
Cell-Specific Immune Regulation by Glucocorticoids in Murine Models of Infection and Inflammation


Abstract
:1. Introduction
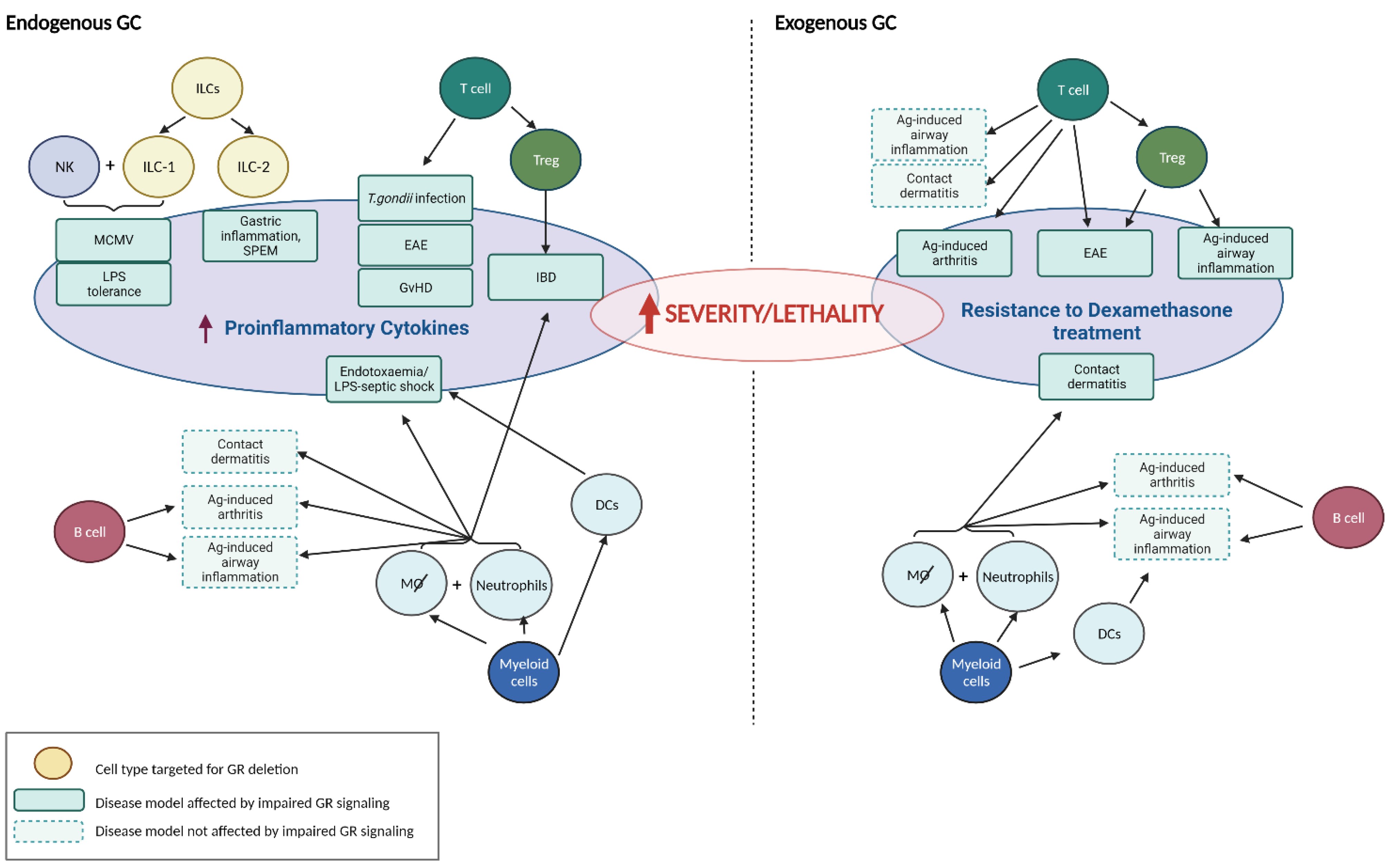
2. GC and Immune Cell Subsets
2.1. GC and Innate Lymphoid Cells

{kind=link}
{kind=link}
| Animal Model | Cells Targeted by GC | Source of GC | Observed Effects Upon Conditional GR Deletion in Targeted Cells | Reference |
|---|---|---|---|---|
| MCMV Infection | NK + ILC-1 | endogenous | splenic hyper-inflammation, survival ↓ | [12] |
| LPS tolerance | NK + ILC-1 | endogenous | loss of LPS tolerance, survival ↓ | [14] |
| Gastric inflammation, SPEM | ILC-2 | endogenous | spontaneous gastric inflammation in ♀, protection by GC (and androgens) * | [16] |
| Polyclonal T cell activation | T cell | endogenous | survival ↓, rescue by COX-2 inhibition | [17] |
| Cecal ligation and puncture (CLP) | T cell | endogenous | survival ↓ | [18] |
| Toxoplasma gondii infection | T cell | endogenous | hyperactive CD4+ T cell response, survival ↓ | [19] |
| Experimental autoimmune encephalomyelitis (EAE) | T cell | endogenous | disease onset earlier, more severe course | [20] |
| Experimental autoimmune encephalomyelitis (EAE) | T cell | exogenous | resistance to DEX treatment, reduced induction of apoptosis in Th17 cells | [20] |
| Allergic airway inflammation | T cell | exogenous | no impact on DEX treatment, airway epithelial cells crucial GC target | [21] |
| Antigen-induced arthritis | T cell | exogenous | resistance to DEX treatment, circulating pro-inflammatory cytokines ↑ | [22] |
| Contact dermatitis | T cell | exogenous | no impact on DEX treatment | [23] |
| Graft-versus-host disease (GvHD) | T cell | endogenous | strongly aggravated clinical disease, accelerated death | [24] |
| Experimental autoimmune encephalomyelitis (EAE) | Foxp3+ T cell | exogenous | resistance to DEX treatment, impaired Treg cell function | [25] |
| Allergic airway inflammation | Foxp3+ T cell | exogenous | resistance to DEX treatment, lung-infiltrating proinflammatory CD4+ T cells ↑ | [25] |
| Experimental Inflammatory Bowel Disease (IBD) | Foxp3+ T cell | endogenous | failure to prevent inflammatory bowel disease, loss of Treg cell phenotype | [26]) |
| Contact dermatitis | macrophages, neutrophils | exogenous | resistance to DEX treatment, massive leukocyte infiltration of the skin | [23] |
