
Molecular Mechanism and Regulation of Autophagy and Its Potential Role in Epilepsy


Abstract
:1. Introduction
2. The Historical Landmarks of Autophagy
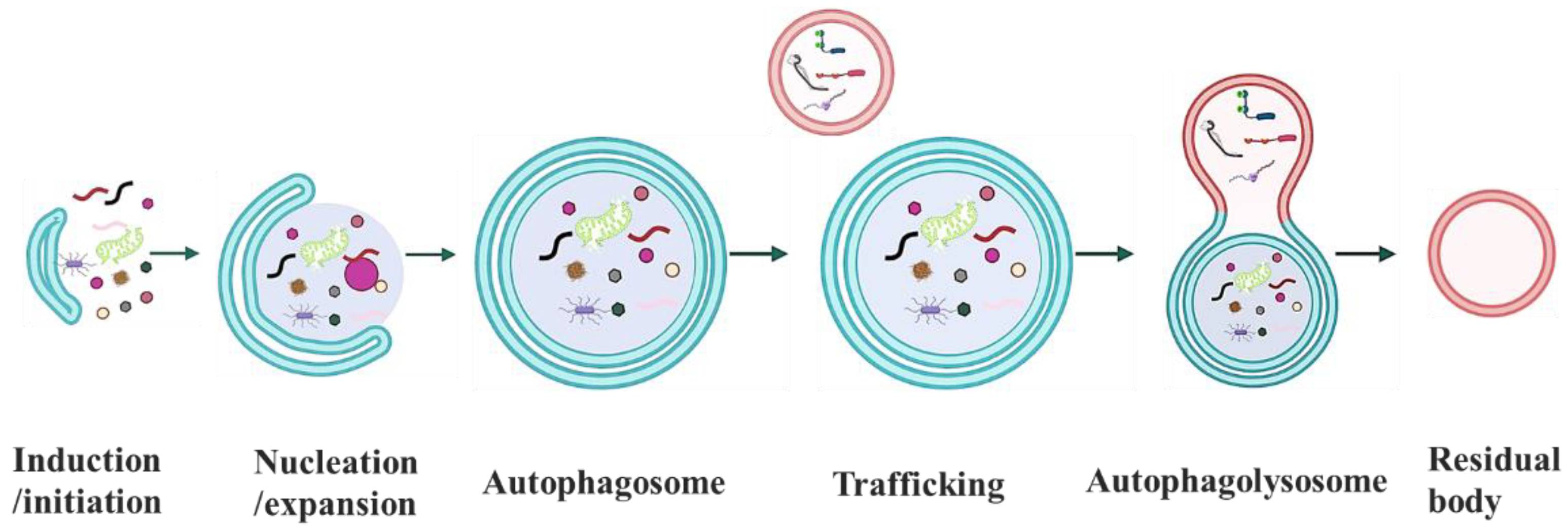
3. The Distinct Types of Autophagy
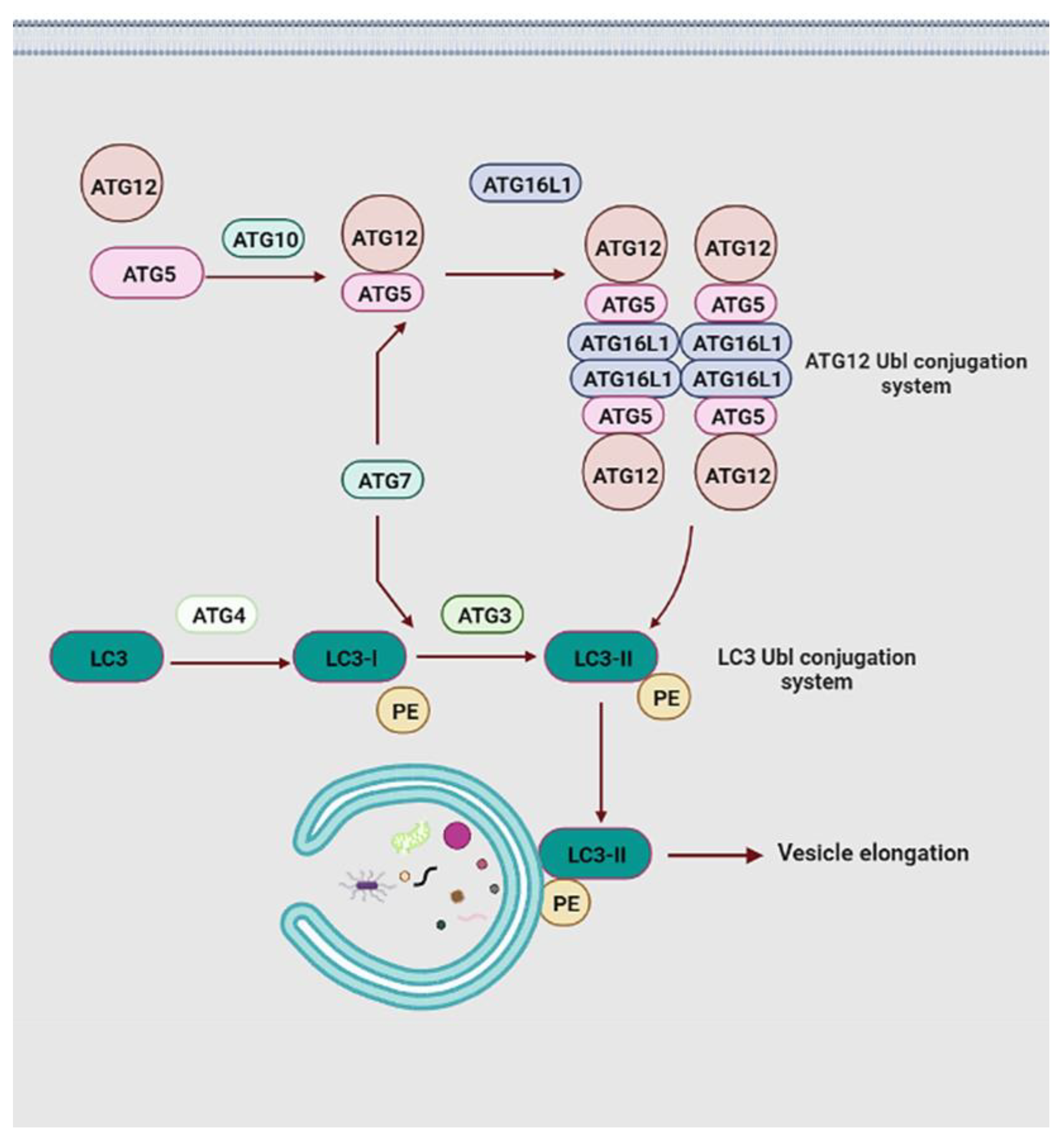
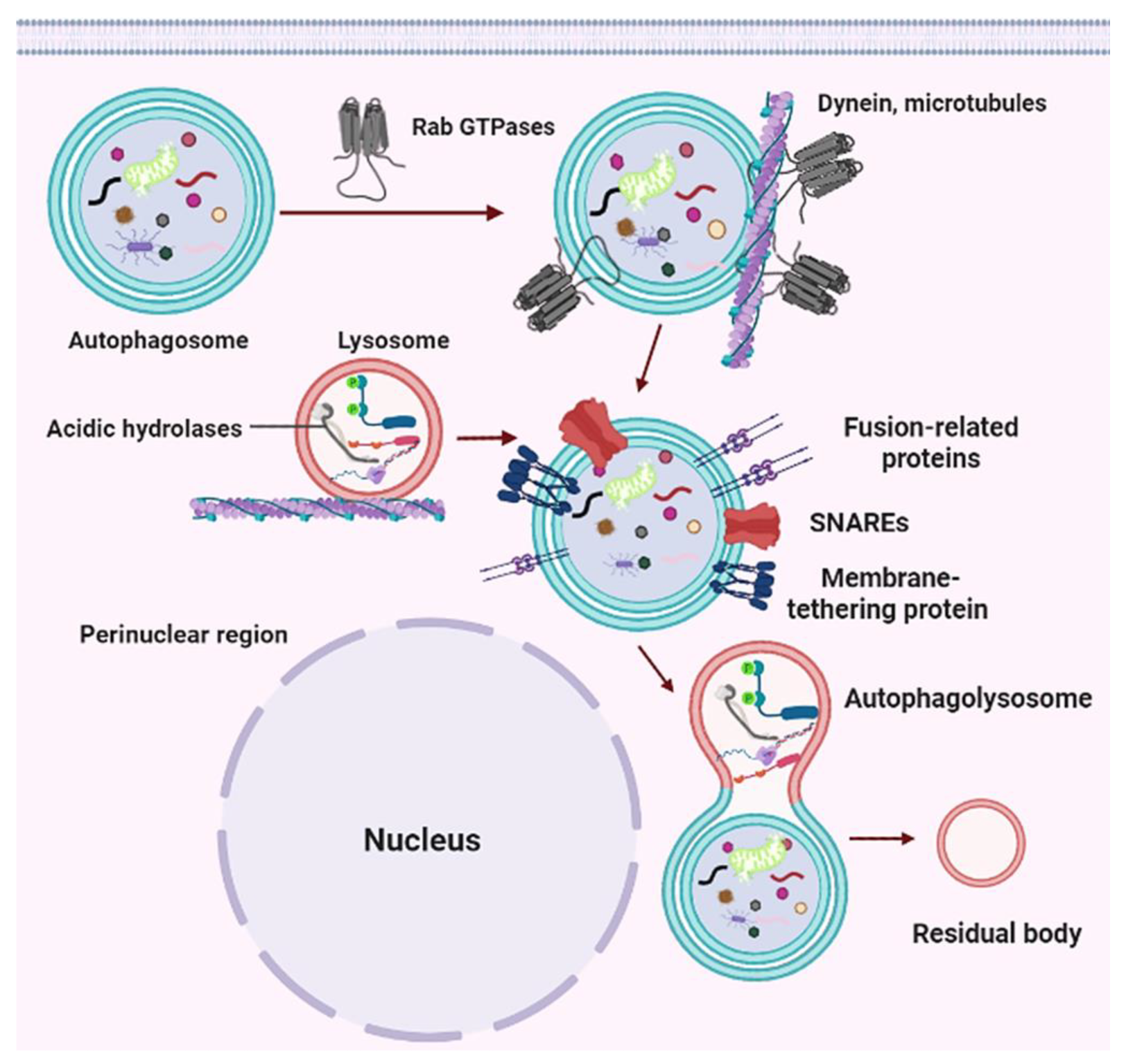
4. The Molecular Machinery of Autophagy
5. The Regulation of Autophagy in Neurons
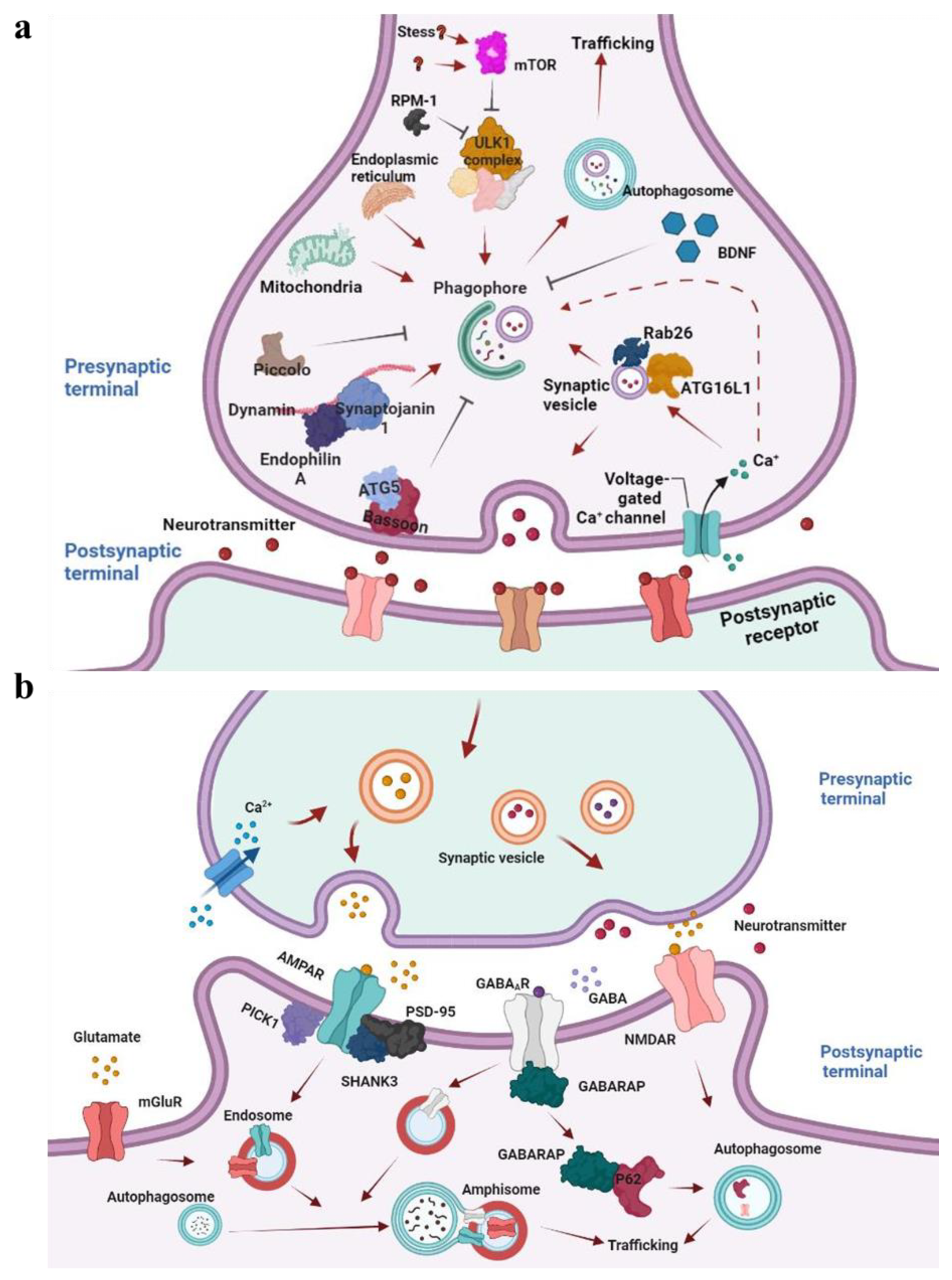
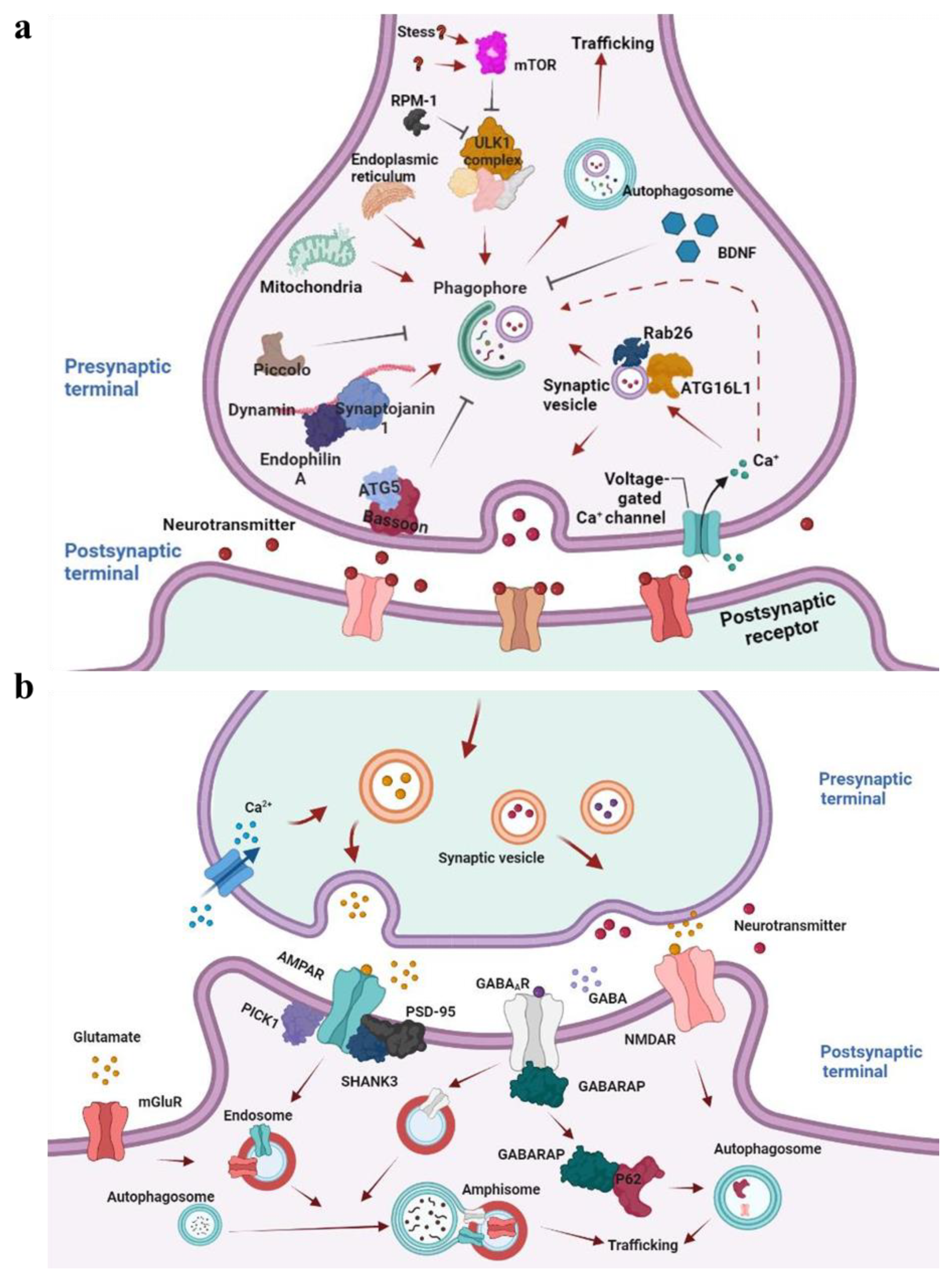
5.1. Autophagy in Axons
5.2. Autophagy in Dendrites
5.3. Autophagy in Soma
5.4. Neuronal Autophagy and Synaptic Plasticities
6. Epilepsy and Autophagy
6.1. Epilepsy
6.2. Neuronal Autophagy and Epilepsy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Models/Patients | Molecular Changes | Pathological Changes | References |
|---|---|---|---|
| Lafora disease mice | Increased Rab5, p62 protein level, decreased LC3-II levels | Generalized stimulus-sensitive tonic-clonic seizures | Puri and Suzuki., 2012 [283]; Criado et al., 2012 [284] |
| Pilocarpine-induced model mice | Increased levels of Beclin 1, ATG5, ATG7 and the ratio of LC3II/I | Epilepsy | Ying et al., 2020 [285] |
| N-ethyl-N-nitrosourea (ENU)-induced mice mutants | Vps15 mutation, decreased LC3-II/LC3-I ratio | Cortical atrophy, dysplasia, and epilepsy | Gstrein et al., 2018 [286] |
| TSC1/PTEN KO mice | mTOR hyperactivation, increased Ulk1 phosphorylation | Epileptogenesis | Yasin et al., 2013 [287] |
| Kainic acid treatment mice | Increased LC3-II levels, elevated ratios of phospho-mTOR/mTOR | Repeated seizures | Shacka et al., 2007 [265] |
| Atg7 KO mice | p62 accumulation | Spontaneous seizures | McMahon et al., 2012 [263] |
| Depdc5 KO mice | Increased mTORC1 signaling | Spontaneous seizures | Yuskaitis et al., 2018 [288] |
| PTEN KO mice + mTOR inhibition | Decreased mTOR activity | Decreased the seizure frequency and death rate | Kwon et al., 2003 [289] |
| Pilocarpine-induced model rats | Increased LC3-II/LC3-I ratio and beclin1 level | Status epilepticus | Cao et al., 2009 [290] |
| Kainic acid treatment rats | mTOR activation | Status epilepticus | Macias et al., 2013 [291] |
| Kainic acid treatment rats + rapamycin | Decreased mTOR activity | Reduced epilepsy | Zeng et al., 2009 [292] |
| Pilocarpine-induced model rats | mTOR activation | Status epilepticus | Buckmaster et al., 2009 [279] |
| Pilocarpine-induced model rats + rapamycin | Decreased mTOR activity | Reduced seizure activity | Huang et al., 2010 [293] |
| Infantile spams/West syndrome rats | mTORC1 pathway overactivation | Spasms, epileptic encephalopathies | Raffo et al., 2011 [294] |
| VPS15 mutation in humans | p62 accumulation | Cortical atrophy, late-onset epilepsy | Gstrein et al., 2018 [286] |
| Beta-propeller protein-associated neurodegeneration patients | De novo mutation in WDR45 | Developmental and epileptic encephalopathies | Carvill et al., 2018 [295] |
| Autosomal dominant lateral temporal epilepsy patients | Reelin mutation | Epilepsy | Dazzo and Nobile., 2022 [260] |
| Vici syndrome patients | EPG5 mutation | Severe seizure disorder, progressive neurodegeneration | Byrne et al., 2016 [296] |
| Pediatric-onset ataxias patients | SNX14 mutation | Progressive cerebellar neurodegeneration, developmental delay, intellectual disability, and seizures | Akizu et al., 2015 [297] |
| Ohtahara syndrome patients | DMXL2 mutation | Intractable seizures and profound developmental disability | Esposito et al., 2019 [298] |
| Children with TBCK p.R126X mutations | Increased LC3-II/LC3-I ratio | Focal and generalized seizures | Ortiz-González et al., 2018 [299] |
| Epilepsy patients | ATG5 gene variant, ATP6V1A/ ATP6AP2 mutation, increased Beclin1 expression | Late-onset epilepsy, temporal lobe epilepsy | Zhang et al., 2021 [300]; Van Damme et al., 2020 [301]; Hirose et al., 2019 [302]; Yang et al., 2022 [303] |
| Focal cortical dysplasia in childhood | mTOR activation, p62 accumulation, TSC1/TSC2 mutation | Epilepsy | Yasin et al., 2013 [287] |
| Human TSC patients | Increased in Ulk1 phosphorylation, p62 accumulation | Cognitive dysfunction, early-onset, intractable epilepsy | McMahon et al., 2012 [263] |
| Hippocampal neuronal culture model of acquired epilepsy | Elevated LC3-II/LC3-I ratio | Acquired epilepsy | Xie et al., 2020 [269] |
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pohl, C.; Dikic, I. Cellular quality control by the ubiquitin-proteasome system and autophagy. Science 2019, 366, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Nakatogawa, H. Mechanisms governing autophagosome biogenesis. Nat. Rev. Mol. Cell Biol. 2020, 21, 439–458. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Biological functions of autophagy genes: A disease perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef]
- Luo, L. Architectures of neuronal circuits. Science 2021, 373, eabg7285. [Google Scholar] [CrossRef]
- Li, S.; Sheng, Z.H. Energy matters: Presynaptic metabolism and the maintenance of synaptic transmission. Nat. Rev. Neurosci. 2022, 23, 4–22. [Google Scholar] [CrossRef]
- Decet, M.; Verstreken, P. Presynaptic autophagy and the connection with neurotransmission. Front. Cell Dev. Biol. 2021, 9, 790721. [Google Scholar] [CrossRef]
- Yamamoto, A.; Yue, Z. Autophagy and its normal and pathogenic states in the brain. Annu Rev. Neurosci. 2014, 37, 55–78. [Google Scholar] [CrossRef]
- Fleming, A.; Rubinsztein, D.C. Autophagy in neuronal development and plasticity. Trends Neurosci. 2020, 43, 767–779. [Google Scholar] [CrossRef]
- Leidal, A.M.; Levine, B.; Debnath, J. Autophagy and the cell biology of age-related disease. Nat. Cell Biol. 2018, 20, 1338–1348. [Google Scholar] [CrossRef]
- Lv, M.; Ma, Q. Autophagy and epilepsy. Adv. Exp. Med. Biol 2020, 1207, 163–169. [Google Scholar]
- Bejarano, E.; Rodríguez-Navarro, J.A. Autophagy and amino acid metabolism in the brain: Implications for epilepsy. Amino Acids 2015, 47, 2113–2126. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.L., Jr. Cellular differentiation in the kidneys of newborn mice studies with the electron microscope. J. Biophys. Biochem. Cytol. 1957, 3, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Ashford, T.P.; Porter, K.R. Cytoplasmic components in hepatic cell lysosomes. J. Cell Biol. 1962, 12, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Hruban, Z.; Spargo, B.; Swift, H.; Wissler, R.W.; Kleinfeld, R.G. Focal cytoplasmic degradation. Am. J. Pathol. 1963, 42, 657–683. [Google Scholar] [PubMed]
- De Duve, C. The lysosome. Sci. Am. 1963, 208, 64–72. [Google Scholar] [CrossRef] [PubMed]
- De Duve, C.; Wattiaux, R. Functions of lysosomes. Annu. Rev. Physiol. 1966, 28, 435–492. [Google Scholar] [CrossRef]
- Deter, R.L.; Baudhuin, P.; De Duve, C. Participation of lysosomes in cellular autophagy induced in rat liver by glucagon. J. Cell Biol. 1967, 35, C11–C16. [Google Scholar] [CrossRef]
- Clarke, P.G. Developmental cell death: Morphological diversity and multiple mechanisms. Anat. Embryol. 1990, 181, 195–213. [Google Scholar] [CrossRef]
- Takeshige, K.; Baba, M.; Tsuboi, S.; Noda, T.; Ohsumi, Y. Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J. Cell Biol. 1992, 119, 301–311. [Google Scholar] [CrossRef]
- Sukada, M.; Ohsumi, Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 1993, 333, 169–174. [Google Scholar] [CrossRef]
- Thumm, M.; Egner, R.; Koch, B.; Schlumpberger, M.; Straub, M.; Veenhuis, M.; Wolf, D.H. Isolation of autophagocytosis mutants of Saccharomyces cerevisiae. FEBS Lett. 1994, 349, 275–280. [Google Scholar] [CrossRef]
- Sakai, Y.; Koller, A.; Rangell, L.K.; Keller, G.A.; Subramani, S. Peroxisome degradation by microautophagy in Pichia pastoris: Identification of specific steps and morphological intermediates. J. Cell Biol. 1998, 141, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Cregg, J.M.; Dunn, W.A., Jr.; Emr, S.D.; Sakai, Y.; Sandoval, I.V.; Sibirny, A.; Subramani, S.; Thumm, M.; Veenhuis, M.; et al. A unified nomenclature for yeast autophagy-related genes. Dev. Cell 2003, 5, 539–545. [Google Scholar] [CrossRef]
- Funakoshi, T.; Matsuura, A.; Noda, T.; Ohsumi, Y. Analyses of APG13 gene involved in autophagy in yeast, Saccharomyces cerevisiae. Gene 1997, 192, 207–213. [Google Scholar] [CrossRef]
- Mizushima, N.; Noda, T.; Yoshimori, T.; Tanaka, Y.; Ishii, T.; George, M.D.; Klionsky, D.J.; Ohsumi, M.; Ohsumi, Y. A protein conjugation system essential for autophagy. Nature 1998, 395, 395–398. [Google Scholar] [CrossRef]
- Mizushima, N.; Sugita, H.; Yoshimori, T.; Ohsumi, Y. A new protein conjugation system in human. The counterpart of the yeast Apg12p conjugation system essential for autophagy. J. Biol. Chem. 1998, 273, 33889–33892. [Google Scholar] [CrossRef]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef]
