
Antioxidant Systems, lncRNAs, and Tunneling Nanotubes in Cell Death Rescue from Cigarette Smoke Exposure


{kind=link}
{kind=link}
Abstract
1. Introduction
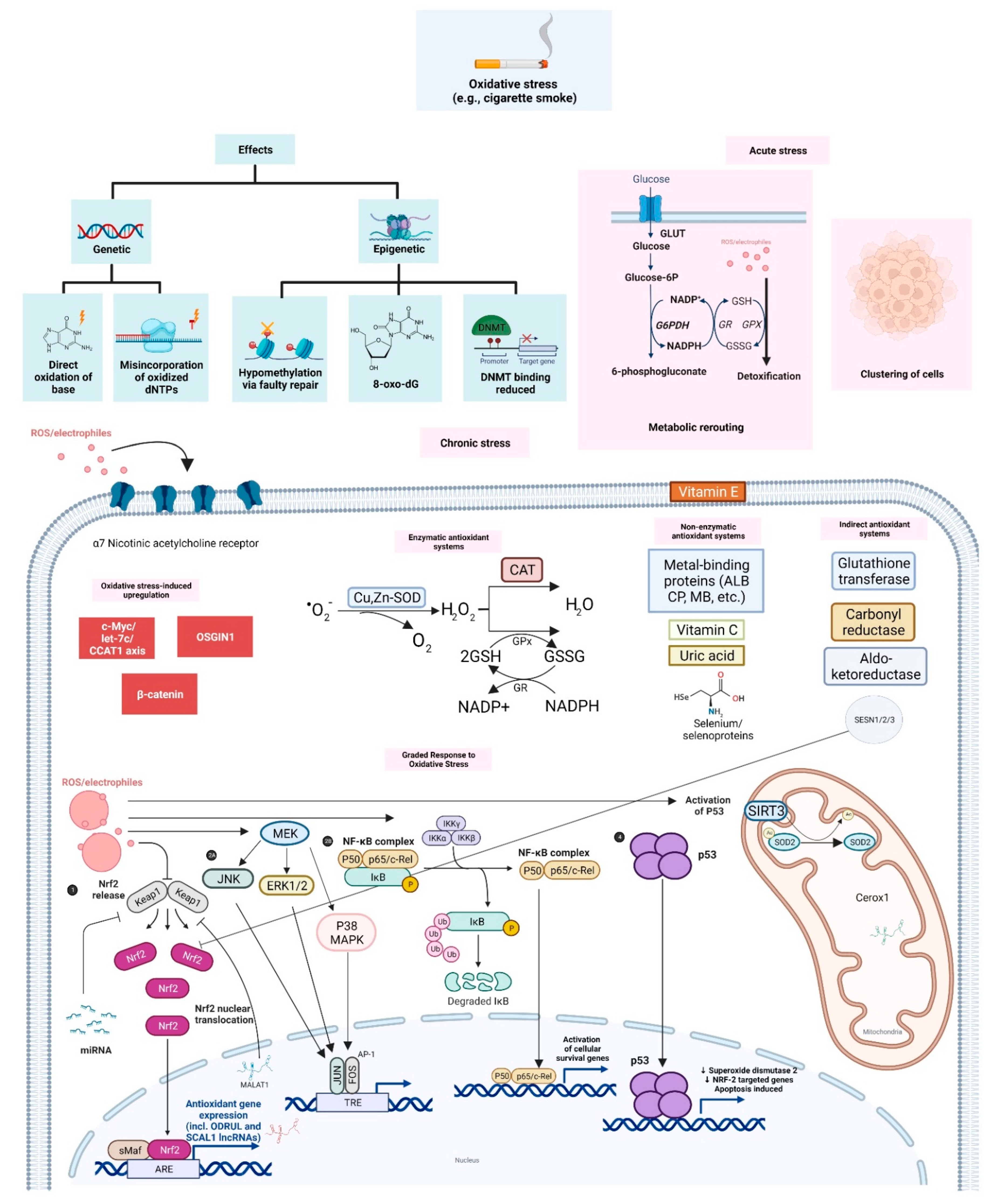
2. Cigarette Smoke and Oxidative Stress
2.1. Genetic and Epigenetic Effects
2.2. Adaptive Responses to Acute and Chronic Oxidative Stress
2.2.1. Graded Adaptive Responses: Multi-Level Transcriptional Control
2.2.2. Oxidative Stress-Upregulated Oncogenes
2.2.3. Upregulated Receptors in Oxidative Stress Response
3. ROS Scavengers: Enzymatic, Non-Enzymatic and Indirect Antioxidant Systems
3.1. Enzymatic Antioxidant Systems
3.2. Non-Enzymatic Antioxidants
3.3. Indirect Antioxidants
4. Long Non-Coding RNAs and Oxidative Stress
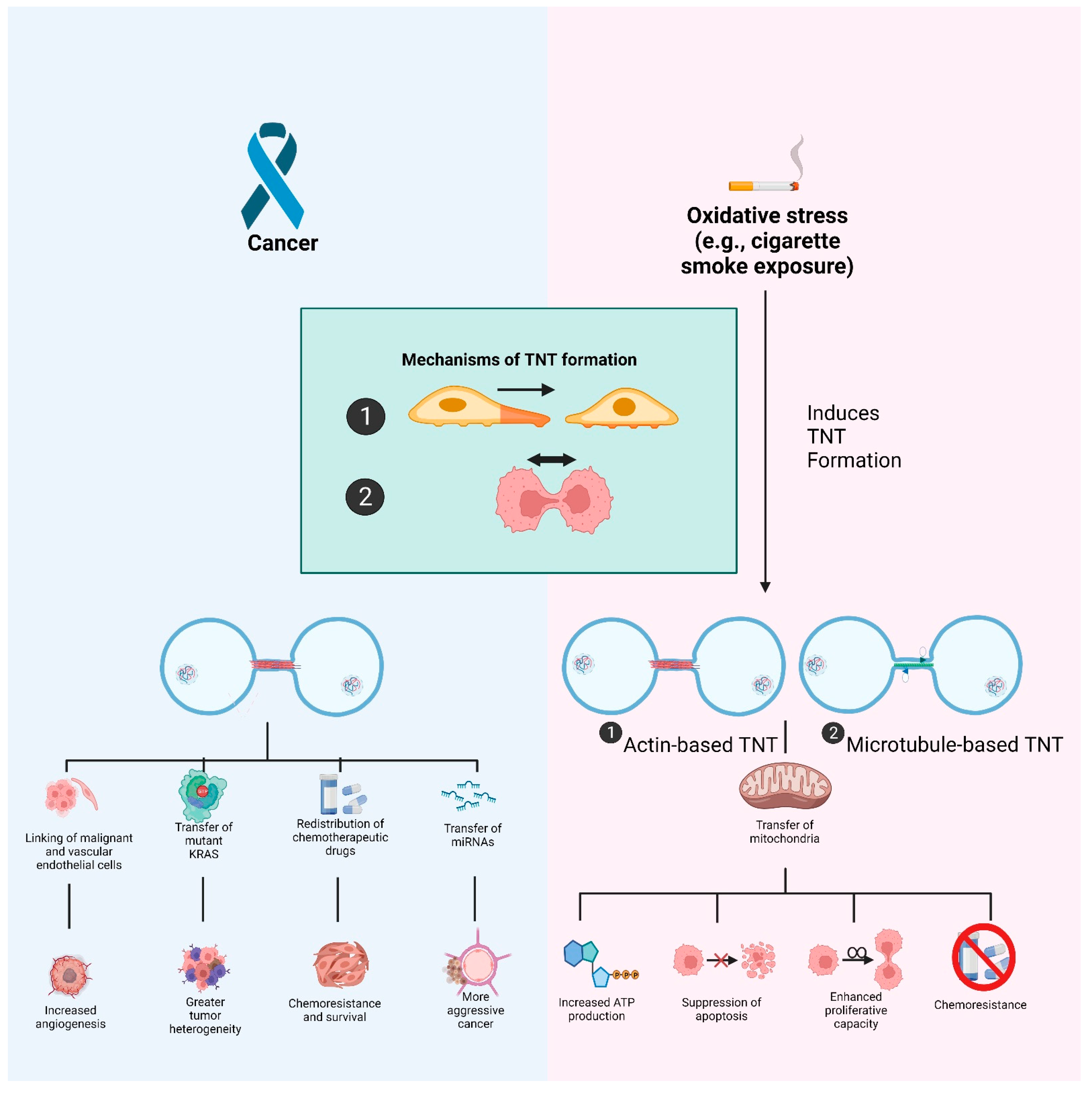
5. Oxidative Stress-Induced Formation of Tunneling Nanotubes
5.1. Tunneling Nanotubes: Biogenesis and Their Role in Malignancies
5.2. TNTs and Oxidative Stress
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine, 5th ed.; Oxford University Press: USA, 2015. [Google Scholar]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef]
- Aggarwal, V.; Tuli, H.; Varol, A.; Thakral, F.; Yerer, M.; Sak, K.; Varol, M.; Jain, A.; Khan, M.; Sethi, G. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules 2019, 9, 735. [Google Scholar] [CrossRef]
- Yang, H.; Villani, R.M.; Wang, H.; Simpson, M.J.; Roberts, M.S.; Tang, M.; Liang, X. The Role of Cellular Reactive Oxygen Species in Cancer Chemotherapy. J. Exp. Clin. Cancer Res. 2018, 37, 266. [Google Scholar] [CrossRef] [PubMed]
- Boldogh, I. ROS Generated by Pollen NADPH Oxidase Provide a Signal That Augments Antigen-Induced Allergic Airway Inflammation. J. Clin. Investig. 2005, 115, 2169–2179. [Google Scholar] [CrossRef] [PubMed]
- DeLeo, F.R.; Allen, L.A.; Apicella, M.; Nauseef, W.M. NADPH Oxidase Activation and Assembly during Phagocytosis. J. Immunol. 1999, 163, 6732–6740. [Google Scholar] [PubMed]
- Elsayed, N.M.; Omaye, S.T.; Klain, G.J.; Korte, D.W. Free Radical-Mediated Lung Response to the Monofunctional Sulfur Mustard Butyl 2-Chloroethyl Sulfide after Subcutaneous Injection. Toxicology 1992, 72, 153–165. [Google Scholar] [CrossRef]
- Obata, T.; Yamanaka, Y.; Kinemuchi, H.; Oreland, L. Release of Dopamine by Perfusion with 1-Methyl-4-Phenylpyridinium Ion (MPP+) into the Striatum Is Associated with Hydroxyl Free Radical Generation. Brain Res. 2001, 906, 170–175. [Google Scholar] [CrossRef]
- Pathak, M.A.; Stratton, K. Free Radicals in Human Skin before and after Exposure to Light. Arch. Biochem. Biophys. 1968, 123, 468–476. [Google Scholar] [CrossRef]
- Pryor, W.A.; Dooley, M.M.; Church, D.F. Mechanisms of Cigarette Smoke Toxicity: The Inactivation of Human α-1-Proteinase Inhibitor by Nitric Oxide/Isoprene Mixtures in Air. Chem.-Biol. Interact. 1985, 54, 171–183. [Google Scholar] [CrossRef]
- Pryor, W.A.; Church, D.F.; Evans, M.D.; Rice, W.Y.; Hayes, J.R. A Comparison of the Free Radical Chemistry of Tobacco-Burning Cigarettes and Cigarettes That Only Heat Tobacco. Free. Radic. Biol. Med. 1990, 8, 275–279. [Google Scholar] [CrossRef]
- Cheng, K.C.; Cahill, D.S.; Kasai, H.; Nishimura, S.; Loeb, L.A. 8-Hydroxyguanine, an Abundant Form of Oxidative DNA Damage, Causes G-T and A-C Substitutions. J. Biol. Chem. 1992, 267, 166–172. [Google Scholar] [CrossRef]
- Salehi, F.; Behboudi, H.; Kavoosi, G.; Ardestani, S.K. Oxidative DNA Damage Induced by ROS-Modulating Agents with the Ability to Target DNA: A Comparison of the Biological Characteristics of Citrus Pectin and Apple Pectin. Sci. Rep. 2018, 8, 13902. [Google Scholar] [CrossRef] [PubMed]
- Cannan, W.J.; Tsang, B.P.; Wallace, S.S.; Pederson, D.S. Nucleosomes Suppress the Formation of Double-Strand DNA Breaks during Attempted Base Excision Repair of Clustered Oxidative Damages. J. Biol. Chem. 2014, 289, 19881–19893. [Google Scholar] [CrossRef]
- Sedletska, Y.; Radicella, J.P.; Sage, E. Replication Fork Collapse Is a Major Cause of the High Mutation Frequency at Three-Base Lesion Clusters. Nucleic Acids Res. 2013, 41, 9339–9348. [Google Scholar] [CrossRef] [PubMed]
- Kotsantis, P.; Petermann, E.; Boulton, S.J. Mechanisms of Oncogene-Induced Replication Stress: Jigsaw Falling into Place. Cancer Discov. 2018, 8, 537–555. [Google Scholar] [CrossRef] [PubMed]
- Xiang, H.; Ramil, C.P.; Hai, J.; Zhang, C.; Wang, H.; Watkins, A.A.; Afshar, R.; Georgiev, P.; Sze, M.A.; Song, X.S.; et al. Cancer-Associated Fibroblasts Promote Immunosuppression by Inducing ROS-Generating Monocytic MDSCs in Lung Squamous Cell Carcinoma. Cancer Immunol. Res. 2020, 8, 436–450. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, S.; Zhao, Y.; Ma, P.; Cao, Y.; Liu, C.; Zhang, X.; Wang, W.; Chen, L.; Li, Y. Cancer-Associated Fibroblasts Promote the Migration and Invasion of Gastric Cancer Cells via Activating IL-17a/JAK2/STAT3 Signaling. Ann. Transl. Med. 2020, 8, 877. [Google Scholar] [CrossRef]
- Aceto, N.; Bardia, A.; Miyamoto, D.T.; Donaldson, M.C.; Wittner, B.S.; Spencer, J.A.; Yu, M.; Pely, A.; Engstrom, A.; Zhu, H.; et al. Circulating Tumor Cell Clusters Are Oligoclonal Precursors of Breast Cancer Metastasis. Cell 2014, 158, 1110–1122. [Google Scholar] [CrossRef]
- Donato, C.; Kunz, L.; Castro-Giner, F.; Paasinen-Sohns, A.; Strittmatter, K.; Szczerba, B.M.; Scherrer, R.; Di Maggio, N.; Heusermann, W.; Biehlmaier, O.; et al. Hypoxia Triggers the Intravasation of Clustered Circulating Tumor Cells. Cell Rep. 2020, 32, 108105. [Google Scholar] [CrossRef]
- Labuschagne, C.F.; Cheung, E.C.; Blagih, J.; Domart, M.-C.; Vousden, K.H. Cell Clustering Promotes a Metabolic Switch That Supports Metastatic Colonization. Cell Metab. 2019, 30, 720–734. [Google Scholar] [CrossRef]
- Szczerba, B.M.; Castro-Giner, F.; Vetter, M.; Krol, I.; Gkountela, S.; Landin, J.; Scheidmann, M.C.; Donato, C.; Scherrer, R.; Singer, J.; et al. Neutrophils Escort Circulating Tumour Cells to Enable Cell Cycle Progression. Nature 2019, 566, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Padmanaban, V.; Krol, I.; Suhail, Y.; Szczerba, B.M.; Aceto, N.; Bader, J.S.; Ewald, A.J. E-Cadherin Is Required for Metastasis in Multiple Models of Breast Cancer. Nature 2019, 573, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, W.; Zhu, W.; Dong, J.; Cheng, Y.; Yin, Z.; Shen, F. Mechanisms and Functions of Long Non-Coding RNAs at Multiple Regulatory Levels. IJMS 2019, 20, 5573. [Google Scholar] [CrossRef] [PubMed]
- Rustom, A. The Missing Link: Does Tunnelling Nanotube-Based Supercellularity Provide a New Understanding of Chronic and Lifestyle Diseases? Open Biol. 2016, 6, 160057. [Google Scholar] [CrossRef]
- Pryor, W.A. Cigarette Smoke Radicals and the Role of Free Radicals in Chemical Carcinogenicity. Environ. Health Perspect. 1997, 105, 875–882. [Google Scholar] [CrossRef]
- Besaratinia, A.; Pfeifer, G.P. Second-Hand Smoke and Human Lung Cancer. Lancet Oncol. 2008, 9, 657–666. [Google Scholar] [CrossRef]
- Goel, R.; Bitzer, Z.T.; Reilly, S.M.; Foulds, J.; Muscat, J.; Elias, R.J.; Richie, J.P. Influence of Smoking Puff Parameters and Tobacco Varieties on Free Radicals Yields in Cigarette Mainstream Smoke. Chem. Res. Toxicol. 2018, 31, 325–331. [Google Scholar] [CrossRef]
- Dizdaroglu, M. Oxidatively Induced DNA Damage and Its Repair in Cancer. Mutat. Res. Rev. Mutat. Res. 2015, 763, 212–245. [Google Scholar] [CrossRef]
- Cadet, J.; Davies, K.J.A.; Medeiros, M.H.; Di Mascio, P.; Wagner, J.R. Formation and Repair of Oxidatively Generated Damage in Cellular DNA. Free. Radic. Biol. Med. 2017, 107, 13–34. [Google Scholar] [CrossRef]
- Nohmi, T.; Kim, S.-R.; Yamada, M. Modulation of Oxidative Mutagenesis and Carcinogenesis by Polymorphic Forms of Human DNA Repair Enzymes. Mutat. Res. Fundam. Mol. Mech. Mutagenesis 2005, 591, 60–73. [Google Scholar] [CrossRef]
