
The Functional Role and Regulatory Mechanism of Bromodomain-Containing Protein 9 in Human Uterine Leiomyosarcoma

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Tissues and Immunohistochemistry
2.2. Cells and Reagents
2.3. Proliferation Assay
2.4. RNA Extraction and Gene Expression
2.5. Protein Extraction and Western Blot
2.6. RNA-Sequencing
2.7. Transcriptome Profiles Analysis
2.7.1. Transcriptome Data Analysis
2.7.2. Integrated Bioinformatics Analysis
2.8. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
2.9. Statistical Analysis
3. Results
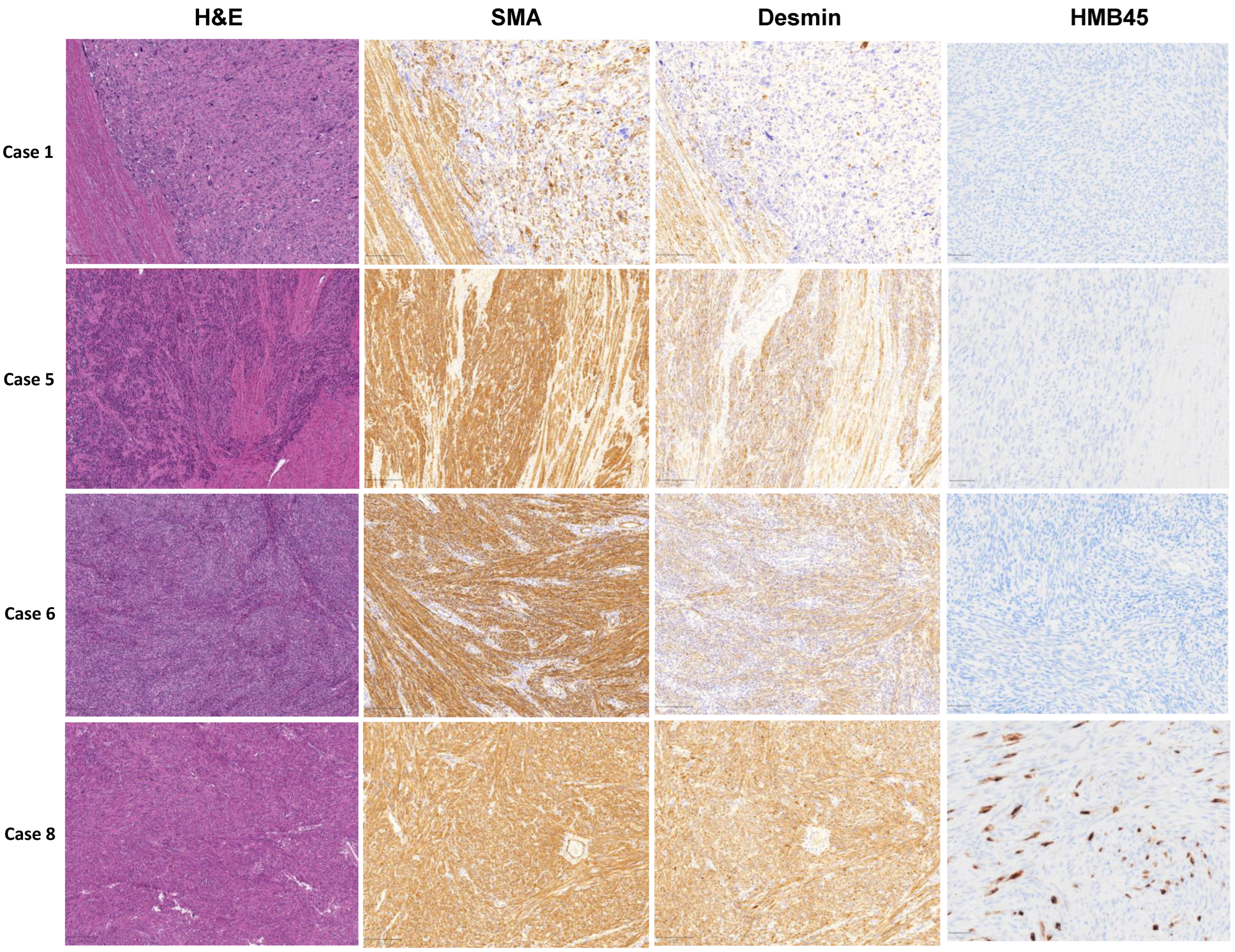
3.1. Pathological Examination
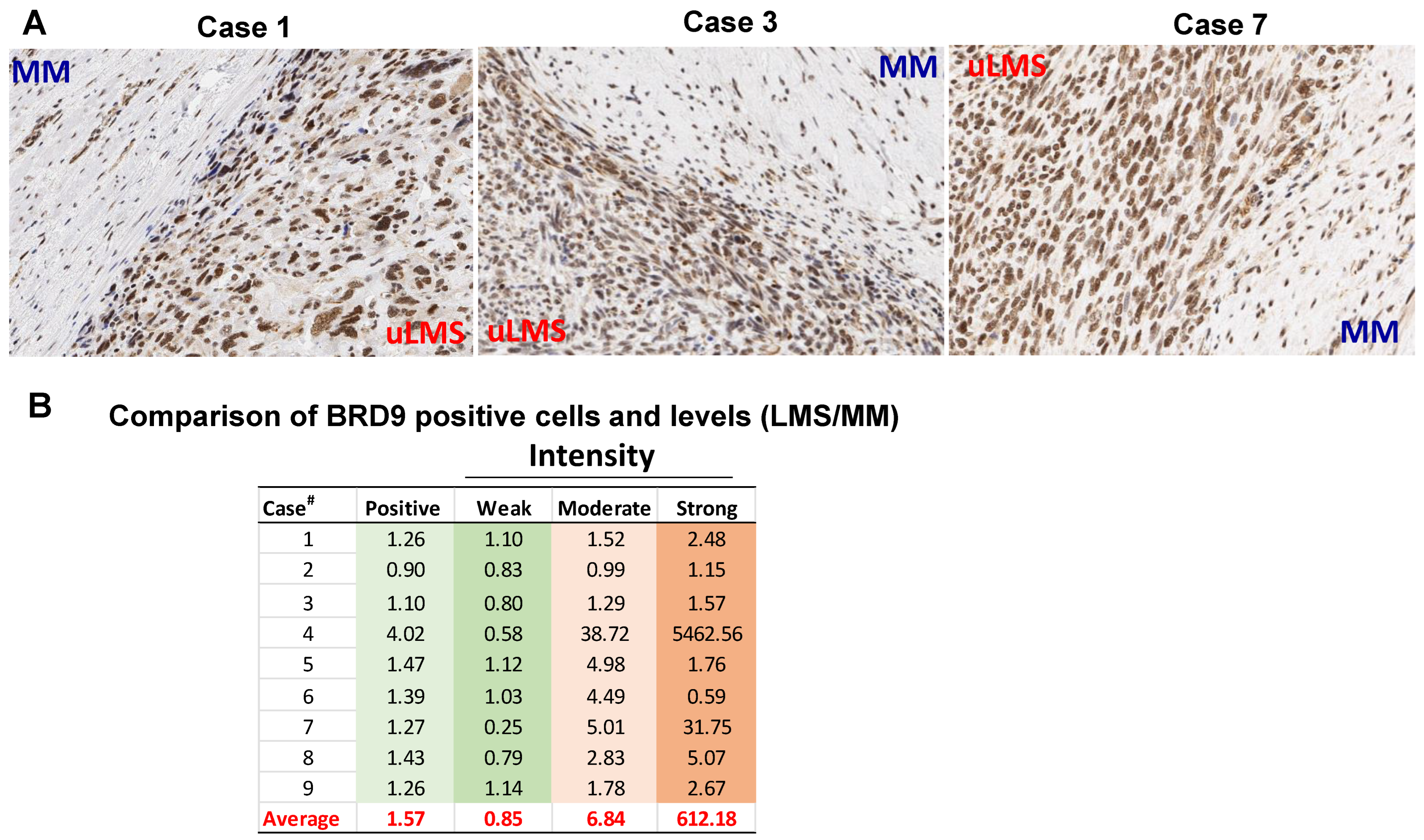
3.2. The BRD9 Expression Is Upregulated in uLMS Tissues Compared to Adjacent Myometrium from Women with uLMS
3.3. BRD9 Levels Are Upregulated in uLMS Cell Lines
3.4. Inhibition of BRD9 Decreased uLMS Cell Proliferation
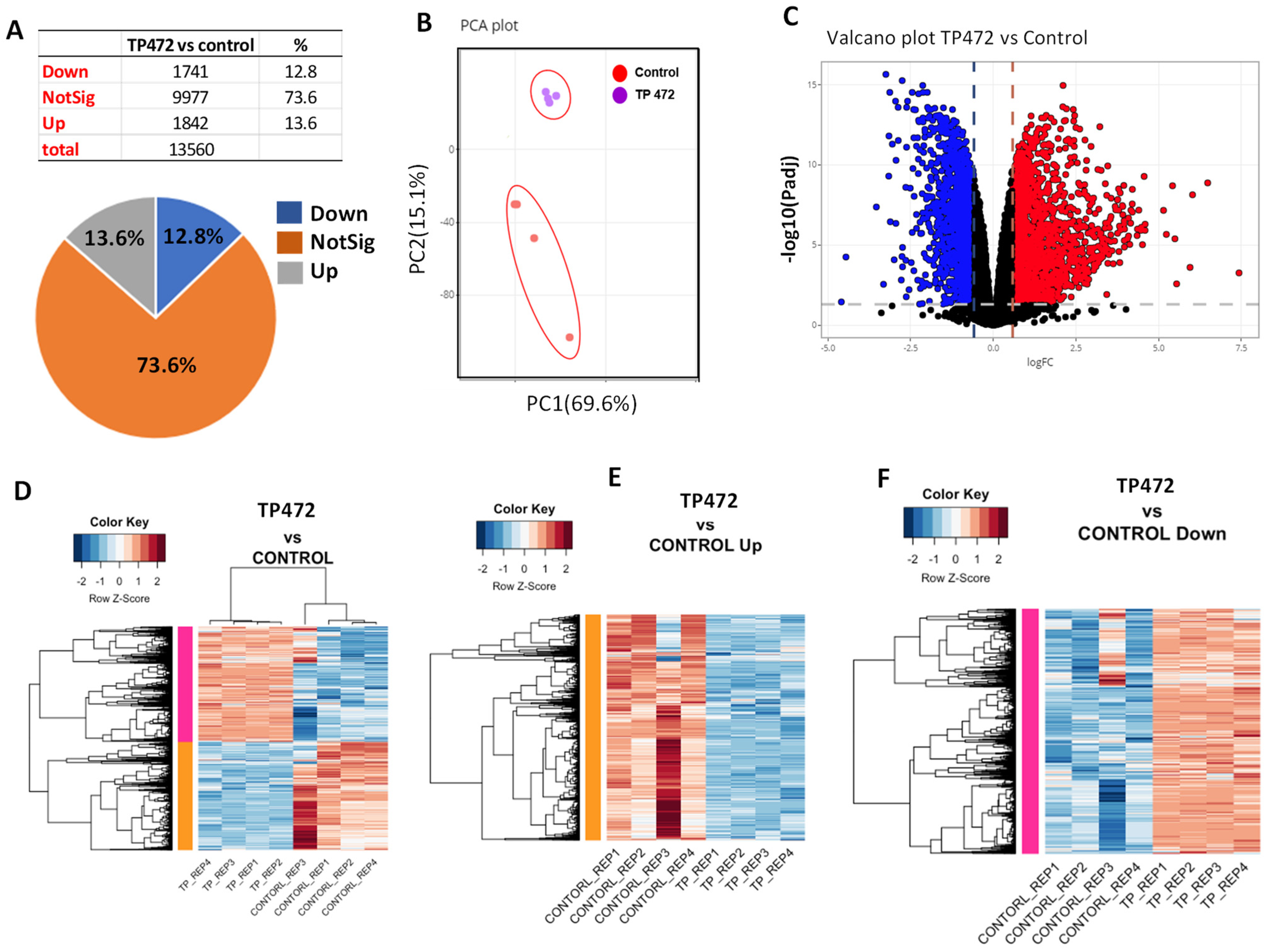
3.5. BRD9 Inhibition Causes Extensive Changes in the uLMS Cell Transcriptome
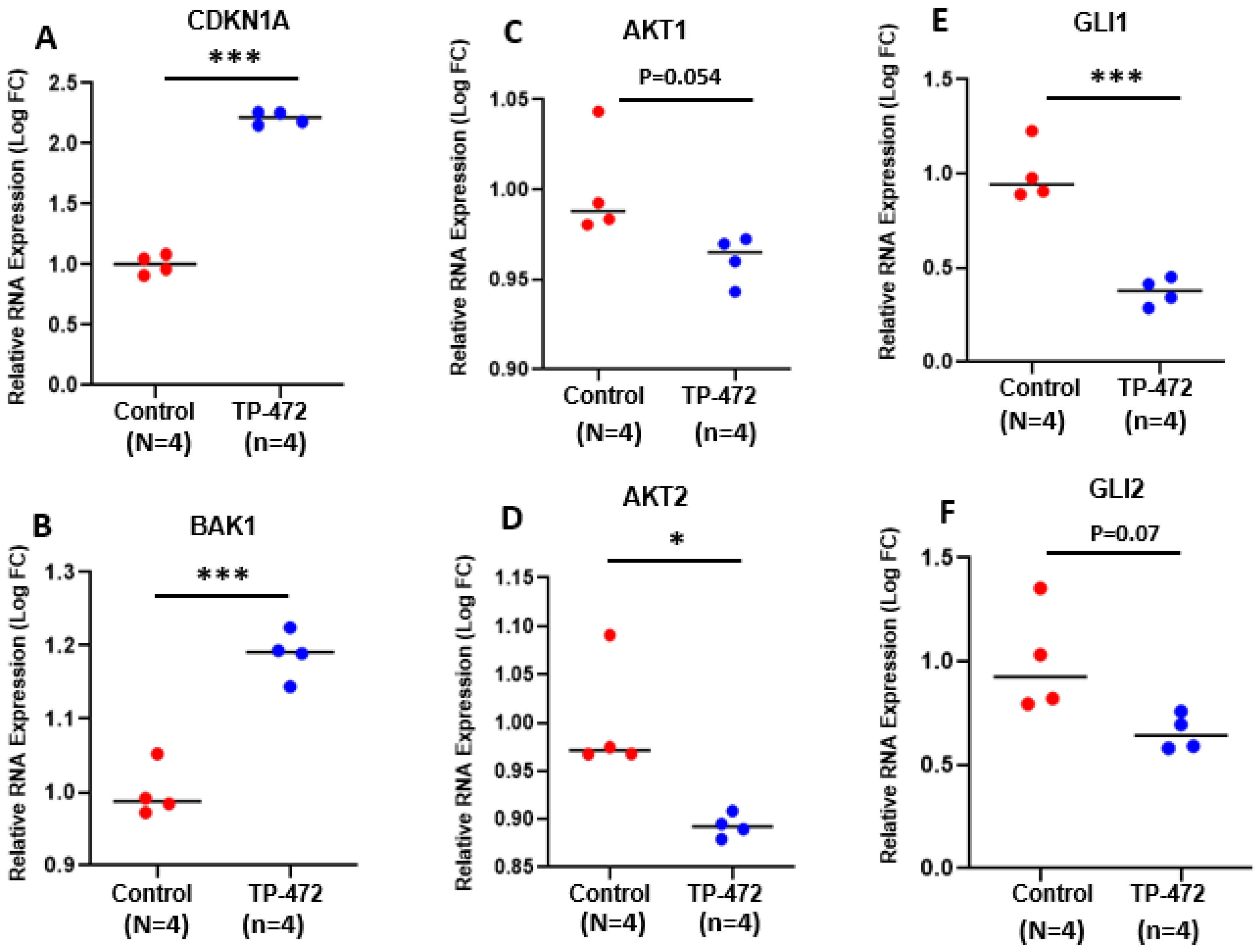
3.5.1. Pathway Analysis of DEGs upon TP-472 Treatment
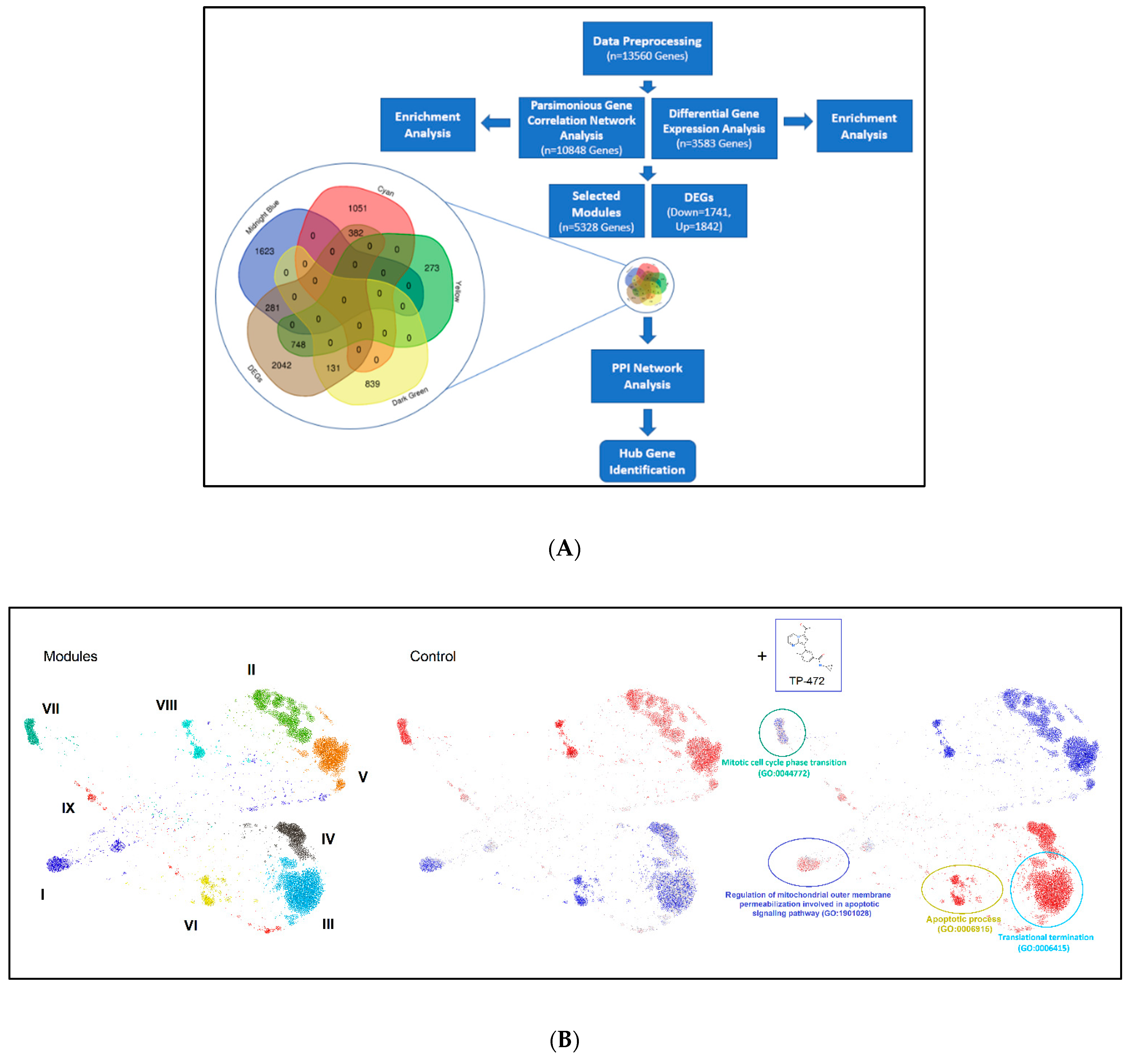
3.5.2. Parsimonious Gene Correlation Network Analysis (PGCNA)
3.5.3. Inhibition of BRD9 Altered the Gene Expression Correlating to Histone Modifications
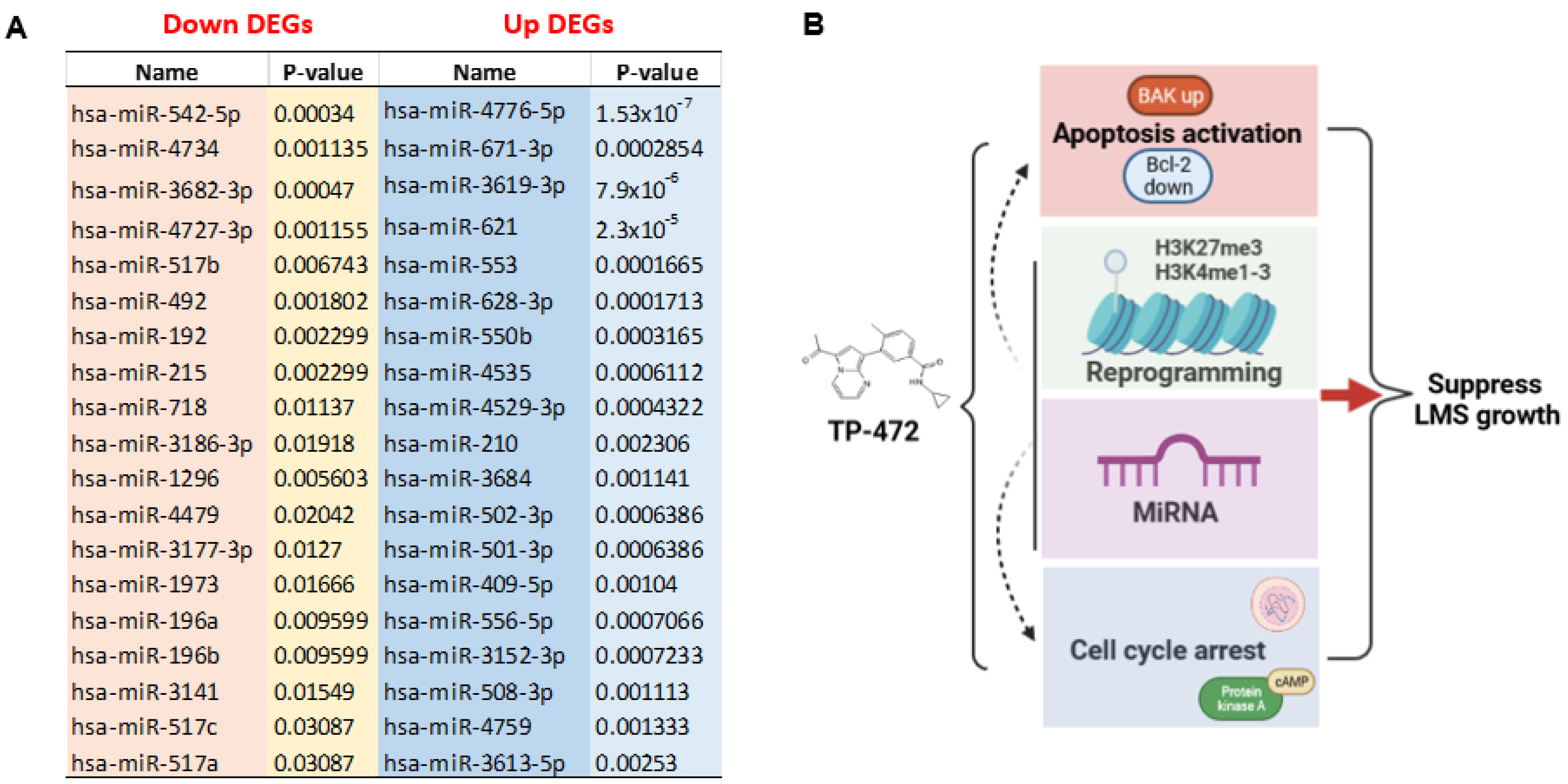
3.5.4. Inhibition of BRD9 Altered Gene Expression Correlating to miRNA Regulation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- D’Angelo, E.; Prat, J. Uterine sarcomas: A review. Gynecol. Oncol. 2010, 116, 131–139. [Google Scholar]
- Seagle, B.L.; Sobecki-Rausch, J.; Strohl, A.E.; Shilpi, A.; Grace, A.; Shahabi, S. Prognosis and treatment of uterine leiomyosarcoma: A National Cancer Database study. Gynecol. Oncol. 2017, 145, 61–70. [Google Scholar]
- Hensley, M.L.; Blessing, J.A.; Mannel, R.; Rose, P.G. Fixed-dose rate gemcitabine plus docetaxel as first-line therapy for metastatic uterine leiomyosarcoma: A Gynecologic Oncology Group phase II trial. Gynecol. Oncol. 2008, 109, 329–334. [Google Scholar]
- Gadducci, A.; Landoni, F.; Sartori, E.; Zola, P.; Maggino, T.; Lissoni, A.; Bazzurini, L.; Arisio, R.; Romagnolo, C.; Cristofani, R. Uterine leiomyosarcoma: Analysis of treatment failures and survival. Gynecol. Oncol. 1996, 62, 25–32. [Google Scholar]
- Garcia, N.; Ulin, M.; Ali, M.; Al-Hendy, A.; Carvalho, K.C.; Yang, Q. Evaluation of Hedgehog Pathway Inhibitors as a Therapeutic Option for Uterine Leiomyosarcoma Using the Xenograft Model. Reprod. Sci. 2021, 29, 781–790. [Google Scholar]
- Yang, Q.; Ciebiera, M.; Bariani, M.V.; Ali, M.; Elkafas, H.; Boyer, T.G.; Al-Hendy, A. Comprehensive Review of Uterine Fibroids: Developmental Origin, Pathogenesis, and Treatment. Endocr. Rev. 2021. [Google Scholar] [CrossRef]
- Zuccala, E. Epigenetics: Misdirecting methylation to drive oncogenesis. Nat. Rev. Cancer 2016, 16, 410. [Google Scholar]
- Wong, C.C.; Qian, Y.; Yu, J. Interplay between epigenetics and metabolism in oncogenesis: Mechanisms and therapeutic approaches. Oncogene 2017, 36, 3359–3374. [Google Scholar]