- Mizushima, N.; Yamamoto, A.; Hatano, M.; Kobayashi, Y.; Kabeya, Y.; Suzuki, K.; Tokuhisa, T.; Ohsumi, Y.; Yoshimori, T. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J. Cell Biol. 2001, 152, 657–668. [Google Scholar] [CrossRef]
- Petiot, A.; Ogier-Denis, E.; Blommaart, E.F.; Meijer, A.J.; Codogno, P. Distinct classes of phosphatidylinositol 3’-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J. Biol. Chem. 2000, 275, 992–998. [Google Scholar] [CrossRef]
- Ravikumar, B.; Duden, R.; Rubinsztein, D.C. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet. 2002, 11, 1107–1117. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Vacher, C.; Berger, Z.; Davies, J.E.; Luo, S.; Oroz, L.G.; Scaravilli, F.; Easton, D.F.; Duden, R.; O’Kane, C.J.; et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 2004, 36, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. Methods for monitoring autophagy. Int. J. Biochem. Cell Biol. 2004, 36, 2491–2502. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Kuma, A. Autophagosomes in GFP-LC3 transgenic mice. Methods Mol. Biol. 2008, 445, 119–124. [Google Scholar] [PubMed]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef]
- Gutierrez, M.G.; Master, S.S.; Singh, S.B.; Taylor, G.A.; Colombo, M.I.; Deretic, V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 2004, 119, 753–766. [Google Scholar] [CrossRef]
- Paludan, C.; Schmid, D.; Landthaler, M.; Vockerodt, M.; Kube, D.; Tuschl, T.; Münz, C. Endogenous MHC Class II processing of a viral nuclear antigen after autophagy. Science 2005, 307, 593–596. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef]
- Lemasters, J.J. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 2005, 8, 3–5. [Google Scholar] [CrossRef]
- Kim, I.; Rodriguez-Enriquez, S.; Lemasters, J.J. Selective degradation of mitochondria by mitophagy. Arch. Biochem. Biophys. 2007, 462, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Bernales, S.; Schuck, S.; Walter, P. ER-phagy: Selective autophagy of the endoplasmic reticulum. Autophagy 2007, 3, 285–287. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.E.; Hayashi, Y.K.; Bonne, G.; Arimura, T.; Noguchi, S.; Nonaka, I.; Nishino, I. Autophagic degradation of nuclear components in mammalian cells. Autophagy 2009, 5, 795–804. [Google Scholar] [CrossRef] [PubMed]
- Tuttle, D.L.; Lewin, A.S.; Dunn, W.A., Jr. Selective autophagy of peroxisomes in methylotrophic yeasts. Eur J. Cell Biol. 1993, 60, 283–290. [Google Scholar]
- Tuttle, D.L.; Dunn, W.A., Jr. Divergent modes of autophagy in the methylotrophic yeast Pichia pastoris. J Cell Sci 1995, 108, 25–35. [Google Scholar] [CrossRef]
- Dunn, W.A., Jr.; Cregg, J.M.; Kiel, J.A.; van der Klei, I.J.; Oku, M.; Sakai, Y.; Sibirny, A.A.; Stasyk, O.V.; Veenhuis, M. Pexophagy: The selective autophagy of peroxisomes. Autophagy 2005, 1, 75–83. [Google Scholar] [CrossRef]
- Maejima, I.; Takahashi, A.; Omori, H.; Kimura, T.; Takabatake, Y.; Saitoh, T.; Yamamoto, A.; Hamasaki, M.; Noda, T.; Isaka, Y.; et al. Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. EMBO J. 2013, 32, 2336–2347. [Google Scholar] [CrossRef]
- Kraft, C.; Deplazes, A.; Sohrmann, M.; Peter, M. Mature ribosomes are selectively degraded upon starvation by an autophagy pathway requiring the Ubp3p/Bre5p ubiquitin protease. Nat. Cell Biol 2008, 10, 602–610. [Google Scholar] [CrossRef]
- Fujioka, Y.; Alam, J.M.; Noshiro, D.; Mouri, K.; Ando, T.; Okada, Y.; May, A.I.; Knorr, R.L.; Suzuki, K.; Ohsumi, Y.; et al. Phase separation organizes the site of autophagosome formation. Nature 2020, 578, 301–305. [Google Scholar] [CrossRef]
- Ponpuak, M.; Mandell, M.A.; Kimura, T.; Chauhan, S.; Cleyrat, C.; Deretic, V. Secretory autophagy. Curr. Opin. Cell Biol. 2015, 35, 106–116. [Google Scholar] [CrossRef]
- Codogno, P.; Mehrpour, M.; Proikas-Cezanne, T. Canonical and non-canonical autophagy: Variations on a common theme of self-eating? Nat. Rev. Mol. Cell Biol 2011, 13, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Noda, N.N.; Mizushima, N. Atg101: Not Just an Accessory Subunit in the Autophagy-initiation Complex. Cell Struct. Funct. 2016, 41, 13–20. [Google Scholar] [CrossRef]
- Ohsumi, Y. Historical landmarks of autophagy research. Cell Res. 2014, 24, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Melia, T.J.; Lystad, A.H.; Simonsen, A. Autophagosome biogenesis: From membrane growth to closure. J. Cell Biol. 2020, 219, e202002085. [Google Scholar] [CrossRef]
- Nguyen, J.A.; Yates, R.M. Better Together: Current Insights Into Phagosome-Lysosome Fusion. Front. Immunol 2021, 12, 636078. [Google Scholar] [CrossRef]
- Leon, C.; Bouchecareilh, M. The Autophagy Pathway: A Critical Route in the Disposal of Alpha 1-Antitrypsin Aggregates That Holds Many Mysteries. Int J. Mol. Sci. 2021, 22, 1875. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Massey, A.C.; Cuervo, A.M. Lysosome membrane lipid microdomains: Novel regulators of chaperone-mediated autophagy. EMBO J. 2006, 25, 3921–3933. [Google Scholar] [CrossRef]
- Massey, A.C.; Kaushik, S.; Sovak, G.; Kiffin, R.; Cuervo, A.M. Consequences of the selective blockage of chaperone-mediated autophagy. Proc. Natl. Acad. Sci. USA 2006, 103, 5805–5810. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Mau, T.; O’Brien, M.; Garg, S.; Yung, R. Impaired autophagy activity is linked to elevated ER-stress and inflammation in aging adipose tissue. Aging (Albany NY) 2016, 8, 2525–2537. [Google Scholar] [CrossRef]
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2018, 14, 207–215. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef] [PubMed]
- Stolz, A.; Ernst, A.; Dikic, I. Cargo recognition and trafficking in selective autophagy. Nat. Cell Biol 2014, 16, 495–501. [Google Scholar] [CrossRef]
- Chiang, H.L.; Terlecky, S.R.; Plant, C.P.; Dice, J.F. A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science 1989, 246, 382–385. [Google Scholar] [CrossRef] [PubMed]
- Tekirdag, K.; Cuervo, A.M. Chaperone-mediated autophagy and endosomal microautophagy: Joint by a chaperone. J. Biol. Chem. 2018, 293, 5414–5424. [Google Scholar] [CrossRef]
- Klionsky, D.J. Autophagy: From phenomenology to molecular understanding in less than a decade. Nat. Rev. Mol. Cell Biol. 2007, 8, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef] [PubMed]
- Ohsumi, Y. Molecular dissection of autophagy: Two ubiquitin-like systems. Nat. Rev. Mol. Cell Biol. 2001, 2, 211–216. [Google Scholar] [CrossRef]
- Kunz, J.B.; Schwarz, H.; Mayer, A. Determination of four sequential stages during microautophagy in vitro. J. Biol. Chem. 2004, 279, 9987–9996. [Google Scholar] [CrossRef]
- Li, W.; He, P.; Huang, Y.; Li, Y.F.; Lu, J.; Li, M.; Kurihara, H.; Luo, Z.; Meng, T.; Onishi, M.; et al. Selective autophagy of intracellular organelles: Recent research advances. Theranostics 2021, 11, 222–256. [Google Scholar] [CrossRef]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R.; et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011, 331, 456–461. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, H.; Zhang, D.; Luo, W.; Liu, R.; Xu, D.; Diao, L.; Liao, L.; Liu, Z. Phosphorylation of ULK1 affects autophagosome fusion and links chaperone-mediated autophagy to macroautophagy. Nat. Commun. 2018, 9, 3492. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, N.; Sasaki, T.; Iemura, S.; Natsume, T.; Hara, T.; Mizushima, N. Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy 2009, 5, 973–979. [Google Scholar] [CrossRef]
- Hara, T.; Takamura, A.; Kishi, C.; Iemura, S.; Natsume, T.; Guan, J.L.; Mizushima, N. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J. Cell Biol. 2008, 181, 497–510. [Google Scholar] [CrossRef]
- Zhou, C.; Ma, K.; Gao, R.; Mu, C.; Chen, L.; Liu, Q.; Luo, Q.; Feng, D.; Zhu, Y.; Chen, Q. Regulation of mATG9 trafficking by Src- and ULK1-mediated phosphorylation in basal and starvation-induced autophagy. Cell Res. 2017, 27, 184–201. [Google Scholar] [CrossRef]
- Ma, B.; Cao, W.; Li, W.; Gao, C.; Qi, Z.; Zhao, Y.; Du, J.; Xue, H.; Peng, J.; Wen, J.; et al. Dapper1 promotes autophagy by enhancing the Beclin1-Vps34-Atg14L complex formation. Cell Res. 2014, 24, 912–924. [Google Scholar] [CrossRef]
- Sawa-Makarska, J.; Baumann, V.; Coudevylle, N.; von Bülow, S.; Nogellova, V.; Abert, C.; Schuschnig, M.; Graef, M.; Hummer, G.; Martens, S. Reconstitution of autophagosome nucleation defines Atg9 vesicles as seeds for membrane formation. Science 2020, 369, eaaz7714. [Google Scholar] [CrossRef]
- Reggiori, F.; Shintani, T.; Nair, U.; Klionsky, D.J. Atg9 cycles between mitochondria and the pre-autophagosomal structure in yeasts. Autophagy 2005, 1, 101–109. [Google Scholar] [CrossRef]
