
NF-κB p65 Attenuates Cardiomyocyte PGC-1α Expression in Hypoxia


Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Cell Culture and Treatments
2.3. Basic RNA-seq Data Processing
2.4. Gene Expression Analysis
2.5. Reactive Oxygen Species Assay
2.6. Mitochondrial Membrane Potential Assay
2.7. Mitochondrial Respiration Assay
2.8. Cell Viability Assay
2.9. Luciferase Assay and Mutagenesis
2.10. qPCR
2.11. Western Blotting
2.12. Electrophoretic Mobility Shift Assay
2.13. Statistical Analysis
3. Results
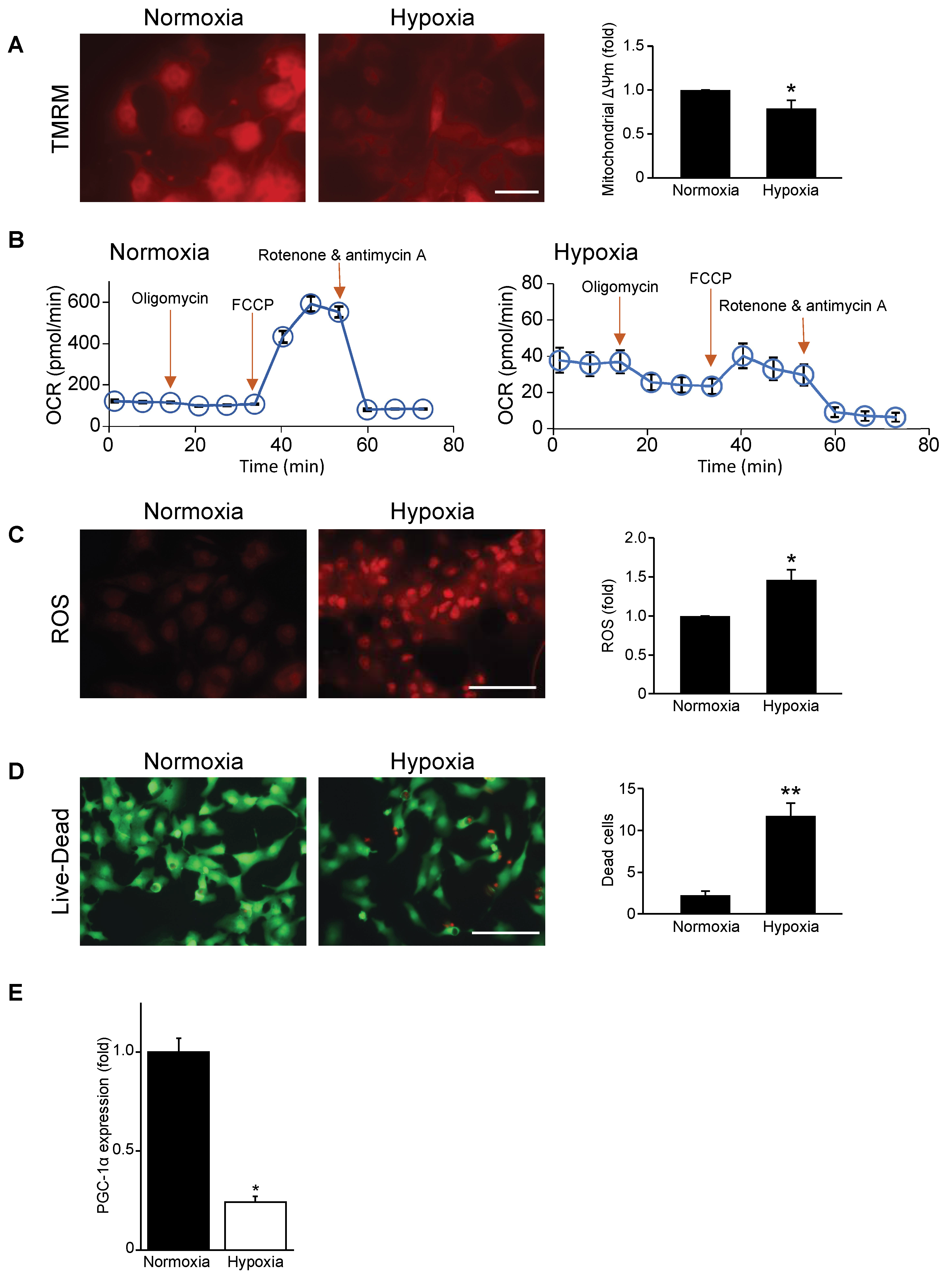
3.1. Mitochondrial Function, Respiration, and Cell Viability Are Impaired during Hypoxia
3.2. Mitochondrial Biogenesis Factor PGC-1α Is Repressed in Hypoxic Cardiac Myocytes
3.3. Hypoxia Increases Nuclear NF-κB p65 and PGC-1α Promoter Binding
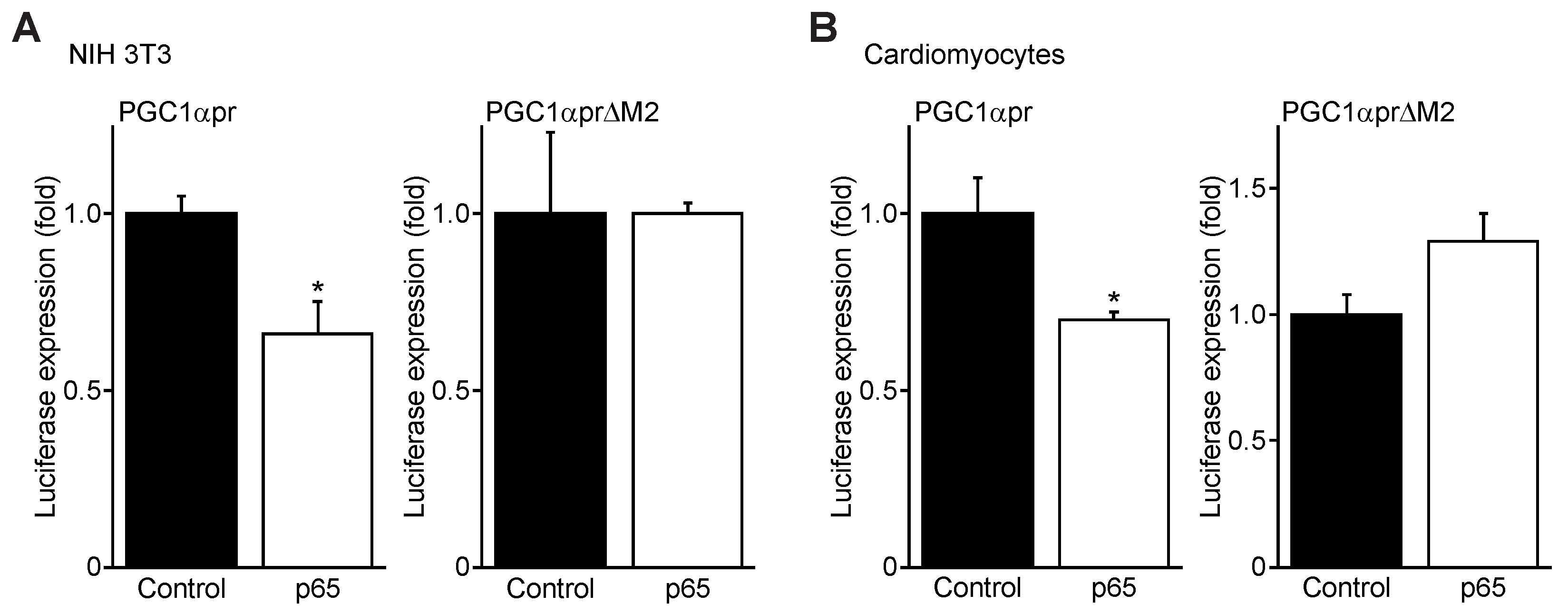
3.4. NFκB p65 Represses PGC1α Promoter Transactivation
3.5. TNFα Represses PGC-1α Expression and Induces p65 Binding to the PGC-1α Promoter M2 Site
3.6. Bioinformatics of Cardiac-Restricted p65 Gene Knockout in Mice
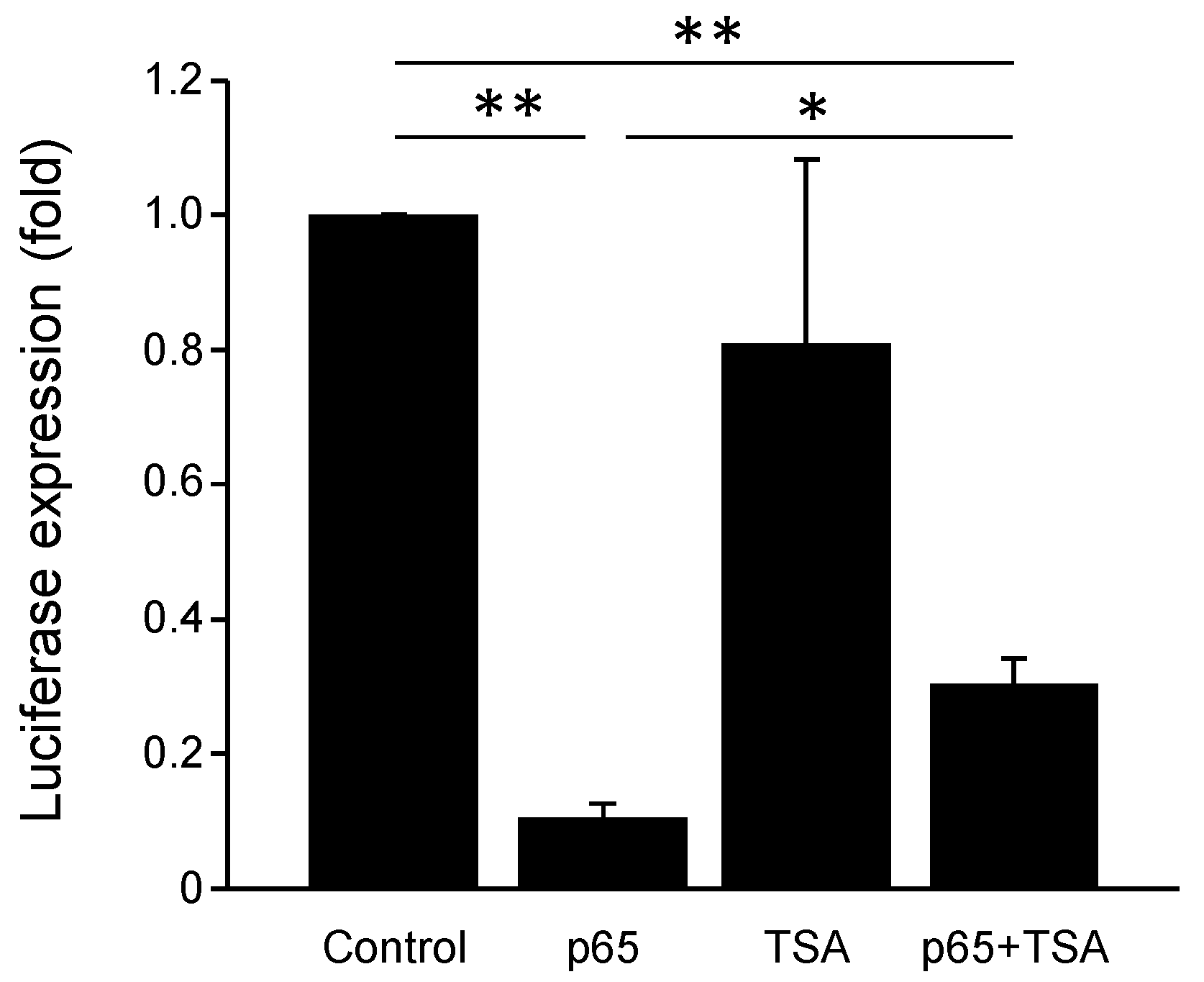
3.7. PGC-1α Promoter Is Repressed by NF-κB and Chromatin Remodeling Histone Deacetylases
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Riehle, C.; Abel, E.D. PGC-1 proteins and heart failure. Trends Cardiovasc. Med. 2012, 22, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Guardia, D.; Palomer, X.; Coll, T.; Davidson, M.M.; Chan, T.O.; Feldman, A.M.; Laguna, J.C.; Vazquez-Carrera, M. The p65 subunit of NF-kappaB binds to PGC-1alpha, linking inflammation and metabolic disturbances in cardiac cells. Cardiovasc. Res. 2010, 87, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Dhingra, R.; Shaw, J.A.; Aviv, Y.; Kirshenbaum, L.A. Dichotomous actions of NF-kappaB signaling pathways in heart. J. Cardiovasc. Transl. Res. 2010, 3, 344–354. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal. Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed]