- Yang, Q.W.; Liu, S.; Tian, Y.; Salwen, H.R.; Chlenski, A.; Weinstein, J.; Cohn, S.L. Methylation-associated silencing of the thrombospondin-1 gene in human neuroblastoma. Cancer Res. 2003, 63, 6299–6310. [Google Scholar]
- Yang, Q.; Zage, P.; Kagan, D.; Tian, Y.; Seshadri, R.; Salwen, H.R.; Liu, S.; Chlenski, A.; Cohn, S.L. Association of epigenetic inactivation of RASSF1A with poor outcome in human neuroblastoma. Clin. Cancer Res. 2004, 10, 8493–8500. [Google Scholar]
- Yang, Q.; Tian, Y.; Ostler, K.R.; Chlenski, A.; Guerrero, L.J.; Salwen, H.R.; Godley, L.A.; Cohn, S.L. Epigenetic alterations differ in phenotypically distinct human neuroblastoma cell lines. BMC Cancer 2010, 10, 286. [Google Scholar]
- Jones, P.A. Epigenetics in carcinogenesis and cancer prevention. Ann. N. Y. Acad. Sci. 2003, 983, 213–219. [Google Scholar]
- Kanwal, R.; Gupta, S. Epigenetics and cancer. J. Appl. Physiol. 2010, 109, 598–605. [Google Scholar]
- Khare, S.; Verma, M. Epigenetics of colon cancer. Methods Mol. Biol. 2012, 863, 177–185. [Google Scholar]
- Kim, W.J.; Kim, Y.J. Epigenetics of bladder cancer. Methods Mol. Biol. 2012, 863, 111–118. [Google Scholar]
- Laird, P.W. Cancer epigenetics. Hum. Mol. Genet. 2005, 14, R65–R76. [Google Scholar]
- Yang, Q.; Mas, A.; Diamond, M.P.; Al-Hendy, A. The Mechanism and Function of Epigenetics in Uterine Leiomyoma Development. Reprod. Sci. 2016, 23, 163–175. [Google Scholar]
- Jain, A.K.; Barton, M.C. Bromodomain Histone Readers and Cancer. J. Mol. Biol. 2017, 429, 2003–2010. [Google Scholar]
- Fujisawa, T.; Filippakopoulos, P. Functions of bromodomain-containing proteins and their roles in homeostasis and cancer. Nat. Rev. Mol. Cell Biol. 2017, 18, 246–262. [Google Scholar]
- Crawford, N.P.; Alsarraj, J.; Lukes, L.; Walker, R.C.; Officewala, J.S.; Yang, H.H.; Lee, M.P.; Ozato, K.; Hunter, K.W. Bromodomain 4 activation predicts breast cancer survival. Proc. Natl. Acad. Sci. USA 2008, 105, 6380–6385. [Google Scholar]
- Leal, A.S.; Liu, P.; Krieger-Burke, T.; Ruggeri, B.; Liby, K.T. The Bromodomain Inhibitor, INCB057643, Targets Both Cancer Cells and the Tumor Microenvironment in Two Preclinical Models of Pancreatic Cancer. Cancers 2020, 13, 96. [Google Scholar]
- Kregel, S.; Malik, R.; Asangani, I.A.; Wilder-Romans, K.; Rajendiran, T.; Xiao, L.; Vo, J.N.; Soni, T.; Cieslik, M.; Fernadez-Salas, E.; et al. Functional and Mechanistic Interrogation of BET Bromodomain Degraders for the Treatment of Metastatic Castration-resistant Prostate Cancer. Clin. Cancer Res. 2019, 25, 4038–4048. [Google Scholar]
- Kato, F.; Fiorentino, F.P.; Alibes, A.; Perucho, M.; Sanchez-Cespedes, M.; Kohno, T.; Yokota, J. MYCL is a target of a BET bromodomain inhibitor, JQ1, on growth suppression efficacy in small cell lung cancer cells. Oncotarget 2016, 7, 77378–77388. [Google Scholar]
