
Fibronectin Functions as a Selective Agonist for Distinct Toll-like Receptors in Triple-Negative Breast Cancer


{kind=link}
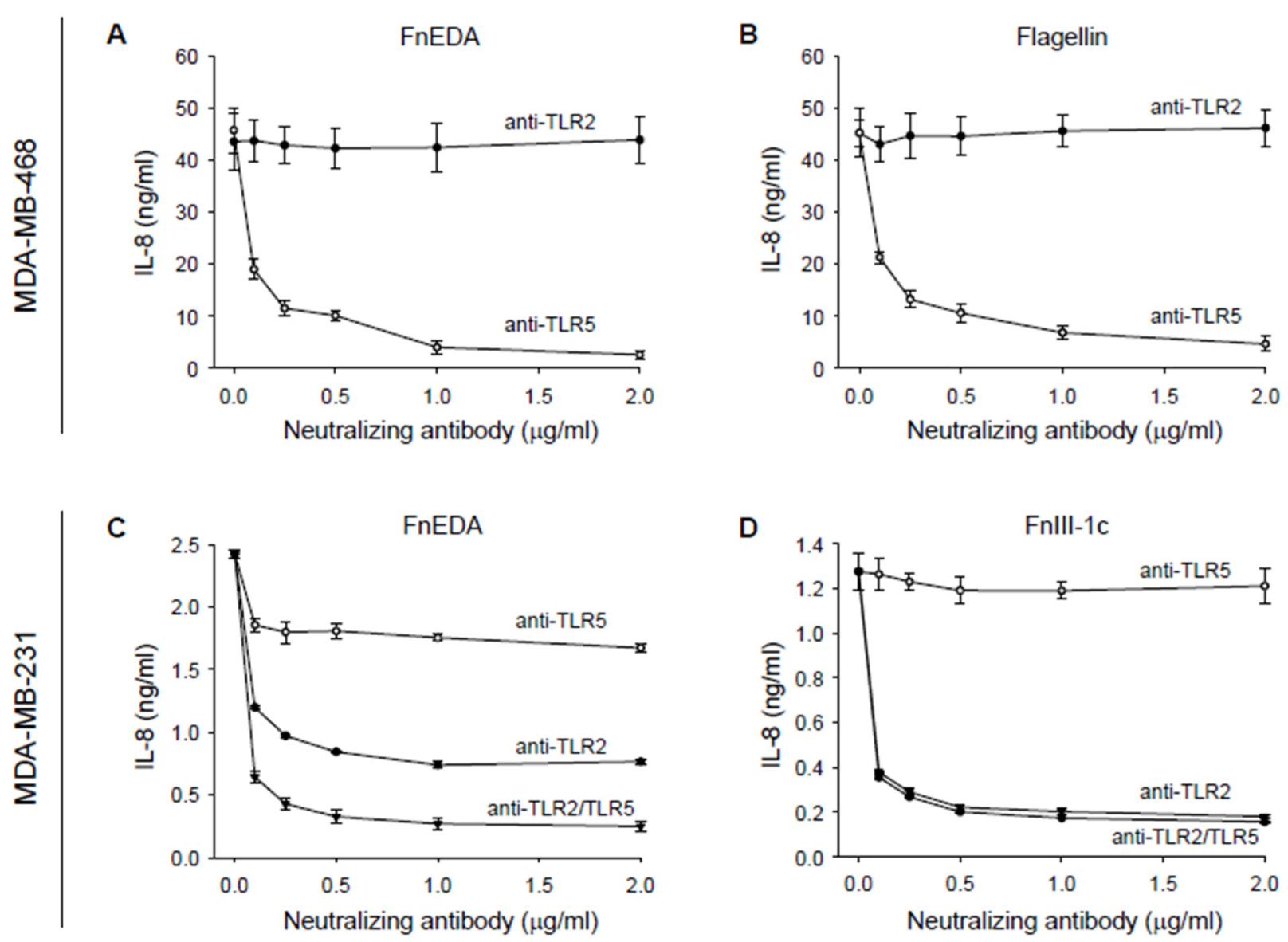{kind=link}
{kind=link}
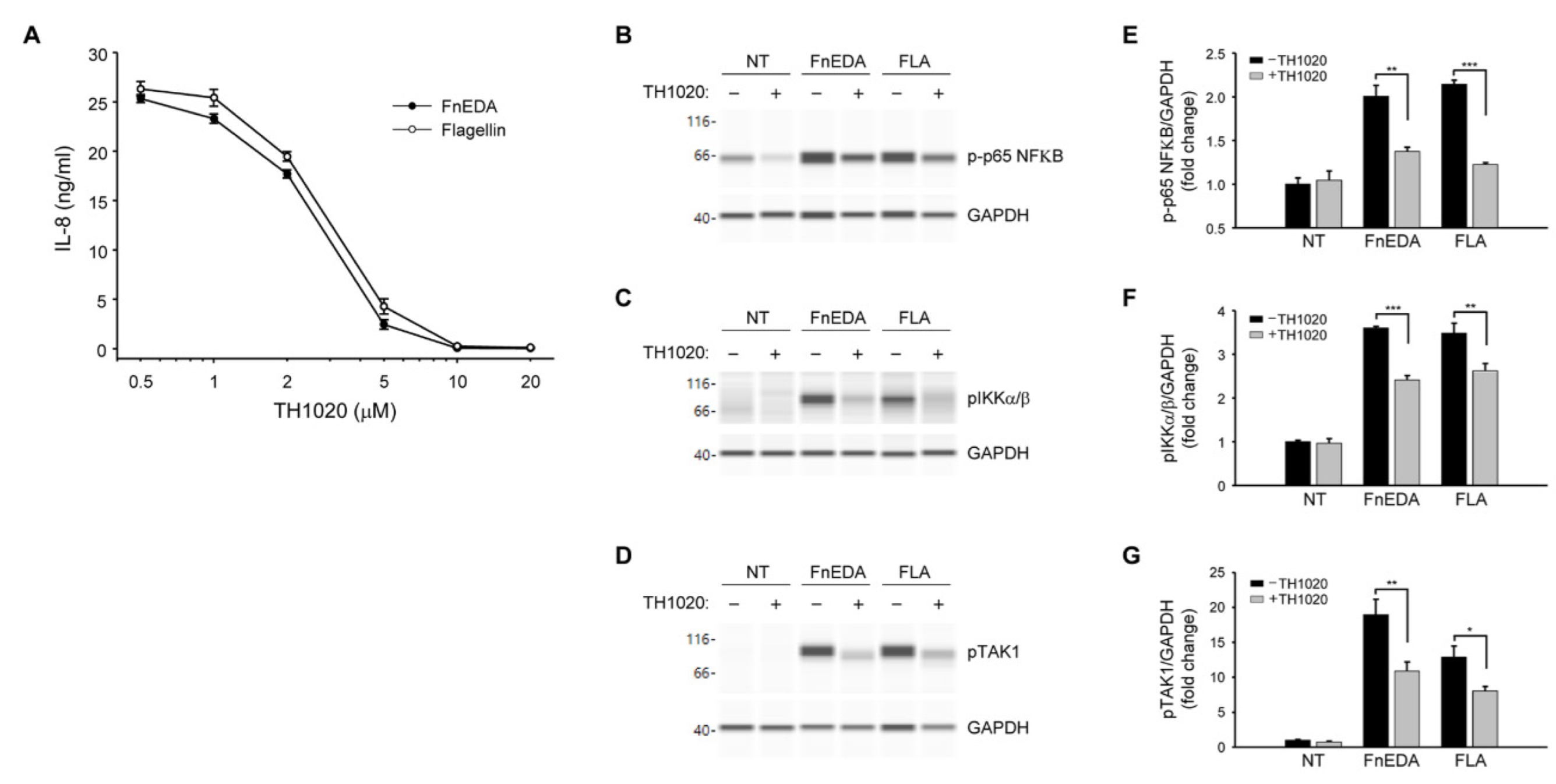{kind=link}
{kind=link}
{kind=link}
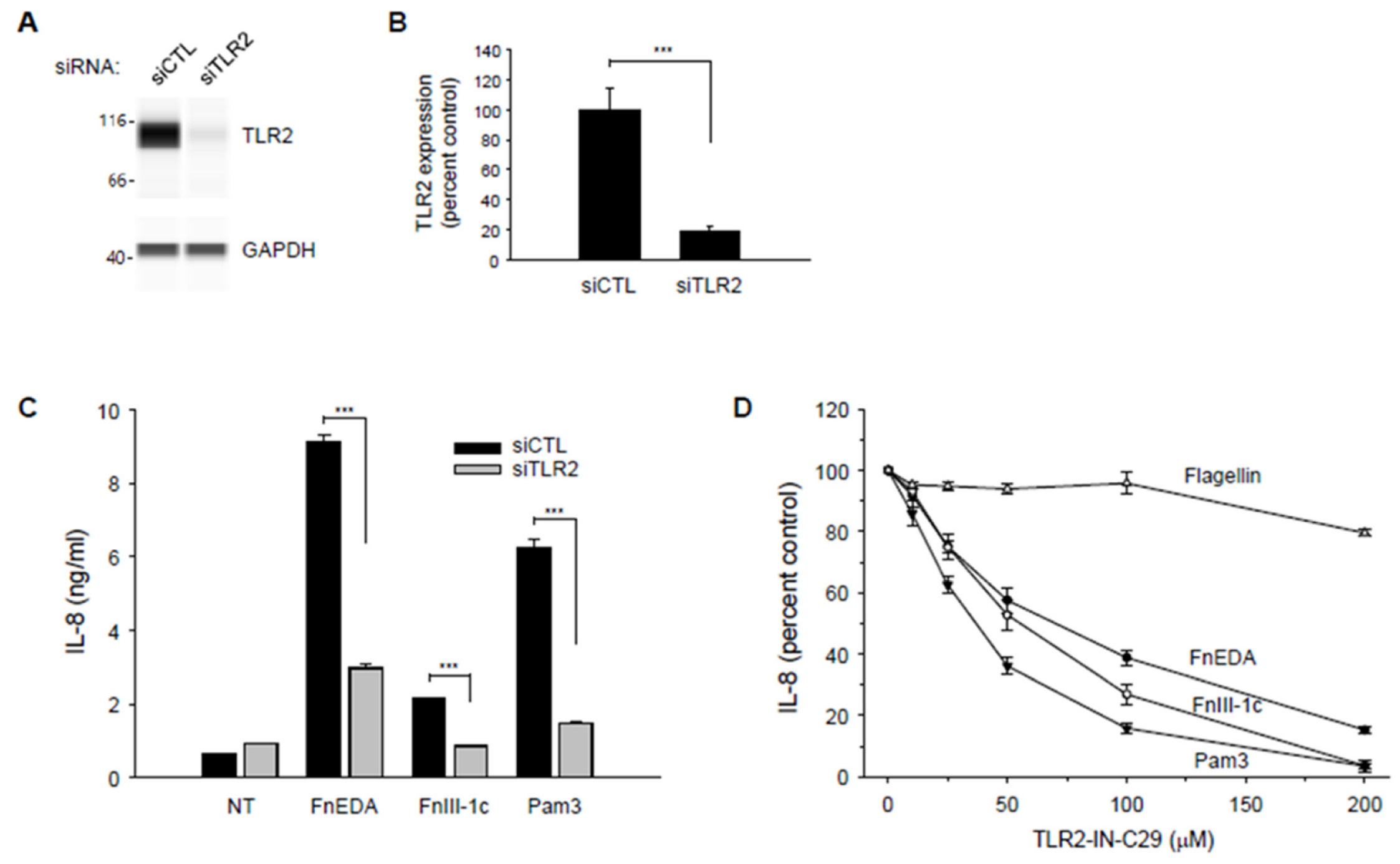{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Antibodies and Reagents
2.2. Cell Culture
2.3. Cell Treatments
2.4. Protein Analysis Using the Wes-ProteinSimple System
2.5. Suppression of TLR5 and TLR2 Expression
2.6. Statistical Analysis
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Manjunath, M.; Choudhary, B. Triple-negative breast cancer: A run-through of features, classification and current therapies. Oncol. Lett. 2021, 22, 512. [Google Scholar] [CrossRef] [PubMed]
- Broders-Bondon, F.; Nguyen Ho-Bouldoires, T.H.; Fernandez-Sanchez, M.E.; Farge, E. Mechanotransduction in tumor progression: The dark side of the force. J. Cell Biol. 2018, 217, 1571–1587. [Google Scholar] [CrossRef] [PubMed]
- Watson, A.W.; Grant, A.D.; Parker, S.S.; Hill, S.; Whalen, M.B.; Chakrabarti, J.; Harman, M.W.; Roman, M.R.; Forte, B.L.; Gowan, C.C.; et al. Breast stiffness instructs bone metastasis via maintenance of mechanical conditioning. Cell Rep. 2021, 35, 109293. [Google Scholar] [CrossRef] [PubMed]
- Kwon, A.; Chae, I.H.; You, E.; Kim, S.H.; Ahn, S.-Y.; Lee, O.-J.; Park, Z.-Y.; Rhee, S.; Huh, Y.H.; Song, W.K. Extra domain A-containing fibronectin expression in Spin90-deficient fibroblasts mediates cancer-stroma interaction and promotes breast cancer progression. J. Cell. Physiol. 2020, 235, 4494–4507. [Google Scholar] [CrossRef]
- Berger, A.J.; Renner, C.M.; Hale, I.; Yang, X.; Ponik, S.M.; Weisman, P.S.; Masters, K.S.; Kreeger, P.K. Scaffold stiffness influences breast cancer cell invasion via EGFR-linked Mena upregulation and matrix remodeling. Matrix Biol. 2020, 85, 80–93. [Google Scholar] [CrossRef]