| Allergic airway inflammation | macrophages, neutrophils | exogenous | no impact on DEX treatment, airway epithelial cells crucial GC target | [21] |
| Endotoxaemia | macrophages; neutrophils? | endogenous | increased circulating pro-inflammatory cytokines, survival ↓ | [27] |
| Antigen-induced arthritis | macrophages, neutrophils | exogenous | no impact on DEX treatment | [22] |
| DSS-induced colitis | macrophages, neutrophils | endogenous | failure to resolve inflammation, increased cytokine expression in colon | [28] |
| Myocardial infarction | macrophages | endogenous | impaired post-ischemic angiogenesis, reduced cardiac function, survival ↓ | [29] |
| Endotoxaemia | dendritic cell | endogenous | increased circulating pro-inflammatory cytokines, survival ↓ | [13] |
| Allergic airway inflammation | dendritic cell | exogenous | no impact on DEX treatment, airway epithelial cells crucial GC target | [21] |
| Antigen-induced arthritis | dendritic cell | exogenous | no impact on DEX treatment | [22] |
| Antigen-induced arthritis | B cell | exogenous | no impact on DEX treatment | [22] |
| Allergic airway inflammation | B cell | exogenous | no impact on DEX treatment, airway epithelial cells crucial GC target | [21] |
2.2. GC and T Cells
2.3. GC and Myeloid Cells
2.4. GC and B Cells
2.5. GC and Other Immune Cells
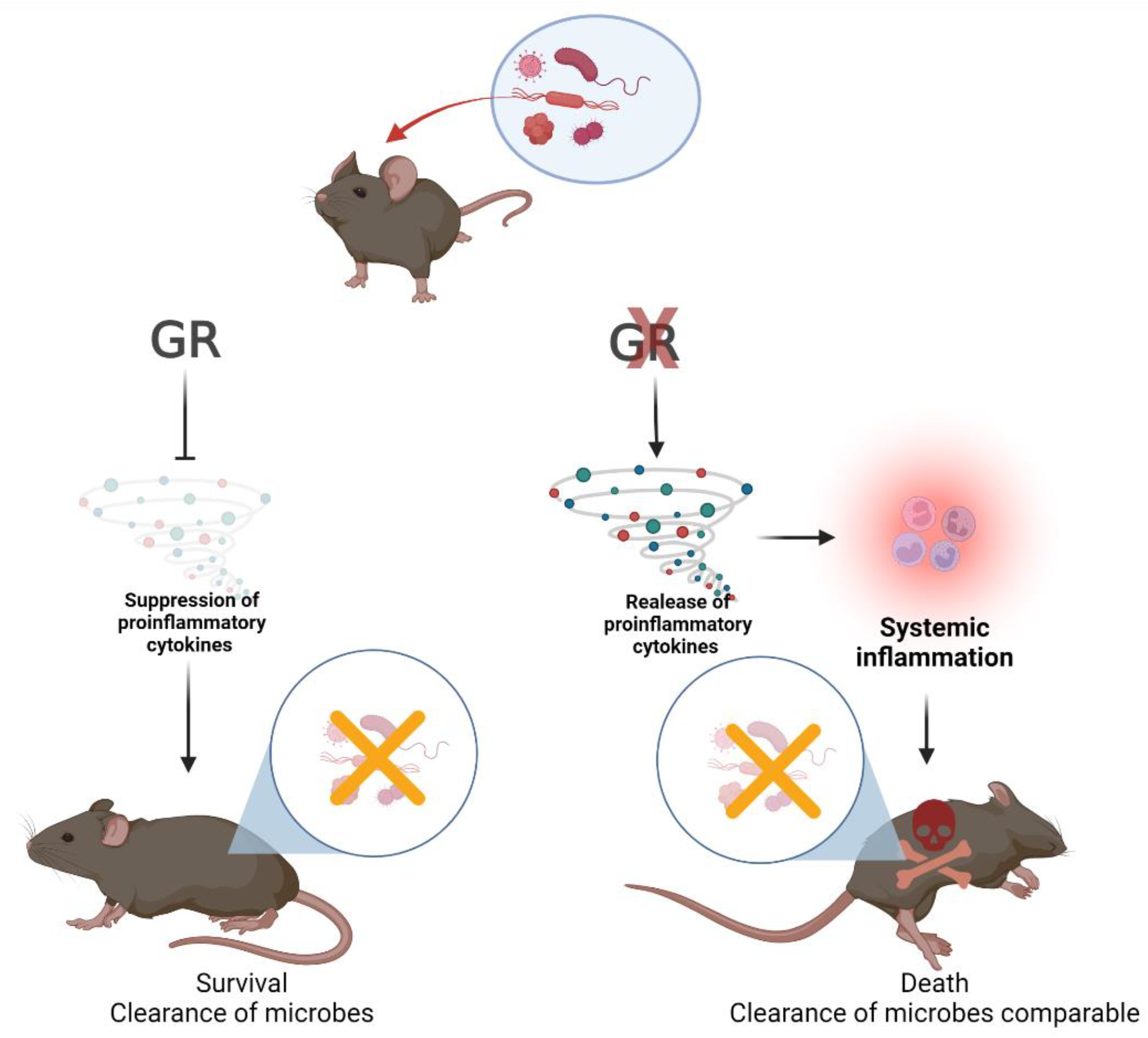
2.6. Endogenous GC and Microbial Clearance
2.7. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Sapolsky, R.M.; Romero, L.M.; Munck, A.U. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr. Rev. 2000, 21, 55–89. [Google Scholar] [PubMed] [Green Version]
- Cain, D.W.; Cidlowski, J.A. Immune regulation by glucocorticoids. Nat. Rev. Immunol. 2017, 17, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Taves, M.D.; Ashwell, J.D. Glucocorticoids in t cell development, differentiation and function. Nat. Rev. Immunol. 2021, 21, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Lechner, O.; Wiegers, G.J.; Oliveira-Dos-Santos, A.J.; Dietrich, H.; Recheis, H.; Waterman, M.; Boyd, R.; Wick, G. Glucocorticoid production in the murine thymus. Eur. J. Immunol. 2000, 30, 337–346. [Google Scholar] [CrossRef]
- Taves, M.D.; Plumb, A.W.; Korol, A.M.; Van Der Gugten, J.G.; Holmes, D.T.; Abraham, N.; Soma, K.K. Lymphoid organs of neonatal and adult mice preferentially produce active glucocorticoids from metabolites, not precursors. Brain. Behav. Immun. 2016, 57, 271–281. [Google Scholar] [CrossRef]
