
Elevated NF-κB/SHh/GLI1 Signature Denotes a Worse Prognosis and Represent a Novel Potential Therapeutic Target in Advanced Prostate Cancer

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Prostate Adenocarcinoma Tissue Samples
2.2. Cell Lines
2.3. Lentiviral Production and Infections
2.4. RNA Extraction and Quantitative Real Time Polymerase-Chain Reaction (qRT-PCR)
2.5. Immunohistochemistry
2.6. Western Blot
2.7. Viability
2.8. Evaluation of NF-κB Activity
2.9. Luciferase Assay
2.10. Analysis on TCGA Dataset
2.11. Synergism Analysis
2.12. Statistical Analysis
3. Results
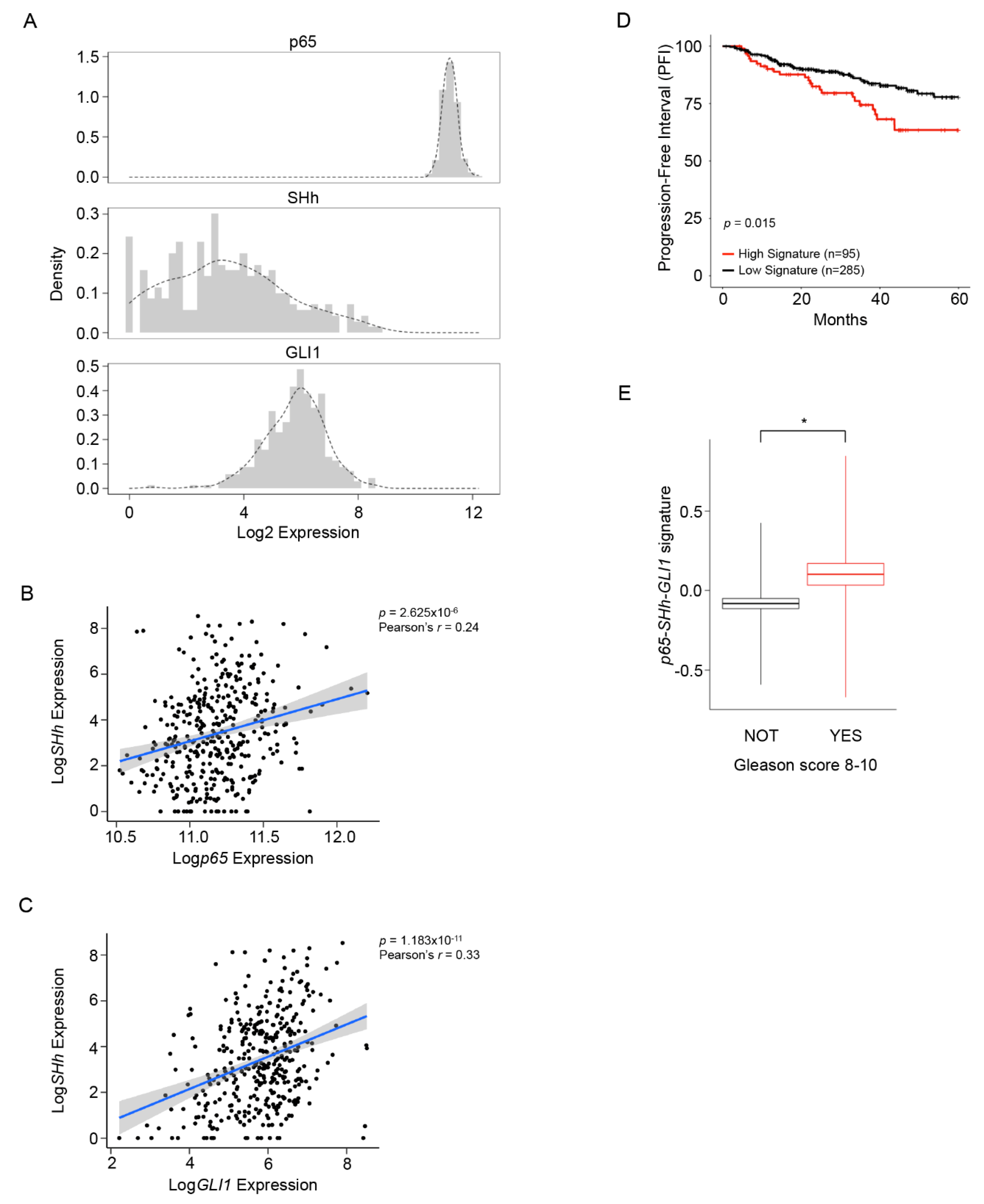
3.1. NF-κB/SHh/GLI1 Signature Expression Correlates with Poor Clinical Outcome in PCa
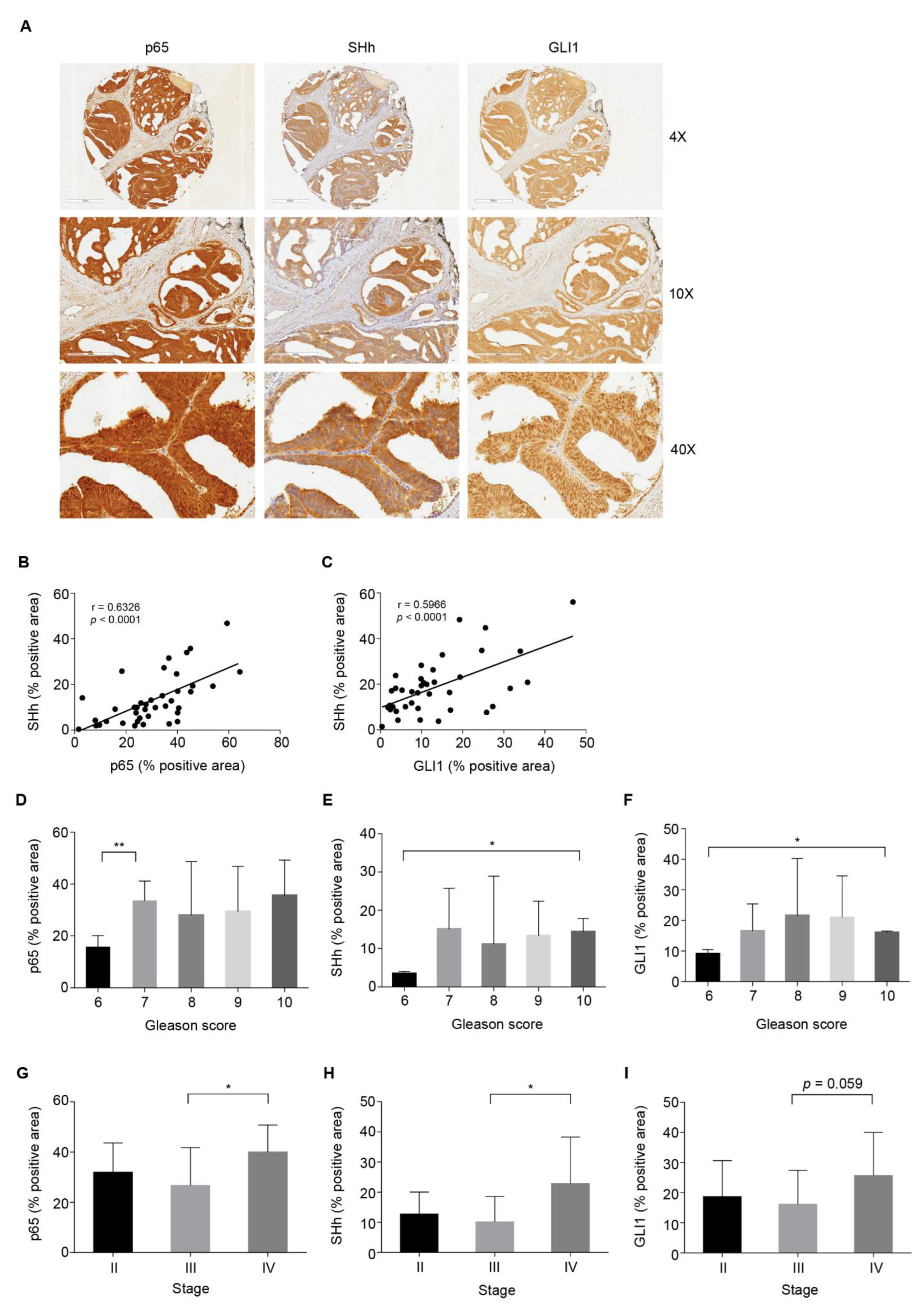
3.2. High NF-κB Activity Strongly Correlates with GLI1 Expression in PCa
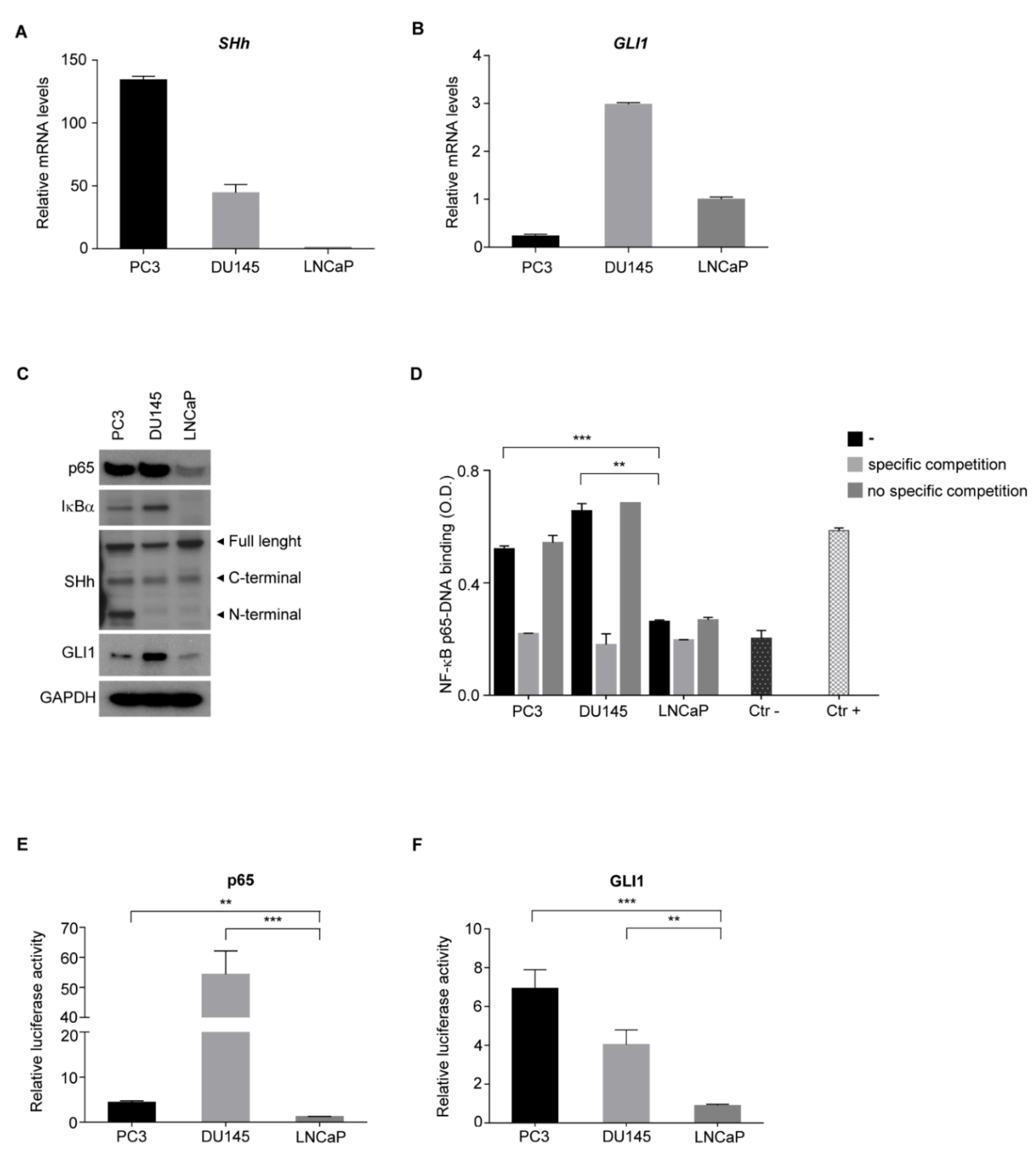
3.3. NF-κB and SHh Pathways Are Activated in Androgen-Independent PCa Cell Lines
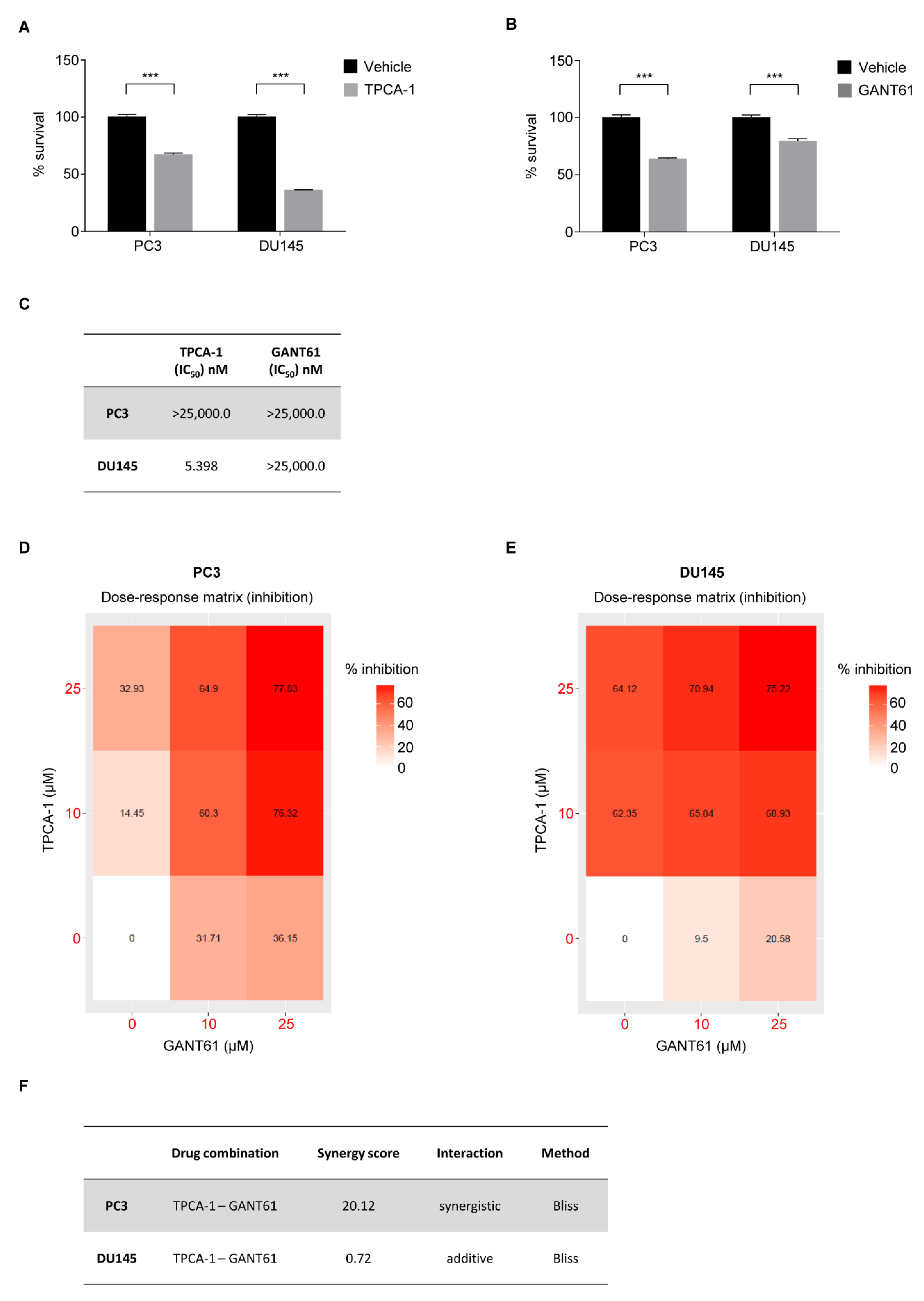
3.4. AI PCa Cells Rely on NF-κB/GLI1 Activity for Survival
3.5. Pharmacological Inhibition of NF-κB and GLI1 Reduces Cell Survival
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Phua, T.J. The Etiology and Pathophysiology Genesis of Benign Prostatic Hyperplasia and Prostate Cancer: A New Perspective. Medicines 2021, 8, 30. [Google Scholar] [CrossRef] [PubMed]
- Feldman, B.J.; Feldman, D. The Development of Androgen-Independent Prostate Cancer. Nat. Rev. Cancer 2001, 1, 34–45. [Google Scholar] [CrossRef] [PubMed]
- De La Taille, A.; Vacherot, F.; Salomon, L.; Druel, C.; Gil Diez De Medina, S.; Abbou, C.; Buttyan, R.; Chopin, D. Hormone-Refractory Prostate Cancer: A Multi-Step and Multi-Event Process. Prostate Cancer Prostatic Dis. 2001, 4, 204–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roth, J.A.; Gulati, R.; Gore, J.L.; Cooperberg, M.R.; Etzioni, R. Economic Analysis of Prostate-Specific Antigen Screening and Selective Treatment Strategies. JAMA Oncol. 