- Smith, M.L.; Gourdon, D.; Little, W.C.; Kubow, K.E.; Eguiluz, R.A.; Luna-Morris, S.; Vogel, V. Force-induced unfolding of fibronectin in the extracellular matrix of living cells. PLoS Biol. 2007, 5, 2243–2254. [Google Scholar] [CrossRef]
- Klotzsch, E.; Smith, M.L.; Kubow, K.E.; Muntwyler, S.; Little, W.C.; Beyeler, F.; Gourdon, D.; Nelson, B.J.; Vogel, V. Fibronectin forms the most extensible biological fibers displaying switchable force-exposed cryptic binding sites. Proc. Natl. Acad. Sci. USA 2009, 106, 18267–18272. [Google Scholar] [CrossRef]
- Cao, L.; Nicosia, J.; Larouche, J.; Zhang, Y.; Bachman, H.; Brown, A.C.; Holmgren, L.; Barker, T.H. Detection of an integrin-binding mechanoswitch within fibronectin during tissue formation and fibrosis. ACS Nano 2017, 11, 7110–7117. [Google Scholar] [CrossRef]
- Chandler, E.M.; Saunders, M.P.; Yoon, C.J.; Gourdon, D.; Fischbach, C. Adipose progenitor cells increase fibronectin matrix strain and unfolding in breast tumors. Phys. Biol. 2011, 8, 015008. [Google Scholar] [CrossRef]
- Wang, K.; Andresen Eguiluz, R.C.; Wu, F.; Seo, B.R.; Fischbach, C.; Gourdon, D. Stiffening and unfolding of early deposited-fibronectin increase proangiogenic factor secretion by breast cancer-associated stromal cells. Biomaterials 2015, 54, 63–71. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Midwood, K.S.; Yin, H.; Varga, J. Toll-like receptor-4 signaling drives persistent fibroblast activation and prevents fibrosis resolution in scleroderma. Adv. Wound Care 2017, 6, 356–369. [Google Scholar] [CrossRef]
- Li, D.; Wu, M. Pattern recognition receptors in health and disease. Signal Transduct. Target Ther. 2021, 6, 291. [Google Scholar] [CrossRef] [PubMed]