- Hidaka, K.; Yamada, M.; Kamiya, H.; Masutani, C.; Harashima, H.; Hanaoka, F.; Nohmi, T. Specificity of Mutations Induced by Incorporation of Oxidized DNTPs into DNA by Human DNA Polymerase η. DNA Repair 2008, 7, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Dutta, A.; Yang, C.; Sengupta, S.; Mitra, S.; Hegde, M.L. New Paradigms in the Repair of Oxidative Damage in Human Genome: Mechanisms Ensuring Repair of Mutagenic Base Lesions during Replication and Involvement of Accessory Proteins. Cell. Mol. Life Sci. 2015, 72, 1679–1698. [Google Scholar] [CrossRef] [PubMed]
- Caliri, A.W.; Tommasi, S.; Bates, S.E.; Besaratinia, A. Spontaneous and Photosensitization-Induced Mutations in Primary Mouse Cells Transitioning through Senescence and Immortalization. J. Biol. Chem. 2020, 295, 9974–9985. [Google Scholar] [CrossRef] [PubMed]
- Poetsch, A.R. The Genomics of Oxidative DNA Damage, Repair, and Resulting Mutagenesis. Comput. Struct. Biotechnol. J. 2020, 18, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Künzi, L.; Holt, G.E. Cigarette Smoke Activates the Parthanatos Pathway of Cell Death in Human Bronchial Epithelial Cells. Cell Death Discov. 2019, 5, 127. [Google Scholar] [CrossRef] [PubMed]
- Ziech, D.; Franco, R.; Pappa, A.; Panayiotidis, M.I. Reactive Oxygen Species (ROS)––Induced Genetic and Epigenetic Alterations in Human Carcinogenesis. Mutat. Res. Fundam. Mol. Mech. Mutagenesis 2011, 711, 167–173. [Google Scholar] [CrossRef]
- Alnajjar, K.S.; Sweasy, J.B. A New Perspective on Oxidation of DNA Repair Proteins and Cancer. DNA Repair 2019, 76, 60–69. [Google Scholar] [CrossRef]
- Weitzman, S.A.; Turk, P.W.; Milkowski, D.H.; Kozlowski, K. Free Radical Adducts Induce Alterations in DNAcytosine Methylation. Proc. Natl. Acad. Sci. USA 1994, 91, 1261–1264. [Google Scholar] [CrossRef]
- Kuchino, Y.; Mori, F.; Kasai, H.; Inoue, H.; Iwai, S.; Miura, K.; Ohtsuka, E.; Nishimura, S. Misreading of DNA Templates Containing 8-Hydroxydeoxyguanosine at the Modified Base and at Adjacent Residues. Nature 1987, 327, 77–79. [Google Scholar] [CrossRef]
- Lee, K.W.K.; Pausova, Z. Cigarette Smoking and DNA Methylation. Front. Genet. 2013, 4. [Google Scholar] [CrossRef]
- Takiguchi, M.; Achanzar, W.E.; Qu, W.; Li, G.; Waalkes, M.P. Effects of Cadmium on DNA-(Cytosine-5) Methyltransferase Activity and DNA Methylation Status during Cadmium-Induced Cellular Transformation. Exp. Cell Res. 2003, 286, 355–365. [Google Scholar] [CrossRef]
- Lee, Y.-W.; Broday, L.; Costa, M. Effects of Nickel on DNA Methyltransferase Activity and Genomic DNA Methylation Levels. Mutat. Res. Genet. Toxicol. Environ. Mutagenesis 1998, 415, 213–218. [Google Scholar] [CrossRef]
- Coulter, J.B.; O’Driscoll, C.M.; Bressler, J.P. Hydroquinone Increases 5-Hydroxymethylcytosine Formation through Ten Eleven Translocation 1 (TET1) 5-Methylcytosine Dioxygenase. J. Biol. Chem. 2013, 288, 28792–28800. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Li, N.; Guo, C.; Li, X.; Qian, Y.; Yang, Y.; Wei, Y. The Global DNA and RNA Methylation and Their Reversal in Lung under Different Concentration Exposure of Ambient Air Particulate Matter in Mice. Ecotoxicol. Environ. Saf. 2019, 172, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Ringh, M.V.; Hagemann-Jensen, M.; Needhamsen, M.; Kular, L.; Breeze, C.E.; Sjöholm, L.K.; Slavec, L.; Kullberg, S.; Wahlström, J.; Grunewald, J.; et al. Tobacco Smoking Induces Changes in True DNA Methylation, Hydroxymethylation and Gene Expression in Bronchoalveolar Lavage Cells. EBioMedicine 2019, 46, 290–304. [Google Scholar] [CrossRef]
- Kuehne, A.; Emmert, H.; Soehle, J.; Winnefeld, M.; Fischer, F.; Wenck, H.; Gallinat, S.; Terstegen, L.; Lucius, R.; Hildebrand, J.; et al. Acute Activation of Oxidative Pentose Phosphate Pathway as First-Line Response to Oxidative Stress in Human Skin Cells. Mol. Cell 2015, 59, 359–371. [Google Scholar] [CrossRef]
- Sakamoto, K.; Iwasaki, K.; Sugiyama, H.; Tsuji, Y. Role of the Tumor Suppressor PTEN in Antioxidant Responsive Element-Mediated Transcription and Associated Histone Modifications. MBoC 2009, 20, 1606–1617. [Google Scholar] [CrossRef]
- van der Reest, J.; Lilla, S.; Zheng, L.; Zanivan, S.; Gottlieb, E. Proteome-Wide Analysis of Cysteine Oxidation Reveals Metabolic Sensitivity to Redox Stress. Nat. Commun. 2018, 9, 1581. [Google Scholar] [CrossRef]
- Talhout, R.; Schulz, T.; Florek, E.; Van Benthem, J.; Wester, P.; Opperhuizen, A. Hazardous Compounds in Tobacco Smoke. Ijerph 2011, 8, 613–628. [Google Scholar] [CrossRef]
- Wang, R.; Li, S.; Wen, W.; Zhang, J. Multi-Omics Analysis of the Effects of Smoking on Human Tumors. Front. Mol. Biosci. 2021, 8, 704910. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 Regulatory Network Provides an Interface between Redox and Intermediary Metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef] [PubMed]
- Xiao, G.G.; Wang, M.; Li, N.; Loo, J.A.; Nel, A.E. Use of Proteomics to Demonstrate a Hierarchical Oxidative Stress Response to Diesel Exhaust Particle Chemicals in a Macrophage Cell Line. J. Biol. Chem. 2003, 278, 50781–50790. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef] [PubMed]
- Thai, P.; Statt, S.; Chen, C.H.; Liang, E.; Campbell, C.; Wu, R. Characterization of a Novel Long Noncoding RNA, SCAL1, Induced by Cigarette Smoke and Elevated in Lung Cancer Cell Lines. Am. J. Respir. Cell Mol. Biol. 2013, 49, 204–211. [Google Scholar] [CrossRef]
- Cruz, C.R.V.; Ferrer, J.L.M.; Garcia, R.L. Concomitant and Decoupled Effects of Cigarette Smoke and SCAL1 Upregulation on Oncogenic Phenotypes and ROS Detoxification in Lung Adenocarcinoma Cells. Sci Rep. 2021, 11, 18345. [Google Scholar] [CrossRef]
- Chhunchha, B.; Kubo, E.; Singh, D.P. Sulforaphane-Induced Klf9/Prdx6 Axis Acts as a Molecular Switch to Control Redox Signaling and Determines Fate of Cells. Cells 2019, 8, 1159. [Google Scholar] [CrossRef]
- Zucker, S.N.; Fink, E.E.; Bagati, A.; Mannava, S.; Bianchi-Smiraglia, A.; Bogner, P.N.; Wawrzyniak, J.A.; Foley, C.; Leonova, K.I.; Grimm, M.J.; et al. Nrf2 Amplifies Oxidative Stress via Induction of Klf9. Mol. Cell 2014, 53, 916–928. [Google Scholar] [CrossRef]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 Represses Nuclear Activation of Antioxidant Responsive Elements by Nrf2 through Binding to the Amino-Terminal Neh2 Domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef]
- Ma, Q. Role of Nrf2 in Oxidative Stress and Toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef]
- Harder, B.; Jiang, T.; Wu, T.; Tao, S.; de la Vega, M.R.; Tian, W.; Chapman, E.; Zhang, D.D. Molecular Mechanisms of Nrf2 Regulation and How These Influence Chemical Modulation for Disease Intervention. Biochem. Soc. Trans. 2015, 43, 680–686. [Google Scholar] [CrossRef] [PubMed]
- Ohta, T.; Iijima, K.; Miyamoto, M.; Nakahara, I.; Tanaka, H.; Ohtsuji, M.; Suzuki, T.; Kobayashi, A.; Yokota, J.; Sakiyama, T.; et al. Loss of Keap1 Function Activates Nrf2 and Provides Advantages for Lung Cancer Cell Growth. Cancer Res. 2008, 68, 1303–1309. [Google Scholar] [CrossRef] [PubMed]
- Bagga, S.; Bracht, J.; Hunter, S.; Massirer, K.; Holtz, J.; Eachus, R.; Pasquinelli, A.E. Regulation by Let-7 and Lin-4 MiRNAs Results in Target MRNA Degradation. Cell 2005, 122, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Djuranovic, S.; Nahvi, A.; Green, R. MiRNA-Mediated Gene Silencing by Translational Repression Followed by MRNA Deadenylation and Decay. Science 2012, 336, 237–240. [Google Scholar] [CrossRef]
- Papp, D.; Lenti, K.; Módos, D.; Fazekas, D.; Dúl, Z.; Türei, D.; Földvári-Nagy, L.; Nussinov, R.; Csermely, P.; Korcsmáros, T. The NRF2-Related Interactome and Regulome Contain Multifunctional Proteins and Fine-Tuned Autoregulatory Loops. FEBS Lett. 2012, 586, 1795–1802. [Google Scholar] [CrossRef] [PubMed]
- Narasimhan, M.; Patel, D.; Vedpathak, D.; Rathinam, M.; Henderson, G.; Mahimainathan, L. Identification of Novel MicroRNAs in Post-Transcriptional Control of Nrf2 Expression and Redox Homeostasis in Neuronal, SH-SY5Y Cells. PLoS ONE 2012, 7, e51111. [Google Scholar] [CrossRef]
- Eades, G.; Yang, M.; Yao, Y.; Zhang, Y.; Zhou, Q. MiR-200a Regulates Nrf2 Activation by Targeting Keap1 MRNA in Breast Cancer Cells. J. Biol. Chem. 2011, 286, 40725–40733. [Google Scholar] [CrossRef]
