
Extracellular Vesicles, Inflammation, and Cardiovascular Disease


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
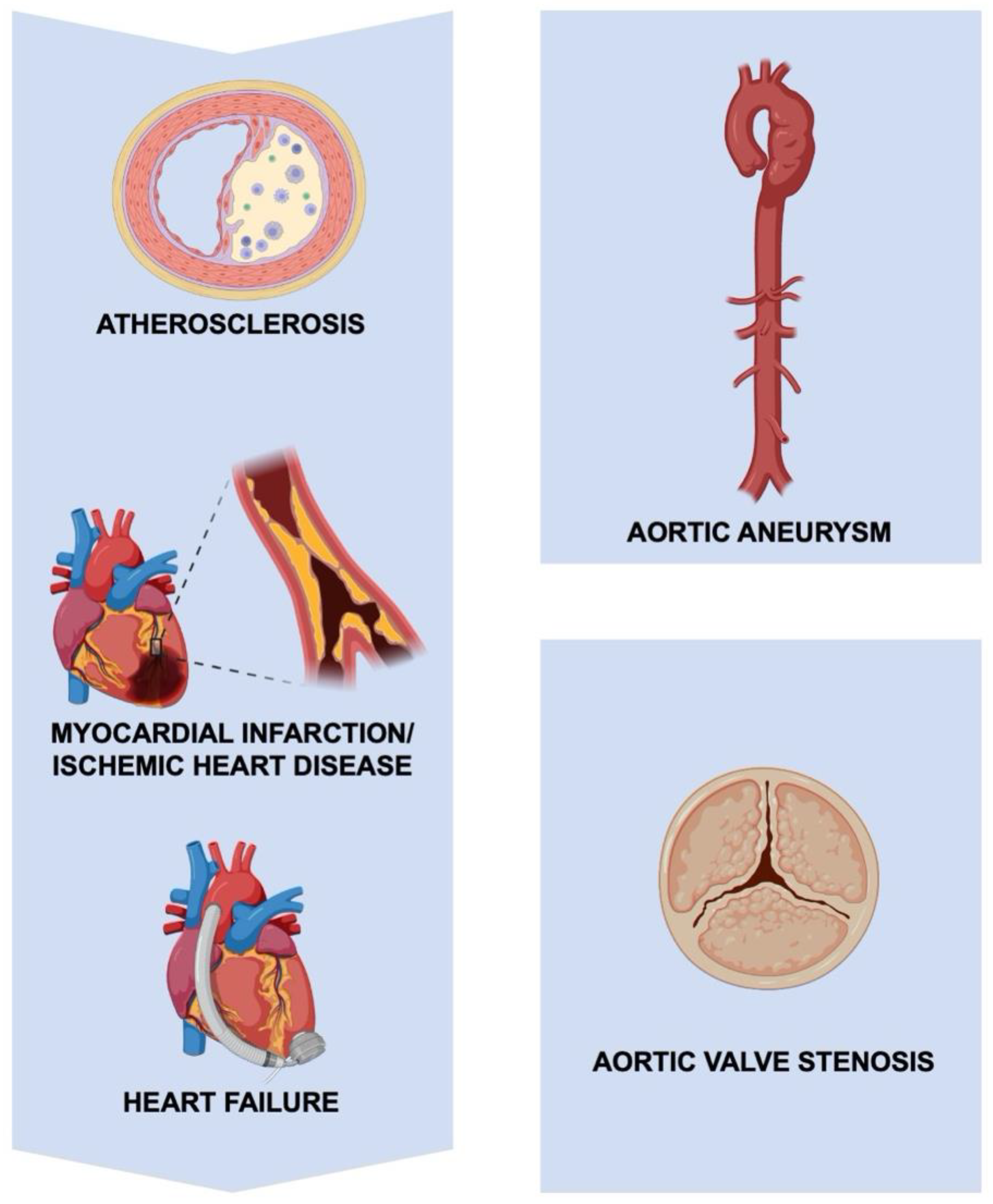
2. The Role of EVs and Inflammation in Specific Cardiovascular Disorders
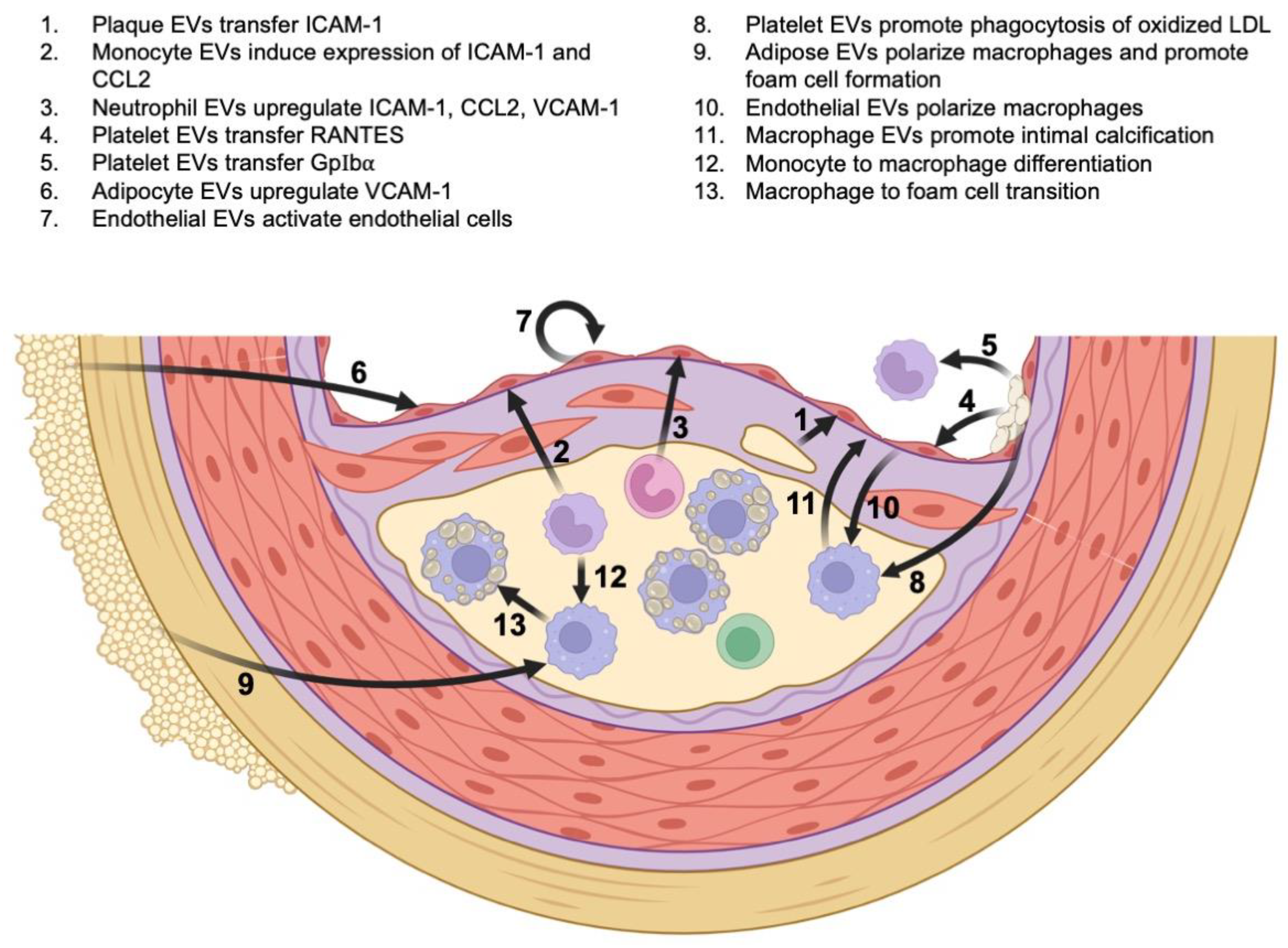
2.1. The Role of EVs and Inflammation in Atherosclerosis
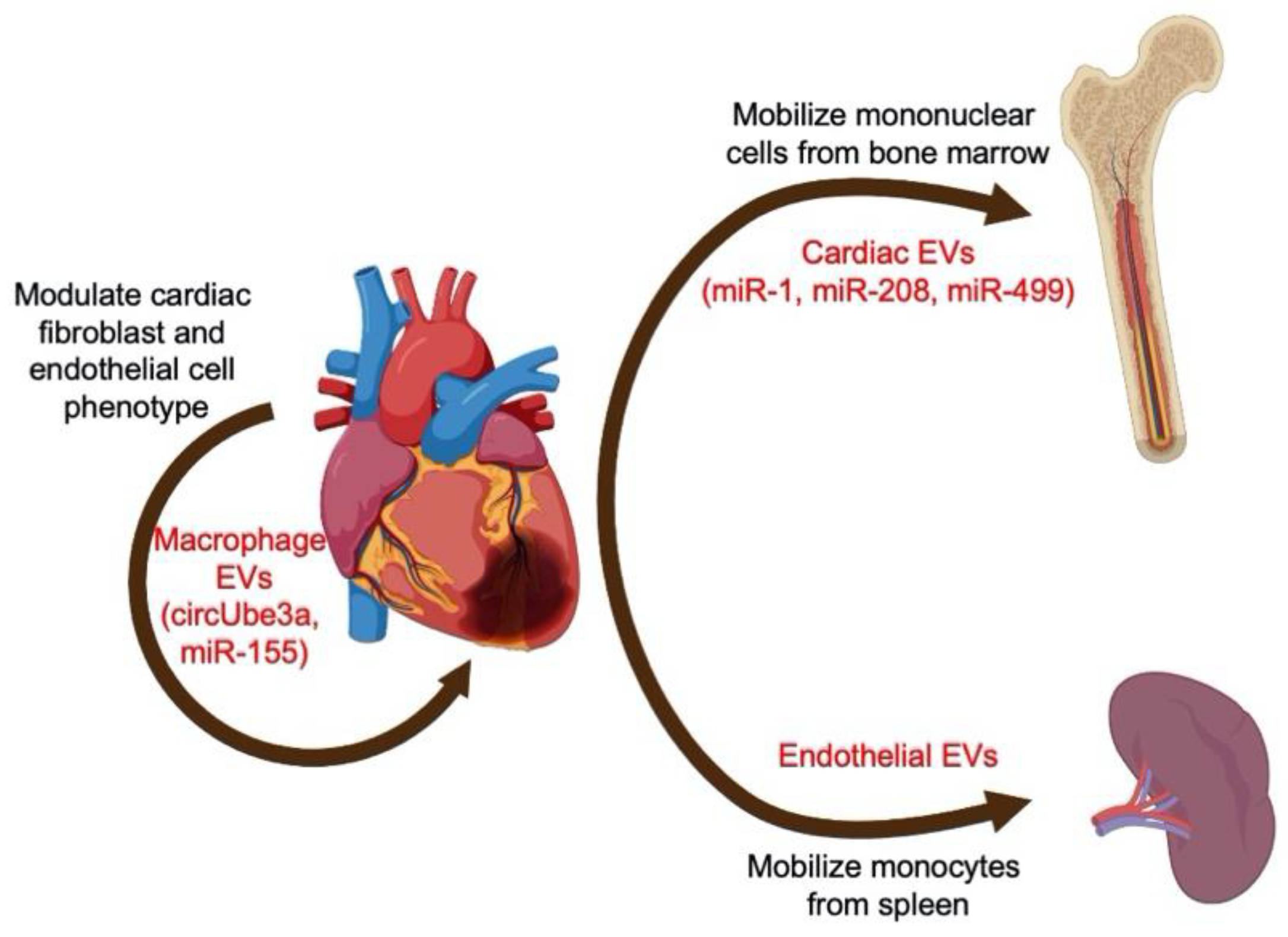
2.2. The Role of EVs and Inflammation in Myocardial Infarction and Ischemic Heart Disease
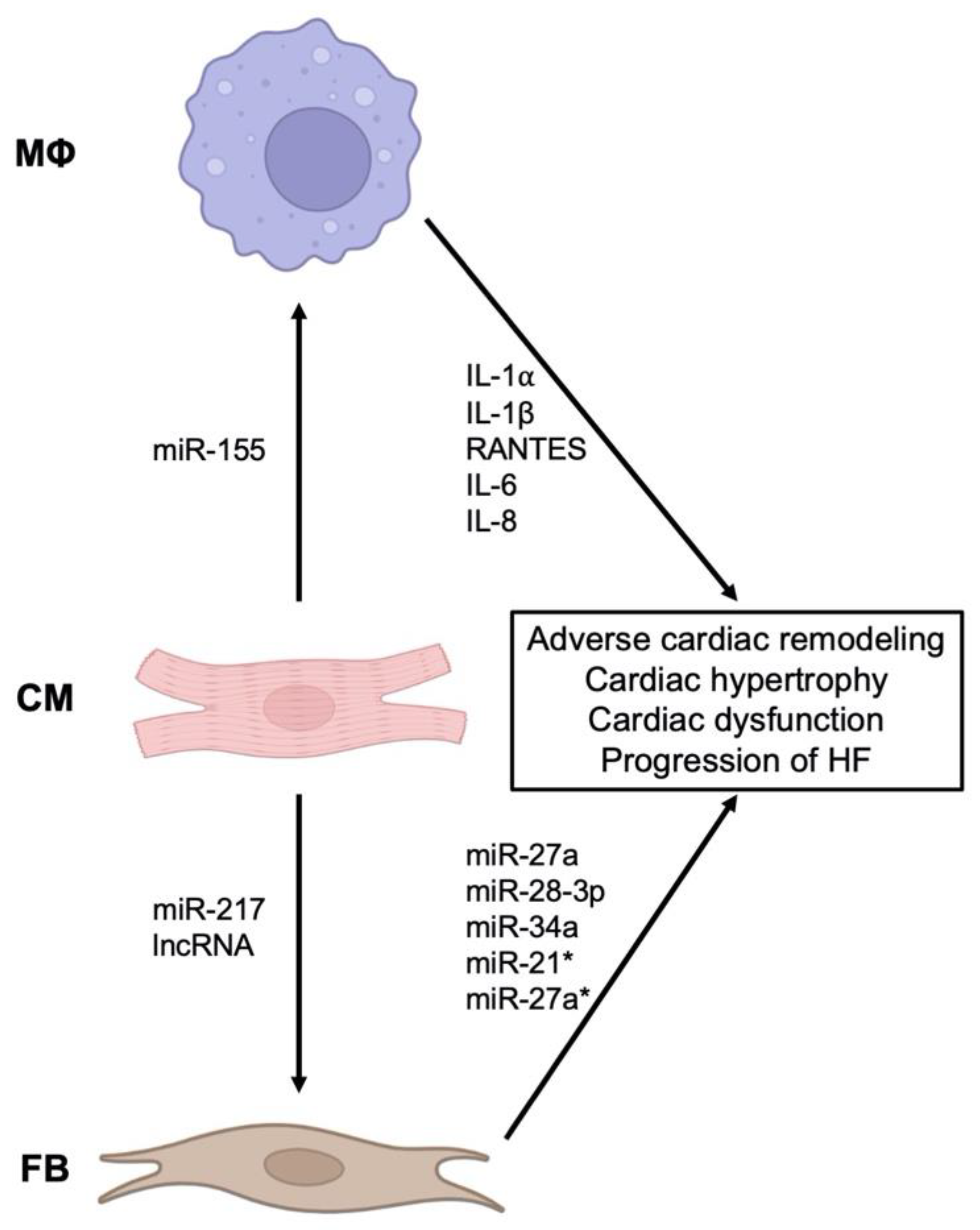
2.3. The Role of EVs and Inflammation in Heart Failure
2.4. The Role of EVs and Inflammation in Aneurysmal and Valvular Pathology of the Aorta
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- WHO. Cardiovascular Diseases (CVDs) Fact Sheet. 2021. Available online: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed on 18 May 2022).
- Lancellotti, P.; Marechal, P.; Donis, N.; Oury, C. Inflammation, cardiovascular disease, and cancer: A common link with far-reaching implications. Eur. Heart J. 2019, 40, 3910–3912. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Cushman, M.; Stampfer, M.J.; Tracy, R.P.; Hennekens, C.H. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N. Engl. J. Med. 1997, 336, 973–979. [Google Scholar] [CrossRef] [PubMed]
- Willerson, J.T.; Ridker, P.M. Inflammation as a cardiovascular risk factor. Circulation 2004, 109, II2–II10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Everett, B.M.; Cornel, J.H.; Lainscak, M.; Anker, S.D.; Abbate, A.; Thuren, T.; Libby, P.; Glynn, R.J.; Ridker, P.M. Anti-Inflammatory Therapy With Canakinumab for the Prevention of Hospitalization for Heart Failure. Circulation 2019, 139, 1289–1299. [Google Scholar] [CrossRef]
- Thery, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [Green Version]
- Denzer, K.; van Eijk, M.; Kleijmeer, M.J.; Jakobson, E.; de Groot, C.; Geuze, H.J. Follicular dendritic cells carry MHC class II-expressing microvesicles at their surface. J. Immunol. 2000, 165, 1259–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoorvogel, W.; Oorschot, V.; Geuze, H.J. A novel class of clathrin-coated vesicles budding from endosomes. J. Cell Biol. 1996, 132, 21–33. [Google Scholar] [CrossRef] [Green Version]
- Cocucci, E.; Meldolesi, J. Ectosomes and exosomes: Shedding the confusion between extracellular vesicles. Trends Cell Biol. 2015, 25, 364–372. [Google Scholar] [CrossRef]
- Stein, J.M.; Luzio, J.P. Ectocytosis caused by sublytic autologous complement attack on human neutrophils. The sorting of endogenous plasma-membrane proteins and lipids into shed vesicles. Biochem. J. 1991, 274 Pt 2, 381–386. [Google Scholar] [CrossRef]
- Robbins, P.D.; Dorronsoro, A.; Booker, C.N. Regulation of chronic inflammatory and immune processes by extracellular vesicles. J. Clin. Investig. 2016, 126, 1173–1180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robbins, P.D.; Morelli, A.E. Regulation of immune responses by extracellular vesicles. Nat. Rev. Immunol. 2014, 14, 195–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen, F.; Nickenig, G.; Werner, N. Extracellular Vesicles in Cardiovascular Disease: Potential Applications in Diagnosis, Prognosis, and Epidemiology. Circ. Res. 2017, 120, 1649–1657. [Google Scholar] [CrossRef] [PubMed]
- Voukalis, C.; Shantsila, E.; Lip, G.Y.H. Microparticles and cardiovascular diseases. Ann. Med. 2019, 51, 193–223. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgozoglu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Primers 2019, 5, 56. [Google Scholar] [CrossRef]