- Roh, J.S.; Sohn, D.H. Damage-associated molecular patterns in inflammatory diseases. Immune Netw. 2018, 18, e27. [Google Scholar] [CrossRef] [PubMed]
- Leifer, C.A.; Medvedev, A.E. Molecular mechanisms of regulation of Toll-like receptor signaling. J. Leukoc. Biol. 2016, 100, 927–941. [Google Scholar] [CrossRef]
- Turner, N.A. Inflammatory and fibrotic responses of cardiac fibroblasts to myocardial damage associated molecular patterns (DAMPs). J. Mol. Cell Cardiol. 2016, 94, 189–200. [Google Scholar] [CrossRef] [PubMed]
- De Haan, J.J.; Smeets, M.B.; Pasterkamp, G.; Arslan, F. Danger signals in the initiation of the inflammatory response after myocardial infarction. Mediat. Inflamm. 2013, 2013, 206039. [Google Scholar] [CrossRef]
- Fitzgerald, K.A.; Kagan, J.C. Toll-like receptors and the control of immunity. Cell 2020, 180, 1044–1066. [Google Scholar] [CrossRef]
- McKeown-Longo, P.J.; Higgins, P.J. Integration of canonical and noncanonical pathways in TLR4 signaling: Complex regulation of the wound in TLR4 signaling: Complex regulation. Adv. Wound Care 2017, 6, 320–329. [Google Scholar] [CrossRef]
- Gupta, S.; Tsoporis, J.N.; Jia, S.-H.; dos Santos, C.C.; Parker, T.G.; Marshall, J.C. Toll-like receptors, associated biochemical signaling networks, and S100 ligands. Shock 2021, 56, 167–177. [Google Scholar] [CrossRef]
- Zhou, R.; Liu, L.; Wang, Y. Viral proteins recognized by different TLRs. J. Med. Virol. 2021, 93, 6116–6123. [Google Scholar] [CrossRef]
- McFadden, J.P.; Baker, B.S.; Powles, A.V.; Fry, L. Psoriasis and extra domain A fibronectin loops. Brit. J. Dermatol. 2010, 163, 5–11. [Google Scholar] [CrossRef]