- Irwin, M.W.; Mak, S.; Mann, D.L.; Qu, R.; Penninger, J.M.; Yan, A.; Dawood, F.; Wen, W.H.; Shou, Z.; Liu, P. Tissue expression and immunolocalization of tumor necrosis factor-alpha in postinfarction dysfunctional myocardium. Circulation 1999, 99, 1492–1498. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, N.; Wada, H.; Kanda, T.; Niwa, T.; Yamada, Y.; Saito, K.; Fujiwara, H.; Sekikawa, K.; Seishima, M. Improved myocardial ischemia/reperfusion injury in mice lacking tumor necrosis factor-alpha. J. Am. Coll. Cardiol. 2002, 39, 1229–1235. [Google Scholar] [CrossRef]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef]
- Morishita, R.; Sugimoto, T.; Aoki, M.; Kida, I.; Tomita, N.; Moriguchi, A.; Maeda, K.; Sawa, Y.; Kaneda, Y.; Higaki, J.; et al. In vivo transfection of cis element “decoy” against nuclear factor- kappaB binding site prevents myocardial infarction. Nat. Med. 1997, 3, 894–899. [Google Scholar] [CrossRef]
- Kawano, S.; Kubota, T.; Monden, Y.; Tsutsumi, T.; Inoue, T.; Kawamura, N.; Tsutsui, H.; Sunagawa, K. Blockade of NF-kappaB improves cardiac function and survival after myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H1337–H1344. [Google Scholar] [CrossRef]
- Hickson-Bick, D.L.; Jones, C.; Buja, L.M. Stimulation of mitochondrial biogenesis and autophagy by lipopolysaccharide in the neonatal rat cardiomyocyte protects against programmed cell death. J. Mol. Cell Cardiol. 2008, 44, 411–418. [Google Scholar] [CrossRef]
- Gang, H.; Shaw, J.; Dhingra, R.; Davie, J.R.; Kirshenbaum, L.A. Epigenetic regulation of canonical TNFalpha pathway by HDAC1 determines survival of cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1662–H1669. [Google Scholar] [CrossRef][Green Version]