2016, 2, 890–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuzick, J.; Berney, D.M.; Fisher, G.; Mesher, D.; Møller, H.; Reid, J.E.; Perry, M.; Park, J.; Younus, A.; Gutin, A.; et al. Prognostic Value of a Cell Cycle Progression Signature for Prostate Cancer Death in a Conservatively Managed Needle Biopsy Cohort. Br. J. Cancer 2012, 106, 1095–1099. [Google Scholar] [CrossRef] [Green Version]
- Tonry, C.; Finn, S.; Armstrong, J.; Pennington, S.R. Clinical Proteomics for Prostate Cancer: Understanding Prostate Cancer Pathology and Protein Biomarkers for Improved Disease Management. Clin. Proteom. 2020, 17, 41. [Google Scholar] [CrossRef]
- Garg, R.; Blando, J.; Perez, C.J.; Wang, H.; Benavides, F.J.; Kazanietz, M.G. Activation of Nuclear Factor KB (NF-KB) in Prostate Cancer Is Mediated by Protein Kinase C Epsilon (PKCepsilon). J. Biol. Chem. 2012, 287, 37570–37582. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez-Uzquiza, A.; Lopez-Haber, C.; Jernigan, D.L.; Fatatis, A.; Kazanietz, M.G. PKCε Is an Essential Mediator of Prostate Cancer Bone Metastasis. Mol. Cancer Res. 2015, 13, 1336–1346. [Google Scholar] [CrossRef] [Green Version]
- Longoni, N.; Sarti, M.; Albino, D.; Civenni, G.; Malek, A.; Ortelli, E.; Pinton, S.; Mello-Grand, M.; Ostano, P.; D’Ambrosio, G.; et al. ETS Transcription Factor ESE1/ELF3 Orchestrates a Positive Feedback Loop That Constitutively Activates NF-KB and Drives Prostate Cancer Progression. Cancer Res. 2013, 73, 4533–4547. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Castranova, V.; Shi, X. New Insights into the Role of Nuclear Factor-KB in Cell Growth Regulation. Am. J. Pathol. 2001, 159, 387–397. [Google Scholar] [CrossRef]
- Hayden, M.; Ghosh, S. Signaling to NF-{kappa}B. Genes Dev. 2004, 18, 2195–2224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oeckinghaus, A.; Ghosh, S. The NF- B Family of Transcription Factors and Its Regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, T.D.; Herscovitch, M. Inhibitors of NF-KB Signaling: 785 and Counting. Oncogene 2006, 25, 6887–6899. [Google Scholar] [CrossRef] [Green Version]