- Legein, B.; Temmerman, L.; Biessen, E.A.; Lutgens, E. Inflammation and immune system interactions in atherosclerosis. Cell Mol. Life Sci. 2013, 70, 3847–3869. [Google Scholar] [CrossRef]
- Konkoth, A.; Saraswat, R.; Dubrou, C.; Sabatier, F.; Leroyer, A.S.; Lacroix, R.; Duchez, A.C.; Dignat-George, F. Multifaceted role of extracellular vesicles in atherosclerosis. Atherosclerosis 2021, 319, 121–131. [Google Scholar] [CrossRef]
- Leroyer, A.S.; Isobe, H.; Leseche, G.; Castier, Y.; Wassef, M.; Mallat, Z.; Binder, B.R.; Tedgui, A.; Boulanger, C.M. Cellular origins and thrombogenic activity of microparticles isolated from human atherosclerotic plaques. J. Am. Coll. Cardiol. 2007, 49, 772–777. [Google Scholar] [CrossRef] [Green Version]
- Buffolo, F.; Monticone, S.; Camussi, G.; Aikawa, E. Role of Extracellular Vesicles in the Pathogenesis of Vascular Damage. Hypertension 2022, 79, 863–873. [Google Scholar] [CrossRef]
- Rautou, P.E.; Leroyer, A.S.; Ramkhelawon, B.; Devue, C.; Duflaut, D.; Vion, A.C.; Nalbone, G.; Castier, Y.; Leseche, G.; Lehoux, S.; et al. Microparticles from human atherosclerotic plaques promote endothelial ICAM-1-dependent monocyte adhesion and transendothelial migration. Circ. Res. 2011, 108, 335–343. [Google Scholar] [CrossRef] [Green Version]
- Tang, N.; Sun, B.; Gupta, A.; Rempel, H.; Pulliam, L. Monocyte exosomes induce adhesion molecules and cytokines via activation of NF-kappaB in endothelial cells. FASEB J. 2016, 30, 3097–3106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez, I.; Ward, B.; Souilhol, C.; Recarti, C.; Ariaans, M.; Johnston, J.; Burnett, A.; Mahmoud, M.; Luong, L.A.; West, L.; et al. Neutrophil microvesicles drive atherosclerosis by delivering miR-155 to atheroprone endothelium. Nat. Commun. 2020, 11, 214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chimen, M.; Evryviadou, A.; Box, C.L.; Harrison, M.J.; Hazeldine, J.; Dib, L.H.; Kuravi, S.J.; Payne, H.; Price, J.M.J.; Kavanagh, D.; et al. Appropriation of GPIbalpha from platelet-derived extracellular vesicles supports monocyte recruitment in systemic inflammation. Haematologica 2020, 105, 1248–1261. [Google Scholar] [CrossRef]
- Mause, S.F.; von Hundelshausen, P.; Zernecke, A.; Koenen, R.R.; Weber, C. Platelet microparticles: A transcellular delivery system for RANTES promoting monocyte recruitment on endothelium. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1512–1518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuravi, S.J.; Harrison, P.; Rainger, G.E.; Nash, G.B. Ability of Platelet-Derived Extracellular Vesicles to Promote Neutrophil-Endothelial Cell Interactions. Inflammation 2019, 42, 290–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wadey, R.M.; Connolly, K.D.; Mathew, D.; Walters, G.; Rees, D.A.; James, P.E. Inflammatory adipocyte-derived extracellular vesicles promote leukocyte attachment to vascular endothelial cells. Atherosclerosis 2019, 283, 19–27. [Google Scholar] [CrossRef] [Green Version]
- Huber, J.; Vales, A.; Mitulovic, G.; Blumer, M.; Schmid, R.; Witztum, J.L.; Binder, B.R.; Leitinger, N. Oxidized membrane vesicles and blebs from apoptotic cells contain biologically active oxidized phospholipids that induce monocyte-endothelial interactions. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 101–107. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.H.; Fu, Y.C.; Zhang, D.W.; Yin, K.; Tang, C.K. Foam cells in atherosclerosis. Clin. Chim. Acta 2013, 424, 245–252. [Google Scholar] [CrossRef] [Green Version]