- Valverde, D.P.; Yu, S.; Boggavarapu, V.; Kumar, N.; Lees, J.A.; Walz, T.; Reinisch, K.M.; Melia, T.J. ATG2 transports lipids to promote autophagosome biogenesis. J. Cell Biol. 2019, 218, 1787–1798. [Google Scholar] [CrossRef] [PubMed]
- Krick, R.; Henke, S.; Tolstrup, J.; Thumm, M. Dissecting the localization and function of Atg18, Atg21 and Ygr223c. Autophagy 2008, 4, 896–910. [Google Scholar] [CrossRef] [PubMed]
- Hill, S.M.; Wrobel, L.; Rubinsztein, D.C. Post-translational modifications of Beclin 1 provide multiple strategies for autophagy regulation. Cell Death Differ. 2019, 26, 617–629. [Google Scholar] [CrossRef]
- Zhong, Y.; Wang, Q.J.; Li, X.; Yan, Y.; Backer, J.M.; Chait, B.T.; Heintz, N.; Yue, Z. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat. Cell Biol. 2009, 11, 468–476. [Google Scholar] [CrossRef]
- Russell, R.C.; Tian, Y.; Yuan, H.; Park, H.W.; Chang, Y.Y.; Kim, J.; Kim, H.; Neufeld, T.P.; Dillin, A.; Guan, K.L. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 2013, 15, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Dooley, H.C.; Razi, M.; Polson, H.E.; Girardin, S.E.; Wilson, M.I.; Tooze, S.A. WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12-5-16L1. Mol. Cell 2014, 55, 238–252. [Google Scholar] [CrossRef]
- Diao, J.; Liu, R.; Rong, Y.; Zhao, M.; Zhang, J.; Lai, Y.; Zhou, Q.; Wilz, L.M.; Li, J.; Vivona, S.; et al. ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature 2015, 520, 563–566. [Google Scholar] [CrossRef]
- Ranaghan, M.J.; Durney, M.A.; Mesleh, M.F.; McCarren, P.R.; Garvie, C.W.; Daniels, D.S.; Carey, K.L.; Skepner, A.P.; Levine, B.; Perez, J.R. The Autophagy-Related Beclin-1 Protein Requires the Coiled-Coil and BARA Domains To Form a Homodimer with Submicromolar Affinity. Biochemistry 2017, 56, 6639–6651. [Google Scholar] [CrossRef]
- Fernández, Á.F.; Sebti, S.; Wei, Y.; Zou, Z.; Shi, M.; McMillan, K.L.; He, C.; Ting, T.; Liu, Y.; Chiang, W.C.; et al. Disruption of the beclin 1-BCL2 autophagy regulatory complex promotes longevity in mice. Nature 2018, 558, 136–140. [Google Scholar] [CrossRef]
- Houtman, J.; Freitag, K.; Gimber, N.; Schmoranzer, J.; Heppner, F.L.; Jendrach, M. Beclin1-driven autophagy modulates the inflammatory response of microglia via NLRP3. EMBO J. 2019, 38, e99430. [Google Scholar] [CrossRef]
- Huang, X.; Li, Y.; Shou, L.; Li, L.; Chen, Z.; Ye, X.; Qian, W. The molecular mechanisms underlying BCR/ABL degradation in chronic myeloid leukemia cells promoted by Beclin1-mediated autophagy. Cancer Manag. Res. 2019, 11, 5197–5208. [Google Scholar] [CrossRef]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef]
- Lee, E.F.; Perugini, M.A.; Pettikiriarachchi, A.; Evangelista, M.; Keizer, D.W.; Yao, S.; Fairlie, W.D. The BECN1 N-terminal domain is intrinsically disordered. Autophagy 2016, 12, 460–471. [Google Scholar] [CrossRef] [PubMed]
- Itakura, E.; Kishi, C.; Inoue, K.; Mizushima, N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol. Biol. Cell 2008, 19, 5360–5372. [Google Scholar] [CrossRef]
- Kim, Y.M.; Jung, C.H.; Seo, M.; Kim, E.K.; Park, J.M.; Bae, S.S.; Kim, D.H. mTORC1 phosphorylates UVRAG to negatively regulate autophagosome and endosome maturation. Mol. Cell 2015, 57, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Zhang, J.; Fan, W.; Wong, K.N.; Ding, X.; Chen, S.; Zhong, Q. The RUN domain of rubicon is important for hVps34 binding, lipid kinase inhibition, and autophagy suppression. J. Biol. Chem. 2011, 286, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, K.; Saitoh, T.; Tabata, K.; Omori, H.; Satoh, T.; Kurotori, N.; Maejima, I.; Shirahama-Noda, K.; Ichimura, T.; Isobe, T.; et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat. Cell Biol. 2009, 11, 385–396. [Google Scholar] [CrossRef]
- Geng, J.; Klionsky, D.J. The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. ‘Protein modifications: Beyond the usual suspects’ review series. EMBO Rep. 2008, 9, 859–864. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Noda, N.N.; Yamamoto, H.; Shima, T.; Kumeta, H.; Kobashigawa, Y.; Akada, R.; Ohsumi, Y.; Agaki, F. Structural insights into Atg10-mediated formation of the autophagy-essential Atg12-Atg5 conjugate. Structure 2012, 20, 1244–1254. [Google Scholar] [CrossRef]
- Fujita, N.; Itoh, T.; Omori, H.; Fukuda, M.; Noda, T.; Yoshimori, T. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol. Biol. Cell 2008, 19, 2092–2100. [Google Scholar] [CrossRef]
- Lystad, A.H.; Carlsson, S.R.; de la Ballina, L.R.; Kauffman, K.J.; Nag, S.; Yoshimori, T.; Melia, T.J.; Simonsen, A. Distinct functions of ATG16L1 isoforms in membrane binding and LC3B lipidation in autophagy-related processes. Nat. Cell Biol. 2019, 21, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Hanada, T.; Ohsumi, Y. Structure-function relationship of Atg12, a ubiquitin-like modifier essential for autophagy. Autophagy 2005, 1, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Kharaziha, P.; Panaretakis, T. Dynamics of Atg5-Atg12-Atg16L1 Aggregation and Deaggregation. Methods Enzymol. 2017, 587, 247–255. [Google Scholar]
- Kabeya, Y.; Mizushima, N.; Yamamoto, A.; Oshitani-Okamoto, S.; Ohsumi, Y.; Yoshimori, T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J. Cell Sci. 2004, 117, 2805–2812. [Google Scholar] [CrossRef]
- Lystad, A.H.; Carlsson, S.R.; Simonsen, A. Toward the function of mammalian ATG12-ATG5-ATG16L1 complex in autophagy and related processes. Autophagy 2019, 15, 1485–1486. [Google Scholar] [CrossRef] [PubMed]
- Jatana, N.; Ascher, D.B.; Pires, D.E.V.; Gokhale, R.S.; Thukral, L. Human LC3 and GABARAP subfamily members achieve functional specificity via specific structural modulations. Autophagy 2020, 16, 239–255. [Google Scholar] [CrossRef] [PubMed]
- Weidberg, H.; Shvets, E.; Shpilka, T.; Shimron, F.; Shinder, V.; Elazar, Z. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J. 2010, 29, 1792–1802. [Google Scholar] [CrossRef]
- Bjørkøy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Overvatn, A.; Stenmark, H.; Johansen, T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef]
- Thurston, T.L.; Ryzhakov, G.; Bloor, S.; von Muhlinen, N.; Randow, F. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat. Immunol. 2009, 10, 1215–1221. [Google Scholar] [CrossRef]
- Lin, X.; Li, S.; Zhao, Y.; Ma, X.; Zhang, K.; He, X.; Wang, Z. Interaction domains of p62: A bridge between p62 and selective autophagy. DNA Cell Biol 2013, 32, 220–227. [Google Scholar] [CrossRef]
- Lamark, T.; Svenning, S.; Johansen, T. Regulation of selective autophagy: The p62/SQSTM1 paradigm. Essays Biochem. 2017, 61, 609–624. [Google Scholar] [PubMed]
- Zhao, Y.G.; Codogno, P.; Zhang, H. Machinery, regulation and pathophysiological implications of autophagosome maturation. Nat. Rev. Mol. Cell Biol. 2021, 22, 733–750. [Google Scholar] [CrossRef]
- Jahreiss, L.; Menzies, F.M.; Rubinsztein, D.C. The itinerary of autophagosomes: From peripheral formation to kiss-and-run fusion with lysosomes. Traffic 2008, 9, 574–587. [Google Scholar] [CrossRef] [PubMed]
- Stenmark, H. Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol. 2009, 10, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, M.G.; Munafó, D.B.; Berón, W.; Colombo, M.I. Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J. Cell Sci 2004, 117, 2687–2697. [Google Scholar] [CrossRef] [PubMed]
- Hegedűs, K.; Takáts, S.; Boda, A.; Jipa, A.; Nagy, P.; Varga, K.; Kovács, A.L.; Juhász, G. The Ccz1-Mon1-Rab7 module and Rab5 control distinct steps of autophagy. Mol. Biol. Cell 2016, 27, 3132–3142. [Google Scholar] [CrossRef]
- Takáts, S.; Boda, A.; Csizmadia, T.; Juhász, G. Small GTPases controlling autophagy-related membrane traffic in yeast and metazoans. Small GTPases 2018, 9, 465–471. [Google Scholar] [CrossRef]
- Kimura, S.; Noda, T.; Yoshimori, T. Dynein-dependent movement of autophagosomes mediates efficient encounters with lysosomes. Cell Struct. Funct. 2008, 33, 109–122. [Google Scholar] [CrossRef]
- Pu, J.; Guardia, C.M.; Keren-Kaplan, T.; Bonifacino, J.S. Mechanisms and functions of lysosome positioning. J. Cell Sci. 2016, 129, 4329–4339. [Google Scholar] [CrossRef]
- Cantalupo, G.; Alifano, P.; Roberti, V.; Bruni, C.B.; Bucci, C. Rab-interacting lysosomal protein (RILP): The Rab7 effector required for transport to lysosomes. EMBO J. 2001, 20, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Rocha, N.; Kuijl, C.; van der Kant, R.; Janssen, L.; Houben, D.; Janssen, H.; Zwart, W.; Neefjes, J. Cholesterol sensor ORP1L contacts the ER protein VAP to control Rab7-RILP-p150 Glued and late endosome positioning. J. Cell Biol. 2009, 185, 1209–1225. [Google Scholar] [CrossRef] [PubMed]
