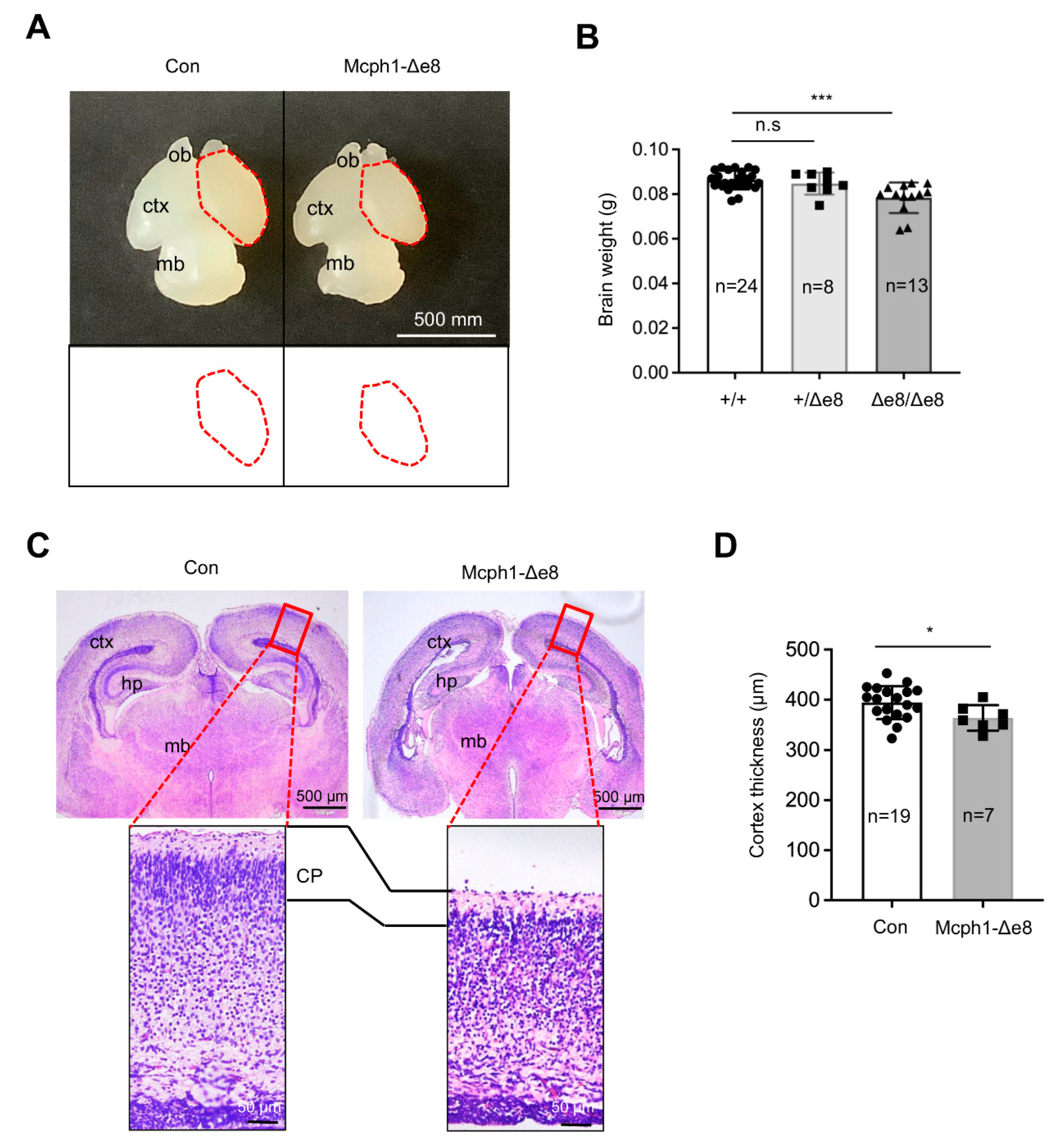
3.2. Mcph1-Δe8 Mice Develop Microcephaly
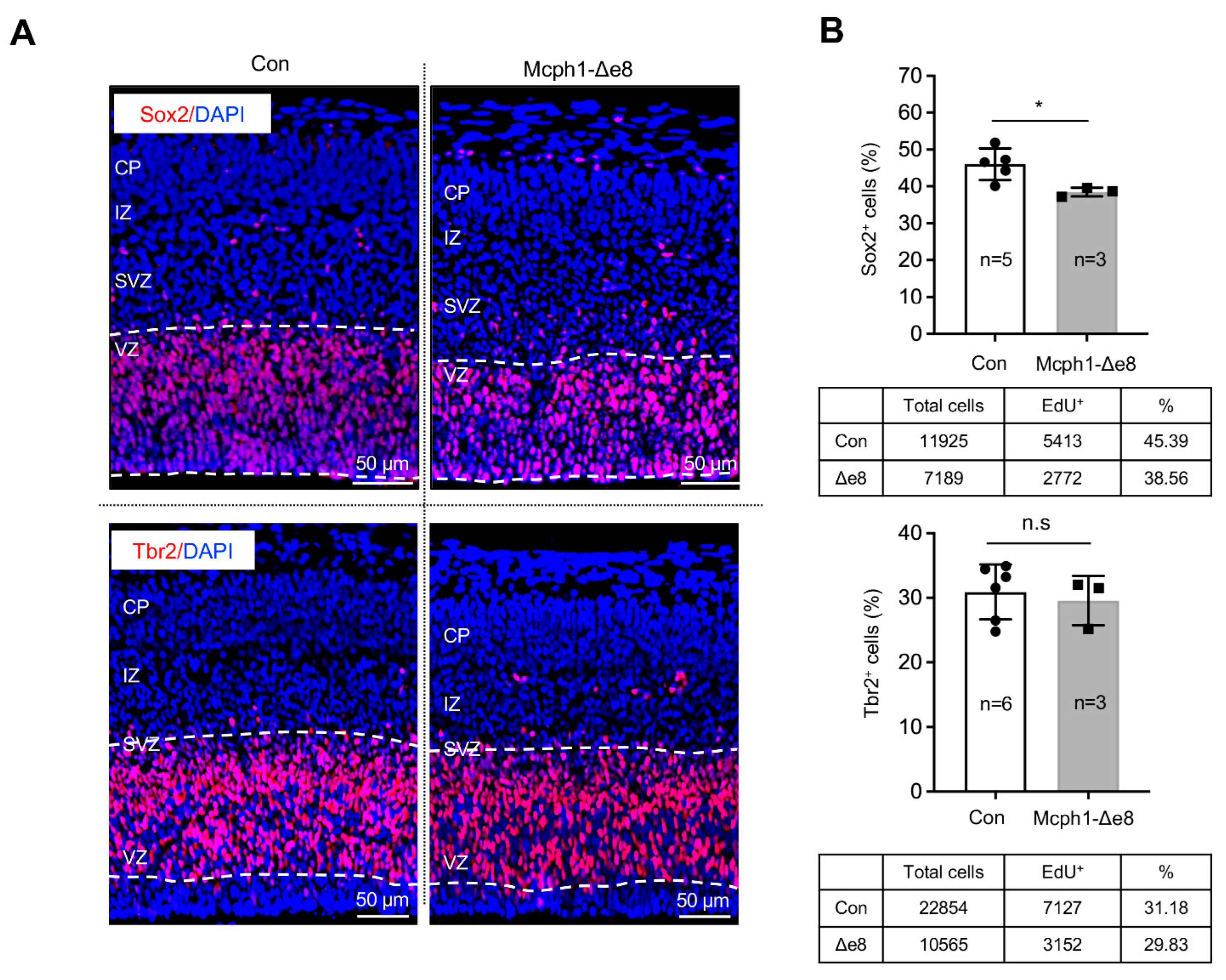
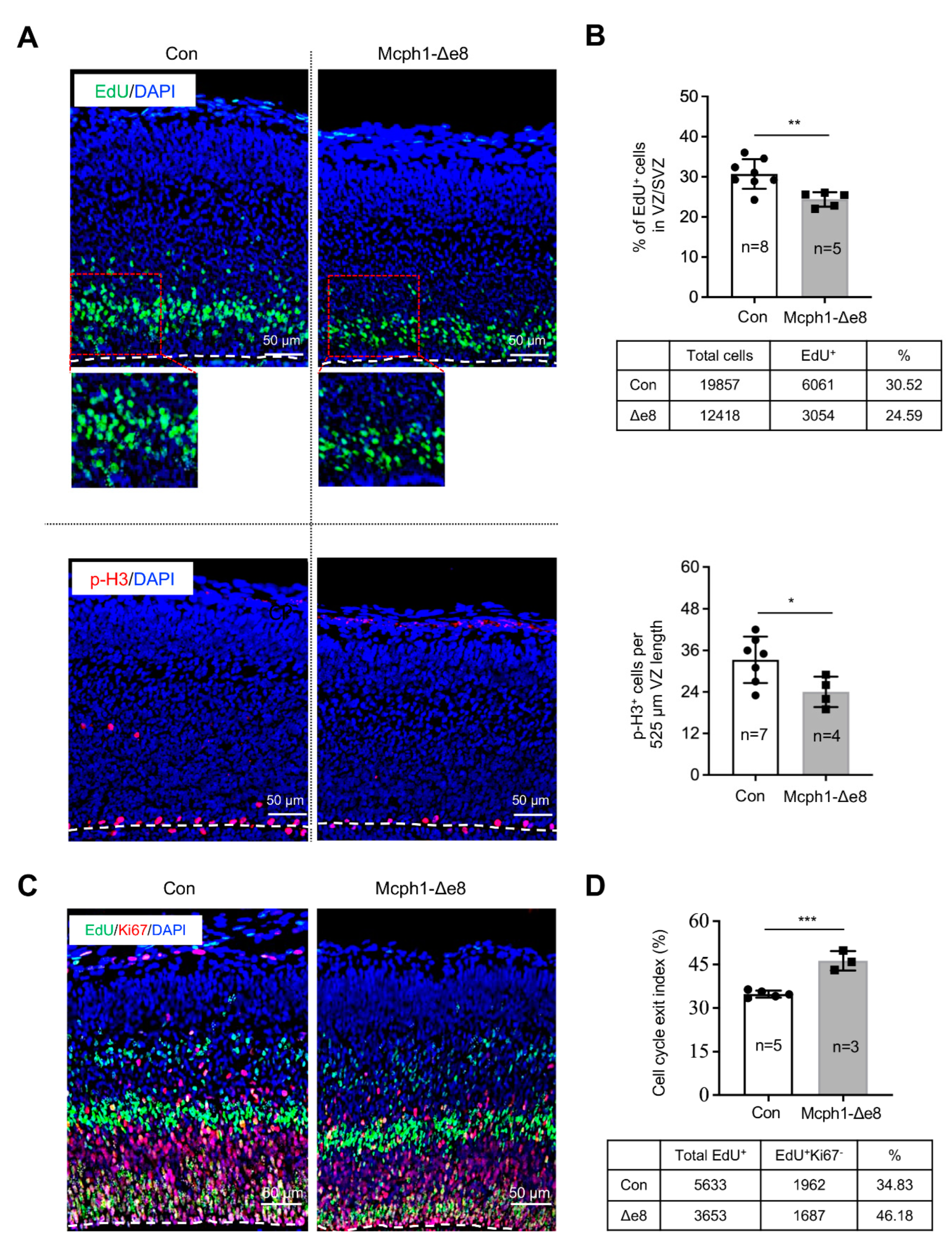
3.3. Mcph1-Δe8 Neuroprogenitors Have Self-Renewal Defect
3.4. Mcph1-Δe8 Neuroprogenitors Are Prone to Differentiate Prematurely
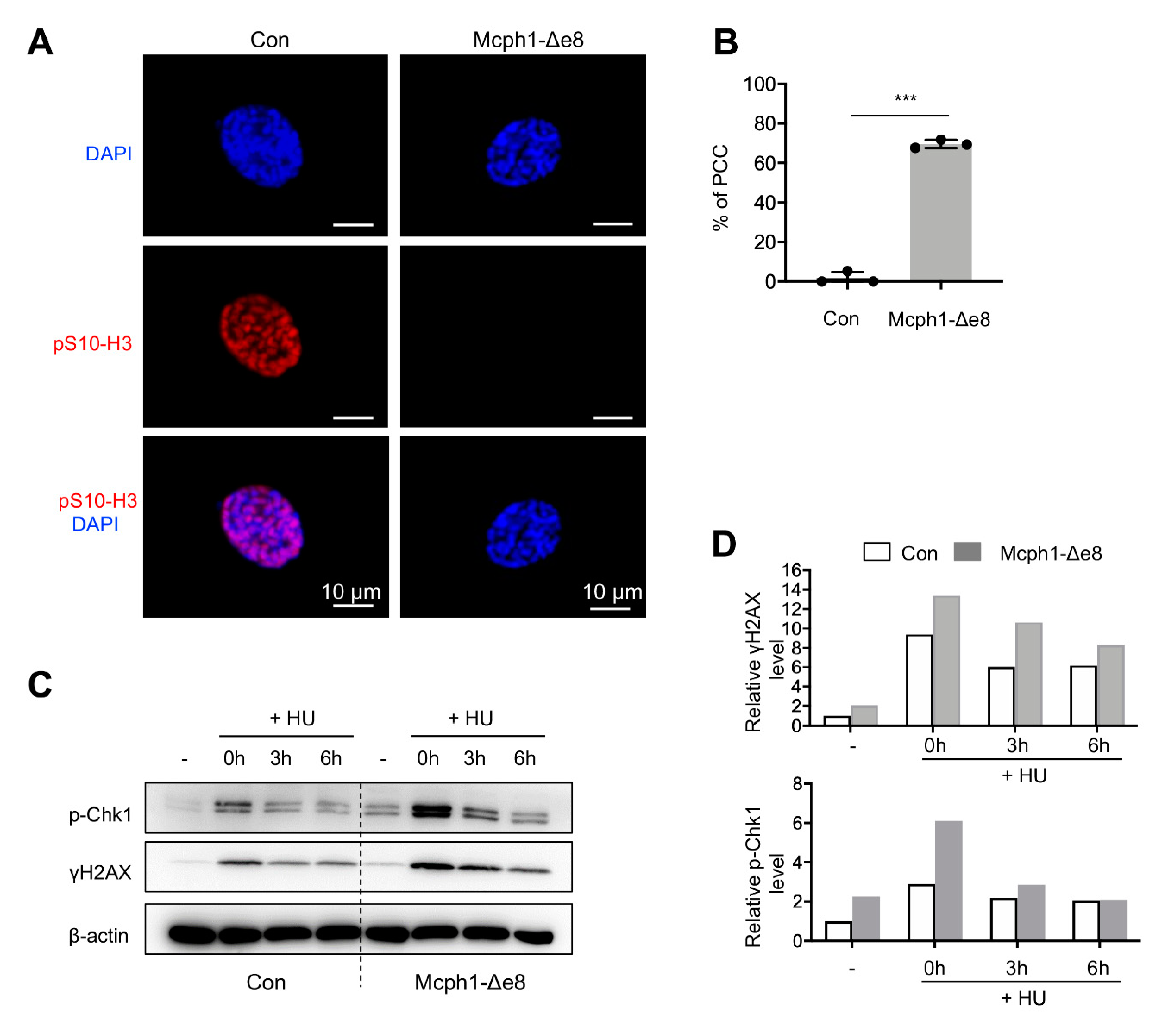
3.5. Mcph1-Δe8 Mutation Renders Cells to PCC and Malfunctional DDR
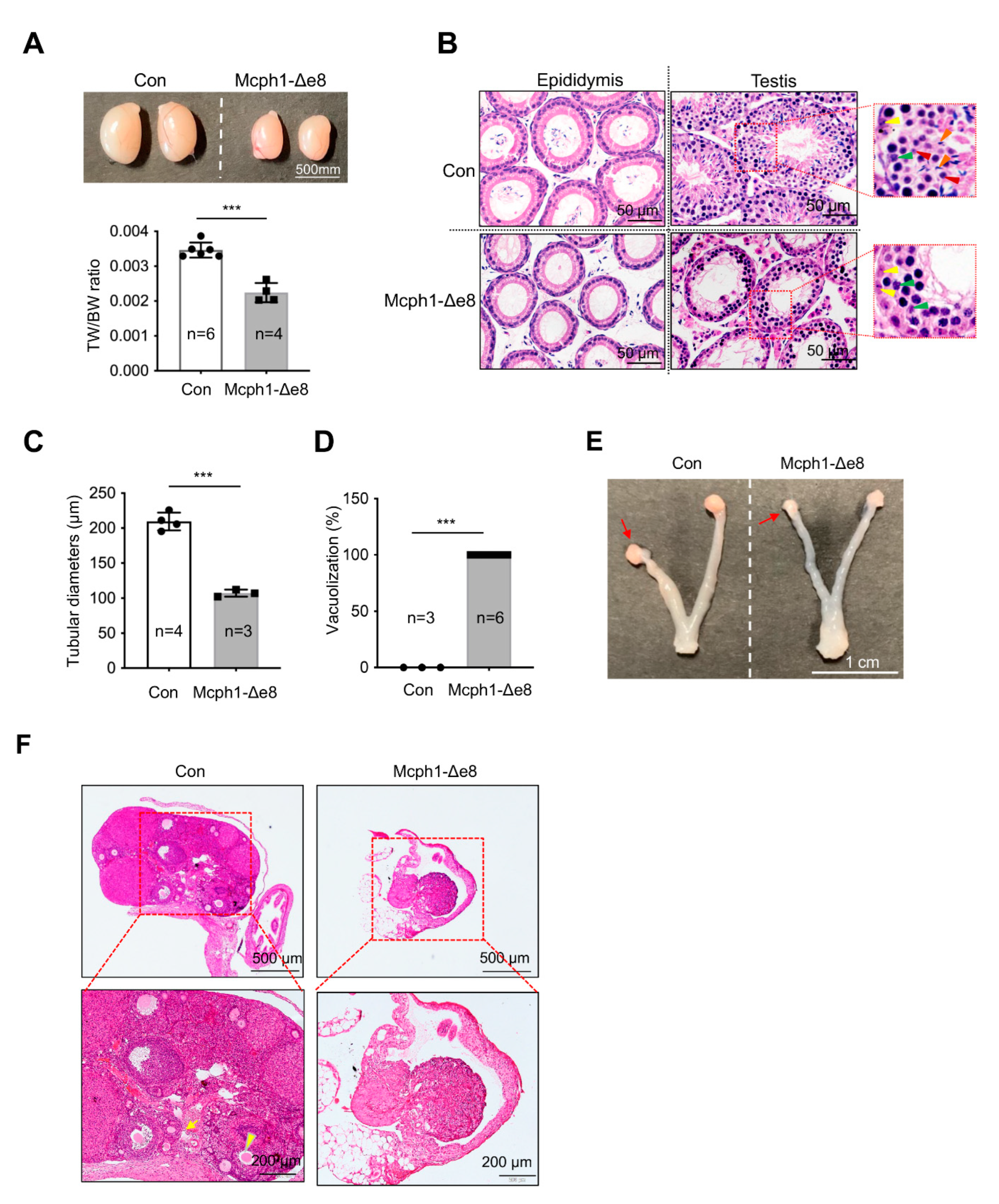
3.6. Defects in Gonad Development in Mcph1-Δe8 Mice
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jackson, A.P.; Eastwood, H.; Bell, S.M.; Adu, J.; Toomes, C.; Carr, I.M.; Roberts, E.; Hampshire, D.J.; Crow, Y.J.; Mighell, A.J.; et al. Identification of microcephalin, a protein implicated in determining the size of the human brain. Am. J. Hum. Genet. 2002, 71, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Markandaya, M.; Girimaji, S.C. Primary microcephaly: Microcephalin and ASPM determine the size of the human brain. J. Biosci. 2002, 27, 629–632. [Google Scholar] [CrossRef] [PubMed]
- Woods, C.G.; Bond, J.; Enard, W. Autosomal recessive primary microcephaly (MCPH): A review of clinical, molecular, and evolutionary findings. Am. J. Hum. Genet. 2005, 76, 717–728. [Google Scholar] [CrossRef]