- Cheng, X.; Ku, C.-H.; Siow, R.C.M. Regulation of the Nrf2 Antioxidant Pathway by MicroRNAs: New Players in Micromanaging Redox Homeostasis. Free. Radic. Biol. Med. 2013, 64, 4–11. [Google Scholar] [CrossRef]
- Sen, R.; Baltimore, D. Multiple Nuclear Factors Interact with the Immunoglobulin Enhancer Sequences. Cell 1986, 46, 705–716. [Google Scholar] [CrossRef]
- Flohé, L.; Brigelius-Flohé, R.; Saliou, C.; Traber, M.G.; Packer, L. Redox Regulation of NF-Kappa B Activation. Free. Radic. Biol. Med. 1997, 22, 1115–1126. [Google Scholar] [CrossRef]
- May, M.J.; Ghosh, S. Signal Transduction through NF-ΚB. Immunol. Today 1998, 19, 80–88. [Google Scholar] [CrossRef]
- Senftleben, U.; Karin, M. The IKK/NF-KappaB Pathway. Crit. Care Med. 2002, 30, S18–S26. [Google Scholar] [CrossRef] [PubMed]
- Djavaheri-Mergny, M.; Javelaud, D.; Wietzerbin, J.; Besançon, F. NF-ΚB Activation Prevents Apoptotic Oxidative Stress via an Increase of Both Thioredoxin and MnSOD Levels in TNFα-Treated Ewing Sarcoma Cells. FEBS Lett. 2004, 578, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Reuther-Madrid, J.Y.; Kashatus, D.; Chen, S.; Li, X.; Westwick, J.; Davis, R.J.; Earp, H.S.; Wang, C.-Y.; Baldwin, A.S. The P65/RelA Subunit of NF-ΚB Suppresses the Sustained, Antiapoptotic Activity of Jun Kinase Induced by Tumor Necrosis Factor. Mol. Cell Biol. 2002, 22, 8175–8183. [Google Scholar] [CrossRef]
- Tang, F.; Tang, G.; Xiang, J.; Dai, Q.; Rosner, M.R.; Lin, A. The Absence of NF-ΚB-Mediated Inhibition of c-Jun N-Terminal Kinase Activation Contributes to Tumor Necrosis Factor Alpha-Induced Apoptosis. Mol. Cell Biol. 2002, 22, 8571–8579. [Google Scholar] [CrossRef]
- Morgan, M.J.; Kim, Y.-S.; Liu, Z. TNFα and Reactive Oxygen Species in Necrotic Cell Death. Cell Res. 2008, 18, 343–349. [Google Scholar] [CrossRef]
- Papachristou, D.J.; Batistatou, A.; Sykiotis, G.P.; Varakis, I.; Papavassiliou, A.G. Activation of the JNK–AP-1 Signal Transduction Pathway Is Associated with Pathogenesis and Progression of Human Osteosarcomas. Bone 2003, 32, 364–371. [Google Scholar] [CrossRef]
- Angel, P.; Karin, M. The Role of Jun, Fos and the AP-1 Complex in Cell-Proliferation and Transformation. Biochim. Biophys. Acta (BBA)-Rev. Cancer 1991, 1072, 129–157. [Google Scholar] [CrossRef]
- Chang, L.; Karin, M. Mammalian MAP Kinase Signalling Cascades. Nature 2001, 410, 37–40. [Google Scholar] [CrossRef]
- Xanthoudakis, S.; Miao, G.; Wang, F.; Pan, Y.C.; Curran, T. Redox Activation of Fos-Jun DNA Binding Activity Is Mediated by a DNA Repair Enzyme. EMBO J. 1992, 11, 3323–3335. [Google Scholar] [CrossRef]
- Zhong, C.-Y.; Zhou, Y.-M.; Douglas, G.C.; Witschi, H.; Pinkerton, K.E. MAPK/AP-1 Signal Pathway in Tobacco Smoke-Induced Cell Proliferation and Squamous Metaplasia in the Lungs of Rats. Carcinogenesis 2005, 26, 2187–2195. [Google Scholar] [CrossRef] [PubMed]
- Iles, K.E.; Dickinson, D.A.; Watanabe, N.; Iwamoto, T.; Forman, H.J. AP-1 Activation through Endogenous H2O2 Generation by Alveolar Macrophages. Free. Radic. Biol. Med. 2002, 32, 1304–1313. [Google Scholar] [CrossRef]
- Semenza, G.L.; Wang, G.L. A Nuclear Factor Induced by Hypoxia via de Novo Protein Synthesis Binds to the Human Erythropoietin Gene Enhancer at a Site Required for Transcriptional Activation. Mol. Cell. Biol. 1992, 12, 5447–5454. [Google Scholar] [CrossRef] [PubMed]
- Waypa, G.B.; Schumacker, P.T. O2 Sensing in Hypoxic Pulmonary Vasoconstriction: The Mitochondrial Door Re-Opens. Respir. Physiol. Neurobiol. 2002, 132, 81–91. [Google Scholar] [CrossRef]
- Turrens, J.F. Mitochondrial Formation of Reactive Oxygen Species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef]
- Chandel, N.S.; Maltepe, E.; Goldwasser, E.; Mathieu, C.E.; Simon, M.C.; Schumacker, P.T. Mitochondrial Reactive Oxygen Species Trigger Hypoxia-Induced Transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 11715–11720. [Google Scholar] [CrossRef]
- Duranteau, J.; Chandel, N.S.; Kulisz, A.; Shao, Z.; Schumacker, P.T. Intracellular Signaling by Reactive Oxygen Species during Hypoxia in Cardiomyocytes. J. Biol. Chem. 1998, 273, 11619–11624. [Google Scholar] [CrossRef]
- Marshall, C.; Mamary, A.J.; Verhoeven, A.J.; Marshall, B.E. Pulmonary Artery NADPH-Oxidase Is Activated in Hypoxic Pulmonary Vasoconstriction. Am. J. Respir Cell Mol. Biol 1996, 15, 633–644. [Google Scholar] [CrossRef]
- Fukuda, R.; Hirota, K.; Fan, F.; Jung, Y.D.; Ellis, L.M.; Semenza, G.L. Insulin-like Growth Factor 1 Induces Hypoxia-Inducible Factor 1-Mediated Vascular Endothelial Growth Factor Expression, Which Is Dependent on MAP Kinase and Phosphatidylinositol 3-Kinase Signaling in Colon Cancer Cells. J. Biol. Chem. 2002, 277, 38205–38211. [Google Scholar] [CrossRef]
- Görlach, A.; Diebold, I.; Schini-Kerth, V.B.; Berchner-Pfannschmidt, U.; Roth, U.; Brandes, R.P.; Kietzmann, T.; Busse, R. Thrombin Activates the Hypoxia-Inducible Factor-1 Signaling Pathway in Vascular Smooth Muscle Cells: Role of the P22(Phox)-Containing NADPH Oxidase. Circ. Res. 2001, 89, 47–54. [Google Scholar] [CrossRef]
- Blouin, C.C.; Pagé, E.L.; Soucy, G.M.; Richard, D.E. Hypoxic Gene Activation by Lipopolysaccharide in Macrophages: Implication of Hypoxia-Inducible Factor 1α. Blood 2004, 103, 1124–1130. [Google Scholar] [CrossRef] [PubMed]
- Haddad, J.J.; Land, S.C. A Non-Hypoxic, ROS-Sensitive Pathway Mediates TNF-α-Dependent Regulation of HIF-1α. FEBS Lett. 2001, 505, 269–274. [Google Scholar] [CrossRef]
- Richard, D.E. Non-Hypoxic Pathway Mediates the Induction of Hypoxia Inducible Factor 1 Alpha (HIF-1 Alpha ) in Vascular Smooth Muscle Cells. J. Biol. Chem. 2000, 275, 26765–26771. [Google Scholar] [CrossRef]
- Daijo, H.; Hoshino, Y.; Kai, S.; Suzuki, K.; Nishi, K.; Matsuo, Y.; Harada, H.; Hirota, K. Cigarette Smoke Reversibly Activates Hypoxia-Inducible Factor 1 in a Reactive Oxygen Species-Dependent Manner. Sci. Rep. 2016, 6, 34424. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Mett, I.; Bhunia, A.K.; Bowman, J.; Perez, M.; Zhang, L.; Gandjeva, A.; Zhen, L.; Chukwueke, U.; Mao, T.; et al. Rtp801, a Suppressor of MTOR Signaling, Is an Essential Mediator of Cigarette Smoke–Induced Pulmonary Injury and Emphysema. Nat. Med. 2010, 16, 767–773. [Google Scholar] [CrossRef] [PubMed]
- Forrester, K.; Ambs, S.; Lupold, S.E.; Kapust, R.B.; Spillare, E.A.; Weinberg, W.C.; Felley-Bosco, E.; Wang, X.W.; Geller, D.A.; Tzeng, E.; et al. Nitric Oxide-Induced P53 Accumulation and Regulation of Inducible Nitric Oxide Synthase Expression by Wild-Type P53. Proc. Natl. Acad. Sci. USA 1996, 93, 2442–2447. [Google Scholar] [CrossRef] [PubMed]
- Subbaramaiah, K.; Altorki, N.; Chung, W.J.; Mestre, J.R.; Sampat, A.; Dannenberg, A.J. Inhibition of Cyclooxygenase-2 Gene Expression by P53. J. Biol. Chem. 1999, 274, 10911–10915. [Google Scholar] [CrossRef]
- Drane, P.; Bravard, A.; Bouvard, V.; May, E. Reciprocal Down-Regulation of P53 and SOD2 Gene Expression–Implication in P53 Mediated Apoptosis. Oncogene 2001, 20, 430–439. [Google Scholar] [CrossRef]
- Faraonio, R.; Vergara, P.; Di Marzo, D.; Pierantoni, M.G.; Napolitano, M.; Russo, T.; Cimino, F. P53 Suppresses the Nrf2-Dependent Transcription of Antioxidant Response Genes. J. Biol. Chem. 2006, 281, 39776–39784. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of Apoptosis Signalling Pathways by Reactive Oxygen Species. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Wang, T.; Wu, Y.; Yuan, Z.; Dong, J.; Li, X.; An, J.; Liao, Z.; Zhang, X.; Xu, D.; et al. WNT/β-Catenin Signaling Regulates Cigarette Smoke-Induced Airway Inflammation via the PPARδ/P38 Pathway. Lab. Invest. 2016, 96, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, J.; Jiang, Y.; Shi, Y.; Zhu, J.; Xie, C.; Geng, S.; Wu, J.; Zhang, Q.; Wang, X.; et al. Wnt/β-Catenin Modulates Chronic Tobacco Smoke Exposure-Induced Acquisition of Pulmonary Cancer Stem Cell Properties and Diallyl Trisulfide Intervention. Toxicol. Lett. 2018, 291, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Zou, Y.; Zhao, Z.; Li, B.; Ran, P. Nicotine-Induced Epithelial-Mesenchymal Transition via Wnt/β-Catenin Signaling in Human Airway Epithelial Cells. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2013, 304, L199–L209. [Google Scholar] [CrossRef] [PubMed]