- Feng, C.; Chen, Q.; Fan, M.; Guo, J.; Liu, Y.; Ji, T.; Zhu, J.; Zhao, X. Platelet-derived microparticles promote phagocytosis of oxidized low-density lipoprotein by macrophages, potentially enhancing foam cell formation. Ann. Transl. Med. 2019, 7, 477. [Google Scholar] [CrossRef]
- Xie, Z.; Wang, X.; Liu, X.; Du, H.; Sun, C.; Shao, X.; Tian, J.; Gu, X.; Wang, H.; Tian, J.; et al. Adipose-Derived Exosomes Exert Proatherogenic Effects by Regulating Macrophage Foam Cell Formation and Polarization. J. Am. Heart Assoc. 2018, 7, e007442. [Google Scholar] [CrossRef]
- Huang, C.; Han, J.; Wu, Y.; Li, S.; Wang, Q.; Lin, W.; Zhu, J. Exosomal MALAT1 derived from oxidized low-density lipoprotein-treated endothelial cells promotes M2 macrophage polarization. Mol. Med. Rep. 2018, 18, 509–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, S.; Wu, C.; Xiao, J.; Li, D.; Sun, Z.; Li, M. Endothelial extracellular vesicles modulate the macrophage phenotype: Potential implications in atherosclerosis. Scand. J. Immunol. 2018, 87, e12648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordts, P.L.S.M.; Esko, J.D. The heparan sulfate proteoglycan grip on hyperlipidemia and atherosclerosis. Matrix Biol. 2018, 71–72, 262–282. [Google Scholar] [CrossRef] [PubMed]
- Tsiantoulas, D.; Eslami, M.; Obermayer, G.; Clement, M.; Smeets, D.; Mayer, F.J.; Kiss, M.G.; Enders, L.; Weißer, J.; Göderle, L. APRIL limits atherosclerosis by binding to heparan sulfate proteoglycans. Nature 2021, 597, 92–96. [Google Scholar] [CrossRef]
- Zhou, Y.; Xie, Q.; Pan, S.; Wu, J.; Wang, X.; Cao, Z.; Wang, M.; Zha, L.; Zhou, M.; Li, Q.; et al. Small extracellular vesicles containing LDLRQ722* protein reconstructed the lipid metabolism via heparan sulfate proteoglycans and clathrin-mediated endocytosis. Clin. Transl. Med. 2022, 12, e773. [Google Scholar] [CrossRef]
- Wight, T.N. A Role for proteoglycans in vascular disease. Matrix Biol. 2018, 71–72, 396–420. [Google Scholar] [CrossRef]
- Rogers, M.A.; Aikawa, E. Cardiovascular calcification: Artificial intelligence and big data accelerate mechanistic discovery. Nat. Rev. Cardiol. 2019, 16, 261–274. [Google Scholar] [CrossRef]
- New, S.E.; Goettsch, C.; Aikawa, M.; Marchini, J.F.; Shibasaki, M.; Yabusaki, K.; Libby, P.; Shanahan, C.M.; Croce, K.; Aikawa, E. Macrophage-derived matrix vesicles: An alternative novel mechanism for microcalcification in atherosclerotic plaques. Circ. Res. 2013, 113, 72–77. [Google Scholar] [CrossRef]
- Kawakami, R.; Katsuki, S.; Travers, R.; Romero, D.C.; Becker-Greene, D.; Passos, L.S.A.; Higashi, H.; Blaser, M.C.; Sukhova, G.K.; Buttigieg, J.; et al. S100A9-RAGE Axis Accelerates Formation of Macrophage-Mediated Extracellular Vesicle Microcalcification in Diabetes Mellitus. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 1838–1853. [Google Scholar] [CrossRef]
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.Z.; Benjamin, E.J.; Benziger, C.P.; et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990–2019: Update From the GBD 2019 Study. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar] [CrossRef]
- Frangogiannis, N.G.; Smith, C.W.; Entman, M.L. The inflammatory response in myocardial infarction. Cardiovasc. Res. 2002, 53, 31–47. [Google Scholar] [CrossRef]
- Nahrendorf, M.; Pittet, M.J.; Swirski, F.K. Monocytes: Protagonists of infarct inflammation and repair after myocardial infarction. Circulation 2010, 121, 2437–2445. [Google Scholar] [CrossRef] [PubMed]