- Phan, T.S.; Merk, V.M.; Brunner, T. Extra-adrenal glucocorticoid synthesis at epithelial barriers. Genes Immun. 2019, 20, 627–640. [Google Scholar] [CrossRef]
- Cima, I.; Corazza, N.; Dick, B.; Fuhrer, A.; Herren, S.; Jakob, S.; Ayuni, E.; Mueller, C.; Brunner, T. Intestinal epithelial cells synthesize glucocorticoids and regulate t cell activation. J. Exp. Med. 2004, 200, 1635–1646. [Google Scholar] [CrossRef] [Green Version]
- Rocamora-Reverte, L.; Reichardt, H.M.; Villunger, A.; Wiegers, G. T-cell autonomous death induced by regeneration of inert glucocorticoid metabolites. Cell Death Dis. 2017, 8, e2948. [Google Scholar] [CrossRef] [Green Version]
- Shimba, A.; Cui, G.; Tani-Ichi, S.; Ogawa, M.; Abe, S.; Okazaki, F.; Kitano, S.; Miyachi, H.; Yamada, H.; Hara, T.; et al. Glucocorticoids drive diurnal oscillations in t cell distribution and responses by inducing interleukin-7 receptor and cxcr4. Immunity 2018, 48, 286–298 e286. [Google Scholar] [CrossRef] [Green Version]
- Shimba, A.; Ikuta, K. Glucocorticoids regulate circadian rhythm of innate and adaptive immunity. Front. Immunol. 2020, 11, 2143. [Google Scholar] [CrossRef]
- Vivier, E.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.J.; Mebius, R.E.; et al. Innate lymphoid cells: 10 years on. Cell 2018, 174, 1054–1066. [Google Scholar] [CrossRef] [Green Version]
- Quatrini, L.; Wieduwild, E.; Escaliere, B.; Filtjens, J.; Chasson, L.; Laprie, C.; Vivier, E.; Ugolini, S. Endogenous glucocorticoids control host resistance to viral infection through the tissue-specific regulation of pd-1 expression on nk cells. Nat. Immunol. 2018, 19, 954–962. [Google Scholar] [CrossRef] [PubMed]
- Li, C.C.; Munitic, I.; Mittelstadt, P.R.; Castro, E.; Ashwell, J.D. Suppression of dendritic cell-derived il-12 by endogenous glucocorticoids is protective in lps-induced sepsis. PLoS Biol. 2015, 13, e1002269. [Google Scholar] [CrossRef] [PubMed]
- Quatrini, L.; Wieduwild, E.; Guia, S.; Bernat, C.; Glaichenhaus, N.; Vivier, E.; Ugolini, S. Host resistance to endotoxic shock requires the neuroendocrine regulation of group 1 innate lymphoid cells. J. Exp. Med. 2017, 214, 3531–3541. [Google Scholar] [CrossRef] [PubMed]
- Busada, J.T.; Ramamoorthy, S.; Cain, D.W.; Xu, X.; Cook, D.N.; Cidlowski, J.A. Endogenous glucocorticoids prevent gastric metaplasia by suppressing spontaneous inflammation. J. Clin. Investig. 2019, 129, 1345–1358. [Google Scholar] [CrossRef] [Green Version]