- Pankiv, S.; Alemu, E.A.; Brech, A.; Bruun, J.A.; Lamark, T.; Overvatn, A.; Bjørkøy, G.; Johansen, T. FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport. J. Cell Biol. 2010, 188, 253–269. [Google Scholar] [CrossRef]
- Cardoso, C.M.; Groth-Pedersen, L.; Høyer-Hansen, M.; Kirkegaard, T.; Corcelle, E.; Andersen, J.S.; Jäättelä, M.; Nylandsted, J. Depletion of kinesin 5B affects lysosomal distribution and stability and induces peri-nuclear accumulation of autophagosomes in cancer cells. PLoS ONE 2009, 4, e4424. [Google Scholar] [CrossRef] [PubMed]
- Korolchuk, V.I.; Saiki, S.; Lichtenberg, M.; Siddiqi, F.H.; Roberts, E.A.; Imarisio, S.; Jahreiss, L.; Sarkar, S.; Futter, M.; Menzies, F.M.; et al. Lysosomal positioning coordinates cellular nutrient responses. Nat. Cell Biol. 2011, 13, 453–460. [Google Scholar] [CrossRef]
- Fu, M.M.; Nirschl, J.J.; Holzbaur, E.L.F. LC3 binding to the scaffolding protein JIP1 regulates processive dynein-driven transport of autophagosomes. Dev. Cell 2014, 29, 577–590. [Google Scholar] [CrossRef]
- Jiang, P.; Nishimura, T.; Sakamaki, Y.; Itakura, E.; Hatta, T.; Natsume, T.; Mizushima, N. The HOPS complex mediates autophagosome-lysosome fusion through interaction with syntaxin 17. Mol. Biol. Cell 2014, 25, 1327–1337. [Google Scholar] [CrossRef]
- Reggiori, F.; Ungermann, C. Autophagosome Maturation and Fusion. J. Mol. Biol. 2017, 429, 486–496. [Google Scholar] [CrossRef]
- Wickner, W.; Schekman, R. Membrane fusion. Nat. Struct. Mol. Biol. 2008, 15, 658–664. [Google Scholar] [CrossRef]
- Bröcker, C.; Kuhlee, A.; Gatsogiannis, C.; Balderhaar, H.J.; Hönscher, C.; Engelbrecht-Vandré, S.; Ungermann, C.; Raunser, S. Molecular architecture of the multisubunit homotypic fusion and vacuole protein sorting (HOPS) tethering complex. Proc. Natl. Acad. Sci. USA 2012, 109, 1991–1996. [Google Scholar] [CrossRef]
- Balderhaar, H.J.; Ungermann, C. CORVET and HOPS tethering complexes - coordinators of endosome and lysosome fusion. J. Cell Sci. 2013, 126, 1307–1316. [Google Scholar] [CrossRef] [PubMed]
- Pols, M.S.; ten Brink, C.; Gosavi, P.; Oorschot, V.; Klumperman, J. The HOPS proteins hVps41 and hVps39 are required for homotypic and heterotypic late endosome fusion. Traffic 2013, 14, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Akbar, M.A.; Ray, S.; Krämer, H. The SM protein Car/Vps33A regulates SNARE-mediated trafficking to lysosomes and lysosome-related organelles. Mol. Biol. Cell 2009, 20, 1705–1714. [Google Scholar] [CrossRef]
- Takáts, S.; Pircs, K.; Nagy, P.; Varga, Á.; Kárpáti, M.; Hegedűs, K.; Kramer, H.; Kovács, A.L.; Sass, M.; Juhász, G. Interaction of the HOPS complex with Syntaxin 17 mediates autophagosome clearance in Drosophila. Mol. Biol. Cell 2014, 25, 1338–1354. [Google Scholar] [CrossRef]
- Fujita, N.; Huang, W.; Lin, T.H.; Groulx, J.F.; Jean, S.; Nguyen, J.; Kuchitsu, Y.; Koyama-Honda, I.; Mizushima, N.; Fukuda, M.; et al. Genetic screen in Drosophila muscle identifies autophagy-mediated T-tubule remodeling and a Rab2 role in autophagy. Elife 2017, 6, e23367. [Google Scholar] [CrossRef] [PubMed]
- Khatter, D.; Raina, V.B.; Dwivedi, D.; Sindhwani, A.; Bahl, S.; Sharma, M. The small GTPase Arl8b regulates assembly of the mammalian HOPS complex on lysosomes. J. Cell Sci. 2015, 128, 1746–1761. [Google Scholar] [CrossRef] [PubMed]
- Jia, R.; Guardia, C.M.; Pu, J.; Chen, Y.; Bonifacino, J.S. BORC coordinates encounter and fusion of lysosomes with autophagosomes. Autophagy 2017, 13, 1648–1663. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Zhao, Y.G.; Wang, X.; Xu, L.; Miao, L.; Feng, D.; Chen, Q.; Kovács, A.L.; Fan, D.; Zhang, H. Mice deficient in Epg5 exhibit selective neuronal vulnerability to degeneration. J. Cell Biol. 2013, 200, 731–741. [Google Scholar] [CrossRef]
- Ogawa, M.; Yoshikawa, Y.; Kobayashi, T.; Mimuro, H.; Fukumatsu, M.; Kiga, K.; Piao, Z.; Ashida, H.; Yoshida, M.; Kakuta, S.; et al. A Tecpr1-dependent selective autophagy pathway targets bacterial pathogens. Cell Host Microbe 2011, 9, 376–389. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, L.; Lak, B.; Li, J.; Jokitalo, E.; Wang, Y. GRASP55 Senses Glucose Deprivation through O-GlcNAcylation to Promote Autophagosome-Lysosome Fusion. Dev. Cell 2018, 45, 245–261.e6. [Google Scholar] [CrossRef]
- Terawaki, S.; Camosseto, V.; Prete, F.; Wenger, T.; Papadopoulos, A.; Rondeau, C.; Combes, A.; Rodriguez Rodrigues, C.; Vu Manh, T.P.; Fallet, M.; et al. RUN and FYVE domain-containing protein 4 enhances autophagy and lysosome tethering in response to Interleukin-4. J. Cell Biol. 2015, 210, 1133–1152. [Google Scholar] [CrossRef] [PubMed]
- Stroupe, C.; Collins, K.M.; Fratti, R.A.; Wickner, W. Purification of active HOPS complex reveals its affinities for phosphoinositides and the SNARE Vam7p. EMBO J. 2006, 25, 1579–1589. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Teng, J.; Chen, J. New insights regarding SNARE proteins in autophagosome-lysosome fusion. Autophagy 2021, 17, 2680–2688. [Google Scholar] [CrossRef]
- Südhof, T.C.; Rothman, J.E. Membrane fusion: Grappling with SNARE and SM proteins. Science 2009, 323, 474–477. [Google Scholar] [CrossRef]
- Baker, R.W.; Hughson, F.M. Chaperoning SNARE assembly and disassembly. Nat. Rev. Mol. Cell Biol. 2016, 17, 465–479. [Google Scholar] [CrossRef]
- Lőrincz, P.; Juhász, G. Autophagosome-Lysosome Fusion. J. Mol. Biol. 2020, 432, 2462–2482. [Google Scholar] [CrossRef] [PubMed]
- Dilcher, M.; Köhler, B.; von Mollard, G.F. Genetic interactions with the yeast Q-SNARE VTI1 reveal novel functions for the R-SNARE YKT6. J. Biol. Chem. 2001, 276, 34537–34544. [Google Scholar] [CrossRef] [PubMed]
- Darsow, T.; Rieder, S.E.; Emr, S.D. A multispecificity syntaxin homologue, Vam3p, essential for autophagic and biosynthetic protein transport to the vacuole. J. Cell Biol. 1997, 138, 517–529. [Google Scholar] [CrossRef]
- Fischer von Mollard, G.; Stevens, T.H. The Saccharomyces cerevisiae v-SNARE Vti1p is required for multiple membrane transport pathways to the vacuole. Mol. Biol. Cell 1999, 10, 1719–1732. [Google Scholar] [CrossRef]
- Ishihara, N.; Hamasaki, M.; Yokota, S.; Suzuki, K.; Kamada, Y.; Kihara, A.; Yoshimori, T.; Noda, T.; Ohsumi, Y. Autophagosome requires specific early Sec proteins for its formation and NSF/SNARE for vacuolar fusion. Mol. Biol. Cell 2001, 12, 3690–3702. [Google Scholar] [CrossRef]
- Saleeb, R.S.; Kavanagh, D.M.; Dun, A.R.; Dalgarno, P.A.; Duncan, R.R. A VPS33A-binding motif on syntaxin 17 controls autophagy completion in mammalian cells. J. Biol Chem. 2019, 294, 4188–4201. [Google Scholar] [CrossRef] [PubMed]
- Itakura, E.; Kishi-Itakura, C.; Mizushima, N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 2012, 151, 1256–1269. [Google Scholar] [CrossRef] [PubMed]
- Takáts, S.; Nagy, P.; Varga, Á.; Pircs, K.; Kárpáti, M.; Varga, K.; Kovács, A.L.; Hegedűs, K.; Juhász, G. Autophagosomal Syntaxin17-dependent lysosomal degradation maintains neuronal function in Drosophila. J. Cell Biol. 2013, 201, 531–539. [Google Scholar] [CrossRef]
- Takáts, S.; Glatz, G.; Szenci, G.; Boda, A.; Horváth, G.V.; Hegedűs, K.; Kovács, A.L.; Juhász, G. Non-canonical role of the SNARE protein Ykt6 in autophagosome-lysosome fusion. PLoS Genet. 2018, 14, e1007359. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T.; Jiang, P.; Nakano, S.; Sakamaki, Y.; Yamamoto, H.; Mizushima, N. Autophagosomal YKT6 is required for fusion with lysosomes independently of syntaxin 17. J. Cell Biol. 2018, 217, 2633–2645. [Google Scholar] [CrossRef]
- Zhao, Y.G.; Zhang, H. Autophagosome maturation: An epic journey from the ER to lysosomes. J. Cell Biol. 2019, 218, 757–770. [Google Scholar] [CrossRef]
- Zhou, Y.; Peskett, T.R.; Landles, C.; Warner, J.B.; Sathasivam, K.; Smith, E.J.; Chen, S.; Wetzel, R.; Lashuel, H.A.; Bates, G.P.; et al. Correlative light and electron microscopy suggests that mutant huntingtin dysregulates the endolysosomal pathway in presymptomatic Huntington’s disease. Acta Neuropathol. Commun. 2021, 9, 70. [Google Scholar] [CrossRef]
- Yoshii, S.R.; Mizushima, N. Monitoring and Measuring Autophagy. Int. J. Mol. Sci. 2017, 18, 1865. [Google Scholar] [CrossRef]
- Zhang, X.J.; Chen, S.; Huang, K.X.; Le, W.D. Why should autophagic flux be assessed? Acta Pharmacol. Sin. 2013, 34, 595–599. [Google Scholar] [CrossRef]
- Ueno, T.; Komatsu, M. Monitoring Autophagy Flux and Activity: Principles and Applications. Bioessays 2020, 42, e2000122. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition)1. Autophagy 2021, 17, 1–382. [Google Scholar] [PubMed]
- Kalil, K.; Dent, E.W. Branch management: Mechanisms of axon branching in the developing vertebrate CNS. Nat. Rev. Neurosci. 2014, 15, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.A. Neuronal autophagy: A housekeeper or a fighter in neuronal cell survival? Exp. Neurobiol. 2012, 21, 1–8. [Google Scholar] [CrossRef]