- Loyer, X.; Zlatanova, I.; Devue, C.; Yin, M.; Howangyin, K.Y.; Klaihmon, P.; Guerin, C.L.; Kheloufi, M.; Vilar, J.; Zannis, K.; et al. Intra-Cardiac Release of Extracellular Vesicles Shapes Inflammation Following Myocardial Infarction. Circ. Res. 2018, 123, 100–106. [Google Scholar] [CrossRef]
- Almeida Paiva, R.; Martins-Marques, T.; Jesus, K.; Ribeiro-Rodrigues, T.; Zuzarte, M.; Silva, A.; Reis, L.; da Silva, M.; Pereira, P.; Vader, P.; et al. Ischaemia alters the effects of cardiomyocyte-derived extracellular vesicles on macrophage activation. J. Cell Mol. Med. 2019, 23, 1137–1151. [Google Scholar] [CrossRef] [Green Version]
- Cheng, M.; Yang, J.; Zhao, X.; Zhang, E.; Zeng, Q.; Yu, Y.; Yang, L.; Wu, B.; Yi, G.; Mao, X.; et al. Circulating myocardial microRNAs from infarcted hearts are carried in exosomes and mobilise bone marrow progenitor cells. Nat. Commun. 2019, 10, 959. [Google Scholar] [CrossRef] [PubMed]
- Akbar, N.; Digby, J.E.; Cahill, T.J.; Tavare, A.N.; Corbin, A.L.; Saluja, S.; Dawkins, S.; Edgar, L.; Rawlings, N.; Ziberna, K.; et al. Endothelium-derived extracellular vesicles promote splenic monocyte mobilization in myocardial infarction. JCI Insight 2017, 2, e93344. [Google Scholar] [CrossRef] [Green Version]
- Qiao, S.; Zhang, W.; Yin, Y.; Wei, Z.; Chen, F.; Zhao, J.; Sun, X.; Mu, D.; Xie, J.; Xu, B. Extracellular vesicles derived from Kruppel-Like Factor 2-overexpressing endothelial cells attenuate myocardial ischemia-reperfusion injury by preventing Ly6C(high) monocyte recruitment. Theranostics 2020, 10, 11562–11579. [Google Scholar] [CrossRef] [PubMed]
- Cambier, L.; de Couto, G.; Ibrahim, A.; Echavez, A.K.; Valle, J.; Liu, W.; Kreke, M.; Smith, R.R.; Marban, L.; Marban, E. Y RNA fragment in extracellular vesicles confers cardioprotection via modulation of IL-10 expression and secretion. EMBO Mol. Med. 2017, 9, 337–352. [Google Scholar] [CrossRef]
- de Couto, G.; Gallet, R.; Cambier, L.; Jaghatspanyan, E.; Makkar, N.; Dawkins, J.F.; Berman, B.P.; Marban, E. Exosomal MicroRNA Transfer Into Macrophages Mediates Cellular Postconditioning. Circulation 2017, 136, 200–214. [Google Scholar] [CrossRef]
- Akhmerov, A.; Marban, E. COVID-19 and the Heart. Circ. Res. 2020, 126, 1443–1455. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Li, C.; Zhao, R.; Qiu, Z.; Shen, C.; Wang, Z.; Liu, W.; Zhang, W.; Ge, J.; Shi, B. CircUbe3a from M2 macrophage-derived small extracellular vesicles mediates myocardial fibrosis after acute myocardial infarction. Theranostics 2021, 11, 6315–6333. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhang, C.; Liu, L.; Xi, A.; Chen, B.; Li, Y.; Du, J. Macrophage-Derived mir-155-Containing Exosomes Suppress Fibroblast Proliferation and Promote Fibroblast Inflammation during Cardiac Injury. Mol. Ther. 2017, 25, 192–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Chen, J.; Shi, J.; Zhou, W.; Wang, L.; Fang, W.; Zhong, Y.; Chen, X.; Chen, Y.; Sabri, A.; et al. M1-like macrophage-derived exosomes suppress angiogenesis and exacerbate cardiac dysfunction in a myocardial infarction microenvironment. Basic Res. Cardiol. 2020, 115, 22. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Gao, W.; Yuan, J.; Wu, C.; Yao, K.; Zhang, L.; Ma, L.; Zhu, J.; Zou, Y.; Ge, J. Exosomes derived from dendritic cells improve cardiac function via activation of CD4(+) T lymphocytes after myocardial infarction. J. Mol. Cell Cardiol. 2016, 91, 123–133. [Google Scholar] [CrossRef]