- Faivre, E.J.; McDaniel, K.F.; Albert, D.H.; Mantena, S.R.; Plotnik, J.P.; Wilcox, D.; Zhang, L.; Bui, M.H.; Sheppard, G.S.; Wang, L.; et al. Selective inhibition of the BD2 bromodomain of BET proteins in prostate cancer. Nature 2020, 578, 306–310. [Google Scholar]
- Zhu, X.; Liao, Y.; Tang, L. Targeting BRD9 for Cancer Treatment: A New Strategy. Onco. Targets Ther. 2020, 13, 13191–13200. [Google Scholar]
- Bell, C.M.; Raffeiner, P.; Hart, J.R.; Vogt, P.K. PIK3CA Cooperates with KRAS to Promote MYC Activity and Tumorigenesis via the Bromodomain Protein BRD9. Cancers 2019, 11, 1634. [Google Scholar]
- Huang, H.; Wang, Y.; Li, Q.; Fei, X.; Ma, H.; Hu, R. miR-140-3p functions as a tumor suppressor in squamous cell lung cancer by regulating BRD9. Cancer Lett. 2019, 446, 81–89. [Google Scholar]
- Del Gaudio, N.; Di Costanzo, A.; Liu, N.Q.; Conte, L.; Migliaccio, A.; Vermeulen, M.; Martens, J.H.A.; Stunnenberg, H.G.; Nebbioso, A.; Altucci, L. BRD9 binds cell type-specific chromatin regions regulating leukemic cell survival via STAT5 inhibition. Cell Death Dis. 2019, 10, 338. [Google Scholar]
- Care, M.A.; Westhead, D.R.; Tooze, R.M. Parsimonious Gene Correlation Network Analysis (PGCNA): A tool to define modular gene co-expression for refined molecular stratification in cancer. NPJ Syst. Biol. Appl. 2019, 5, 13. [Google Scholar]
- Jacomy, M.; Venturini, T.; Heymann, S.; Bastian, M. ForceAtlas2, a continuous graph layout algorithm for handy network visualization designed for the Gephi software. PLoS ONE 2014, 9, e98679. [Google Scholar]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar]
- Supek, F.; Bosnjak, M.; Skunca, N.; Smuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar]
- Yang, Q.; Elam, L.; Laknaur, A.; Gavrilova-Jordan, L.; Lue, J.; Diamond, M.P.; Al-Hendy, A. Altered DNA repair genes in human uterine fibroids are epigenetically regulated via EZH2 histone methyltransferase. Fertil. Steril. 2015, 104, e72. [Google Scholar]
- Mason, L.D.; Chava, S.; Reddi, K.K.; Gupta, R. The BRD9/7 Inhibitor TP-472 Blocks Melanoma Tumor Growth by Suppressing ECM-Mediated Oncogenic Signaling and Inducing Apoptosis. Cancers 2021, 13, 5516. [Google Scholar]
- Nitulescu, G.M.; Van De Venter, M.; Nitulescu, G.; Ungurianu, A.; Juzenas, P.; Peng, Q.; Olaru, O.T.; Grădinaru, D.; Tsatsakis, A.; Tsoukalas, D.; et al. The Akt pathway in oncology therapy and beyond (Review). Int. J. Oncol. 2018, 53, 2319–2331. [Google Scholar]
- Costa, R.L.B.; Han, H.S.; Gradishar, W.J. Targeting the PI3K/AKT/mTOR pathway in triple-negative breast cancer: A review. Breast Cancer Res. Treat. 2018, 169, 397–406. [Google Scholar]
- Schrump, D.S.; Hong, J.A.; Nguyen, D.M. Utilization of chromatin remodeling agents for lung cancer therapy. Cancer J. 2007, 13, 56–64. [Google Scholar]
- Qi, J. Bromodomain and extraterminal domain inhibitors (BETi) for cancer therapy: Chemical modulation of chromatin structure. Cold Spring Harb. Perspect. Biol. 2014, 6, a018663. [Google Scholar]
- Kaur, J.; Daoud, A.; Eblen, S.T. Targeting Chromatin Remodeling for Cancer Therapy. Curr. Mol. Pharmacol. 2019, 12, 215–229. [Google Scholar]