- Busada, J.T.; Peterson, K.N.; Khadka, S.; Xu, X.; Oakley, R.H.; Cook, D.N.; Cidlowski, J.A. Glucocorticoids and androgens protect from gastric metaplasia by suppressing group 2 innate lymphoid cell activation. Gastroenterology 2021, 161, 637–652.e634. [Google Scholar] [CrossRef]
- Brewer, J.A.; Khor, B.; Vogt, S.K.; Muglia, L.M.; Fujiwara, H.; Haegele, K.E.; Sleckman, B.P.; Muglia, L.J. T-cell glucocorticoid receptor is required to suppress cox-2-mediated lethal immune activation. Nat. Med. 2003, 9, 1318–1322. [Google Scholar] [CrossRef]
- Guo, L.; Zheng, Z.; Ai, J.; Howatt, D.A.; Mittelstadt, P.R.; Thacker, S.; Daugherty, A.; Ashwell, J.D.; Remaley, A.T.; Li, X.A. Scavenger receptor bi and high-density lipoprotein regulate thymocyte apoptosis in sepsis. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 966–975. [Google Scholar] [CrossRef] [Green Version]
- Kugler, D.G.; Mittelstadt, P.R.; Ashwell, J.D.; Sher, A.; Jankovic, D. Cd4+ t cells are trigger and target of the glucocorticoid response that prevents lethal immunopathology in toxoplasma infection. J. Exp. Med. 2013, 210, 1919–1927. [Google Scholar] [CrossRef]
- Wüst, S.; van den Brandt, J.; Tischner, D.; Kleiman, A.; Tuckermann, J.P.; Gold, R.; Luhder, F.; Reichardt, H.M. Peripheral t cells are the therapeutic targets of glucocorticoids in experimental autoimmune encephalomyelitis. J. Immunol. 2008, 180, 8434–8443. [Google Scholar] [CrossRef] [Green Version]
- Klassen, C.; Karabinskaya, A.; Dejager, L.; Vettorazzi, S.; Van Moorleghem, J.; Luhder, F.; Meijsing, S.H.; Tuckermann, J.P.; Bohnenberger, H.; Libert, C.; et al. Airway epithelial cells are crucial targets of glucocorticoids in a mouse model of allergic asthma. J. Immunol. 2017, 199, 48–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baschant, U.; Frappart, L.; Rauchhaus, U.; Bruns, L.; Reichardt, H.M.; Kamradt, T.; Brauer, R.; Tuckermann, J.P. Glucocorticoid therapy of antigen-induced arthritis depends on the dimerized glucocorticoid receptor in t cells. Proc. Natl. Acad. Sci. USA 2011, 108, 19317–19322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuckermann, J.P.; Kleiman, A.; Moriggl, R.; Spanbroek, R.; Neumann, A.; Illing, A.; Clausen, B.E.; Stride, B.; Forster, I.; Habenicht, A.J.; et al. Macrophages and neutrophils are the targets for immune suppression by glucocorticoids in contact allergy. J. Clin. Investig. 2007, 117, 1381–1390. [Google Scholar] [CrossRef] [PubMed]