- Wang, Y.; Dembowsky, K.; Chevalier, E.; Stuve, P.; Korf-Klingebiel, M.; Lochner, M.; Napp, L.C.; Frank, H.; Brinkmann, E.; Kanwischer, A.; et al. C-X-C Motif Chemokine Receptor 4 Blockade Promotes Tissue Repair After Myocardial Infarction by Enhancing Regulatory T Cell Mobilization and Immune-Regulatory Function. Circulation 2019, 139, 1798–1812. [Google Scholar] [CrossRef]
- Feng, G.; Bajpai, G.; Ma, P.; Koenig, A.; Bredemeyer, A.; Lokshina, I.; Lai, L.; Forster, I.; Leuschner, F.; Kreisel, D.; et al. CCL17 Aggravates Myocardial Injury by Suppressing Recruitment of Regulatory T Cells. Circulation 2022, 145, 765–782. [Google Scholar] [CrossRef]
- Xia, N.; Lu, Y.; Gu, M.; Li, N.; Liu, M.; Jiao, J.; Zhu, Z.; Li, J.; Li, D.; Tang, T.; et al. A Unique Population of Regulatory T Cells in Heart Potentiates Cardiac Protection From Myocardial Infarction. Circulation 2020, 142, 1956–1973. [Google Scholar] [CrossRef]
- Akhmerov, A.; Rogers, R.; de Couto, G.; Valle, J.; Li, L.; Ibrahim, A.; Sanchez, L.; Zhang, R.; Lin, Y.N.; Liu, W.; et al. Regulatory T cell activation, proliferation, and reprogramming induced by extracellular vesicles. J. Heart Lung Transpl. 2021, 40, 1387–1395. [Google Scholar] [CrossRef]
- Bozkurt, B.; Coats, A.J.S.; Tsutsui, H.; Abdelhamid, C.M.; Adamopoulos, S.; Albert, N.; Anker, S.D.; Atherton, J.; Bohm, M.; Butler, J.; et al. Universal definition and classification of heart failure: A report of the Heart Failure Society of America, Heart Failure Association of the European Society of Cardiology, Japanese Heart Failure Society and Writing Committee of the Universal Definition of Heart Failure: Endorsed by the Canadian Heart Failure Society, Heart Failure Association of India, Cardiac Society of Australia and New Zealand, and Chinese Heart Failure Association. Eur. J. Heart Fail. 2021, 23, 352–380. [Google Scholar]
- GBD 2017 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1789–1858. [Google Scholar] [CrossRef] [Green Version]
- Savarese, G.; Becher, P.M.; Lund, L.H.; Seferovic, P.; Rosano, G.M.C.; Coats, A. Global burden of heart failure: A comprehensive and updated review of epidemiology. Cardiovasc. Res. 2022, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Tsao, C.W.; Aday, A.W.; Almarzooq, Z.I.; Alonso, A.; Beaton, A.Z.; Bittencourt, M.S.; Boehme, A.K.; Buxton, A.E.; Carson, A.P.; Commodore-Mensah, Y.; et al. Heart Disease and Stroke Statistics-2022 Update: A Report From the American Heart Association. Circulation 2022, 145, e153–e639. [Google Scholar] [CrossRef] [PubMed]
- Owan, T.E.; Hodge, D.O.; Herges, R.M.; Jacobsen, S.J.; Roger, V.L.; Redfield, M.M. Trends in prevalence and outcome of heart failure with preserved ejection fraction. N. Engl. J. Med. 2006, 355, 251–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akhmerov, A.; Ramzy, D. Mechanical Circulatory Support in Right Ventricular Failure. Interv. Cardiol. Clin. 2021, 10, 185–194. [Google Scholar] [CrossRef]
- Matsumoto, S.; Sakata, Y.; Suna, S.; Nakatani, D.; Usami, M.; Hara, M.; Kitamura, T.; Hamasaki, T.; Nanto, S.; Kawahara, Y.; et al. Circulating p53-responsive microRNAs are predictive indicators of heart failure after acute myocardial infarction. Circ. Res. 2013, 113, 322–326. [Google Scholar] [CrossRef] [Green Version]
- Nie, X.; Fan, J.; Li, H.; Yin, Z.; Zhao, Y.; Dai, B.; Dong, N.; Chen, C.; Wang, D.W. miR-217 Promotes Cardiac Hypertrophy and Dysfunction by Targeting PTEN. Mol. Ther. Nucleic Acids 2018, 12, 254–266. [Google Scholar] [CrossRef] [Green Version]