- Rabinovich-Nikitin, I.; Rasouli, M.; Reitz, C.J.; Posen, I.; Margulets, V.; Dhingra, R.; Khatua, T.N.; Thliveris, J.A.; Martino, T.A.; Kirshenbaum, L.A. Mitochondrial autophagy and cell survival is regulated by the circadian Clock gene in cardiac myocytes during ischemic stress. Autophagy 2021, 17, 3794–3812. [Google Scholar] [CrossRef] [PubMed]
- Friedman, H.M.; Yee, A.; Diggelmann, H.; Hastings, J.C.; Tal-Singer, R.; Seidel-Dugan, C.A.; Eisenberg, R.J.; Cohen, G.H. Use of a glucocorticoid-inducible promoter for expression of herpes simplex virus type 1 glycoprotein gC1, a cytotoxic protein in mammalian cells. Mol. Cell Biol. 1989, 9, 2303–2314. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Bourgey, M.; Dali, R.; Eveleigh, R.; Chen, K.C.; Letourneau, L.; Fillon, J.; Michaud, M.; Caron, M.; Sandoval, J.; Lefebvre, F.; et al. GenPipes: An open-source framework for distributed and scalable genomic analyses. Gigascience 2019, 8, giz037. [Google Scholar] [CrossRef] [PubMed]
- Huber, W.; Carey, V.J.; Gentleman, R.; Anders, S.; Carlson, M.; Carvalho, B.S.; Bravo, H.C.; Davis, S.; Gatto, L.; Girke, T.; et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 2015, 12, 115–121. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef]
- Czubryt, M.P.; McAnally, J.; Fishman, G.I.; Olson, E.N. Regulation of peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PGC-1alpha) and mitochondrial function by MEF2 and HDAC5. Proc. Natl. Acad. Sci. USA 2003, 100, 1711–1716. [Google Scholar] [CrossRef]