- Theiss-Suennemann, J.; Jorss, K.; Messmann, J.J.; Reichardt, S.D.; Montes-Cobos, E.; Luhder, F.; Tuckermann, J.P.; H, A.W.; Dressel, R.; Grone, H.J.; et al. Glucocorticoids attenuate acute graft-versus-host disease by suppressing the cytotoxic capacity of cd8(+) t cells. J. Pathol. 2015, 235, 646–655. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Nguyen, Q.T.; Lee, J.; Lee, S.H.; Janocha, A.; Kim, S.; Le, H.T.; Dvorina, N.; Weiss, K.; Cameron, M.J.; et al. Anti-inflammatory roles of glucocorticoids are mediated by foxp3(+) regulatory t cells via a mir-342-dependent mechanism. Immunity 2020, 53, 581–596.e585. [Google Scholar] [CrossRef]
- Rocamora-Reverte, L.; Tuzlak, S.; von Raffay, L.; Tisch, M.; Fiegl, H.; Drach, M.; Reichardt, H.M.; Villunger, A.; Tischner, D.; Wiegers, G.J. Glucocorticoid receptor-deficient foxp3(+) regulatory t cells fail to control experimental inflammatory bowel disease. Front. Immunol. 2019, 10, 472. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Brown, D.E.; Brewer, J.A.; Vogt, S.K.; Muglia, L.J. Macrophage glucocorticoid receptors regulate toll-like receptor 4-mediated inflammatory responses by selective inhibition of p38 map kinase. Blood 2007, 109, 4313–4319. [Google Scholar] [CrossRef] [Green Version]
- Meers, G.K.; Bohnenberger, H.; Reichardt, H.M.; Luhder, F.; Reichardt, S.D. Impaired resolution of dss-induced colitis in mice lacking the glucocorticoid receptor in myeloid cells. PLoS ONE 2018, 13, e0190846. [Google Scholar] [CrossRef]
- Galuppo, P.; Vettorazzi, S.; Hovelmann, J.; Scholz, C.J.; Tuckermann, J.P.; Bauersachs, J.; Fraccarollo, D. The glucocorticoid receptor in monocyte-derived macrophages is critical for cardiac infarct repair and remodeling. FASEB J. 2017, 31, 5122–5132. [Google Scholar] [CrossRef] [Green Version]
- Seshadri, S.; Pope, R.L.; Zenewicz, L.A. Glucocorticoids inhibit group 3 innate lymphocyte il-22 production. J. Immunol. 2018, 201, 1267–1274. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Xia, P.; Chen, Y.; Qu, Y.; Xiong, Z.; Ye, B.; Du, Y.; Tian, Y.; Yin, Z.; Xu, Z.; et al. Regulatory innate lymphoid cells control innate intestinal inflammation. Cell 2017, 171, 201–216.e218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuzlak, S.; Dejean, A.S.; Iannacone, M.; Quintana, F.J.; Waisman, A.; Ginhoux, F.; Korn, T.; Becher, B. Repositioning th cell polarization from single cytokines to complex help. Nat. Immunol. 