- Behrens, J.; von Kries, J.P.; Kühl, M.; Bruhn, L.; Wedlich, D.; Grosschedl, R.; Birchmeier, W. Functional Interaction of β-Catenin with the Transcription Factor LEF-1. Nature 1996, 382, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Essers, M.A.G.; de Vries-Smits, L.M.M.; Barker, N.; Polderman, P.E.; Burgering, B.M.T.; Korswagen, H.C. Functional Interaction Between SS-Catenin and FOXO in Oxidative Stress Signaling. Science 2005, 308, 1181–1184. [Google Scholar] [CrossRef]
- Klotz, L.-O.; Sánchez-Ramos, C.; Prieto-Arroyo, I.; Urbánek, P.; Steinbrenner, H.; Monsalve, M. Redox Regulation of FoxO Transcription Factors. Redox Biol. 2015, 6, 51–72. [Google Scholar] [CrossRef]
- Arnold, R.S.; Shi, J.; Murad, E.; Whalen, A.M.; Sun, C.Q.; Polavarapu, R.; Parthasarathy, S.; Petros, J.A.; Lambeth, J.D. Hydrogen Peroxide Mediates the Cell Growth and Transformation Caused by the Mitogenic Oxidase Nox1. Proc. Natl. Acad. Sci. USA 2001, 98, 5550–5555. [Google Scholar] [CrossRef]
- Funato, Y.; Michiue, T.; Asashima, M.; Miki, H. The Thioredoxin-Related Redox-Regulating Protein Nucleoredoxin Inhibits Wnt–β-Catenin Signalling through Dishevelled. Nat. Cell Biol. 2006, 8, 501–508. [Google Scholar] [CrossRef]
- Korswagen, H.C. Regulation of the Wnt/β-Catenin Pathway by Redox Signaling. Dev. Cell 2006, 10, 687–688. [Google Scholar] [CrossRef]
- Vikram, A.; Kim, Y.-R.; Kumar, S.; Naqvi, A.; Hoffman, T.A.; Kumar, A.; Miller, F.J.; Kim, C.-S.; Irani, K. Canonical Wnt Signaling Induces Vascular Endothelial Dysfunction via P66 Shc -Regulated Reactive Oxygen Species. ATVB 2014, 34, 2301–2309. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Qi, H.; Luo, F.; Xu, H.; Ling, M.; Qin, Y.; Yang, P.; Liu, X.; Yang, Q.; Xue, J.; et al. Feedback Circuitry via Let-7c between LncRNA CCAT1 and c-Myc Is Involved in Cigarette Smoke Extract-Induced Malignant Transformation of HBE Cells. Oncotarget 2017, 8, 19285–19297. [Google Scholar] [CrossRef]
- Iwakawa, R.; Kohno, T.; Kato, M.; Shiraishi, K.; Tsuta, K.; Noguchi, M.; Ogawa, S.; Yokota, J. MYC Amplification as a Prognostic Marker of Early-Stage Lung Adenocarcinoma Identified by Whole Genome Copy Number Analysis. Clin. Cancer Res. 2011, 17, 1481–1489. [Google Scholar] [CrossRef] [PubMed]
- Wolfer, A.; Wittner, B.S.; Irimia, D.; Flavin, R.J.; Lupien, M.; Gunawardane, R.N.; Meyer, C.A.; Lightcap, E.S.; Tamayo, P.; Mesirov, J.P.; et al. MYC Regulation of a “Poor-Prognosis” Metastatic Cancer Cell State. Proc. Natl. Acad. Sci. USA 2010, 107, 3698–3703. [Google Scholar] [CrossRef] [PubMed]
- Seo, A.N.; Yang, J.M.; Kim, H.; Jheon, S.; Kim, K.; Lee, C.T.; Jin, Y.; Yun, S.; Chung, J.-H.; Paik, J.H. Clinicopathologic and Prognostic Significance of C-MYC Copy Number Gain in Lung Adenocarcinomas. Br. J. Cancer 2014, 110, 2688–2699. [Google Scholar] [CrossRef]
- Lewis, L.M.; Edwards, M.C.; Meyers, Z.R.; Talbot, C.C.; Hao, H.; Blum, D.; Reproducibility Project: Cancer Biology; Iorns, E.; Tsui, R.; Denis, A.; et al. Replication Study: Transcriptional Amplification in Tumor Cells with Elevated c-Myc. eLife 2018, 7, e30274. [Google Scholar] [CrossRef]
- Nissan, A.; Stojadinovic, A.; Mitrani-Rosenbaum, S.; Halle, D.; Grinbaum, R.; Roistacher, M.; Bochem, A.; Dayanc, B.E.; Ritter, G.; Gomceli, I.; et al. Colon Cancer Associated Transcript-1: A Novel RNA Expressed in Malignant and Pre-Malignant Human Tissues. Int. J. Cancer 2012, 130, 1598–1606. [Google Scholar] [CrossRef]
- Yang, F.; Xue, X.; Bi, J.; Zheng, L.; Zhi, K.; Gu, Y.; Fang, G. Long Noncoding RNA CCAT1, Which Could Be Activated by c-Myc, Promotes the Progression of Gastric Carcinoma. J. Cancer Res. Clin. Oncol 2013, 139, 437–445. [Google Scholar] [CrossRef]
- Deng, L.; Yang, S.-B.; Xu, F.-F.; Zhang, J.-H. Long Noncoding RNA CCAT1 Promotes Hepatocellular Carcinoma Progression by Functioning as Let-7 Sponge. J. Exp. Clin. Cancer Res. 2015, 34, 18. [Google Scholar] [CrossRef]
- Ozawa, T.; Matsuyama, T.; Toiyama, Y.; Takahashi, N.; Ishikawa, T.; Uetake, H.; Yamada, Y.; Kusunoki, M.; Calin, G.; Goel, A. CCAT1 and CCAT2 Long Noncoding RNAs, Located within the 8q.24.21 ‘Gene Desert’, Serve as Important Prognostic Biomarkers in Colorectal Cancer. Ann. Oncol. 2017, 28, 1882–1888. [Google Scholar] [CrossRef]
- Shang, A.; Wang, W.; Gu, C.; Chen, W.; Lu, W.; Sun, Z.; Li, D. Long Non-Coding RNA CCAT1 Promotes Colorectal Cancer Progression by Regulating MiR-181a-5p Expression. Aging 2020, 12, 8301–8320. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhou, H.; Strulovici-Barel, Y.; Al-Hijji, M.; Ou, X.; Salit, J.; Walters, M.S.; Staudt, M.R.; Kaner, R.J.; Crystal, R.G. Role of OSGIN1 in Mediating Smoking-Induced Autophagy in the Human Airway Epithelium. Autophagy 2017, 13, 1205–1220. [Google Scholar] [CrossRef] [PubMed]
- Ong, C.K.; Ng, C.Y.; Leong, C.; Ng, C.P.; Foo, K.T.; Tan, P.H.; Huynh, H. Genomic Structure of Human OKL38 Gene and Its Differential Expression in Kidney Carcinogenesis. J. Biol. Chem. 2004, 279, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Brennan, M.S.; Matos, M.F.; Richter, K.E.; Li, B.; Scannevin, R.H. The NRF2 Transcriptional Target, OSGIN1, Contributes to Monomethyl Fumarate-Mediated Cytoprotection in Human Astrocytes. Sci. Rep. 2017, 7, 42054. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Chen, W.; Yanes, R.; Lee, S.; Berliner, J.A. OKL38 Is an Oxidative Stress Response Gene Stimulated by Oxidized Phospholipids. J. Lipid Res. 2007, 48, 709–715. [Google Scholar] [CrossRef]
- Schuller, H.M.; Orloff, M. Tobacco-Specific Carcinogenic Nitrosamines. Biochem. Pharmacol. 1998, 55, 1377–1384. [Google Scholar] [CrossRef]
- Zhang, C.; Ding, X.-P.; Zhao, Q.-N.; Yang, X.-J.; An, S.-M.; Wang, H.; Xu, L.; Zhu, L.; Chen, H.-Z. Role of A7-Nicotinic Acetylcholine Receptor in Nicotine-Induced Invasion and Epithelial-to-Mesenchymal Transition in Human Non-Small Cell Lung Cancer Cells. Oncotarget 2016, 7, 59199–59208. [Google Scholar] [CrossRef]
- Xu, L.; Deng, X. Tobacco-Specific Nitrosamine 4-(Methylnitrosamino)-1-(3-Pyridyl)-1-Butanone Induces Phosphorylation of μ- and m-Calpain in Association with Increased Secretion, Cell Migration, and Invasion. J. Biol. Chem. 2004, 279, 53683–53690. [Google Scholar] [CrossRef]
- Heeschen, C.; Weis, M.; Aicher, A.; Dimmeler, S.; Cooke, J.P. A Novel Angiogenic Pathway Mediated by Non-Neuronal Nicotinic Acetylcholine Receptors. J. Clin. Invest. 2002, 110, 527–536. [Google Scholar] [CrossRef]
- Song, P.; Sekhon, H.S.; Jia, Y.; Keller, J.A.; Blusztajn, J.K.; Mark, G.P.; Spindel, E.R. Acetylcholine Is Synthesized by and Acts as an Autocrine Growth Factor for Small Cell Lung Carcinoma. Cancer Res. 2003, 63, 214–221. [Google Scholar]
- Govind, A.P.; Vezina, P.; Green, W.N. Nicotine-Induced Upregulation of Nicotinic Receptors: Underlying Mechanisms and Relevance to Nicotine Addiction. Biochem. Pharmacol. 2009, 78, 756–765. [Google Scholar] [CrossRef] [PubMed]
- Lam, D.C.; Girard, L.; Ramirez, R.; Chau, W.; Suen, W.; Sheridan, S.; Tin, V.P.C.; Chung, L.; Wong, M.P.; Shay, J.W.; et al. Expression of Nicotinic Acetylcholine Receptor Subunit Genes in Non–Small-Cell Lung Cancer Reveals Differences between Smokers and Nonsmokers. Cancer Res. 2007, 67, 4638–4647. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.C.; Perry, H.E.; Lau, J.K.; Jones, D.V.; Pulliam, J.F.; Thornhill, B.A.; Crabtree, C.M.; Luo, H.; Chen, Y.C.; Dasgupta, P. Nicotine Induces the Up-Regulation of the A7-Nicotinic Receptor (A7-NAChR) in Human Squamous Cell Lung Cancer Cells via the Sp1/GATA Protein Pathway. J. Biol. Chem. 2013, 288, 33049–33059. [Google Scholar] [CrossRef] [PubMed]