- Crawley, O.; Grill, B. Autophagy in axonal and presynaptic development. Curr. Opin. Neurobiol. 2021, 69, 139–148. [Google Scholar] [CrossRef]
- Xilouri, M.; Stefanis, L. Autophagy in the central nervvous system: Implications for neurodegenerative disorders. CNS Neurol. Disord. Drug Targets 2010, 9, 701–719. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A.; Cataldo, A.M. Lysosomal system pathways: Genes to neurodegeneration in Alzheimer’s disease. J. Alzheimers Dis. 2006, 9, 277–289. [Google Scholar] [CrossRef]
- Gallo, G. The cytoskeletal and signaling mechanisms of axon collateral branching. Dev. Neurobiol. 2011, 71, 201–220. [Google Scholar] [CrossRef]
- Hernandez, D.; Torres, C.A.; Setlik, W.; Cebrian, C.; Mosharov, E.V.; Tang, G.; Cheng, H.C.; Kholodilov, N.; Yarygina, O.; Burke, R.E.; et al. Regulation of presynaptic neurotransmission by macroautophagy. Neuron 2012, 74, 277–284. [Google Scholar] [CrossRef]
- Boland, B.; Nixon, R.A. Neuronal macroautophagy: From development to degeneration. Mol. Aspects Med. 2006, 27, 503–519. [Google Scholar] [CrossRef]
- Cronin, J.; Dudek, F.E. Chronic seizures and collateral sprouting of dentate mossy fibers after kainic acid treatment in rats. Brain Res. 1988, 474, 181–184. [Google Scholar] [CrossRef]
- Sutula, T.; He, X.X.; Cavazos, J.; Scott, G. Synaptic reorganization in the hippocampus induced by abnormal functional activity. Science 1988, 239, 1147–1150. [Google Scholar] [CrossRef] [PubMed]
- Babb, T.L.; Kupfer, W.R.; Pretorius, J.K.; Crandall, P.H.; Levesque, M.F. Synaptic reorganization by mossy fibers in human epileptic fascia dentata. Neuroscience 1991, 42, 351–363. [Google Scholar] [CrossRef]
- Mello, L.E.; Cavalheiro, E.A.; Tan, A.M.; Kupfer, W.R.; Pretorius, J.K.; Babb, T.L.; Finch, D.M. Circuit mechanisms of seizures in the pilocarpine model of chronic epilepsy: Cell loss and mossy fiber sprouting. Epilepsia 1993, 34, 985–995. [Google Scholar] [CrossRef]
- Desguerre, I.; Hully, M.; Rio, M.; Nabbout, R. Mitochondrial disorders and epilepsy. Rev. Neurol. 2014, 170, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Armijo, J.A.; Shushtarian, M.; Valdizan, E.M.; Cuadrado, A.; de las Cuevas, I.; Adín, J. Ion channels and epilepsy. Curr. Pharm. Des. 2005, 11, 1975–2003. [Google Scholar] [CrossRef] [PubMed]
- Spitzer, N.C. Activity-dependent neurotransmitter respecification. Nat. Rev. Neurosci. 2012, 13, 94–106. [Google Scholar] [CrossRef] [PubMed]
- Faust, T.E.; Gunner, G.; Schafer, D.P. Mechanisms governing activity-dependent synaptic pruning in the developing mammalian CNS. Nat. Rev. Neurosci. 2021, 22, 657–673. [Google Scholar] [CrossRef] [PubMed]
- Hollenbeck, P.J. Products of endocytosis and autophagy are retrieved from axons by regulated retrograde organelle transport. J. Cell Biol. 1993, 121, 305–315. [Google Scholar] [CrossRef]
- Mizushima, N.; Yamamoto, A.; Matsui, M.; Yoshimori, T.; Ohsumi, Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol. Biol. Cell 2004, 15, 1101–1111. [Google Scholar] [CrossRef]
- Stavoe, A.K.; Hill, S.E.; Hall, D.H.; Colón-Ramos, D.A. KIF1A/UNC-104 Transports ATG-9 to Regulate Neurodevelopment and Autophagy at Synapses. Dev. Cell 2016, 38, 171–185. [Google Scholar] [CrossRef]
- Neisch, A.L.; Neufeld, T.P.; Hays, T.S. A STRIPAK complex mediates axonal transport of autophagosomes and dense core vesicles through PP2A regulation. J. Cell Biol. 2017, 216, 441–461. [Google Scholar] [CrossRef] [PubMed]
- Hill, S.E.; Kauffman, K.J.; Krout, M.; Richmond, J.E.; Melia, T.J.; Colón-Ramos, D.A. Maturation and Clearance of Autophagosomes in Neurons Depends on a Specific Cysteine Protease Isoform, ATG-4.2. Dev. Cell 2019, 49, 251–266.e8. [Google Scholar] [CrossRef] [PubMed]
- Stavoe, A.K.H.; Holzbaur, E.L.F. Autophagy in Neurons. Annu. Rev. Cell Dev. Biol. 2019, 35, 477–500. [Google Scholar] [CrossRef]
- Joo, J.H.; Wang, B.; Frankel, E.; Ge, L.; Xu, L.; Iyengar, R.; Li-Harms, X.; Wright, C.; Shaw, T.I.; Lindsten, T.; et al. The Noncanonical Role of ULK/ATG1 in ER-to-Golgi Trafficking Is Essential for Cellular Homeostasis. Mol. Cell 2016, 62, 491–506. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Wang, Q.J.; Holstein, G.R.; Friedrich, V.L., Jr.; Iwata, J.; Kominami, E.; Chait, B.T.; Tanaka, K.; Yue, Z. Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc. Natl. Acad. Sci. USA 2007, 104, 14489–14494. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.C.; Wang, C.; Peng, X.; Gan, B.; Guan, J.L. Neural-specific deletion of FIP200 leads to cerebellar degeneration caused by increased neuronal death and axon degeneration. J. Biol. Chem. 2010, 285, 3499–3509. [Google Scholar] [CrossRef]
- Kuijpers, M.; Kochlamazashvili, G.; Stumpf, A.; Puchkov, D.; Swaminathan, A.; Lucht, M.T.; Krause, E.; Maritzen, T.; Schmitz, D.; Haucke, V. Neuronal Autophagy Regulates Presynaptic Neurotransmission by Controlling the Axonal Endoplasmic Reticulum. Neuron 2021, 109, 299–313.e9. [Google Scholar] [CrossRef]
- Lüningschrör, P.; Binotti, B.; Dombert, B.; Heimann, P.; Perez-Lara, A.; Slotta, C.; Thau-Habermann, N.R.; von Collenberg, C.; Karl, F.; Damme, M.; et al. Plekhg5-regulated autophagy of synaptic vesicles reveals a pathogenic mechanism in motoneuron disease. Nat. Commun. 2017, 8, 678. [Google Scholar] [CrossRef]
- Binotti, B.; Pavlos, N.J.; Riedel, D.; Wenzel, D.; Vorbrüggen, G.; Schalk, A.M.; Kühnel, K.; Boyken, J.; Erck, C.; Martens, H.; et al. The GTPase Rab26 links synaptic vesicles to the autophagy pathway. Elife 2015, 4, e05597. [Google Scholar] [CrossRef]
- Pimentel-Muiños, F.X.; Boada-Romero, E. Selective autophagy against membranous compartments: Canonical and unconventional purposes and mechanisms. Autophagy 2014, 10, 397–407. [Google Scholar] [CrossRef]
- Nikoletopoulou, V.; Tavernarakis, N. Regulation and Roles of Autophagy at Synapses. Trends Cell Biol. 2018, 28, 646–661. [Google Scholar] [CrossRef] [PubMed]
- Verstreken, P.; Koh, T.W.; Schulze, K.L.; Zhai, R.G.; Hiesinger, P.R.; Zhou, Y.; Mehta, S.Q.; Cao, Y.; Roos, J.; Bellen, H.J. Synaptojanin is recruited by endophilin to promote synaptic vesicle uncoating. Neuron 2003, 40, 733–748. [Google Scholar] [CrossRef]
- Soukup, S.F.; Verstreken, P. EndoA/Endophilin-A creates docking stations for autophagic proteins at synapses. Autophagy 2017, 13, 971–972. [Google Scholar] [CrossRef]
- Milosevic, I.; Giovedi, S.; Lou, X.; Raimondi, A.; Collesi, C.; Shen, H.; Paradise, S.; O’Toole, E.; Ferguson, S.; Cremona, O.; et al. Recruitment of endophilin to clathrin-coated pit necks.s is required for efficient vesicle uncoating after fission. Neuron 2011, 72, 587–601. [Google Scholar] [CrossRef]
- Hohendahl, A.; Talledge, N.; Galli, V.; Shen, P.S.; Humbert, F.; De Camilli, P.; Frost, A.; Roux, A. Structural inhibition of dynamin-mediated membrane fission by endophilin. Elife 2017, 6, e26856. [Google Scholar] [CrossRef]
- Ramachandran, R.; Schmid, S.L. The dynamin superfamily. Curr. Biol. 2018, 28, R411–R416. [Google Scholar] [CrossRef] [PubMed]
- Pan, P.Y.; Zhu, J.; Rizvi, A.; Zhu, X.; Tanaka, H.; Dreyfus, C.F. Synaptojanin1 deficiency upregulates basal autophagosome formation in astrocytes. J. Biol. Chem. 2021, 297, 100873. [Google Scholar] [CrossRef] [PubMed]
- George, A.A.; Hayden, S.; Stanton, G.R.; Brockerhoff, S.E. Arf6 and the 5’phosphatase of synaptojanin 1 regulate autophagy in cone photoreceptors. Bioessays 2016, 38 (Suppl. S1), S119–S135. [Google Scholar] [CrossRef]
- Vanhauwaert, R.; Kuenen, S.; Masius, R.; Bademosi, A.; Manetsberger, J.; Schoovaerts, N.; Bounti, L.; Gontcharenko, S.; Swerts, J.; Vilain, S.; et al. The SAC1 domain in synaptojanin is required for autophagosome maturation at presynaptic terminals. EMBO J. 2017, 36, 1392–1411. [Google Scholar] [CrossRef]
- Cao, M.; Park, D.; Wu, Y.; De Camilli, P. Absence of Sac2/INPP5F enhances the phenotype of a Parkinson’s disease mutation of synaptojanin 1. Proc. Natl. Acad. Sci. USA 2020, 117, 12428–12434. [Google Scholar] [CrossRef]
- Murdoch, J.D.; Rostosky, C.M.; Gowrisankaran, S.; Arora, A.S.; Soukup, S.F.; Vidal, R.; Capece, V.; Freytag, S.; Fischer, A.; Verstreken, P.; et al. Endophilin-A Deficiency Induces the Foxo3a-Fbxo32 Network in the Brain and Causes Dysregulation of Autophagy and the Ubiquitin-Proteasome System. Cell Rep. 2016, 17, 1071–1086. [Google Scholar] [CrossRef] [PubMed]
- Cullup, T.; Kho, A.L.; Dionisi-Vici, C.; Brandmeier, B.; Smith, F.; Urry, Z.; Simpson, M.A.; Yau, S.; Bertini, E.; McClelland, V.; et al. Recessive mutations in EPG5 cause Vici syndrome, a multisystem disorder with defective autophagy. Nat. Genet. 2013, 45, 83–87. [Google Scholar] [CrossRef]