- Shukla, S.; MacLennan, G.T.; Fu, P.; Patel, J.; Marengo, S.R.; Resnick, M.I.; Gupta, S. Nuclear Factor-KappaB/P65 (Rel A) Is Constitutively Activated in Human Prostate Adenocarcinoma and Correlates with Disease Progression. Neoplasia 2004, 6, 390–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verzella, D.; Fischietti, M.; Capece, D.; Vecchiotti, D.; Del Vecchio, F.; Cicciarelli, G.; Mastroiaco, V.; Tessitore, A.; Alesse, E.; Zazzeroni, F. Targeting the NF-KB Pathway in Prostate Cancer: A Promising Therapeutic Approach? Curr. Drug Targets 2016, 17, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, A.S.J. Series Introduction: The Transcription Factor NF-KappaB and Human Disease. J. Clin. Investig. 2001, 107, 3–6. [Google Scholar] [CrossRef] [Green Version]
- Suh, J.; Payvandi, F.; Edelstein, L.C.; Amenta, P.S.; Zong, W.-X.; Gélinas, C.; Rabson, A.B. Mechanisms of Constitutive NF-KappaB Activation in Human Prostate Cancer Cells. Prostate 2002, 52, 183–200. [Google Scholar] [CrossRef]
- Gasparian, A.V.; Yao, Y.J.; Lü, J.; Yemelyanov, A.Y.; Lyakh, L.A.; Slaga, T.J.; Budunova, I. V Selenium Compounds Inhibit I Kappa B Kinase (IKK) and Nuclear Factor-Kappa B (NF-Kappa B) in Prostate Cancer Cells. Mol. Cancer Ther. 2002, 1, 1079–1087. [Google Scholar]
- Lessard, L.; Mes-Masson, A.-M.; Lamarre, L.; Wall, L.; Lattouf, J.-B.; Saad, F. NF-Kappa B Nuclear Localization and Its Prognostic Significance in Prostate Cancer. BJU Int. 2003, 91, 417–420. [Google Scholar] [CrossRef]
- Bariwal, J.; Kumar, V.; Dong, Y.; Mahato, R.I. Design of Hedgehog Pathway Inhibitors for Cancer Treatment. Med. Res. Rev. 2019, 39, 1137–1204. [Google Scholar] [CrossRef] [PubMed]
- Berman, D.M.; Karhadkar, S.S.; Hallahan, A.R.; Pritchard, J.I.; Eberhart, C.G.; Watkins, D.N.; Chen, J.K.; Cooper, M.K.; Taipale, J.; Olson, J.M.; et al. Medulloblastoma Growth Inhibition by Hedgehog Pathway Blockade. Science 2002, 297, 1559–1561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thayer, S.P.; Di Magliano, M.P.; Heiser, P.W.; Nielsen, C.M.; Roberts, D.J.; Lauwers, G.Y.; Qi, Y.P.; Gysin, S.; Fernández-del