2021, 22, 1210–1217. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Mikami, N.; Wing, J.B.; Tanaka, A.; Ichiyama, K.; Ohkura, N. Regulatory t cells and human disease. Annu. Rev. Immunol. 2020, 38, 541–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engler, J.B.; Kursawe, N.; Solano, M.E.; Patas, K.; Wehrmann, S.; Heckmann, N.; Luhder, F.; Reichardt, H.M.; Arck, P.C.; Gold, S.M.; et al. Glucocorticoid receptor in t cells mediates protection from autoimmunity in pregnancy. Proc. Natl. Acad. Sci. USA 2017, 114, E181–E190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yakimchuk, K.; Chen, L.; Hasni, M.S.; Okret, S.; Jondal, M. The selective impact of transgenically expressed glucocorticoid receptor on t cells. Autoimmunity 2015, 48, 117–124. [Google Scholar] [CrossRef]
- van den Brandt, J.; Luhder, F.; McPherson, K.G.; de Graaf, K.L.; Tischner, D.; Wiehr, S.; Herrmann, T.; Weissert, R.; Gold, R.; Reichardt, H.M. Enhanced glucocorticoid receptor signaling in t cells impacts thymocyte apoptosis and adaptive immune responses. Am. J. Pathol. 2007, 170, 1041–1053. [Google Scholar] [CrossRef] [Green Version]
- Acharya, N.; Madi, A.; Zhang, H.; Klapholz, M.; Escobar, G.; Dulberg, S.; Christian, E.; Ferreira, M.; Dixon, K.O.; Fell, G.; et al. Endogenous glucocorticoid signaling regulates cd8(+) t cell differentiation and development of dysfunction in the tumor microenvironment. Immunity 2020, 53, 658–671.e656. [Google Scholar] [CrossRef]
- Merk, V.M.; Phan, T.S.; Brunner, T. Regulation of tissue immune responses by local glucocorticoids at epithelial barriers and their impact on interorgan crosstalk. Front. Immunol. 2021, 12, 672808. [Google Scholar] [CrossRef]
- Yin, X.; Chen, S.; Eisenbarth, S.C. Dendritic cell regulation of t helper cells. Annu. Rev. Immunol. 2021, 39, 759–790. [Google Scholar] [CrossRef]
- Bassler, K.; Schulte-Schrepping, J.; Warnat-Herresthal, S.; Aschenbrenner, A.C.; Schultze, J.L. The myeloid cell compartment-cell by cell. Annu. Rev. Immunol. 2019, 37, 269–293. [Google Scholar] [CrossRef]