- Ramjiawan, A.; Bagchi, R.A.; Blant, A.; Albak, L.; Cavasin, M.A.; Horn, T.R.; McKinsey, T.A.; Czubryt, M.P. Roles of histone deacetylation and AMP kinase in regulation of cardiomyocyte PGC-1alpha gene expression in hypoxia. Am. J. Physiol. Cell Physiol. 2013, 304, C1064–C1072. [Google Scholar] [CrossRef] [PubMed]
- Ramjiawan, A.; Bagchi, R.A.; Albak, L.; Czubryt, M.P. Mechanism of cardiomyocyte PGC-1alpha gene regulation by ERRalpha. Biochem. Cell Biol. 2013, 91, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Baetz, D.; Regula, K.M.; Ens, K.; Shaw, J.; Kothari, S.; Yurkova, N.; Kirshenbaum, L.A. Nuclear factor- kappaB -mediated cell survival involves transcriptional silencing of the mitochondrial death gene BNIP3 in ventricular myocytes. Circulation 2005, 112, 3777–3785. [Google Scholar] [CrossRef] [PubMed]
- Dhingra, R.; Guberman, M.; Rabinovich-Nikitin, I.; Gerstein, J.; Margulets, V.; Gang, H.; Madden, N.; Thliveris, J.; Kirshenbaum, L.A. Impaired NF- kappaB signalling underlies cyclophilin D-mediated mitochondrial permeability transition pore opening in doxorubicin cardiomyopathy. Cardiovasc. Res. 2020, 116, 1161–1174. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Javadov, S.; Margreiter, R.; Grimm, M.; Hagenbuchner, J.; Ausserlechner, M.J. The Role of Mitochondria in the Mechanisms of Cardiac Ischemia-Reperfusion Injury. Antioxidants 2019, 8, 454. [Google Scholar] [CrossRef]
- Rowe, G.C.; Jiang, A.; Arany, Z. PGC-1 coactivators in cardiac development and disease. Circ. Res. 2010, 107, 825–838. [Google Scholar] [CrossRef]
- Fiordelisi, A.; Iaccarino, G.; Morisco, C.; Coscioni, E.; Sorriento, D. NFkappaB is a Key Player in the Crosstalk between Inflammation and Cardiovascular Diseases. Int. J. Mol. Sci. 2019, 20, 1599. [Google Scholar] [CrossRef]