Castillo, C.; Yajnik, V.; et al. Hedgehog Is an Early and Late Mediator of Pancreatic Cancer Tumorigenesis. Nature 2003, 425, 851–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watkins, D.N.; Berman, D.M.; Burkholder, S.G.; Wang, B.; Beachy, P.A.; Baylin, S.B. Hedgehog Signalling within Airway Epithelial Progenitors and in Small-Cell Lung Cancer. Nature 2003, 422, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Lees, C.; Howie, S.; Sartor, R.B.; Satsangi, J. The Hedgehog Signalling Pathway in the Gastrointestinal Tract: Implications for Development, Homeostasis, and Disease. Gastroenterology 2005, 129, 1696–1710. [Google Scholar] [CrossRef]
- Varjosalo, M.; Taipale, J. Hedgehog: Functions and Mechanisms. Genes Dev. 2008, 22, 2454–2472. [Google Scholar] [CrossRef] [Green Version]
- Nüsslein-Volhard, C.; Wieschaus, E. Mutations Affecting Segment Number and Polarity in Drosophila. Nature 1980, 287, 795–801. [Google Scholar] [CrossRef]
- Sheng, T.; Li, C.; Zhang, X.; Chi, S.; He, N.; Chen, K.; McCormick, F.; Gatalica, Z.; Xie, J. Activation of the Hedgehog Pathway in Advanced Prostate Cancer. Mol. Cancer 2004, 3, 29. [Google Scholar] [CrossRef] [Green Version]
- Azoulay, S.; Terry, S.; Chimingqi, M.; Sirab, N.; Faucon, H.; Gil Diez de Medina, S.; Moutereau, S.; Maillé, P.; Soyeux, P.; Abbou, C.; et al. Comparative Expression of Hedgehog Ligands at Different Stages of Prostate Carcinoma Progression. J. Pathol. 2008, 216, 460–470. [Google Scholar] [CrossRef]
- Kim, T.-J.; Lee, J.Y.; Hwang, T.-K.; Kang, C.S.; Choi, Y.-J. Hedgehog Signaling Protein Expression and Its Association with Prognostic Parameters in Prostate Cancer: A Retrospective Study from the View Point of New 2010 Anatomic Stage/Prognostic Groups. J. Surg. Oncol. 2011, 104, 472–479. [Google Scholar] [CrossRef]