- Goren, Y.; Kushnir, M.; Zafrir, B.; Tabak, S.; Lewis, B.S.; Amir, O. Serum levels of microRNAs in patients with heart failure. Eur. J. Heart Fail. 2012, 14, 147–154. [Google Scholar] [CrossRef]
- Qiao, L.; Hu, S.; Liu, S.; Zhang, H.; Ma, H.; Huang, K.; Li, Z.; Su, T.; Vandergriff, A.; Tang, J.; et al. microRNA-21-5p dysregulation in exosomes derived from heart failure patients impairs regenerative potential. J. Clin. Investig. 2019, 129, 2237–2250. [Google Scholar] [CrossRef]
- Biemmi, V.; Milano, G.; Ciullo, A.; Cervio, E.; Burrello, J.; Dei Cas, M.; Paroni, R.; Tallone, T.; Moccetti, T.; Pedrazzini, G.; et al. Inflammatory extracellular vesicles prompt heart dysfunction via TRL4-dependent NF-kappaB activation. Theranostics 2020, 10, 2773–2790. [Google Scholar] [CrossRef]
- Yu, H.; Qin, L.; Peng, Y.; Bai, W.; Wang, Z. Exosomes Derived From Hypertrophic Cardiomyocytes Induce Inflammation in Macrophages via miR-155 Mediated MAPK Pathway. Front. Immunol. 2020, 11, 606045. [Google Scholar] [CrossRef]
- Lindner, D.; Zietsch, C.; Tank, J.; Sossalla, S.; Fluschnik, N.; Hinrichs, S.; Maier, L.; Poller, W.; Blankenberg, S.; Schultheiss, H.P.; et al. Cardiac fibroblasts support cardiac inflammation in heart failure. Basic Res. Cardiol. 2014, 109, 428. [Google Scholar] [CrossRef]
- Tian, C.; Gao, L.; Zimmerman, M.C.; Zucker, I.H. Myocardial infarction-induced microRNA-enriched exosomes contribute to cardiac Nrf2 dysregulation in chronic heart failure. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H928–H939. [Google Scholar] [CrossRef] [PubMed]
- Bang, C.; Batkai, S.; Dangwal, S.; Gupta, S.K.; Foinquinos, A.; Holzmann, A.; Just, A.; Remke, J.; Zimmer, K.; Zeug, A.; et al. Cardiac fibroblast-derived microRNA passenger strand-enriched exosomes mediate cardiomyocyte hypertrophy. J. Clin. Investig. 2014, 124, 2136–2146. [Google Scholar] [CrossRef]
- Tian, C.; Hu, G.; Gao, L.; Hackfort, B.T.; Zucker, I.H. Extracellular vesicular MicroRNA-27a* contributes to cardiac hypertrophy in chronic heart failure. J. Mol. Cell Cardiol. 2020, 143, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Yang, J.; Sun, J.; Qin, G. Extracellular vesicles in cardiovascular disease: Biological functions and therapeutic implications. Pharmacol. Ther. 2022, 233, 108025. [Google Scholar] [CrossRef] [PubMed]
- Ailawadi, S.; Wang, X.; Gu, H.; Fan, G.C. Pathologic function and therapeutic potential of exosomes in cardiovascular disease. Biochim. Biophys. Acta 2015, 1852, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenneweg, F.; Bang, C.; Xiao, K.; Boulanger, C.M.; Loyer, X.; Mazlan, S.; Schroen, B.; Hermans-Beijnsberger, S.; Foinquinos, A.; Hirt, M.N.; et al. Long Noncoding RNA-Enriched Vesicles Secreted by Hypoxic Cardiomyocytes Drive Cardiac Fibrosis. Mol. Ther. Nucleic Acids 2019, 18, 363–374. [Google Scholar] [CrossRef] [Green Version]
- Ashton, H.A.; Buxton, M.J.; Day, N.E.; Kim, L.G.; Marteau, T.M.; Scott, R.A.; Thompson, S.G.; Walker, N.M.; Multicentre Aneurysm Screening Study Group. The Multicentre Aneurysm Screening Study (MASS) into the effect of abdominal aortic aneurysm screening on mortality in men: A randomised controlled trial. Lancet 2002, 360, 1531–1539. [Google Scholar]
- Norman, P.E.; Jamrozik, K.; Lawrence-Brown, M.M.; Le, M.T.; Spencer, C.A.; Tuohy, R.J.; Parsons, R.W.; Dickinson, J.A. Population based randomised controlled trial on impact of screening on mortality from abdominal aortic aneurysm. BMJ 2004, 329, 1259. [Google Scholar] [CrossRef] [Green Version]