- Cyster, J.G.; Allen, C.D.C. B cell responses: Cell interaction dynamics and decisions. Cell 2019, 177, 524–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruver-Yates, A.L.; Quinn, M.A.; Cidlowski, J.A. Analysis of glucocorticoid receptors and their apoptotic response to dexamethasone in male murine b cells during development. Endocrinology 2014, 155, 463–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cain, D.W.; Bortner, C.D.; Diaz-Jimenez, D.; Petrillo, M.G.; Gruver-Yates, A.; Cidlowski, J.A. Murine glucocorticoid receptors orchestrate b cell migration selectively between bone marrow and blood. J. Immunol. 2020, 205, 619–629. [Google Scholar] [CrossRef]
- Ribot, J.C.; Lopes, N.; Silva-Santos, B. Gammadelta t cells in tissue physiology and surveillance. Nat. Rev. Immunol. 2021, 21, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Jarillo-Luna, A.; Rivera-Aguilar, V.; Martinez-Carrillo, B.E.; Barbosa-Cabrera, E.; Garfias, H.R.; Campos-Rodriguez, R. Effect of restraint stress on the population of intestinal intraepithelial lymphocytes in mice. Brain Behav. Immun. 2008, 22, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Pellicci, D.G.; Koay, H.F.; Berzins, S.P. Thymic development of unconventional t cells: How nkt cells, mait cells and gammadelta t cells emerge. Nat. Rev. Immunol. 2020, 20, 756–770. [Google Scholar] [CrossRef] [PubMed]
- Rudak, P.T.; Choi, J.; Parkins, K.M.; Summers, K.L.; Jackson, D.N.; Foster, P.J.; Skaro, A.I.; Leslie, K.; McAlister, V.C.; Kuchroo, V.K.; et al. Chronic stress physically spares but functionally impairs innate-like invariant t cells. Cell Rep. 2021, 35, 108979. [Google Scholar] [CrossRef]
- Roggero, E.; Perez, A.R.; Tamae-Kakazu, M.; Piazzon, I.; Nepomnaschy, I.; Besedovsky, H.O.; Bottasso, O.A.; del Rey, A. Endogenous glucocorticoids cause thymus atrophy but are protective during acute trypanosoma cruzi infection. J. Endocrinol. 2006, 190, 495–503. [Google Scholar] [CrossRef] [Green Version]
- Ruzek, M.C.; Pearce, B.D.; Miller, A.H.; Biron, C.A. Endogenous glucocorticoids protect against cytokine-mediated lethality during viral infection. J. Immunol. 1999, 162, 3527–3533. [Google Scholar]
- Vandermosten, L.; Pham, T.T.; Knoops, S.; De Geest, C.; Lays, N.; Van der Molen, K.; Kenyon, C.J.; Verma, M.; Chapman, K.E.; Schuit, F.; et al. Adrenal hormones mediate disease tolerance in malaria. Nat. Commun. 2018, 9, 4525. [Google Scholar] [CrossRef] [Green Version]
- Jamieson, A.M.; Yu, S.; Annicelli, C.H.; Medzhitov, R. Influenza virus-induced glucocorticoids compromise innate host defense against a secondary bacterial infection. Cell Host Microbe 2010, 7, 103–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergthorsdottir, R.; Leonsson-Zachrisson, M.; Oden, A.; Johannsson, G. Premature mortality in patients with addison’s disease: A population-based study. J. Clin. Endocrinol. Metab. 2006, 91, 4849–4853. [Google Scholar] [CrossRef] [PubMed]