- Sanchez, P.; Hernández, A.M.; Stecca, B.; Kahler, A.J.; DeGueme, A.M.; Barrett, A.; Beyna, M.; Datta, M.W.; Datta, S.; Ruiz i Altaba, A. Inhibition of Prostate Cancer Proliferation by Interference with SONIC HEDGEHOG-GLI1 Signaling. Proc. Natl. Acad. Sci. USA 2004, 101, 12561–12566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karhadkar, S.S.; Bova, G.S.; Abdallah, N.; Dhara, S.; Gardner, D.; Maitra, A.; Isaacs, J.T.; Berman, D.M.; Beachy, P.A. Hedgehog Signalling in Prostate Regeneration, Neoplasia and Metastasis. Nature 2004, 431, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Kasperczyk, H.; Baumann, B.; Debatin, K.-M.; Fulda, S. Characterization of Sonic Hedgehog as a Novel NF-KappaB Target Gene That Promotes NF-KappaB-Mediated Apoptosis Resistance and Tumor Growth In Vivo. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2009, 23, 21–33. [Google Scholar] [CrossRef]
- Moretti, M.; Di Francesco, B.; Nolfi, M.D.V.; Angrisani, A.; De Smaele, E. Methods for Modulating the Pathway of NF-$κ$B Using Short Hairpin RNA (ShRNA)Short Hairpin RNA (ShRNA). In NF-$κ$B Transcription Factors: Methods and Protocols; Franzoso, G., Zazzeroni, F., Eds.; Springer US: New York, NY, USA, 2021; pp. 95–107. ISBN 978-1-0716-1669-7. [Google Scholar]
- Vecchiotti, D.; Verzella, D.; Capece, D.; Cornice, J.; Nolfi, M.D.V.; Di Francesco, B.; Franzoso, G.; Alesse, E.; Zazzeroni, F. Immunohistochemical Analysis of Expression, Phosphorylation, and Nuclear Translocation of NF-KB Proteins in Human Tissues. Methods Mol. Biol. 2021, 2366, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Bankhead, P.; Loughrey, M.B.; Fernández, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G.; et al. QuPath: Open Source Software for Digital Pathology Image Analysis. Sci. Rep. 2017, 7, 16878. [Google Scholar] [CrossRef] [Green Version]
- Fischietti, M.; Fratini, E.; Verzella, D.; Vecchiotti, D.; Capece, D.; Di Francesco, B.; Esposito, G.; Balata, M.; Ioannuci, L.; Sykes, P.; et al. Low Radiation Environment Switches the Overgrowth-Induced Cell Apoptosis Toward Autophagy. Front. Public Health 2021, 8, 594789. [Google Scholar] [CrossRef]