- Desir, J.; Cassart, M.; David, P.; Van Bogaert, P.; Abramowicz, M. Primary microcephaly with ASPM mutation shows simplified cortical gyration with antero-posterior gradient pre- and post-natally. Am. J. Med. Genet. Part A 2008, 146, 1439–1443. [Google Scholar] [CrossRef]
- Siskos, N.; Stylianopoulou, E.; Skavdis, G.; Grigoriou, M.E. Molecular Genetics of Microcephaly Primary Hereditary: An Overview. Brain Sci. 2021, 11, 581. [Google Scholar] [CrossRef] [PubMed]
- Jean, F.; Stuart, A.; Tarailo-Graovac, M. Dissecting the Genetic and Etiological Causes of Primary Microcephaly. Front. Neurol. 2020, 11, 570830. [Google Scholar] [CrossRef]
- Kristofova, M.; Ori, A.; Wang, Z.Q. Multifaceted Microcephaly-Related Gene MCPH1. Cells 2022, 11, 275. [Google Scholar] [CrossRef]
- Trimborn, M.; Bell, S.M.; Felix, C.; Rashid, Y.; Jafri, H.; Griffiths, P.D.; Neumann, L.M.; Krebs, A.; Reis, A.; Sperling, K.; et al. Mutations in microcephalin cause aberrant regulation of chromosome condensation. Am. J. Hum. Genet. 2004, 75, 261–266. [Google Scholar] [CrossRef]