- Erichsen, M.M.; Lovas, K.; Fougner, K.J.; Svartberg, J.; Hauge, E.R.; Bollerslev, J.; Berg, J.P.; Mella, B.; Husebye, E.S. Normal overall mortality rate in addison’s disease, but young patients are at risk of premature death. Eur. J. Endocrinol. 2009, 160, 233–237. [Google Scholar] [CrossRef] [Green Version]
- Tresoldi, A.S.; Sumilo, D.; Perrins, M.; Toulis, K.A.; Prete, A.; Reddy, N.; Wass, J.A.H.; Arlt, W.; Nirantharakumar, K. Increased infection risk in addison’s disease and congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 2020, 105, 418–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munck, A.; Guyre, P.M.; Holbrook, N.J. Physiological functions of glucocorticoids in stress and their relation to pharmacological actions. Endocr. Rev. 1984, 5, 25–44. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Papadopoulou, P.; de Wit, B.; d’Engelbronner, J.C.; van Hage, P.; Kros, A.; Schaaf, M.J.M. Two types of liposomal formulations improve the therapeutic ratio of prednisolone phosphate in a zebrafish model for inflammation. Cells 2022, 11, 671. [Google Scholar] [CrossRef] [PubMed]
- Reichardt, S.D.; Amouret, A.; Muzzi, C.; Vettorazzi, S.; Tuckermann, J.P.; Luhder, F.; Reichardt, H.M. The role of glucocorticoids in inflammatory diseases. Cells 2021, 10, 2921. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rocamora-Reverte, L.; Villunger, A.; Wiegers, G.J. Cell-Specific Immune Regulation by Glucocorticoids in Murine Models of Infection and Inflammation. Cells 2022, 11, 2126. https://doi.org/10.3390/cells11142126
Rocamora-Reverte L, Villunger A, Wiegers GJ. Cell-Specific Immune Regulation by Glucocorticoids in Murine Models of Infection and Inflammation. Cells. 2022; 11(14):2126. https://doi.org/10.3390/cells11142126
Chicago/Turabian StyleRocamora-Reverte, Lourdes, Andreas Villunger, and G. Jan Wiegers. 2022. "Cell-Specific Immune Regulation by Glucocorticoids in Murine Models of Infection and Inflammation" Cells 11, no. 14: 2126. https://doi.org/10.3390/cells11142126
APA StyleRocamora-Reverte, L., Villunger, A., & Wiegers, G. J. (2022). Cell-Specific Immune Regulation by Glucocorticoids in Murine Models of Infection and Inflammation. Cells, 11(14), 2126. https://doi.org/10.3390/cells11142126