- Antonucci, L.; Di Magno, L.; D’Amico, D.; Manni, S.; Serrao, S.M.; Pastena, F.D.I.; Bordone, R.; Yurtsever, Z.N.; Caimano, M.; Petroni, M.; et al. Mitogen-Activated Kinase Kinase Kinase 1 Inhibits Hedgehog Signaling and Medulloblastoma Growth through GLI1 Phosphorylation. Int. J. Oncol. 2019, 54, 505–514. [Google Scholar] [CrossRef]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173, 400–416.e11. [Google Scholar] [CrossRef] [Green Version]
- Ianevski, A.; Giri, A.K.; Aittokallio, T. SynergyFinder 2.0: Visual Analytics of Multi-Drug Combination Synergies. Nucleic Acids Res. 2020, 48, W488–W493. [Google Scholar] [CrossRef]
- Ianevski, A.; Giri, A.K.; Gautam, P.; Kononov, A.; Potdar, S.; Saarela, J.; Wennerberg, K.; Aittokallio, T. Prediction of Drug Combination Effects with a Minimal Set of Experiments. Nat. Mach. Intell. 2019, 1, 568–577. [Google Scholar] [CrossRef]
- Wang, X.; Peng, H.; Huang, Y.; Kong, W.; Cui, Q.; Du, J. Post-Translational Modifications of IκBA: The State of the Art. Front. Cell Dev. Biol. 2020, 8, 574706. [Google Scholar] [CrossRef] [PubMed]
- Kotulak-Chrzaszcz, A.; Kmieć, Z.; Wierzbicki, P.M. Sonic Hedgehog Signaling Pathway in Gynecological and Genitourinary Cancer (Review). Int. J. Mol. Med. 2021, 47, 106. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Huang, S.Y.; Li, K.K.W.; Li, Y.H.; Hsu, W.H.; Zhang, G.J.; Chang, C.J.; Yang, J.Y. Dual Degradation Signals Destruct GLI1: AMPK Inhibits GLI1 through β-TrCP-Mediated Proteasome Degradation. Oncotarget 2017, 8, 49869–49881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atwood, S.X.; Li, M.; Lee, A.; Tang, J.Y.; Oro, A.E. GLI activation by atypical protein kinase C ι/λ regulates the growth of basal cell carcinomas. Nature 2013, 494, 484–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietrobono, S.; Gagliardi, S.; Stecca, B. Non-Canonical Hedgehog Signaling Pathway in Cancer: Activation of GLI Transcription Factors beyond Smoothened. Front. Genet. 2019, 10, 556. [Google Scholar] [CrossRef] [Green Version]