- Pfau, R.B.; Thrush, D.L.; Hamelberg, E.; Bartholomew, D.; Botes, S.; Pastore, M.; Tan, C.; del Gaudio, D.; Gastier-Foster, J.M.; Astbury, C. MCPH1 deletion in a newborn with severe microcephaly and premature chromosome condensation. Eur. J. Med. Genet. 2013, 56, 609–613. [Google Scholar] [CrossRef]
- Trimborn, M.; Schindler, D.; Neitzel, H.; Hirano, T. Misregulated chromosome condensation in MCPH1 primary microcephaly is mediated by condensin II. Cell Cycle 2006, 5, 322–326. [Google Scholar]
- Barbelanne, M.; Tsang, W.Y. Molecular and cellular basis of autosomal recessive primary microcephaly. BioMed Res. Int. 2014, 2014, 547986. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhou, Z.W.; Wang, Z.Q. The DNA damage response molecule MCPH1 in brain development and beyond. Acta Biochim. Biophys. Sin. 2016, 48, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Schneble-Lohnert, N.; Kristofova, M.; Qing, X.; Labisch, J.; Hofmann, S.; Ehrenberg, S.; Sannai, M.; Jorss, T.; Ori, A.; et al. The N-terminal BRCT domain determines MCPH1 function in brain development and fertility. Cell Death Dis. 2021, 12, 143. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Gao, H.; Lin, S.Y.; Peng, G.; Huang, X.; Zhang, P.; Goss, J.A.; Brunicardi, F.C.; Multani, A.S.; Chang, S.; et al. BRIT1/MCPH1 is essential for mitotic and meiotic recombination DNA repair and maintaining genomic stability in mice. PLoS Genet. 2010, 6, e1000826. [Google Scholar] [CrossRef]
- Jeffers, L.J.; Coull, B.J.; Stack, S.J.; Morrison, C.G. Distinct BRCT domains in Mcph1/Brit1 mediate ionizing radiation-induced focus formation and centrosomal localization. Oncogene 2008, 27, 139–144. [Google Scholar] [CrossRef][Green Version]
- Wood, J.L.; Liang, Y.; Li, K.; Chen, J. Microcephalin/MCPH1 associates with the Condensin II complex to function in homologous recombination repair. J. Biol. Chem. 2008, 283, 29586–29592. [Google Scholar] [CrossRef]
- Manke, I.A.; Lowery, D.M.; Nguyen, A.; Yaffe, M.B. BRCT repeats as phosphopeptide-binding modules involved in protein targeting. Science 2003, 302, 636–639. [Google Scholar] [CrossRef]
- Wood, J.L.; Singh, N.; Mer, G.; Chen, J. MCPH1 functions in an H2AX-dependent but MDC1-independent pathway in response to DNA damage. J. Biol. Chem. 2007, 282, 35416–35423. [Google Scholar] [CrossRef]