- Hung, R.J.; McKay, J.D.; Gaborieau, V.; Boffetta, P.; Hashibe, M.; Zaridze, D.; Mukeria, A.; Szeszenia-Dabrowska, N.; Lissowska, J.; Rudnai, P.; et al. A Susceptibility Locus for Lung Cancer Maps to Nicotinic Acetylcholine Receptor Subunit Genes on 15q25. Nature 2008, 452, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Lu, X.; Qiu, F.; Fang, W.; Zhang, L.; Huang, D.; Xie, C.; Zhong, N.; Ran, P.; Zhou, Y.; et al. Duplicated Copy of CHRNA7 Increases Risk and Worsens Prognosis of COPD and Lung Cancer. Eur. J. Hum. Genet. 2015, 23, 1019–1024. [Google Scholar] [CrossRef] [PubMed]
- Lam, D.C.-L.; Luo, S.Y.; Fu, K.-H.; Lui, M.M.-S.; Chan, K.-H.; Wistuba, I.I.; Gao, B.; Tsao, S.-W.; Ip, M.S.-M.; Minna, J.D. Nicotinic Acetylcholine Receptor Expression in Human Airway Correlates with Lung Function. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2016, 310, L232–L239. [Google Scholar] [CrossRef]
- Gahring, L.C.; Myers, E.J.; Dunn, D.M.; Weiss, R.B.; Rogers, S.W. Lung Epithelial Response to Cigarette Smoke and Modulation by the Nicotinic Alpha 7 Receptor. PLoS ONE 2017, 12, e0187773. [Google Scholar] [CrossRef]
- Nguyen, H.D.; Liao, Y.-C.; Ho, Y.-S.; Chen, L.-C.; Chang, H.-W.; Cheng, T.-C.; Liu, D.; Lee, W.-R.; Shen, S.-C.; Wu, C.-H.; et al. The A9 Nicotinic Acetylcholine Receptor Mediates Nicotine-Induced PD-L1 Expression and Regulates Melanoma Cell Proliferation and Migration. Cancers 2019, 11, 1991. [Google Scholar] [CrossRef]
- Messenheimer, D.J.; Jensen, S.M.; Afentoulis, M.E.; Wegmann, K.W.; Feng, Z.; Friedman, D.J.; Gough, M.J.; Urba, W.J.; Fox, B.A. Timing of PD-1 Blockade Is Critical to Effective Combination Immunotherapy with Anti-OX40. Clin. Cancer Res. 2017, 23, 6165–6177. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, B.; Zhu, J.; Wang, D.; Wang, Y. Nicotine-Mediated Autophagy of Vascular Smooth Muscle Cell Accelerates Atherosclerosis via NAChRs/ROS/NF-ΚB Signaling Pathway. Atherosclerosis 2019, 284, 1–10. [Google Scholar] [CrossRef]
- Kim, C.S.; Choi, J.S.; Joo, S.Y.; Bae, E.H.; Ma, S.K.; Lee, J.; Kim, S.W. Nicotine-Induced Apoptosis in Human Renal Proximal Tubular Epithelial Cells. PLoS ONE 2016, 11, e0152591. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhang, H.; Qi, W.; Zhang, Y.; Li, J.; Li, Z.; Lin, Y.; Bai, X.; Liu, X.; Chen, X.; et al. Nicotine Promotes Atherosclerosis via ROS-NLRP3-Mediated Endothelial Cell Pyroptosis. Cell Death Dis. 2018, 9, 171. [Google Scholar] [CrossRef]
- Li, Y.; King, M.A.; Meyer, E.M. A7 Nicotinic Receptor-Mediated Protection against Ethanol-Induced Oxidative Stress and Cytotoxicity in PC12 Cells. Brain Res. 2000, 861, 165–167. [Google Scholar] [CrossRef]
- Guan, Z. Dual Effects of Nicotine on Oxidative Stress and Neuroprotection in PC12 Cells. Neurochem. Int. 2003, 43, 243–249. [Google Scholar] [CrossRef]
- Patel, H.; McIntire, J.; Ryan, S.; Dunah, A.; Loring, R. Anti-Inflammatory Effects of Astroglial A7 Nicotinic Acetylcholine Receptors Are Mediated by Inhibition of the NF-ΚB Pathway and Activation of the Nrf2 Pathway. J. Neuroinflammation 2017, 14, 192. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.-L.; Nordberg, A.; Xiu, J.; Guan, Z.-Z. The Consequences of Reducing Expression of the A7 Nicotinic Receptor by RNA Interference and of Stimulating Its Activity with an A7 Agonist in SH-SY5Y Cells Indicate That This Receptor Plays a Neuroprotective Role in Connection with the Pathogenesis of Alzheimer’s Disease. Neurochem. Int. 2007, 51, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative Stress and Antioxidant Defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef]
- Muftuoglu, M.; Mori, M.P.; Souza-Pinto, N.C. de Formation and Repair of Oxidative Damage in the Mitochondrial DNA. Mitochondrion 2014, 17, 164–181. [Google Scholar] [CrossRef]
- Mirończuk-Chodakowska, I.; Witkowska, A.M.; Zujko, M.E. Endogenous Non-Enzymatic Antioxidants in the Human Body. Adv. Med. Sci. 2018, 63, 68–78. [Google Scholar] [CrossRef]
- Crapo, J.; Tierney, D. Superoxide Dismutase and Pulmonary Oxygen Toxicity. Am. J. Physiol.-Leg. Content 1974, 226, 1401–1407. [Google Scholar] [CrossRef]
- Kinnula, V.L.; Crapo, J.D.; Raivio, K.O. Generation and Disposal of Reactive Oxygen Metabolites in the Lung. Lab. Invest. 1995, 73, 3–19. [Google Scholar] [PubMed]
- McCord, J.M.; Fridovich, I. Superoxide Dismutase. J. Biol. Chem. 1969, 244, 6049–6055. [Google Scholar] [CrossRef]
- Marklund, S.L. Human Copper-Containing Superoxide Dismutase of High Molecular Weight. Proc. Natl. Acad. Sci. USA 1982, 79, 7634–7638. [Google Scholar] [CrossRef]
- Weisiger, R.A.; Fridovich, I. Mitochondrial Superoxide Simutase. Site of Synthesis and Intramitochondrial Localization. J. Biol. Chem. 1973, 248, 4793–4796. [Google Scholar] [CrossRef]
- Zelko, I.N.; Mariani, T.J.; Folz, R.J. Superoxide Dismutase Multigene Family: A Comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) Gene Structures, Evolution, and Expression. Free. Radic. Biol. Med. 2002, 33, 337–349. [Google Scholar] [CrossRef]
- Sheng, Y.; Abreu, I.A.; Cabelli, D.E.; Maroney, M.J.; Miller, A.-F.; Teixeira, M.; Valentine, J.S. Superoxide Dismutases and Superoxide Reductases. Chem. Rev. 2014, 114, 3854–3918. [Google Scholar] [CrossRef]
- Gill, J.G.; Piskounova, E.; Morrison, S.J. Cancer, Oxidative Stress, and Metastasis. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 163–175. [Google Scholar] [CrossRef]
- Kirkman, H.N.; Gaetani, G.F. Mammalian Catalase: A Venerable Enzyme with New Mysteries. Trends Biochem. Sci. 2007, 32, 44–50. [Google Scholar] [CrossRef]
- Brigelius-Flohé, R.; Maiorino, M. Glutathione Peroxidases. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2013, 1830, 3289–3303. [Google Scholar] [CrossRef]
- Elko, E.A.; Cunniff, B.; Seward, D.J.; Chia, S.B.; Aboushousha, R.; van de Wetering, C.; van der Velden, J.; Manuel, A.; Shukla, A.; Heintz, N.H.; et al. Peroxiredoxins and Beyond; Redox Systems Regulating Lung Physiology and Disease. Antioxid. Redox Signal. 2019, 31, 1070–1091. [Google Scholar] [CrossRef]
- Benhar, M. Roles of Mammalian Glutathione Peroxidase and Thioredoxin Reductase Enzymes in the Cellular Response to Nitrosative Stress. Free. Radic. Biol. Med. 2018, 127, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Aquilano, K.; Baldelli, S.; Ciriolo, M.R. Glutathione: New Roles in Redox Signaling for an Old Antioxidant. Front. Pharmacol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Ighodaro, O.M.; Akinloye, O.A. First Line Defence Antioxidants-Superoxide Dismutase (SOD), Catalase (CAT) and Glutathione Peroxidase (GPX): Their Fundamental Role in the Entire Antioxidant Defence Grid. Alex. J. Med. 2018, 54, 287–293. [Google Scholar] [CrossRef]
- Tinggi, U. Selenium: Its Role as Antioxidant in Human Health. Environ. Health Prev. Med. 2008, 13, 102–108. [Google Scholar] [CrossRef]
- Stocks, J.; Gutteridge, J.M.C.; Sharp, R.J.; Dormandy, T.L. The Inhibition of Lipid Autoxidation by Human Serum and Its Relation to Serum Proteins and α-Tocopherol. Clin. Sci. 1974, 47, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Balla, G.; Jacob, H.S.; Balla, J.; Rosenberg, M.; Nath, K.; Apple, F.; Eaton, J.W.; Vercellotti, G.M. Ferritin: A Cytoprotective Antioxidant Strategem of Endothelium. J. Biol. Chem. 1992, 267, 18148–18153. [Google Scholar] [CrossRef]
- Cairo, G.; Tacchini, L.; Pogliaghi, G.; Anzon, E.; Tomasi, A.; Bernelli-Zazzera, A. Induction of Ferritin Synthesis by Oxidative Stress. J. Biol. Chem. 1995, 270, 700–703. [Google Scholar] [CrossRef]
- Gutteridge, J.M.C.; Paterson, S.K.; Segal, A.W.; Halliwell, B. Inhibition of Lipid Peroxidation by the Iron-Binding Protein Lactoferrin. Biochem. J. 1981, 199, 259–261. [Google Scholar] [CrossRef]
- Gutteridge, J.M.C. Antioxidant Properties of Caeruloplasmin towards Iron- and Copper-Dependent Oxygen Radical Formation. FEBS Lett. 1983, 157, 37–40. [Google Scholar] [CrossRef]
- Al-Timimi, D.J.; Dormandy, T.L. The Inhibition of Lipid Autoxidation by Human Caeruloplasmin. Biochem. J. 1977, 168, 283–288. [Google Scholar] [CrossRef]