- Fraiberg, M.; Tamim-Yecheskel, B.C.; Kokabi, K.; Subic, N.; Heimer, G.; Eck, F.; Nalbach, K.; Behrends, C.; Ben-Zeev, B.; Shatz, O.; et al. Lysosomal targeting of autophagosomes by the TECPR domain of TECPR2. Autophagy 2021, 17, 3096–3108. [Google Scholar] [CrossRef] [PubMed]
- Davies, A.K.; Itzhak, D.N.; Edgar, J.R.; Archuleta, T.L.; Hirst, J.; Jackson, L.P.; Robinson, M.S.; Borner, G.H.H. AP-4 vesicles contribute to spatial control of autophagy via RUSC-dependent peripheral delivery of ATG9A. Nat. Commun. 2018, 9, 3958. [Google Scholar] [CrossRef]
- Dragich, J.M.; Kuwajima, T.; Hirose-Ikeda, M.; Yoon, M.S.; Eenjes, E.; Bosco, J.R.; Fox, L.M.; Lystad, A.H.; Oo, T.F.; Yarygina, O.; et al. Autophagy linked FYVE (Alfy/WDFY3) is required for establishing neuronal connectivity in the mammalian brain. Elife 2016, 5, e14810. [Google Scholar] [CrossRef] [PubMed]
- Wojnacki, J.; Nola, S.; Bun, P.; Cholley, B.; Filippini, F.; Pressé, M.T.; Lipecka, J.; Man Lam, S.; N’guyen, J.; Simon, A.; et al. Role of VAMP7-Dependent Secretion of Reticulon 3 in Neurite Growth. Cell Rep. 2020, 33, 108536. [Google Scholar] [CrossRef]
- Sakamoto, K.; Ozaki, T.; Ko, Y.C.; Tsai, C.F.; Gong, Y.; Morozumi, M.; Ishikawa, Y.; Uchimura, K.; Nadanaka, S.; Kitagawa, H.; et al. Glycan sulfation patterns define autophagy flux at axon tip via PTPRσ-cortactin axis. Nat. Chem. Biol. 2019, 15, 699–709. [Google Scholar] [CrossRef]
- Hirst, J.; Irving, C.; Borner, G.H. Adaptor protein complexes AP-4 and AP-5: New players in endosomal trafficking and progressive spastic paraplegia. Traffic 2013, 14, 153–164. [Google Scholar] [CrossRef]
- Yap, C.C.; Murate, M.; Kishigami, S.; Muto, Y.; Kishida, H.; Hashikawa, T.; Yano, R. Adaptor protein complex-4 (AP-4) is expressed in the central nervous system neurons and interacts with glutamate receptor delta2. Mol. Cell Neurosci. 2003, 24, 283–295. [Google Scholar] [CrossRef]
- Matsuda, S.; Miura, E.; Matsuda, K.; Kakegawa, W.; Kohda, K.; Watanabe, M.; Yuzaki, M. Accumulation of AMPA receptors in autophagosomes in neuronal axons lacking adaptor protein AP-4. Neuron 2008, 57, 730–745. [Google Scholar] [CrossRef]
- De Pace, R.; Skirzewski, M.; Damme, M.; Mattera, R.; Mercurio, J.; Foster, A.M.; Cuitino, L.; Jarnik, M.; Hoffmann, V.; Morris, H.D.; et al. Altered distribution of ATG9A and accumulation of axonal aggregates in neurons from a mouse model of AP-4 deficiency syndrome. PLoS Genet. 2018, 14, e100736. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, J.; Miura, E.; Mizushima, N.; Watanabe, M.; Yuzaki, M. Aberrant membranes and double-membrane structures accumulate in the axons of Atg5-null Purkinje cells before neuronal death. Autophagy 2007, 3, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Rodriguez-Navarro, J.A.; Arias, E.; Kiffin, R.; Sahu, S.; Schwartz, G.J.; Cuervo, A.M.; Singh, R. Autophagy in hypothalamic AgRP neurons regulates food intake and energy balance. Cell Metab. 2011, 14, 173–183. [Google Scholar] [CrossRef]
- Chen, Y.; Sawada, O.; Kohno, H.; Le, Y.Z.; Subauste, C.; Maeda, T.; Maeda, A. Autophagy protects the retina from light-induced degeneration. J. Biol. Chem. 2013, 288, 7506–7518. [Google Scholar] [CrossRef]
- Zhou, Z.; Doggett, T.A.; Sene, A.; Apte, R.S.; Ferguson, T.A. Autophagy supports survival and phototransduction protein levels in rod photoreceptors. Cell Death Differ. 2015, 22, 488–498. [Google Scholar] [CrossRef]
- Ban, B.K.; Jun, M.H.; Ryu, H.H.; Jang, D.J.; Ahmad, S.T.; Lee, J.A. Autophagy negatively regulates early axon growth in cortical neurons. Mol Cell Biol 2013, 33, 3907–3919. [Google Scholar] [CrossRef]
- Yang, K.; Yu, B.; Cheng, C.; Cheng, T.; Yuan, B.; Li, K.; Xiao, J.; Qiu, Z.; Zhou, Y. Mir505-3p regulates axonal development via inhibiting the autophagy pathway by targeting Atg12. Autophagy 2017, 13, 1679–1696. [Google Scholar] [CrossRef]
- Nixon, R.A. Endosome function and dysfunction in Alzheimer’s disease and other neurodegenerative diseases. Neurobiol. Aging 2005, 26, 373–382. [Google Scholar] [CrossRef]
- Waites, C.L.; Leal-Ortiz, S.A.; Okerlund, N.; Dalke, H.; . Fejtova, A.; Altrock, W.D.; Gundelfinger, E.D.; Garner, C.C. Bassoon and Piccolo maintain synapse integrity by regulating protein ubiquitination and degradation. EMBO J. 2013, 32, 954–969. [Google Scholar] [CrossRef]
- Okerlund, N.D.; Schneider, K.; Leal-Ortiz, S.; Montenegro-Venegas, C.; Kim, S.A.; Garner, L.C.; Waites, C.L.; Gundelfinger, E.D.; Reimer, R.J.; Garner, C.C. Bassoon Controls Presynaptic Autophagy through Atg5. Neuron 2017, 93, 897–913.e7. [Google Scholar] [CrossRef] [PubMed]
- Crawley, O.; Opperman, K.J.; Desbois, M.; Adrados, I.; Borgen, M.A.; Giles, A.C.; Duckett, D.R.; Grill, B. Autophagy is inhibited by ubiquitin ligase activity in the nervous system. Nat. Commun. 2019, 10, 5017. [Google Scholar] [CrossRef] [PubMed]
- Tyler, W.J.; Pozzo-Miller, L.D. BDNF enhances quantal neurotransmitter release and increases the number of docked vesicles at the active zones of hippocampal excitatory synapses. J. Neurosci 2001, 21, 4249–4258. [Google Scholar] [CrossRef]
- Nikoletopoulou, V.; Sidiropoulou, K.; Kallergi, E.; Dalezios, Y.; Tavernarakis, N. Modulation of Autophagy by BDNF Underlies Synaptic Plasticity. Cell Metab. 2017, 26, 230–242.e5. [Google Scholar] [CrossRef] [PubMed]
- Tavosanis, G. Dendrite enlightenment. Curr. Opin. Neurobiol. 2021, 69, 222–230. [Google Scholar] [CrossRef]
- Clark, S.G.; Graybeal, L.L.; Bhattacharjee, S.; Thomas, C.; Bhattacharya, S.; Cox, D.N. Basal autophagy is required for promoting dendritic terminal branching in Drosophila sensory neurons. PLoS ONE 2018, 13, e0206743. [Google Scholar] [CrossRef] [PubMed]
- Friedman, L.G.; Lachenmayer, M.L.; Wang, J.; He, L.; Poulose, S.M.; Komatsu, M.; Holstein, G.R.; Yue, Z. Disrupted autophagy leads to dopaminergic axon and dendrite degeneration and promotes presynaptic accumulation of α-synuclein and LRRK2 in the brain. J. Neurosci. 2012, 32, 7585–7593. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.; Gudsnuk, K.; Kuo, S.H.; Cotrina, M.L.; Rosoklija, G.; Sosunov, A.; Sonders, M.S.; Kanter, E.; Castagna, C.; Yamamoto, A.; et al. Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 2014, 83, 1131–1143. [Google Scholar] [CrossRef]
- Chen, Z.W.; Chang, C.S.; Leil, T.A.; Olsen, R.W. C-terminal modification is required for GABARAP-mediated GABA(A) receptor trafficking. J. Neurosci. 2007, 27, 6655–6663. [Google Scholar] [CrossRef]
- Rowland, A.M.; Richmond, J.E.; Olsen, J.G.; Hall, D.H.; Bamber, B.A. Presynaptic terminals independently regulate synaptic clustering and autophagy of GABAA receptors in Caenorhabditis elegans. J. Neurosci 2006, 26, 1711–1720. [Google Scholar] [CrossRef]
- Lieberman, O.J.; Sulzer, D. The Synaptic Autophagy Cycle. J. Mol. Biol. 2020, 432, 2589–2604. [Google Scholar] [CrossRef] [PubMed]
- Shehata, M.; Matsumura, H.; Okubo-Suzuki, R.; Ohkawa, N.; Inokuchi, K. Neuronal stimulation induces autophagy in hippocampal neurons that is involved in AMPA receptor degradation after chemical long-term depression. J. Neurosci. 2012, 32, 10413–10422. [Google Scholar] [CrossRef]
- Compans, B.; Camus, C.; Kallergi, E.; Sposini, S.; Martineau, M.; Butler, C.; Kechkar, A.; Klaassen, R.V.; Retailleau, N.; Sejnowski, T.J.; et al. NMDAR-dependent long-term depression is associated with increased short term plasticity through autophagy mediated loss of PSD-95. Nat. Commun. 2021, 12, 2849. [Google Scholar] [CrossRef] [PubMed]
- Maday, S.; Holzbaur, E.L. Compartment-Specific Regulation of Autophagy in Primary Neurons. J. Neurosci. 2016, 36, 5933–5945. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Zakaria, H.M.; Simone, A.; Sheng, Z.H. Spatial parkin translocation and degradation of damaged mitochondria via mitophagy in live cortical neurons. Curr. Biol. 2012, 22, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Devireddy, S.; Liu, A.; Lampe, T.; Hollenbeck, P.J. The Organization of Mitochondrial Quality Control and Life Cycle in the Nervous System In Vivo in the Absence of PINK1. J. Neurosci. 2015, 35, 9391–9401. [Google Scholar] [CrossRef] [PubMed]
- Magee, J.C.; Grienberger, C. Synaptic Plasticity Forms and Functions. Annu. Rev. Neurosci. 2020, 43, 95–117. [Google Scholar] [CrossRef]
- Hegde, A.N. Proteolysis, synaptic plasticity and memory. Neurobiol. Learn. Mem. 2017, 138, 98–110. [Google Scholar] [CrossRef]
- Tomoda, T.; Yang, K.; Sawa, A. Neuronal Autophagy in Synaptic Functions and Psychiatric Disorders. Biol. Psychiatry 2020, 87, 787–796. [Google Scholar] [CrossRef]
- Kim, H.J.; Cho, M.H.; Shim, W.H.; Kim, J.K.; Jeon, E.Y.; Kim, D.H.; Yoon, S.Y. Deficient autophagy in microglia impairs synaptic pruning and causes social behavioral defects. Mol. Psychiatry 2017, 22, 1576–1584. [Google Scholar] [CrossRef]