- Yamashita, D.; Shintomi, K.; Ono, T.; Gavvovidis, I.; Schindler, D.; Neitzel, H.; Trimborn, M.; Hirano, T. MCPH1 regulates chromosome condensation and shaping as a composite modulator of condensin II. J. Cell Biol. 2011, 194, 841–854. [Google Scholar] [CrossRef]
- Pulvers, J.N.; Journiac, N.; Arai, Y.; Nardelli, J. MCPH1: A window into brain development and evolution. Front. Cell. Neurosci. 2015, 9, 92. [Google Scholar] [CrossRef]
- Gruber, R.; Zhou, Z.; Sukchev, M.; Joerss, T.; Frappart, P.O.; Wang, Z.Q. MCPH1 regulates the neuroprogenitor division mode by coupling the centrosomal cycle with mitotic entry through the Chk1-Cdc25 pathway. Nat. Cell Biol. 2011, 13, 1325–1334. [Google Scholar] [CrossRef] [PubMed]
- Naseer, M.I.; Rasool, M.; Abdulkareem, A.A.; Bassiouni, R.I.; Algahtani, H.; Chaudhary, A.G.; Al-Qahtani, M.H. Novel compound heterozygous mutations in MCPH1 gene causes primary microcephaly in Saudi family. Neurosciences 2018, 23, 347–350. [Google Scholar] [CrossRef]
- Khan, N.M.; Masoud, M.S.; Baig, S.M.; Qasim, M.; Chang, J. Identification of Pathogenic Mutations in Primary Microcephaly- (MCPH-) Related Three Genes CENPJ, CASK, and MCPH1 in Consanguineous Pakistani Families. BioMed Res. Int. 2022, 2022, 3769948. [Google Scholar] [CrossRef]