- Magnani, L.; Stoeck, A.; Zhang, X.; Lanczky, A.; Mirabella, A.C.; Wang, T.L.; Gyorffy, B.; Lupien, M. Genome-wide reprogramming of the chromatin landscape underlies endocrine therapy resistance in breast cancer. Proc. Natl. Acad. Sci. USA 2013, 110, E1490–E1499. [Google Scholar]
- Santillan, D.A.; Theisler, C.M.; Ryan, A.S.; Popovic, R.; Stuart, T.; Zhou, M.M.; Alkan, S.; Zeleznik-Le, N.J. Bromodomain and histone acetyltransferase domain specificities control mixed lineage leukemia phenotype. Cancer Res. 2006, 66, 10032–10039. [Google Scholar]
- Dawson, M.A.; Prinjha, R.K.; Dittmann, A.; Giotopoulos, G.; Bantscheff, M.; Chan, W.I.; Robson, S.C.; Chung, C.-w.; Hopf, C.; Savitski, M.M.; et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 2011, 478, 529–533. [Google Scholar]
- Lucas, X.; Gunther, S. Targeting the BET family for the treatment of leukemia. Epigenomics 2014, 6, 153–155. [Google Scholar]
- Belkina, A.C.; Nikolajczyk, B.S.; Denis, G.V. BET protein function is required for inflammation: Brd2 genetic disruption and BET inhibitor JQ1 impair mouse macrophage inflammatory responses. J. Immunol. 2013, 190, 3670–3678. [Google Scholar]
- Qiu, H.; Jackson, A.L.; Kilgore, J.E.; Zhong, Y.; Chan, L.L.; Gehrig, P.A.; Zhou, C.; Bae-Jump, V.L. JQ1 suppresses tumor growth through downregulating LDHA in ovarian cancer. Oncotarget 2015, 6, 6915–6930. [Google Scholar]
- Richter, G.H.S.; Hensel, T.; Schmidt, O.; Saratov, V.; von Heyking, K.; Becker-Dettling, F.; Prexler, C.; Yen, H.-Y.; Steiger, K.; Fulda, S.; et al. Combined Inhibition of Epigenetic Readers and Transcription Initiation Targets the EWS-ETS Transcriptional Program in Ewing Sarcoma. Cancers 2020, 12, 304. [Google Scholar]
- Choi, J.; Manzano, A.; Dong, W.; Bellone, S.; Bonazzoli, E.; Zammataro, L.; Yao, X.; Deshpande, A.; Zaidi, S.; Guglielmi, A.; et al. Integrated mutational landscape analysis of uterine leiomyosarcomas. Proc. Natl. Acad. Sci. USA 2021, 118, e2025182118. [Google Scholar]
- Park, S.G.; Lee, D.; Seo, H.R.; Lee, S.A.; Kwon, J. Cytotoxic activity of bromodomain inhibitor NVS-CECR2-1 on human cancer cells. Sci. Rep. 2020, 10, 16330. [Google Scholar]
- Lu, J.; Qian, C.; Ji, Y.; Bao, Q.; Lu, B. Gene Signature Associated With Bromodomain Genes Predicts the Prognosis of Kidney Renal Clear Cell Carcinoma. Front. Genet. 2021, 12, 643935. [Google Scholar]
- Yamaguchi, M.; Kusunoki, S.; Hirayama, T.; Fujino, K.; Terao, Y.; Itakura, A. Case of leiomyosarcoma arising from subserosal leiomyoma. J. Obs. Gynaecol. Res. 2019, 45, 1944–1947. [Google Scholar]
- Di Giorgio, E.; Dalla, E.; Franforte, E.; Paluvai, H.; Minisini, M.; Trevisanut, M.; Picco, R.; Brancolini, C. Different class IIa HDACs repressive complexes regulate specific epigenetic responses related to cell survival in leiomyosarcoma cells. Nucleic Acids Res. 2020, 48, 646–664. [Google Scholar]
- Moustakim, M.; Clark, P.G.K.; Hay, D.A.; Dixon, D.J.; Brennan, P.E. Chemical probes and inhibitors of bromodomains outside the BET family. Medchemcomm 2016, 7, 2246–2264. [Google Scholar]