- Suematsu, N.; Tsutsui, H.; Wen, J.; Kang, D.; Ikeuchi, M.; Ide, T.; Hayashidani, S.; Shiomi, T.; Kubota, T.; Hamasaki, N.; et al. Oxidative stress mediates tumor necrosis factor-alpha-induced mitochondrial DNA damage and dysfunction in cardiac myocytes. Circulation 2003, 107, 1418–1423. [Google Scholar] [CrossRef]
- Ashburner, B.P.; Westerheide, S.D.; Baldwin, A.S., Jr. The p65 (RelA) subunit of NF- kappaB interacts with the histone deacetylase (HDAC) corepressors HDAC1 and HDAC2 to negatively regulate gene expression. Mol. Cell Biol. 2001, 21, 7065–7077. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence | Use |
|---|---|---|
| PGC1a-M2-fwd | 5′-GCTTTGTCATGTGACTGGGGACTGTAGTAAGACAGGTGCCTTCAG-3′ | EMSA |
| PGC1a-M2-rev | 5′-CTGAAGGCACCTGTCTTACTACAGTCCCCAGTCACATGACAAAGC-3′ | EMSA |
| PGC1a-∆M2-fwd | 5′-GCTTTGTCATGTGACTGGaGctcGTAGTAAGACAGGTGCCTTCAG-3′ | Mut. |
| PGC1a-∆M2-rev | 5′-CTGAAGGCACCTGTCTTACTACgagCtCCAGTCACATGACAAAGC-3′ | Mut. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rabinovich-Nikitin, I.; Blant, A.; Dhingra, R.; Kirshenbaum, L.A.; Czubryt, M.P. NF-κB p65 Attenuates Cardiomyocyte PGC-1α Expression in Hypoxia. Cells 2022, 11, 2193. https://doi.org/10.3390/cells11142193
Rabinovich-Nikitin I, Blant A, Dhingra R, Kirshenbaum LA, Czubryt MP. NF-κB p65 Attenuates Cardiomyocyte PGC-1α Expression in Hypoxia. Cells. 2022; 11(14):2193. https://doi.org/10.3390/cells11142193
Chicago/Turabian StyleRabinovich-Nikitin, Inna, Alexandra Blant, Rimpy Dhingra, Lorrie A. Kirshenbaum, and Michael P. Czubryt. 2022. "NF-κB p65 Attenuates Cardiomyocyte PGC-1α Expression in Hypoxia" Cells 11, no. 14: 2193. https://doi.org/10.3390/cells11142193
APA StyleRabinovich-Nikitin, I., Blant, A., Dhingra, R., Kirshenbaum, L. A., & Czubryt, M. P. (2022). NF-κB p65 Attenuates Cardiomyocyte PGC-1α Expression in Hypoxia. Cells, 11(14), 2193. https://doi.org/10.3390/cells11142193