- Glatigny, M.; Moriceau, S.; Rivagorda, M.; Ramos-Brossier, M.; Nascimbeni, A.C.; Lante, F.; Shanley, M.R.; Boudarene, N.; Rousseaud, A.; Friedman, A.K.; et al. Autophagy Is Required for Memory Formation and Reverses Age-Related Memory Decline. Curr. Biol. 2019, 29, 435–448.e8. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Shang, X.; Zhai, B.; Zhang, H.; Zhang, T. Nicotine alleviates chronic stress-induced anxiety and depressive-like behavior and hippocampal neuropathology via regulating autophagy signaling. Neurochem. Int. 2018, 114, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Porch, M.W.; Court-Vazquez, B.; Bennett, M.V.L.; Zukin, R.S. Activation of autophagy rescues synaptic and cognitive deficits in fragile X mice. Proc. Natl. Acad. Sci. USA 2018, 115, E9707–E9716. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, L.K.; Le Prieult, F.; Felzen, V.; Thal, S.C.; Engelhard, K.; Behl, C.; Mittmann, T. Proteasome and Autophagy-Mediated Impairment of Late Long-Term Potentiation (l-LTP) after Traumatic Brain Injury in the Somatosensory Cortex of Mice. Int. J. Mol. Sci. 2019, 20, 3048. [Google Scholar] [CrossRef]
- Horwood, J.M.; Dufour, F.; Laroche, S.; Davis, S. Signalling mechanisms mediated by the phosphoinositide 3-kinase/Akt cascade in synaptic plasticity and memory in the rat. Eur. J. Neurosci. 2006, 23, 3375–3384. [Google Scholar] [CrossRef]
- Thijs, R.D.; Surges, R.; O’Brien, T.J.; Sander, J.W. Epilepsy in adults. Lancet 2019, 393, 689–701. [Google Scholar] [CrossRef]
- Fiest, K.M.; Sauro, K.M.; Wiebe, S.; Patten, S.B.; Kwon, C.S.; Dykeman, J.; Pringsheim, T.; Lorenzetti, D.L.; Jetté, N. Prevalence and incidence of epilepsy: A systematic review and meta-analysis of international studies. Neurology 2017, 88, 296–303. [Google Scholar] [CrossRef]
- Beretta, S.; Carone, D.; Zanchi, C.; Bianchi, E.; Pirovano, M.; Trentini, C.; Padovano, G.; Colombo, M.; Cereda, D.; Scanziani, S.; et al. PRO-LONG Study Group. Long-term applicabili.ity of the new ILAE definition of epilepsy. Results from the PRO-LONG study. Epilepsia 2017, 58, 1518–1523. [Google Scholar] [CrossRef]
- Fisher, R.S.; Cross, J.H.; French, J.A.; Higurashi, N.; Hirsch, E.; Jansen, F.E.; Lagae, L.; Moshé, S.L.; Peltola, J.; Roulet Perez, E.; et al. Operational classification of seizure types by the International League Against Epilepsy: Position Paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 522–530. [Google Scholar] [CrossRef]
- Balestrini, S.; Arzimanoglou, A.; Blümcke, I.; Scheffer, I.E.; Wiebe, S.; Zelano, J.; Walker, M.C. The aetiologies of epilepsy. Epileptic Disord. 2021, 23, 1–16. [Google Scholar] [CrossRef]
- Fisher, R.S.; Acevedo, C.; Arzimanoglou, A.; Bogacz, A.; Cross, J.H.; Elger, C.E.; Engel, J., Jr.; Forsgren, L.; French, J.A.; Glynn, M.; et al. ILAE official report: A practical clinical definition of epilepsy. Epilepsia 2014, 55, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Helbig, I.; Scheffer, I.E.; Mulley, J.C.; Berkovic, S.F. Navigating the channels and beyond: Unravelling the genetics of the epilepsies. Lancet Neurol. 2008, 7, 231–245. [Google Scholar] [CrossRef]
- Berg, A.T.; Mathern, G.W.; Bronen, R.A.; Fulbright, R.K.; DiMario, F.; Testa, F.M.; Levy, S.R. Frequency, prognosis and surgical treatment of structural abnormalities seen with magnetic resonance imaging in childhood epilepsy. Brain 2009, 132, 2785–2797. [Google Scholar] [CrossRef] [PubMed]
- Vivash, L.; Tostevin, A.; Liu, D.S.; Dalic, L.; Dedeurwaerdere, S.; Hicks, R.J.; Williams, D.A.; Myers, D.E.; O’Brien, T.J. Changes in hippocampal GABAA/cBZR density during limbic epileptogenesis: Relationship to cell loss and mossy fibre sprouting. Neurobiol. Dis. 2011, 41, 227–236. [Google Scholar] [CrossRef]
- Crino, P.B. mTOR: A pathogenic signaling pathway in developmental brain malformations. Trends Mol. Med. 2011, 17, 734–742. [Google Scholar] [CrossRef]
- Nguyen, L.H.; Mahadeo, T.; Bordey, A. mTOR Hyperactivity Levels Influence the Severity of Epilepsy and Associated Neuropathology in an Experimental Model of Tuberous Sclerosis Complex and Focal Cortical Dysplasia. J. Neurosci. 2019, 39, 2762–2773. [Google Scholar] [CrossRef]
- Fernandez, D.; Perl, A. mTOR signaling: A central pathway to pathogenesis in systemic lupus erythematosus? Discov. Med. 2010, 9, 173–178. [Google Scholar]
- Limanaqi, F.; Biagioni, F.; Busceti, C.L.; Fabrizi, C.; Frati, A.; Fornai, F. mTOR-Related Cell-Clearing Systems in Epileptic Seizures, an Update. Int. J. Mol. Sci. 2020, 21, 1642. [Google Scholar] [CrossRef]
- Lasarge, C.L.; Danzer, S.C. Mechanisms regulating neuronal excitability and seizure development following mTOR pathway hyperactivation. Front. Mol. Neurosci. 2014, 7, 18. [Google Scholar] [CrossRef]
- Dazzo, E.; Nobile, C. Epilepsy-causing Reelin mutations result in impaired secretion and intracellular degradation of mutant proteins. Hum. Mol. Genet. 2022, 31, 665–673. [Google Scholar] [CrossRef]
- Giorgi, F.S.; Biagioni, F.; Lenzi, P.; Frati, A.; Fornai, F. The role of autophagy in epileptogenesis and in epilepsy-induced neuronal alterations. J. Neural. Transm. 2015, 122, 849–862. [Google Scholar] [CrossRef] [PubMed]
- Saitsu, H.; Nishimura, T.; Muramatsu, K.; Kodera, H.; Kumada, S.; Sugai, K.; Kasai-Yoshida, E.; Sawaura, N.; Nishida, H.; Hoshino, A.; et al. De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat. Genet. 2013, 45, 445–449. [Google Scholar] [CrossRef] [PubMed]
- McMahon, J.; Huang, X.; Yang, J.; Komatsu, M.; Yue, Z.; Qian, J.; Zhu, X.; Huang, Y. Impaired autophagy in neurons after disinhibition of mammalian target of rapamycin and its contribution to epileptogenesis. J. Neurosci. 2012, 32, 15704–15714. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Han, Y.; Du, J.; Jin, H.; Zhang, J.; Niu, M.; Qin, J. Alterations of apoptosis and autophagy in developing brain of rats with epilepsy: Changes in LC3, P62, Beclin-1 and Bcl-2 levels. Neurosci. Res. 2018, 130, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Shacka, J.J.; Lu, J.; Xie, Z.L.; Uchiyama, Y.; Roth, K.A.; Zhang, J. Kainic acid induces early and transient autophagic stress in mouse hippocampus. Neurosci. Lett. 2007, 414, 57–60. [Google Scholar] [CrossRef]
- Fulceri, F.; Ferrucci, M.; Lazzeri, G.; Paparelli, S.; Bartalucci, A.; Tamburini, I.; Paparelli, A.; Fornai, F. Autophagy activation in glutamate-induced motor neuron loss. Arch. Ital. Biol. 2011, 149, 101–111. [Google Scholar]
- Wong, M. Cleaning up epilepsy and neurodegeneration: The role of autophagy in epileptogenesis. Epilepsy Curr. 2013, 13, 177–178. [Google Scholar] [CrossRef]
- Fassio, A.; Falace, A.; Esposito, A.; Aprile, D.; Guerrini, R.; Benfenati, F. Emerging role of the autophagy/lysosomal degradative pathway in neurodevelopmental disorders with epilepsy. Front. Cell Neurosci. 2020, 14, 39. [Google Scholar] [CrossRef]
- Xie, N.; Li, Y.; Wang, C.; Lian, Y.; Zhang, H.; Li, Y.; Meng, X.; Du, L. FAM134B attenuates seizure-induced apoptosis and endoplasmic reticulum stress in hippocampal neurons by promoting autophagy. Cell Mol. Neurobiol. 2020, 40, 1297–1305. [Google Scholar] [CrossRef]
- Wang, L.; Song, L.F.; Chen, X.Y.; Ma, Y.L.; Suo, J.F.; Shi, J.H.; Chen, G.H. MiR-181b inhibits P38/JNK signaling pathway to attenuate autophagy and apoptosis in juvenile rats with kainic acid-induced epilepsy via targeting TLR4. CNS Neurosci. Ther. 2019, 25, 112–122. [Google Scholar] [CrossRef]
- Sun, J.; Gao, X.; Meng, D.; Xu, Y.; Wang, X.; Gu, X.; Guo, M.; Shao, X.; Yan, H.; Jiang, C.; et al. Antagomirs targeting miroRNA-134 attenuates epilepsy in rats through regulation of oxidative stress, mitochondrial functions and autophagy. Front. Pharmacol. 2017, 8, 524. [Google Scholar] [CrossRef] [PubMed]
- Xiao, D.; Lv, J.; Zheng, Z.; Liu, Y.; Zhang, Y.; Luo, C.; Qi, L.; Qin, B.; Liu, C. Mechanisms of microRNA-142 in mitochondrial autophagy and hippocampal damage in a rat model of epilepsy. Int. J. Mol. Med. 2021, 47, 98. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Chen, H.; Zhong, Y.; Yang, Q.; Wang, X.; Chen, R.; Guo, Y. MicroRNA 223 Targeting ATG16L1 affects microglial autophagy in the kainic acid model of temporal lobe epilepsy. Front. Neurol. 2021, 12, 704550. [Google Scholar] [CrossRef]