- Dey, P.; Li, J.; Zhang, J.; Chaurasiya, S.; Strom, A.; Wang, H.; Liao, W.-T.; Cavallaro, F.; Denz, P.; Bernard, V.; et al. Oncogenic KRAS-Driven Metabolic Reprogramming in Pancreatic Cancer Cells Utilizes Cytokines from the Tumor Microenvironment. Cancer Discov. 2020, 10, 608–625. [Google Scholar]
- Hann, S.R. Role of post-translational modifications in regulating c-Myc proteolysis, transcriptional activity and biological function. Semin. Cancer Biol. 2006, 16, 288–302. [Google Scholar]
- Archer, T.C.; Ehrenberger, T.; Mundt, F.; Gold, M.P.; Krug, K.; Mah, C.K.; Mahoney, E.L.; Daniel, C.J.; LeNail, A.; Ramamoorthy, D.; et al. Proteomics, Post-translational Modifications, and Integrative Analyses Reveal Molecular Heterogeneity within Medulloblastoma Subgroups. Cancer Cell 2018, 34, 396–410. [Google Scholar]
- Tu, W.B.; Helander, S.; Pilstal, R.; Hickman, K.A.; Lourenco, C.; Jurisica, I.; Raught, B.; Wallner, B.; Sunnerhagen, M.; Penn, L.Z. Myc and its interactors take shape. Biochim. Biophys. Acta 2015, 1849, 469–483. [Google Scholar]
- Lourenco, C.; Resetca, D.; Redel, C.; Lin, P.; MacDonald, A.S.; Ciaccio, R.; Kenney, T.M.G.; Wei, Y.; Andrews, D.W.; Sunnerhagen, M.; et al. MYC protein interactors in gene transcription and cancer. Nat. Rev. Cancer 2021, 21, 579–591. [Google Scholar]
- Gazon, H.; Barbeau, B.; Mesnard, J.M.; Peloponese, J.M., Jr. Hijacking of the AP-1 Signaling Pathway during Development of, A.T.L. Front. Microbiol. 2017, 8, 2686. [Google Scholar]
- Langer, S.; Singer, C.F.; Hudelist, G.; Dampier, B.; Kaserer, K.; Vinatzer, U.; Pehamberger, H.; Zielinski, C.; Kubista, E.; Schreibner, M. Jun and Fos family protein expression in human breast cancer: Correlation of protein expression and clinicopathological parameters. Eur. J. Gynaecol. Oncol. 2006, 27, 345–352. [Google Scholar]
- Chinenov, Y.; Kerppola, T.K. Close encounters of many kinds: Fos-Jun interactions that mediate transcription regulatory specificity. Oncogene 2001, 20, 2438–2452. [Google Scholar]
- Han, Z.; Boyle, D.L.; Aupperle, K.R.; Bennett, B.; Manning, A.M.; Firestein, G.S. Jun N-terminal kinase in rheumatoid arthritis. J. Pharm. Exp. Ther. 1999, 291, 124–130. [Google Scholar]
- Aicher, W.K.; Dinkel, A.; Grimbacher, B.; Haas, C.; Seydlitz-Kurzbach, E.V.; Peter, H.H.; Eibel, H. Serum response elements activate and cAMP responsive elements inhibit expression of transcription factor Egr-1 in synovial fibroblasts of rheumatoid arthritis patients. Int. Immunol. 1999, 11, 47–61. [Google Scholar]
- Wang, B.; Guo, H.; Yu, H.; Chen, Y.; Xu, H.; Zhao, G. The Role of the Transcription Factor EGR1 in Cancer. Front. Oncol. 2021, 11, 642547. [Google Scholar]
- Zhang, C.; Chen, L.; Lou, W.; Su, J.; Huang, J.; Liu, A.; Xu, Y.; He, H.; Gao, Y.; Xu, D.; et al. Aberrant activation of m6A demethylase FTO renders HIF2alpha(low/-) clear cell renal cell carcinoma sensitive to BRD9 inhibitors. Sci. Transl. Med. 2021, 13, eabf6045. [Google Scholar]