- Lindholt, J.S.; Juul, S.; Fasting, H.; Henneberg, E.W. Screening for abdominal aortic aneurysms: Single centre randomised controlled trial. BMJ 2005, 330, 750. [Google Scholar] [CrossRef] [Green Version]
- Ashton, H.A.; Gao, L.; Kim, L.G.; Druce, P.S.; Thompson, S.G.; Scott, R.A. Fifteen-year follow-up of a randomized clinical trial of ultrasonographic screening for abdominal aortic aneurysms. Br. J. Surg. 2007, 94, 696–701. [Google Scholar] [CrossRef] [PubMed]
- Von Allmen, R.S.; Powell, J.T. The management of ruptured abdominal aortic aneurysms: Screening for abdominal aortic aneurysm and incidence of rupture. J. Cardiovasc. Surg. 2012, 53, 69–76. [Google Scholar]
- Martin-Ventura, J.L.; Roncal, C.; Orbe, J.; Blanco-Colio, L.M. Role of Extracellular Vesicles as Potential Diagnostic and/or Therapeutic Biomarkers in Chronic Cardiovascular Diseases. Front. Cell Dev. Biol. 2022, 10, 813885. [Google Scholar] [CrossRef]
- Martinez-Pinna, R.; Gonzalez de Peredo, A.; Monsarrat, B.; Burlet-Schiltz, O.; Martin-Ventura, J.L. Label-free quantitative proteomic analysis of human plasma-derived microvesicles to find protein signatures of abdominal aortic aneurysms. Proteom. Clin. Appl. 2014, 8, 620–625. [Google Scholar] [CrossRef]
- Touat, Z.; Lepage, L.; Ollivier, V.; Nataf, P.; Hvass, U.; Labreuche, J.; Jandrot-Perrus, M.; Michel, J.B.; Jondeau, G. Dilation-dependent activation of platelets and prothrombin in human thoracic ascending aortic aneurysm. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 940–946. [Google Scholar] [CrossRef] [Green Version]
- Touat, Z.; Ollivier, V.; Dai, J.; Huisse, M.G.; Bezeaud, A.; Sebbag, U.; Palombi, T.; Rossignol, P.; Meilhac, O.; Guillin, M.C.; et al. Renewal of mural thrombus releases plasma markers and is involved in aortic abdominal aneurysm evolution. Am. J. Pathol. 2006, 168, 1022–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Garcia, C.E.; Burillo, E.; Lindholt, J.S.; Martinez-Lopez, D.; Pilely, K.; Mazzeo, C.; Michel, J.B.; Egido, J.; Garred, P.; Blanco-Colio, L.M.; et al. Association of ficolin-3 with abdominal aortic aneurysm presence and progression. J. Thromb. Haemost. 2017, 15, 575–585. [Google Scholar] [CrossRef] [Green Version]
- Munthe-Fog, L.; Hummelshoj, T.; Honore, C.; Madsen, H.O.; Permin, H.; Garred, P. Immunodeficiency associated with FCN3 mutation and ficolin-3 deficiency. N. Engl. J. Med. 2009, 360, 2637–2644. [Google Scholar] [CrossRef]
- Wang, Y.; Jia, L.; Xie, Y.; Cai, Z.; Liu, Z.; Shen, J.; Lu, Y.; Wang, Y.; Su, S.; Ma, Y.; et al. Involvement of macrophage-derived exosomes in abdominal aortic aneurysms development. Atherosclerosis 2019, 289, 64–72. [Google Scholar] [CrossRef]
- Folkesson, M.; Li, C.; Frebelius, S.; Swedenborg, J.; Wagsater, D.; Williams, K.J.; Eriksson, P.; Roy, J.; Liu, M.L. Proteolytically active ADAM10 and ADAM17 carried on membrane microvesicles in human abdominal aortic aneurysms. Thromb. Haemost. 2015, 114, 1165–1174. [Google Scholar]
- Dang, G.; Li, T.; Yang, D.; Yang, G.; Du, X.; Yang, J.; Miao, Y.; Han, L.; Ma, X.; Song, Y.; et al. T lymphocyte-derived extracellular vesicles aggravate abdominal aortic aneurysm by promoting macrophage lipid peroxidation and migration via pyruvate kinase muscle isozyme 2. Redox Biol. 2022, 50, 102257. [Google Scholar] [CrossRef] [PubMed]
- Petsophonsakul, P.; Furmanik, M.; Forsythe, R.; Dweck, M.; Schurink, G.W.; Natour, E.; Reutelingsperger, C.; Jacobs, M.; Mees, B.; Schurgers, L. Role of Vascular Smooth Muscle Cell Phenotypic Switching and Calcification in Aortic Aneurysm Formation. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1351–1368. [Google Scholar] [CrossRef] [PubMed]