- Su, C.Y.; Huang, G.C.; Chang, Y.C.; Chen, Y.J.; Fang, H.W. Analyzing the Expression of Biomarkers in Prostate Cancer Cell Lines. In Vivo 2021, 35, 1545–1548. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Jin, R.; Yi, Y.; Yull, F.E.; Blackwell, T.S.; Clark, P.E.; Koyama, T.; Smith, J.A.; Matusik, R.J. NF-κB Gene Signature Predicts Prostate Cancer Progression. Cancer Res. 2014, 74, 2763–2772. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Goto, Y.; Sakamoto, R.; Tanaka, K.; Matsubara, E.; Nakamura, M.; Zheng, H.; Lu, J.; Takayanagi, R.; Nomura, M. GLI1, a Crucial Mediator of Sonic Hedgehog Signaling in Prostate Cancer, Functions as a Negative Modulator for Androgen Receptor. Biochem. Biophys. Res. Commun. 2011, 404, 809–815. [Google Scholar] [CrossRef]
- Fan, L.; Pepicelli, C.V.; Dibble, C.C.; Catbagan, W.; Zarycki, J.L.; Laciak, R.; Gipp, J.; Shaw, A.; Lamm, M.L.G.; Munoz, A.; et al. Hedgehog Signaling Promotes Prostate Xenograft Tumor Growth. Endocrinology 2004, 145, 3961–3970. [Google Scholar] [CrossRef] [Green Version]
- Tzelepi, V.; Karlou, M.; Wen, S.; Hoang, A.; Logothetis, C.; Troncoso, P.; Efstathiou, E. Expression of Hedgehog Pathway Components in Prostate Carcinoma Microenvironment: Shifting the Balance towards Autocrine Signalling. Histopathology 2011, 58, 1037–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prescott, J.A.; Cook, S.J. Targeting IKKβ in Cancer: Challenges and Opportunities for the Therapeutic Utilisation of IKKβ Inhibitors. Cells 2018, 7, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonnissen, A.; Isebaert, S.; Haustermans, K. Targeting the Hedgehog Signaling Pathway in Cancer: Beyond Smoothened. Oncotarget 2015, 6, 13899–13913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awasthee, N.; Rai, V.; Chava, S.; Nallasamy, P.; Kunnumakkara, A.B.; Bishayee, A.; Chauhan, S.C.; Challagundla, K.B.; Gupta, S.C. Targeting IκappaB Kinases for Cancer Therapy. Semin. Cancer Biol. 2019, 56, 12–24. [Google Scholar] [CrossRef]
- Gonnissen, A.; Isebaert, S.; McKee, C.M.; Dok, R.; Haustermans, K.; Muschel, R.J. The Hedgehog Inhibitor GANT61 Sensitizes Prostate Cancer Cells to Ionizing Radiation Both In Vitro and In Vivo. Oncotarget 2016, 7, 84286–84298. [Google Scholar] [CrossRef] [Green Version]