- Garcia, N.; Al-Hendy, A.; Baracat, E.C.; Carvalho, K.C.; Yang, Q. Targeting Hedgehog Pathway and DNA Methyltransferases in Uterine Leiomyosarcoma Cells. Cells 2020, 10, 53. [Google Scholar]
- Garcia, N.; Ulin, M.; Al-Hendy, A.; Yang, Q. The Role of Hedgehog Pathway in Female Cancers. J. Cancer Sci. Clin. Ther. 2020, 4, 487–498. [Google Scholar]
- Bulavin, D.V.; Kovalsky, O.; Hollander, M.C.; Fornace, A.J., Jr. Loss of oncogenic H-ras-induced cell cycle arrest and p38 mitogen-activated protein kinase activation by disruption of Gadd45a. Mol. Cell Biol. 2003, 23, 3859–3871. [Google Scholar]
- Bhargava, S.; Patil, V.; Shah, R.A.; Somasundaram, K. IGF2 mRNA binding protein 3 (IMP3) mediated regulation of transcriptome and translatome in glioma cells. Cancer Biol. Ther. 2018, 19, 42–52. [Google Scholar]
- Zhang, R.; Shi, H.; Ren, F.; Zhang, M.; Ji, P.; Wang, W.; Liu, C. The aberrant upstream pathway regulations of CDK1 protein were implicated in the proliferation and apoptosis of ovarian cancer cells. J. Ovarian Res. 2017, 10, 60. [Google Scholar]
- Ying, X.; Che, X.; Wang, J.; Zou, G.; Yu, Q.; Zhang, X. CDK1 serves as a novel therapeutic target for endometrioid endometrial cancer. J. Cancer 2021, 12, 2206–2215. [Google Scholar]
- Gothe, H.J.; Bouwman, B.A.M.; Gusmao, E.G.; Piccinno, R.; Petrosino, G.; Sayols, S.; Drechsel, O.; Minneker, V.; Josipovic, N.; Mizi, A.; et al. Spatial Chromosome Folding and Active Transcription Drive DNA Fragility and Formation of Oncogenic MLL Translocations. Mol. Cell 2019, 75, 267–283. [Google Scholar]
- Chen, T.; Sun, Y.; Ji, P.; Kopetz, S.; Zhang, W. Topoisomerase IIalpha in chromosome instability and personalized cancer therapy. Oncogene 2015, 34, 4019–4031. [Google Scholar]
- Tsai, S.C.; Valkov, N.; Yang, W.M.; Gump, J.; Sullivan, D.; Seto, E. Histone deacetylase interacts directly with DNA topoisomerase II. Nat. Genet. 2000, 26, 349–353. [Google Scholar]
- Ali, M.; Shahin, S.M.; Sabri, N.A.; Al-Hendy, A.; Yang, Q. Activation of beta-Catenin Signaling and its Crosstalk With Estrogen and Histone Deacetylases in Human Uterine Fibroids. J. Clin. Endocrinol. Metab. 2020, 105, e1517–e1535. [Google Scholar]
- Zhao, W.; Xu, Y.; Wang, Y.; Gao, D.; King, J.; Xu, Y.; Liang, F.-S. Investigating crosstalk between H3K27 acetylation and H3K4 trimethylation in CRISPR/dCas-based epigenome editing and gene activation. Sci. Rep. 2021, 11, 15912. [Google Scholar]
- Hasan, N.M.; Sharma, A.; Ruzgar, N.M.; Deshpande, H.; Olino, K.; Khan, S.; Ahuja, N. Epigenetic signatures differentiate uterine and soft tissue leiomyosarcoma. Oncotarget 2021, 12, 1566–1579. [Google Scholar]
- De Carvalho Fischer, C.; Hu, Y.; Morreale, M.; Lin, W.Y.; Wali, A.; Thakar, M.; Karunasena, E.; Sen, R.; Cai, Y.; Murphy, L.; et al. Treatment with epigenetic agents profoundly inhibits tumor growth in leiomyosarcoma. Oncotarget 2018, 9, 19379–19395. [Google Scholar]