- Liu, X.; Zong, W.; Li, T.; Wang, Y.; Xu, X.; Zhou, Z.W.; Wang, Z.Q. The E3 ubiquitin ligase APC/C(C)(dh1) degrades MCPH1 after MCPH1-βTrCP2-Cdc25A-mediated mitotic entry to ensure neurogenesis. EMBO J. 2017, 36, 3666–3681. [Google Scholar] [CrossRef] [PubMed]
- Busino, L.; Donzelli, M.; Chiesa, M.; Guardavaccaro, D.; Ganoth, D.; Dorrello, N.V.; Hershko, A.; Pagano, M.; Draetta, G.F. Degradation of Cdc25A by beta-TrCP during S phase and in response to DNA damage. Nature 2003, 426, 87–91. [Google Scholar] [CrossRef]
- Li, T.; Wang, Z.Q. Point mutation at the Nbs1 Threonine 278 site does not affect mouse development, but compromises the Chk2 and Smc1 phosphorylation after DNA damage. Mech. Ageing Dev. 2011, 132, 382–388. [Google Scholar] [CrossRef]
- Guerra, G.M.; May, D.; Kroll, T.; Koch, P.; Groth, M.; Wang, Z.Q.; Li, T.L.; Grigaravicius, P. Cell Type-Specific Role of RNA Nuclease SMG6 in Neurogenesis. Cells 2021, 10, 3365. [Google Scholar] [CrossRef]
- Alderton, G.K.; Galbiati, L.; Griffith, E.; Surinya, K.H.; Neitzel, H.; Jackson, A.P.; Jeggo, P.A.; O’Driscoll, M. Regulation of mitotic entry by microcephalin and its overlap with ATR signalling. Nat. Cell Biol. 2006, 8, 725–733. [Google Scholar] [CrossRef]