- Hjortnaes, J.; Butcher, J.; Figueiredo, J.L.; Riccio, M.; Kohler, R.H.; Kozloff, K.M.; Weissleder, R.; Aikawa, E. Arterial and aortic valve calcification inversely correlates with osteoporotic bone remodelling: A role for inflammation. Eur. Heart J. 2010, 31, 1975–1984. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Rashdan, N.A.; Zhu, D.; Milne, E.M.; Ajuh, P.; Milne, G.; Helfrich, M.H.; Lim, K.; Prasad, S.; Lerman, D.A.; et al. End stage renal disease-induced hypercalcemia may promote aortic valve calcification via Annexin VI enrichment of valve interstitial cell derived-matrix vesicles. J. Cell Physiol. 2017, 232, 2985–2995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cote, N.; Mahmut, A.; Bosse, Y.; Couture, C.; Page, S.; Trahan, S.; Boulanger, M.C.; Fournier, D.; Pibarot, P.; Mathieu, P. Inflammation is associated with the remodeling of calcific aortic valve disease. Inflammation 2013, 36, 573–581. [Google Scholar] [CrossRef]
- Diehl, P.; Nagy, F.; Sossong, V.; Helbing, T.; Beyersdorf, F.; Olschewski, M.; Bode, C.; Moser, M. Increased levels of circulating microparticles in patients with severe aortic valve stenosis. Thromb. Haemost. 2008, 99, 711–719. [Google Scholar]
- Kaden, J.J.; Dempfle, C.E.; Grobholz, R.; Fischer, C.S.; Vocke, D.C.; Kilic, R.; Sarikoc, A.; Pinol, R.; Hagl, S.; Lang, S.; et al. Inflammatory regulation of extracellular matrix remodeling in calcific aortic valve stenosis. Cardiovasc. Pathol. 2005, 14, 80–87. [Google Scholar] [CrossRef]
- Blaser, M.C.; Aikawa, E. Roles and Regulation of Extracellular Vesicles in Cardiovascular Mineral Metabolism. Front. Cardiovasc. Med. 2018, 5, 187. [Google Scholar] [CrossRef] [Green Version]
- Jansen, F.; Xiang, X.; Werner, N. Role and function of extracellular vesicles in calcific aortic valve disease. Eur. Heart J. 2017, 38, 2714–2716. [Google Scholar] [CrossRef] [Green Version]
- Di Vito, A.; Donato, A.; Presta, I.; Mancuso, T.; Brunetti, F.S.; Mastroroberto, P.; Amorosi, A.; Malara, N.; Donato, G. Extracellular Matrix in Calcific Aortic Valve Disease: Architecture, Dynamic and Perspectives. Int. J. Mol. Sci. 2021, 22, 913. [Google Scholar] [CrossRef]
- Barile, L.; Moccetti, T.; Marban, E.; Vassalli, G. Roles of exosomes in cardioprotection. Eur. Heart J. 2017, 38, 1372–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marban, E. The Secret Life of Exosomes: What Bees Can Teach Us About Next-Generation Therapeutics. J. Am. Coll. Cardiol. 2018, 71, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Marban, E. A mechanistic roadmap for the clinical application of cardiac cell therapies. Nat. Biomed. Eng. 2018, 2, 353–361. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akhmerov, A.; Parimon, T. Extracellular Vesicles, Inflammation, and Cardiovascular Disease. Cells 2022, 11, 2229. https://doi.org/10.3390/cells11142229
Akhmerov A, Parimon T. Extracellular Vesicles, Inflammation, and Cardiovascular Disease. Cells. 2022; 11(14):2229. https://doi.org/10.3390/cells11142229
Chicago/Turabian StyleAkhmerov, Akbarshakh, and Tanyalak Parimon. 2022. "Extracellular Vesicles, Inflammation, and Cardiovascular Disease" Cells 11, no. 14: 2229. https://doi.org/10.3390/cells11142229
APA StyleAkhmerov, A., & Parimon, T. (2022). Extracellular Vesicles, Inflammation, and Cardiovascular Disease. Cells, 11(14), 2229. https://doi.org/10.3390/cells11142229