- Conconi, D.; Redaelli, S.; Lissoni, A.A.; Cilibrasi, C.; Perego, P.; Gautiero, E.; Sala, E.; Paderno, M.; Dalprà, L.; Landoni, F.; et al. Genomic and Epigenomic Profile of Uterine Smooth Muscle Tumors of Uncertain Malignant Potential (STUMPs) Revealed Similarities and Differences with Leiomyomas and Leiomyosarcomas. Int. J. Mol. Sci. 2021, 22, 1580. [Google Scholar]
- de Almeida, B.C.; Dos Anjos, L.G.; Uno, M.; Cunha, I.W.D.; Soares, F.A.; Baiocchi, G.; Baracat, E.C.; Carvalho, K.C. Let-7 miRNA’s Expression Profile and Its Potential Prognostic Role in Uterine Leiomyosarcoma. Cells 2019, 8, 1452. [Google Scholar]
- Gonzalez Dos Anjos, L.; de Almeida, B.C.; Gomes de Almeida, T.; Mourao Lavorato Rocha, A.; De Nardo Maffazioli, G.; Soares, F.A.; Cunha, I.W.D.; Baracat, E.C.; Carvalho, K.C. Could miRNA Signatures be Useful for Predicting Uterine Sarcoma and Carcinosarcoma Prognosis and Treatment? Cancers 2018, 10, 315. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case Number | Type of LMS | Age at Diagnosis | Tumor Size (cm) | Stage | Recurrence | Necrosis | Metastasis | Survival Status |
|---|---|---|---|---|---|---|---|---|
| 1 | Conventional | 58 | 7 | IB | N | focal coagulative necrosis | None | A |
| 2 | Conventional | 58 | 8 | I | Y | extensive hyaline necrosis. focal coagulative necrosis | Lung | D |
| 3 | Conventional | 42 | 7.1 | IB | N | coagulative tumor cell necrosis and hyaline necrosis | Brain and lung | A |
| 4 | Conventional | 62 | 7.9 | IB | Y | focal coagulative necrosis | Lung | A |
| 5 | non-conventional | 55 | 17 | IIIC1 | N | focal coagulative necrosis | Ovary & Lung | D |
| 6 | non-conventional | 54 | 18.8 | IIIB | Y | focal coagulative necrosis | Abdominal wall + Bowel | D |
| 7 | Conventional | 54 | 27 | II | Y | focal coagulative necrosis | Omentum | A |
| 8 | Conventional | 61 | 9.5 | IIIB | N | focal coagulative necrosis | Omentum, Small intestine, Liver | A |
| 9 | Conventional | 42 | 24.6 | IB | N | focal coagulative necrosis | None | D |
| Antibodies | Company | Catalog Number | Source | Application | Dilution |
|---|---|---|---|---|---|
| BRD9 | Cell Signaling | 58906 | Rabbit | WB | 1:1000 |
| Bcl2 | Abcam | ab182858 | Rabbit | WB | 1:1000 |
| β-actin | Sigma | A5316 | Mouse | WB | 1:8000 |
| BRD9 | Abcam | ab259839 | Rabbit | IHC-P | 1:1000 |
| SMA | Dako | M0851 | Mouse | IHC-P | 1:1600 |
| Desmin | Santa Cruz | SC-14026 | Rabbit | IHC-P | 1:800 |
| HMB45 | Dako | M0634 | Mouse | IHC-P | 1:100 |
| Gene Symbol | Primer Sequences | F or R | Assay | Species | Size (bp) | Accession |
|---|---|---|---|---|---|---|
| CDKN1A | CGGAACAAGGAGTCAGACATT | F | q-PCR | Human | 105 | NM_00389.5 |
| CDKN1A | AGTGCCAGGAAAGACAACTAC | R | q-PCR | Human | 105 | NM_00389.5 |
| BAK | AGGGCTTAGGACTTGGTTTG | F | q-PCR | Human | 100 | U16811.1 |
| BAK | GGGATTCCTAGTGGTGTTGATA | R | q-PCR | Human | 100 | U16811.1 |
| 18 S | CACGGACAGGATTGACAGATT | F | q-PCR | Human | 119 | NR_145820 |
| 18 S | GCCAGAGTCTCGTTCGTTATC | R | q-PCR | Human | 119 | NR_145820 |
| Module | Mid Night Blue | Yellow | Cyan | Dark Green |
|---|---|---|---|---|