- Atanasiu, R.L.; Stea, D.; Mateescu, M.A.; Vergely, C.; Dalloz, F.; Briot, F.; Maupoil, V.; Nadeau, R.; Rochette, L. Direct Evidence of Caeruloplasmin Antioxidant Properties. Mol. Cell Biochem. 1998, 189, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Loban, A.; Kime, R.; Powers, H. Iron-Binding Antioxidant Potential of Plasma Albumin. Clin. Sci. 1997, 93, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Marx, G.; Chevion, M. Site-Specific Modification of Albumin by Free Radicals. Reaction with Copper(II) and Ascorbate. Biochem. J. 1986, 236, 397–400. [Google Scholar] [CrossRef] [PubMed]
- Gutteridge, J.M.C. Antioxidant Properties of the Proteins Caeruloplasmin, Albumin and Transferrin. A Study of Their Activity in Serum and Synovial Fluid from Patients with Rheumatoid Arthritis. Biochim. Et Biophys. Acta (BBA)-Protein Struct. Mol. Enzymol. 1986, 869, 119–127. [Google Scholar] [CrossRef]
- Wayner, D.D.M.; Burton, G.W.; Ingold, K.U.; Locke, S. Quantitative Measurement of the Total, Peroxyl Radical-Trapping Antioxidant Capability of Human Blood Plasma by Controlled Peroxidation: The Important Contribution Made by Plasma Proteins. FEBS Lett. 1985, 187, 33–37. [Google Scholar] [CrossRef]
- Flögel, U.; Merx, M.W.; Gödecke, A.; Decking, U.K.M.; Schrader, J. Myoglobin: A Scavenger of Bioactive NO. Proc. Natl. Acad. Sci. USA 2001, 98, 735–740. [Google Scholar] [CrossRef]
- Burton, G.W.; Ingold, K.U. Autoxidation of Biological Molecules. 1. Antioxidant Activity of Vitamin E and Related Chain-Breaking Phenolic Antioxidants in Vitro. J. Am. Chem. Soc. 1981, 103, 6472–6477. [Google Scholar] [CrossRef]
- Burton, G.W.; Joyce, A.; Ingold, K.U. First proof that vitamin e is major lipid-soluble, chain-breaking antioxidant in human blood plasma. Lancet 1982, 320, 327. [Google Scholar] [CrossRef]
- Yu, W.; Simmons-Menchaca, M.; Gapor, A.; Sanders, B.G.; Kline, K. Induction of Apoptosis in Human Breast Cancer Cells by Tocopherols and Tocotrienols. Nutr. Cancer 1999, 33, 26–32. [Google Scholar] [CrossRef]
- Doba, T.; Burton, G.W.; Ingold, K.U. Antioxidant and Co-Antioxidant Activity of Vitamin C. The Effect of Vitamin C, Either Alone or in the Presence of Vitamin E or a Water-Soluble Vitamin E Analogue, upon the Peroxidation of Aqueous Multilamellar Phospholipid Liposomes. Biochim. Biophys. Acta (BBA)-Lipids Lipid Metab. 1985, 835, 298–303. [Google Scholar] [CrossRef]
- Packer, J.E.; Slater, T.F.; Willson, R.L. Direct Observation of a Free Radical Interaction between Vitamin E and Vitamin C. Nature 1979, 278, 737–738. [Google Scholar] [CrossRef] [PubMed]
- Ames, B.N.; Cathcart, R.; Schwiers, E.; Hochstein, P. Uric Acid Provides an Antioxidant Defense in Humans against Oxidant- and Radical-Caused Aging and Cancer: A Hypothesis. Proc. Natl. Acad. Sci. USA 1981, 78, 6858–6862. [Google Scholar] [CrossRef] [PubMed]
- Muraoka, S.; Miura, T. Inhibition by Uric Acid of Free Radicals That Damage Biological Molecules. Pharm. Toxicol 2003, 93, 284–289. [Google Scholar] [CrossRef] [PubMed]
- Kuzkaya, N.; Weissmann, N.; Harrison, D.G.; Dikalov, S. Interactions of Peroxynitrite with Uric Acid in the Presence of Ascorbate and Thiols: Implications for Uncoupling Endothelial Nitric Oxide Synthase. Biochem. Pharmacol. 2005, 70, 343–354. [Google Scholar] [CrossRef]
- Tapiero, H.; Townsend, D.M.; Tew, K.D. The Antioxidant Role of Selenium and Seleno-Compounds. Biomed. Pharmacother. 2003, 57, 134–144. [Google Scholar] [CrossRef]
- Kryukov, G.V.; Castellano, S.; Novoselov, S.V.; Lobanov, A.V.; Zehtab, O.; Guigó, R.; Gladyshev, V.N. Characterization of Mammalian Selenoproteomes. Science 2003, 300, 1439–1443. [Google Scholar] [CrossRef]
- Poeggeler, B.; Reiter, R.J.; Tan, D.-X.; Chen, L.-D.; Manchester, L.C. Melatonin, Hydroxyl Radical-Mediated Oxidative Damage, and Aging: A Hypothesis. J. Pineal Res. 1993, 14, 151–168. [Google Scholar] [CrossRef]
- Stocker, R.; Glazer, A.N.; Ames, B.N. Antioxidant Activity of Albumin-Bound Bilirubin. Proc. Natl. Acad. Sci. USA 1987, 84, 5918–5922. [Google Scholar] [CrossRef]
- Løvaas, E. Antioxidative Effects of Polyamines. J. Am. Oil Chem. Soc. 1991, 68, 353–358. [Google Scholar] [CrossRef]
- Rhee, S.G.; Bae, S.H. The Antioxidant Function of Sestrins Is Mediated by Promotion of Autophagic Degradation of Keap1 and Nrf2 Activation and by Inhibition of MTORC1. Free. Radic. Biol. Med. 2015, 88, 205–211. [Google Scholar] [CrossRef]
- Hayes, J.D.; Flanagan, J.U.; Jowsey, I.R. GLUTATHIONE TRANSFERASES. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 51–88. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Penning, T.M. Aldo-Keto Reductases and Bioactivation/Detoxication. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 263–292. [Google Scholar] [CrossRef] [PubMed]
- Oppermann, U. Carbonyl Reductases: The Complex Relationships of Mammalian Carbonyl- and Quinone-Reducing Enzymes and Their Role in Physiology. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 293–322. [Google Scholar] [CrossRef]
- Rodríguez-Zavala, J.S.; Calleja, L.F.; Moreno-Sánchez, R.; Yoval-Sánchez, B. Role of Aldehyde Dehydrogenases in Physiopathological Processes. Chem. Res. Toxicol. 2019, 32, 405–420. [Google Scholar] [CrossRef] [PubMed]
- Rowland, A.; Miners, J.O.; Mackenzie, P.I. The UDP-Glucuronosyltransferases: Their Role in Drug Metabolism and Detoxification. Int. J. Biochem. Cell Biol. 2013, 45, 1121–1132. [Google Scholar] [CrossRef] [PubMed]
- Kincaid, B.; Bossy-Wetzel, E. Forever Young: SIRT3 a Shield against Mitochondrial Meltdown, Aging, and Neurodegeneration. Front. Aging Neurosci. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Fullwood, M.J. Roles, Functions, and Mechanisms of Long Non-Coding RNAs in Cancer. Genom. Proteom. Bioinform. 2016, 14, 42–54. [Google Scholar] [CrossRef]
- Gutschner, T.; Diederichs, S. The Hallmarks of Cancer: A Long Non-Coding RNA Point of View. RNA Biol. 2012, 9, 703–719. [Google Scholar] [CrossRef]
- Chen, J.; Ke, S.; Zhong, L.; Wu, J.; Tseng, A.; Morpurgo, B.; Golovko, A.; Wang, G.; Cai, J.J.; Ma, X.; et al. Long Noncoding RNA MALAT1 Regulates Generation of Reactive Oxygen Species and the Insulin Responses in Male Mice. Biochem. Pharmacol. 2018, 152, 94–103. [Google Scholar] [CrossRef]
- Zhao, Z.; Hao, W.; Meng, Q.; Du, X.; Lei, S.; Xia, Z. Long Non-Coding RNA MALAT1 Functions as a Mediator in Cardioprotective Effects of Fentanyl in Myocardial Ischemia-Reperfusion Injury: MALAT1 in Myocardial I/R Injury. Cell Biol. Int. 2017, 41, 62–70. [Google Scholar] [CrossRef]
- Yang, L.; Xu, F.; Zhang, M.; Shang, X.-Y.; Xie, X.; Fu, T.; Li, J.-P.; Li, H.-L. Role of LncRNA MALAT-1 in Hypoxia-Induced PC12 Cell Injury via Regulating P38MAPK Signaling Pathway. Neurosci. Lett. 2018, 670, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Zhang, Y.; Liu, W.; Zhu, X.; Xu, D.; Zhao, J.; Cui, Z. Long Non-Coding RNA MALAT1 Protects Human Osteoblasts from Dexamethasone-Induced Injury via Activation of PPM1E-AMPK Signaling. Cell Physiol. Biochem. 2018, 51, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Chen, M.; Li, C.; Xu, M.; Liu, Y.; Cong, M.; Sang, N.; Liu, S. Long Non-Coding RNA MT1DP Shunts the Cellular Defense to Cytotoxicity through Crosstalk with MT1H and RhoC in Cadmium Stress. Cell Discov. 2018, 4, 5. [Google Scholar] [CrossRef] [PubMed]
- Gong, W.; Li, J.; Zhu, G.; Wang, Y.; Zheng, G.; Kan, Q. Chlorogenic Acid Relieved Oxidative Stress Injury in Retinal Ganglion Cells through IncRNA-TUG1/Nrf2. Cell Cycle 2019, 18, 1549–1559. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Zhao, B.; Chen, M.; Liu, Y.; Xu, M.; Wang, Z.; Liu, S.; Zhang, C. Nrf-2-Driven Long Noncoding RNA ODRUL Contributes to Modulating Silver Nanoparticle-Induced Effects on Erythroid Cells. Biomaterials 2017, 130, 14–27. [Google Scholar] [CrossRef]
- Li, W.-W.; Cao, A.-H.; Sun, F.-Y. LncRNA MIAT Stimulates Oxidative Stress in the Hypoxic Pulmonary Hypertension Model by Sponging MiR-29a-5p and Inhibiting Nrf2 Pathway. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 9022–9029. [Google Scholar] [CrossRef]