- Peer, E.; Tesanovic, S.; Aberger, F. Next-Generation Hedgehog/GLI Pathway Inhibitors for Cancer Therapy. Cancers 2019, 11, 538. [Google Scholar] [CrossRef] [Green Version]
- Infante, P.; Malfanti, A.; Quaglio, D.; Balducci, S.; De Martin, S.; Bufalieri, F.; Mastrotto, F.; Basili, I.; Garofalo, M.; Lospinoso Severini, L.; et al. Glabrescione B Delivery by Self-Assembling Micelles Efficiently Inhibits Tumor Growth in Preclinical Models of Hedgehog-Dependent Medulloblastoma. Cancer Lett. 2021, 499, 220–231. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vecchiotti, D.; Verzella, D.; Di Vito Nolfi, M.; D’Andrea, D.; Flati, I.; Di Francesco, B.; Cornice, J.; Alesse, E.; Capece, D.; Zazzeroni, F. Elevated NF-κB/SHh/GLI1 Signature Denotes a Worse Prognosis and Represent a Novel Potential Therapeutic Target in Advanced Prostate Cancer. Cells 2022, 11, 2118. https://doi.org/10.3390/cells11132118
Vecchiotti D, Verzella D, Di Vito Nolfi M, D’Andrea D, Flati I, Di Francesco B, Cornice J, Alesse E, Capece D, Zazzeroni F. Elevated NF-κB/SHh/GLI1 Signature Denotes a Worse Prognosis and Represent a Novel Potential Therapeutic Target in Advanced Prostate Cancer. Cells. 2022; 11(13):2118. https://doi.org/10.3390/cells11132118
Chicago/Turabian StyleVecchiotti, Davide, Daniela Verzella, Mauro Di Vito Nolfi, Daniel D’Andrea, Irene Flati, Barbara Di Francesco, Jessica Cornice, Edoardo Alesse, Daria Capece, and Francesca Zazzeroni. 2022. "Elevated NF-κB/SHh/GLI1 Signature Denotes a Worse Prognosis and Represent a Novel Potential Therapeutic Target in Advanced Prostate Cancer" Cells 11, no. 13: 2118. https://doi.org/10.3390/cells11132118
APA StyleVecchiotti, D., Verzella, D., Di Vito Nolfi, M., D’Andrea, D., Flati, I., Di Francesco, B., Cornice, J., Alesse, E., Capece, D., & Zazzeroni, F. (2022). Elevated NF-κB/SHh/GLI1 Signature Denotes a Worse Prognosis and Represent a Novel Potential Therapeutic Target in Advanced Prostate Cancer. Cells, 11(13), 2118. https://doi.org/10.3390/cells11132118