- Hu, F.; Shao, L.; Zhang, J.; Zhang, H.; Wen, A.; Zhang, G.P. Knockdown of ZFAS1 inhibits hippocampal neurons apoptosis and autophagy by activating the PI3K/AKT pathway via up-regulating miR-421 in epilepsy. Neurochem. Res. 2020, 45, 2433–2441. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Yi, X. Down-regulation of long noncoding RNA MALAT1 protects hippocampal neurons against excessive autophagy and apoptosis via the PI3K/Akt signaling pathway in rats with epilepsy. J. Mol. Neurosci. 2018, 65, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.M.; Zhao, X.F.; Liao, Y.; Mathur, R.; McCallum, S.E.; Mazurkiewicz, J.E.; Adamo, M.A.; Feustel, P.; Belin, S.; Poitelon, Y.; et al. Deficiency of microglial autophagy increases the density of oligodendrocytes and susceptibility to severe forms of seizures. eNeuro 2021, 8, ENEURO.0183-20.2021. [Google Scholar] [CrossRef]
- Peng, J.; Wu, S.; Guo, C.; Guo, K.; Zhang, W.; Liu, R.; Li, J.; Hu, Z. Effect of Ibuprofen on Autophagy of Astrocytes during Pentylenetetrazol-Induced Epilepsy and its Significance: An Experimental Study. Neurochem. Res. 2019, 44, 2566–2576. [Google Scholar] [CrossRef]
- Garyali, P.; Segvich, D.M.; DePaoli-Roach, A.A.; Roach, P.J. Protein degradation and quality control in cells from Laforin and Malin knockout mice. J. Biol. Chem. 2014, 289, 20606–20614. [Google Scholar] [CrossRef]
- Buckmaster, P.S.; Ingram, E.A.; Wen, X. Inhibition of the mammalian target of rapamycin signaling pathway suppresses dentate granule cell axon sprouting in a rodent model of temporal lobe epilepsy. J. Neurosci. 2009, 29, 8259–8269. [Google Scholar] [CrossRef]
- Buckmaster, P.S.; Lew, F.H. Rapamycin suppresses mossy fiber sprouting but not seizure frequency in a mouse model of temporal lobe epilepsy. J. Neurosci. 2011, 31, 2337–2347. [Google Scholar] [CrossRef]
- Wong, M. Mammalian target of rapamycin (mTOR) pathways in neurological diseases. Biomed. J. 2013, 36, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Amegandjin, C.A.; Choudhury, M.; Jadhav, V.; Carriço, J.N.; Quintal, A.; Berryer, M.; Snapyan, M.; Chattopadhyaya, B.; Saghatelyan, A.; Di Cristo, G. Sensitive period for rescuing parvalbumin interneurons connectivity and social behavior deficits caused by TSC1 loss. Nat. Commun. 2021, 12, 3653. [Google Scholar] [CrossRef]
- Puri, R.; Suzuki, T.; Yamakawa, K.; Ganesh, S. Dysfunctions in endosomal-lysosomal and autophagy pathways underlie neuropathology in a mouse model for Lafora disease. Hum. Mol. Genet. 2012, 21, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Criado, O.; Aguado, C.; Gayarre, J.; Duran-Trio, L.; Garcia-Cabrero, A.M.; Vernia, S.; San Millán, B.; Heredia, M.; Romá-Mateo, C.; Mouron, S.; et al. Lafora bodies and neurological defects in malin-deficient mice correlate with impaired autophagy. Hum. Mol. Genet. 2012, 21, 1521–1533. [Google Scholar] [CrossRef] [PubMed]
- Ying, C.; Ying, L.; Yanxia, L.; Le, W.; Lili, C. High mobility group box 1 antibody represses autophagy and alleviates hippocampus damage in pilocarpine-induced mouse epilepsy model. Acta Histochem. 2020, 122, 151485. [Google Scholar] [CrossRef] [PubMed]
- Gstrein, T.; Edwards, A.; Přistoupilová, A.; Leca, I.; Breuss, M.; Pilat-Carotta, S.; Hansen, A.H.; Tripathy, R.; Traunbauer, A.K.; Hochstoeger, T.; et al. Mutations in Vps15 perturb neuronal migration in mice and are associated with neurodevelopmental disease in humans. Nat. Neurosci. 2018, 21, 207–217. [Google Scholar] [CrossRef]
- Yasin, S.A.; Ali, A.M.; Tata, M.; Picker, S.R.; Anderson, G.W.; Latimer-Bowman, E.; Nicholson, S.L.; Harkness, W.; Cross, J.H.; Paine, S.M.; et al. mTOR-dependent abnormalities in autophagy characterize human malformations of cortical development: Evidence from focal cortical dysplasia and tuberous sclerosis. Acta Neuropathol. 2013, 126, 207–218. [Google Scholar] [CrossRef]
- Yuskaitis, C.J.; Jones, B.M.; Wolfson, R.L.; Super, C.E.; Dhamne, S.C.; Rotenberg, A.; Sabatini, D.M.; Sahin, M.; Poduri, A. A mouse model of DEPDC5-related epilepsy: Neuronal loss of Depdc5 causes dysplastic and ectopic neurons, increased mTOR signaling, and seizure susceptibility. Neurobiol. Dis. 2018, 111, 91–101. [Google Scholar] [CrossRef]
- Kwon, C.H.; Zhu, X.; Zhang, J.; Baker, S.J. mTor is required for hypertrophy of Pten-deficient neuronal soma in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 12923–12928. [Google Scholar] [CrossRef]
- Cao, L.; Xu, J.; Lin, Y.; Zhao, X.; Liu, X.; Chi, Z. Autophagy is upregulated in rats with status epilepticus and partly inhibited by Vitamin, E. Biochem. Biophys. Res. Commun. 2009, 379, 949–953. [Google Scholar] [CrossRef]
- Macias, M.; Blazejczyk, M.; Kazmierska, P.; Caban, B.; Skalecka, A.; Tarkowski, B.; Rodo, A.; Konopacki, J.; Jaworski, J. Spatiotemporal characterization of mTOR kinase activity following kainic acid induced status epilepticus and analysis of rat brain response to chronic rapamycin treatment. PLoS ONE 2013, 8, e64455. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.H.; Rensing, N.R.; Wong, M. The mammalian target of rapamycin signaling pathway mediates epileptogenesis in a model of temporal lobe epilepsy. J. Neurosci. 2009, 29, 6964–6972. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Zhang, H.; Yang, J.; Wu, J.; McMahon, J.; Lin, Y.; Cao, Z.; Gruenthal, M.; Huang, Y. Pharmacological inhibition of the mammalian target of rapamycin pathway suppresses acquired epilepsy. Neurobiol. Dis. 2010, 40, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Raffo, E.; Coppola, A.; Ono, T.; Briggs, S.W.; Galanopoulou, A.S. A pulse rapamycin therapy for infantile spasms and associated cognitive decline. Neurobiol Dis 2011, 43, 322–329. [Google Scholar] [CrossRef]
- Carvill, G.L.; Liu, A.; Mandelstam, S.; Schneider, A.; Lacroix, A.; Zemel, M.; McMahon, J.M.; Bello-Espinosa, L.; Mackay, M.; Wallace, G.; et al. Severe infantile onset developmental and epileptic encephalopathy caused by mutations in autophagy gene WDR45. Epilepsia 2018, 59, e5–e13. [Google Scholar] [CrossRef]
- Byrne, S.; Jansen, L.; U-King-Im, J.M.; Siddiqui, A.; Lidov, H.G.; Bodi, I.; Smith, L.; Mein, R.; Cullup, T.; Dionisi-Vici, C.; et al. EPG5-related Vici syndrome: A paradigm of neurodevelopmental disorders with defective autophagy. Brain 2016, 139, 765–781. [Google Scholar] [CrossRef] [PubMed]
- Akizu, N.; Cantagrel, V.; Zaki, M.S.; Al-Gazali, L.; Wang, X.; Rosti, R.O.; Dikoglu, E.; Gelot, A.B.; Rosti, B.; Vaux, K.K.; et al. Biallelic mutations in SNX14 cause a syndromic form of cerebellar atrophy and lysosome-autophagosome dysfunction. Nat. Genet. 2015, 47, 528–534. [Google Scholar] [CrossRef]
- Esposito, A.; Falace, A.; Wagner, M.; Gal, M.; Mei, D.; Conti, V.; Pisano, T.; Aprile, D.; Cerullo, M.S.; De Fusco, A.; et al. Biallelic DMXL2 mutations impair autophagy and cause Ohtahara syndrome with progressive course. Brain 2019, 142, 3876–3891. [Google Scholar] [CrossRef]
- Ortiz-González, X.R.; Tintos-Hernández, J.A.; Keller, K.; Li, X.; Foley, A.R.; Bharucha-Goebel, D.X.; Kessler, S.K.; Yum, S.W.; Crino, P.B.; He, M.; et al. Homozygous boricua TBCK mutation causes neurodegeneration and aberrant autophagy. Ann. Neurol. 2018, 83, 153–165. [Google Scholar] [CrossRef]
- Zhang, Y.X.; Qiao, S.; Cai, M.T.; Lai, Q.L.; Shen, C.H.; Ding, M.P. Association between autophagy-related protein 5 gene polymorphisms and epilepsy in Chinese patients. Neurosci. Lett. 2021, 753, 135870. [Google Scholar] [CrossRef]
- Van Damme, T.; Gardeitchik, T.; Mohamed, M.; Guerrero-Castillo, S.; Freisinger, P.; Guillemyn, B.; Kariminejad, A.; Dalloyaux, D.; van Kraaij, S.; Lefeber, D.J.; et al. Mutations in ATP6V1E1 or ATP6V1A Cause Autosomal-Recessive Cutis Laxa. Am. J. Hum. Genet. 2020, 107, 374. [Google Scholar] [CrossRef] [PubMed]
- Hirose, T.; Cabrera-Socorro, A.; Chitayat, D.; Lemonnier, T.; Féraud, O.; Cifuentes-Diaz, C.; Gervasi, N.; Mombereau, C.; Ghosh, T.; Stoica, L.; et al. ATP6AP2 variant impairs CNS development and neuronal survival to cause fulminant neurodegeneration. J. Clin. Invest. 2019, 129, 2145–2162. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Lin, P.; Jing, W.; Guo, H.; Chen, H.; Chen, Y.; Guo, Y.; Gu, Y.; He, M.; Wu, J.; et al. Beclin1 Deficiency Suppresses Epileptic Seizures. Front. Mol. Neurosci. 2022, 15, 807671. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, H.; Wang, W.; Li, Y. Molecular Mechanism and Regulation of Autophagy and Its Potential Role in Epilepsy. Cells 2022, 11, 2621. https://doi.org/10.3390/cells11172621
Zhu H, Wang W, Li Y. Molecular Mechanism and Regulation of Autophagy and Its Potential Role in Epilepsy. Cells. 2022; 11(17):2621. https://doi.org/10.3390/cells11172621
Chicago/Turabian StyleZhu, Hanxiao, Wei Wang, and Yun Li. 2022. "Molecular Mechanism and Regulation of Autophagy and Its Potential Role in Epilepsy" Cells 11, no. 17: 2621. https://doi.org/10.3390/cells11172621
APA StyleZhu, H., Wang, W., & Li, Y. (2022). Molecular Mechanism and Regulation of Autophagy and Its Potential Role in Epilepsy. Cells, 11(17), 2621. https://doi.org/10.3390/cells11172621