- Muro, A.F.; Moretti, F.A.; Moore, B.B.; Yan, M.; Atrasz, R.G.; Wilke, C.A.; Flaherty, K.R.; Martinez, F.J.; Tsui, J.L.; Sheppard, D.; et al. An essential role for fibronectin extra type III domain A in pulmonary fibrosis. Am. J. Resp. Crit. Care Med. 2008, 177, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Booth, A.J.; Wood, S.C.; Cornett, A.M.; Dreffs, A.A.; Lu, G.; Muro, A.F.; White, E.S.; Bishop, D.K. Recipient-derived EDA fibronectin promotes cardiac allograft fibrosis. J. Pathol. 2012, 226, 609–618. [Google Scholar] [CrossRef] [PubMed]
- McKeown-Longo, P.J.; Higgins, P.J. Hyaluronan, transforming growth factor b, and extra domain A-fibronectin: A fibrotic triad. Adv. Wound Care 2021, 10, 137–152. [Google Scholar] [CrossRef]
- You, R.; Zheng, M.; McKeown-Longo, P.J. The first type III repeat in fibronectin activates an inflammatory pathway in dermal fibroblasts. J. Biol. Chem. 2010, 285, 36255–36259. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Jones, D.M.; Horzempa, C.; Prasad, A.; McKeown-Longo, P.M. The first type III domain of fibronectin is associated with the expression of cytokines within the lung tumor microenvironment. J. Cancer 2011, 2, 478–483. [Google Scholar] [CrossRef] [PubMed]
- Kelsh, R.; You, R.; Horzempa, C.; Zheng, M.; McKeown-Longo, P.J. Regulation of the innate immune response by fibronectin: Synergism between the III-1 and EDA domains. PLoS ONE 2014, 9, e102974. [Google Scholar] [CrossRef]
- Zheng, M.; Ambesi, A.; McKeown-Longo, P.J. Role of TLR4 receptor complex in the regulation of the innate immune response by fibronectin. Cells 2020, 9, 216. [Google Scholar] [CrossRef]
- Cho, C.; Horzempa, C.; Longo, C.M.; Peters, D.M.; Jones, D.M.; McKeown-Longo, P.J. Fibronectin in the tumor microenvironment activates a TLR4-dependent inflammatory response in lung cancer cells. J. Cancer 2020, 11, 3099–3105. [Google Scholar] [CrossRef]
- Yin, L.; Duan, J.-J.; Bian, X.-W.; Yu, S.-C. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res. 2020, 22, 61. [Google Scholar] [CrossRef]