| GO_Biological_Process_2021 | Regulation of mitochondrial outer membrane permeabilization involved in apoptotic signaling pathway (GO:1901028) (1.38 × 10−5) | Apoptotic process (GO:0006915) (2.00 × 10−5) | Translational termination (GO:0006415) (8.54 × 10−31) | Mitotic cell cycle phase transition (GO:0044772) (3.36 × 10−7) |
| MSigDB_Hallmark_2020 | TNF-alpha Signaling via NF-kB (0.000138704) | TNF-alpha Signaling via NF-kB (1.08 × 10−16) | Myc Targets V1 (1.25 × 10−39) | PI3K/AKT/mTOR Signaling (0.031214077) |
| WikiPathway_2021 | MAPK Signaling Pathway WP382 (0.001957412) | Sudden Infant Death Syndrome (SIDS) Susceptibility Pathways WP706 (0.000455634) | G1 to S cell cycle control WP45 (0.037948453) | Cell cycle WP179 (1.90 × 10−5) |
| Reactome_2015 | TRAF6-mediated induction of NFkB and MAP kinases upon TLR7/8 or 9 activation Homo sapiens R-HSA-975138 (0.093688577) | MAPK family signaling cascades Homo sapiens R-HSA-5683057 (0.019875184) | Cyclin E associated events during G1/S transition Homo sapiens R-HSA-69202 (9.09 × 10−10) | Cell Cycle Homo sapiens R-HSA-1640170 (3.50 × 10−11) |
| ENCODE_Histone_Modifications_2015 | H3K4me1 (2.98 × 10−15) | H3K27me3 (4.96 × 10−48) | H3K4me3 (5.19 × 10−10) | H3K4me3 (1.91 × 10−6) |
| TargetScan_microRNA_2017 | hsa-miR-4776-5p MicroRNAs (0.001954789) | hsa-miR-4727-3p (0.001242581) | hsa-miR-1284 (0.021500132) | hsa-miR-3682-3p MicroRNAs (5.49 × 10−14) |
| InterPro_Domains_2019 | NFkappaB IPT domain (0.007433747) | Death domain (0.001693697) | LSM domain (1.28 × 10−9) | Protein kinase domain (4.49 × 10−5) |
| Pfam_Domains_2019 | Pkinase (0.000212328) | Death (0.000791274) | LSM (5.50 × 10−10) | Pkinase (3.62 × 10−5) |
| Jensen_COMPARTMENTS | Neuron part (5.89 × 10−7) | BCL-2 complex (3.34 × 10−8) | Intracellular ribonucleoprotein complex (5.21 × 10−49) | Cellular component (5.48 × 10−22) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Q.; Bariani, M.V.; Falahati, A.; Khosh, A.; Lastra, R.R.; Siblini, H.; Boyer, T.G.; Al-Hendy, A. The Functional Role and Regulatory Mechanism of Bromodomain-Containing Protein 9 in Human Uterine Leiomyosarcoma. Cells 2022, 11, 2160. https://doi.org/10.3390/cells11142160
Yang Q, Bariani MV, Falahati A, Khosh A, Lastra RR, Siblini H, Boyer TG, Al-Hendy A. The Functional Role and Regulatory Mechanism of Bromodomain-Containing Protein 9 in Human Uterine Leiomyosarcoma. Cells. 2022; 11(14):2160. https://doi.org/10.3390/cells11142160
Chicago/Turabian StyleYang, Qiwei, Maria Victoria Bariani, Ali Falahati, Azad Khosh, Ricardo R. Lastra, Hiba Siblini, Thomas G. Boyer, and Ayman Al-Hendy. 2022. "The Functional Role and Regulatory Mechanism of Bromodomain-Containing Protein 9 in Human Uterine Leiomyosarcoma" Cells 11, no. 14: 2160. https://doi.org/10.3390/cells11142160
APA StyleYang, Q., Bariani, M. V., Falahati, A., Khosh, A., Lastra, R. R., Siblini, H., Boyer, T. G., & Al-Hendy, A. (2022). The Functional Role and Regulatory Mechanism of Bromodomain-Containing Protein 9 in Human Uterine Leiomyosarcoma. Cells, 11(14), 2160. https://doi.org/10.3390/cells11142160