- Zhang, D.; Lee, H.; Haspel, J.A.; Jin, Y. Long Noncoding RNA FOXD3-AS1 Regulates Oxidative Stress-Induced Apoptosis via Sponging MicroRNA-150. FASEB J. 2017, 31, 4472–4481. [Google Scholar] [CrossRef]
- Wang, W.-T.; Ye, H.; Wei, P.-P.; Han, B.-W.; He, B.; Chen, Z.-H.; Chen, Y.-Q. LncRNAs H19 and HULC, Activated by Oxidative Stress, Promote Cell Migration and Invasion in Cholangiocarcinoma through a CeRNA Manner. J. Hematol Oncol. 2016, 9, 117. [Google Scholar] [CrossRef]
- Sirey, T.M.; Roberts, K.; Haerty, W.; Bedoya-Reina, O.; Rogatti-Granados, S.; Tan, J.Y.; Li, N.; Heather, L.C.; Carter, R.N.; Cooper, S.; et al. The Long Non-Coding RNA Cerox1 Is a Post Transcriptional Regulator of Mitochondrial Complex I Catalytic Activity. eLife 2019, 8, e45051. [Google Scholar] [CrossRef]
- Crane, F.L.; Hatefi, Y.; Lester, R.L.; Widmer, C. Isolation of a Quinone from Beef Heart Mitochondria. Biochim. Biophys. Acta 1957, 25, 220–221. [Google Scholar] [CrossRef]
- Ren, J.; Wang, A.; Liu, J.; Yuan, Q. Identification and Validation of a Novel Redox-Related LncRNA Prognostic Signature in Lung Adenocarcinoma. Bioengineered 2021, 12, 4331–4348. [Google Scholar] [CrossRef] [PubMed]
- Rustom, A. Nanotubular Highways for Intercellular Organelle Transport. Science 2004, 303, 1007–1010. [Google Scholar] [CrossRef] [PubMed]
- Van Prooyen, N.; Gold, H.; Andresen, V.; Schwartz, O.; Jones, K.; Ruscetti, F.; Lockett, S.; Gudla, P.; Venzon, D.; Franchini, G. Human T-Cell Leukemia Virus Type 1 P8 Protein Increases Cellular Conduits and Virus Transmission. Proc. Natl. Acad. Sci. USA 2010, 107, 20738–20743. [Google Scholar] [CrossRef]
- Ahmad, T.; Mukherjee, S.; Pattnaik, B.; Kumar, M.; Singh, S.; Kumar, M.; Rehman, R.; Tiwari, B.K.; Jha, K.A.; Barhanpurkar, A.P.; et al. Miro1 Regulates Intercellular Mitochondrial Transport & Enhances Mesenchymal Stem Cell Rescue Efficacy. EMBO J. 2014, 33, 994–1010. [Google Scholar] [CrossRef] [PubMed]
- Watkins, S.C.; Salter, R.D. Functional Connectivity between Immune Cells Mediated by Tunneling Nanotubules. Immunity 2005, 23, 309–318. [Google Scholar] [CrossRef]
- Arkwright, P.D.; Luchetti, F.; Tour, J.; Roberts, C.; Ayub, R.; Morales, A.P.; Rodríguez, J.J.; Gilmore, A.; Canonico, B.; Papa, S.; et al. Fas Stimulation of T Lymphocytes Promotes Rapid Intercellular Exchange of Death Signals via Membrane Nanotubes. Cell Res. 2010, 20, 72–88. [Google Scholar] [CrossRef]
- Polak, R.; de Rooij, B.; Pieters, R.; den Boer, M.L. B-Cell Precursor Acute Lymphoblastic Leukemia Cells Use Tunneling Nanotubes to Orchestrate Their Microenvironment. Blood 2015, 126, 2404–2414. [Google Scholar] [CrossRef]
- Omsland, M.; Pise-Masison, C.; Fujikawa, D.; Galli, V.; Fenizia, C.; Parks, R.W.; Gjertsen, B.T.; Franchini, G.; Andresen, V. Inhibition of Tunneling Nanotube (TNT) Formation and Human T-Cell Leukemia Virus Type 1 (HTLV-1) Transmission by Cytarabine. Sci. Rep. 2018, 8, 11118. [Google Scholar] [CrossRef]
- Panasiuk, M.; Rychłowski, M.; Derewońko, N.; Bieńkowska-Szewczyk, K. Tunneling Nanotubes as a Novel Route of Cell-to-Cell Spread of Herpesviruses. J. Virol. 2018, 92, e00090-18. [Google Scholar] [CrossRef]
- Wang, X.; Bukoreshtliev, N.V.; Gerdes, H.-H. Developing Neurons Form Transient Nanotubes Facilitating Electrical Coupling and Calcium Signaling with Distant Astrocytes. PLoS ONE 2012, 7, e47429. [Google Scholar] [CrossRef]
- Wang, X.; Veruki, M.L.; Bukoreshtliev, N.V.; Hartveit, E.; Gerdes, H.-H. Animal Cells Connected by Nanotubes Can Be Electrically Coupled through Interposed Gap-Junction Channels. Proc. Natl. Acad. Sci. USA 2010, 107, 17194–17199. [Google Scholar] [CrossRef]
- Sowinski, S.; Jolly, C.; Berninghausen, O.; Purbhoo, M.A.; Chauveau, A.; Köhler, K.; Oddos, S.; Eissmann, P.; Brodsky, F.M.; Hopkins, C.; et al. Membrane Nanotubes Physically Connect T Cells over Long Distances Presenting a Novel Route for HIV-1 Transmission. Nat. Cell Biol. 2008, 10, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Hase, K.; Kimura, S.; Takatsu, H.; Ohmae, M.; Kawano, S.; Kitamura, H.; Ito, M.; Watarai, H.; Hazelett, C.C.; Yeaman, C.; et al. M-Sec Promotes Membrane Nanotube Formation by Interacting with Ral and the Exocyst Complex. Nat. Cell Biol. 2009, 11, 1427–1432. [Google Scholar] [CrossRef] [PubMed]
- Valdebenito, S.; Audia, A.; Bhat, K.P.L.; Okafo, G.; Eugenin, E.A. Tunneling Nanotubes Mediate Adaptation of Glioblastoma Cells to Temozolomide and Ionizing Radiation Treatment. iScience 2020, 23, 101450. [Google Scholar] [CrossRef] [PubMed]
- Hanna, S.; McCoy-Simandle, K.; Leung, E.; Condeelis, J.; Cox, D. Abstract 969: Tunneling Nanotubes, a Novel Mode of Tumor Cell-Macrophage Communication in Tumor Cell Invasion. J. Cell Sci. 2019, 132, jcs223321. [Google Scholar] [CrossRef]
- Venkatesh, V.S.; Lou, E. Tunneling Nanotubes: A Bridge for Heterogeneity in Glioblastoma and a New Therapeutic Target? Cancer Rep. 2019, 2, e1185. [Google Scholar] [CrossRef]
- Thayanithy, V.; Dickson, E.L.; Steer, C.; Subramanian, S.; Lou, E. Tumor-Stromal Cross Talk: Direct Cell-to-Cell Transfer of Oncogenic MicroRNAs via Tunneling Nanotubes. Transl. Res. 2014, 164, 359–365. [Google Scholar] [CrossRef]
- Lu, J.J.; Yang, W.M.; Li, F.; Zhu, W.; Chen, Z. Tunneling Nanotubes Mediated MicroRNA-155 Intercellular Transportation Promotes Bladder Cancer Cells’ Invasive and Proliferative Capacity. IJN 2019, 14, 9731–9743. [Google Scholar] [CrossRef]
- Burt, R.; Dey, A.; Aref, S.; Aguiar, M.; Akarca, A.; Bailey, K.; Day, W.; Hooper, S.; Kirkwood, A.; Kirschner, K.; et al. Activated Stromal Cells Transfer Mitochondria to Rescue Acute Lymphoblastic Leukemia Cells from Oxidative Stress. Blood 2019, 134, 1415–1429. [Google Scholar] [CrossRef]
- Kato, K.; Nguyen, K.T.; Decker, C.W.; Silkwood, K.H.; Eck, S.M.; Hernandez, J.B.; Garcia, J.; Han, D. Tunneling Nanotube Formation Promotes Survival against 5-fluorouracil in MCF-7 Breast Cancer Cells. FEBS Open Bio. 2022, 12, 203–210. [Google Scholar] [CrossRef]
- Desir, S.; O’Hare, P.; Vogel, R.I.; Sperduto, W.; Sarkari, A.; Dickson, E.L.; Wong, P.; Nelson, A.C.; Fong, Y.; Steer, C.J.; et al. Chemotherapy-Induced Tunneling Nanotubes Mediate Intercellular Drug Efflux in Pancreatic Cancer. Sci. Rep. 2018, 8, 9484. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cui, J.; Sun, X.; Zhang, Y. Tunneling-Nanotube Development in Astrocytes Depends on P53 Activation. Cell Death Differ. 2011, 18, 732–742. [Google Scholar] [CrossRef] [PubMed]
- Gousset, K.; Marzo, L.; Commere, P.-H.; Zurzolo, C. Myo10 Is a Key Regulator of TNT Formation in Neuronal Cells. J. Cell Sci. 2013, 126, 4424–4435. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, Y. Tunneling Nanotubes between Rat Primary Astrocytes and C6 Glioma Cells Alter Proliferation Potential of Glioma Cells. Neurosci. Bull. 2015, 31, 371–378. [Google Scholar] [CrossRef]
- Ranzinger, J.; Rustom, A.; Heide, D.; Morath, C.; Schemmer, P.; Nawroth, P.P.; Zeier, M.; Schwenger, V. The Receptor for Advanced Glycation End-Products (RAGE) Plays a Key Role in the Formation of Nanotubes (NTs) between Peritoneal Mesothelial Cells and in Murine Kidneys. Cell Tissue Res. 2014, 357, 667–679. [Google Scholar] [CrossRef]
- Austefjord, M.W.; Gerdes, H.-H.; Wang, X. Tunneling Nanotubes: Diversity in Morphology and Structure. Commun. Integr. Biol. 2014, 7, e27934. [Google Scholar] [CrossRef]
- Raghu, G.; Striker, L.; Harlan, J.; Gown, A.; Striker, G. Cytoskeletal Changes as an Early Event in Hydrogen Peroxide-Induced Cell Injury: A Study in A549 Cells. Br. J. Exp. Pathol. 1986, 67, 105–112. [Google Scholar]
- Zhao, Y.; Davis, H.W. Hydrogen Peroxide-Induced Cytoskeletal Rearrangement in Cultured Pulmonary Endothelial Cells. J. Cell Physiol. 1998, 174, 370–379. [Google Scholar] [CrossRef]
- Zhu, D.; Tan, K.S.; Zhang, X.; Sun, A.Y.; Sun, G.Y.; Lee, J.C.-M. Hydrogen Peroxide Alters Membrane and Cytoskeleton Properties and Increases Intercellular Connections in Astrocytes. J. Cell Sci. 2005, 118, 3695–3703. [Google Scholar] [CrossRef]