- Rai, R.; Dai, H.; Multani, A.S.; Li, K.; Chin, K.; Gray, J.; Lahad, J.P.; Liang, J.; Mills, G.B.; Meric-Bernstam, F.; et al. BRIT1 regulates early DNA damage response, chromosomal integrity, and cancer. Cancer Cell 2006, 10, 145–157. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Fardaei, M.; Gholami, M.; Miryounesi, M. A case report: Autosomal recessive microcephaly caused by a novel mutation in MCPH1 gene. Gene 2015, 571, 149–150. [Google Scholar] [CrossRef]
- Journiac, N.; Gilabert-Juan, J.; Cipriani, S.; Benit, P.; Liu, X.; Jacquier, S.; Faivre, V.; Delahaye-Duriez, A.; Csaba, Z.; Hourcade, T.; et al. Cell Metabolic Alterations due to Mcph1 Mutation in Microcephaly. Cell Rep. 2020, 31, 107506. [Google Scholar] [CrossRef] [PubMed]
- Trimborn, M.; Ghani, M.; Walther, D.J.; Dopatka, M.; Dutrannoy, V.; Busche, A.; Meyer, F.; Nowak, S.; Nowak, J.; Zabel, C.; et al. Establishment of a mouse model with misregulated chromosome condensation due to defective Mcph1 function. PLoS ONE 2010, 5, e9242. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ingham, N.; Clare, S.; Raisen, C.; Vancollie, V.E.; Ismail, O.; McIntyre, R.E.; Tsang, S.H.; Mahajan, V.B.; Dougan, G.; et al. Mcph1-deficient mice reveal a role for MCPH1 in otitis media. PLoS ONE 2013, 8, e58156. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Zong, W.; Sun, W.; Chen, C.; Wang, Z.-Q.; Li, T. The Central Domain of MCPH1 Controls Development of the Cerebral Cortex and Gonads in Mice. Cells 2022, 11, 2715. https://doi.org/10.3390/cells11172715
Wang Y, Zong W, Sun W, Chen C, Wang Z-Q, Li T. The Central Domain of MCPH1 Controls Development of the Cerebral Cortex and Gonads in Mice. Cells. 2022; 11(17):2715. https://doi.org/10.3390/cells11172715
Chicago/Turabian StyleWang, Yaru, Wen Zong, Wenli Sun, Chengyan Chen, Zhao-Qi Wang, and Tangliang Li. 2022. "The Central Domain of MCPH1 Controls Development of the Cerebral Cortex and Gonads in Mice" Cells 11, no. 17: 2715. https://doi.org/10.3390/cells11172715
APA StyleWang, Y., Zong, W., Sun, W., Chen, C., Wang, Z.-Q., & Li, T. (2022). The Central Domain of MCPH1 Controls Development of the Cerebral Cortex and Gonads in Mice. Cells, 11(17), 2715. https://doi.org/10.3390/cells11172715