- Abounit, S.; Bousset, L.; Loria, F.; Zhu, S.; Chaumont, F.; Pieri, L.; Olivo-Marin, J.; Melki, R.; Zurzolo, C. Tunneling Nanotubes Spread Fibrillar A-synuclein by Intercellular Trafficking of Lysosomes. EMBO J. 2016, 35, 2120–2138. [Google Scholar] [CrossRef]
- Zins, K.; Lucas, T.; Reichl, P.; Abraham, D.; Aharinejad, S. A Rac1/Cdc42 GTPase-Specific Small Molecule Inhibitor Suppresses Growth of Primary Human Prostate Cancer Xenografts and Prolongs Survival in Mice. PLoS ONE 2013, 8, e74924. [Google Scholar] [CrossRef] [PubMed]
- Stankiewicz, T.R.; Linseman, D.A. Rho Family GTPases: Key Players in Neuronal Development, Neuronal Survival, and Neurodegeneration. Front. Cell. Neurosci. 2014, 8, 314. [Google Scholar] [CrossRef] [PubMed]
- Hanna, S.J.; McCoy-Simandle, K.; Miskolci, V.; Guo, P.; Cammer, M.; Hodgson, L.; Cox, D. The Role of Rho-GTPases and Actin Polymerization during Macrophage Tunneling Nanotube Biogenesis. Sci. Rep. 2017, 7, 8547. [Google Scholar] [CrossRef]
- Ozaki, M.; Deshpande, S.S.; Angkeow, P.; Bellan, J.; Lowenstein, C.J.; Dinauer, M.C.; Goldschmidt-Clermont, P.J.; Irani, K. Inhibition of the Rac1 GTPase Protects against Nonlethal Ischemia/Reperfusion-induced Necrosis and Apoptosis in Vivo. FASEB J. 2000, 14, 418–429. [Google Scholar] [CrossRef]
- Poyton, R.O.; Ball, K.A.; Castello, P.R. Mitochondrial Generation of Free Radicals and Hypoxic Signaling. Trends Endocrinol. Metab. 2009, 20, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Marlein, C.R.; Zaitseva, L.; Piddock, R.E.; Robinson, S.D.; Edwards, D.R.; Shafat, M.S.; Zhou, Z.; Lawes, M.; Bowles, K.M.; Rushworth, S.A. NADPH Oxidase-2 Derived Superoxide Drives Mitochondrial Transfer from Bone Marrow Stromal Cells to Leukemic Blasts. Blood 2017, 130, 1649–1660. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, Y.; Yeung, S.C.; Liang, Y.; Liang, X.; Ding, Y.; Ip, M.S.M.; Tse, H.-F.; Mak, J.C.W.; Lian, Q. Mitochondrial Transfer of Induced Pluripotent Stem Cell–Derived Mesenchymal Stem Cells to Airway Epithelial Cells Attenuates Cigarette Smoke–Induced Damage. Am. J. Respir Cell Mol. Biol. 2014, 51, 455–465. [Google Scholar] [CrossRef]
- Mizutani, H.; Tada-Oikawa, S.; Hiraku, Y.; Kojjima, M.; Kawanishi, S. Mechanism of Apoptosis Induced by Doxorubicin through the Generation of Hydrogen Peroxide. Life Sci. 2005, 76, 1439–1453. [Google Scholar] [CrossRef]
- Pasquier, J.; Guerrouahen, B.S.; Al Thawadi, H.; Ghiabi, P.; Maleki, M.; Abu-Kaoud, N.; Jacob, A.; Mirshahi, M.; Galas, L.; Rafii, S.; et al. Preferential Transfer of Mitochondria from Endothelial to Cancer Cells through Tunneling Nanotubes Modulates Chemoresistance. J. Transl. Med. 2013, 11, 94. [Google Scholar] [CrossRef]
- Wang, J.; Liu, X.; Qiu, Y.; Shi, Y.; Cai, J.; Wang, B.; Wei, X.; Ke, Q.; Sui, X.; Wang, Y.; et al. Cell Adhesion-Mediated Mitochondria Transfer Contributes to Mesenchymal Stem Cell-Induced Chemoresistance on T Cell Acute Lymphoblastic Leukemia Cells. J. Hematol Oncol. 2018, 11, 11. [Google Scholar] [CrossRef]
- Sun, X.; Wang, Y.; Zhang, J.; Tu, J.; Wang, X.-J.; Su, X.-D.; Wang, L.; Zhang, Y. Tunneling-Nanotube Direction Determination in Neurons and Astrocytes. Cell Death Dis. 2012, 3, e438. [Google Scholar] [CrossRef] [PubMed]
- Flatmark, K.; Pedersen, K.B.; Nesland, J.M.; Rasmussen, H.; Aamodt, G.; Mikalsen, S.-O.; Bjørnland, K.; Fodstad, Ø.; Maelandsmo, G.M. Nuclear Localization of the Metastasis-Related Protein S100A4 Correlates with Tumour Stage in Colorectal Cancer: Nuclear S100A4 in Colorectal Cancer. J. Pathol. 2003, 200, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Malashkevich, V.N.; Varney, K.M.; Garrett, S.C.; Wilder, P.T.; Knight, D.; Charpentier, T.H.; Ramagopal, U.A.; Almo, S.C.; Weber, D.J.; Bresnick, A.R. Structure of Ca2+ -Bound S100A4 and Its Interaction with Peptides Derived from Nonmuscle Myosin-IIA. Biochemistry 2008, 47, 5111–5126. [Google Scholar] [CrossRef]
- Grigorian, M.; Ambartsumian, N.; Lykkesfeldt, A.E.; Bastholm, L.; Elling, F.; Georgiev, G.; Lukanidin, E. Effect Ofmts1 (S100A4) Expression on the Progression of Human Breast Cancer Cells. Int. J. Cancer 1996, 67, 831–841. [Google Scholar] [CrossRef]
- Hofmann, M.A.; Drury, S.; Fu, C.; Qu, W.; Taguchi, A.; Lu, Y.; Avila, C.; Kambham, N.; Bierhaus, A.; Nawroth, P.; et al. RAGE Mediates a Novel Proinflammatory Axis. Cell 1999, 97, 889–901. [Google Scholar] [CrossRef]
- Reimann, S.; Fink, L.; Wilhelm, J.; Hoffmann, J.; Bednorz, M.; Seimetz, M.; Dessureault, I.; Troesser, R.; Ghanim, B.; Klepetko, W.; et al. Increased S100A4 Expression in the Vasculature of Human COPD Lungs and Murine Model of Smoke-Induced Emphysema. Respir Res. 2015, 16, 127. [Google Scholar] [CrossRef]
- Basta, G.; Lazzerini, G.; Del Turco, S.; Ratto, G.M.; Schmidt, A.M.; De Caterina, R. At Least 2 Distinct Pathways Generating Reactive Oxygen Species Mediate Vascular Cell Adhesion Molecule-1 Induction by Advanced Glycation End Products. ATVB 2005, 25, 1401–1407. [Google Scholar] [CrossRef]
- Luchetti, F.; Canonico, B.; Arcangeletti, M.; Guescini, M.; Cesarini, E.; Stocchi, V.; Degli Esposti, M.; Papa, S. Fas Signalling Promotes Intercellular Communication in T Cells. PLoS ONE 2012, 7, e35766. [Google Scholar] [CrossRef]
- Redondo-Morata, L.; Giannotti, M.I.; Sanz, F. Influence of Cholesterol on the Phase Transition of Lipid Bilayers: A Temperature-Controlled Force Spectroscopy Study. Langmuir 2012, 28, 12851–12860. [Google Scholar] [CrossRef]
- Sisakhtnezhad, S.; Khosravi, L. Emerging Physiological and Pathological Implications of Tunneling Nanotubes Formation between Cells. Eur. J. Cell Biol. 2015, 94, 429–443. [Google Scholar] [CrossRef]
- Wang, X.; Gerdes, H.-H. Transfer of Mitochondria via Tunneling Nanotubes Rescues Apoptotic PC12 Cells. Cell Death Differ. 2015, 22, 1181–1191. [Google Scholar] [CrossRef]
- Hagenbuchner, J.; Ausserlechner, M.J. Mitochondria and FOXO3: Breath or Die. Front. Physiol. 2013, 4, 147. [Google Scholar] [CrossRef] [PubMed]
- Wakeyama, H.; Akiyama, T.; Takahashi, K.; Amano, H.; Kadono, Y.; Nakamura, M.; Oshima, Y.; Itabe, H.; Nakayama, K.I.; Nakayama, K.; et al. Negative Feedback Loop in the Bim-Caspase-3 Axis Regulating Apoptosis and Activity of Osteoclasts. J. Bone Miner. Res. 2007, 22, 1631–1639. [Google Scholar] [CrossRef] [PubMed]
- Lau, W.F.; Das, N.P. In Vitro Modulation of Rat Adipocyte Ghost Membrane Fluidity by Cholesterol Oxysterols. Experientia 1995, 51, 731–737. [Google Scholar] [CrossRef] [PubMed]
- Kultti, A.; Kärnä, R.; Rilla, K.; Nurminen, P.; Koli, E.; Makkonen, K.M.; Si, J.; Tammi, M.I.; Tammi, R.H. Methyl-β-Cyclodextrin Suppresses Hyaluronan Synthesis by Down-Regulation of Hyaluronan Synthase 2 through Inhibition of Akt. J. Biol. Chem. 2010, 285, 22901–22910. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Xian, G.; Li, M.; Zhang, Y.; Yang, M.; Yu, Y.; Lv, H.; Xuan, S.; Lin, Y.; Gao, L. Cholesterol Oxidase from Bordetella Species Promotes Irreversible Cell Apoptosis in Lung Adenocarcinoma by Cholesterol Oxidation. Cell Death Dis. 2014, 5, e1372. [Google Scholar] [CrossRef] [PubMed][Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferrer, J.L.M.; Garcia, R.L. Antioxidant Systems, lncRNAs, and Tunneling Nanotubes in Cell Death Rescue from Cigarette Smoke Exposure. Cells 2022, 11, 2277. https://doi.org/10.3390/cells11152277
Ferrer JLM, Garcia RL. Antioxidant Systems, lncRNAs, and Tunneling Nanotubes in Cell Death Rescue from Cigarette Smoke Exposure. Cells. 2022; 11(15):2277. https://doi.org/10.3390/cells11152277
Chicago/Turabian StyleFerrer, Jose Lorenzo M., and Reynaldo L. Garcia. 2022. "Antioxidant Systems, lncRNAs, and Tunneling Nanotubes in Cell Death Rescue from Cigarette Smoke Exposure" Cells 11, no. 15: 2277. https://doi.org/10.3390/cells11152277
APA StyleFerrer, J. L. M., & Garcia, R. L. (2022). Antioxidant Systems, lncRNAs, and Tunneling Nanotubes in Cell Death Rescue from Cigarette Smoke Exposure. Cells, 11(15), 2277. https://doi.org/10.3390/cells11152277