- Baram, T.; Rubinstein-Achiasaf, L.; Ben-Yaakov, H.; Ben-Baruch, A. Inflammation-driven breast tumor cell plasticity: Stemness/EMT, therapy resistance and dormancy. Front. Oncol. 2021, 10, 614468. [Google Scholar] [CrossRef] [PubMed]
- Liubomirski, Y.; Lerrer, S.; Meshel, T.; Rubinstein-Achiasaf, L.; Morein, D.; Wiemann, S.; Korner, C.; Ben-Baruch, A. Tumor-stroma-inflammation networks promote pro-metastatic chemokines and aggressivness charactertistics in triple-negative breast cancer. Front. Immunol. 2019, 10, 757. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Ma, L.; Wang, D.; Yang, J. Uncarboxylated osteocalcin promotes proliferation and metastasis of MDA-MB-231 cells through TGF-b/SMAD3 signaling pathway. BMC Mol. Cell Biol. 2022, 23, 18. [Google Scholar] [CrossRef] [PubMed]
- Benchama, O.; Tyukhtenko, S.; Malamas, M.S.; Williams, M.K.; Makriyannis, A.; Avraham, H.K. Inhibition of triple negative breast cancer-associated inflammation, tumor growth and brain colonization by targeting monoacylglycerol lipase. Sci. Rep. 2022, 12, 5328. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Ren, W.; Li, Q.; Duan, C.; Lin, X.; Bi, Z.; You, K.; Hu, Q.; Xie, N.; Yu, Y.; et al. LncRNA Ucoo3xsl.1-mediated activation of the NFKkB/IL8 axis promotes progression of triple-negative breast cancer. Cancer Res. 2022, 82, 556–570. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Tacam, M.J.; Chauhan, G.; Cohen, E.N.; Gagliardi, M.; Iles, L.R.; Ueno, N.T.; Battula, V.; Sohn, Y.-K.; Wang, X.; et al. Nonphosphorylatable PEA15 mutant inhibits epithelial-mesenchymal transition in triple-negative breast cancer partly through the regulation of IL-8 expression. Breast Cancer Res. Treat. 2021, 189, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Hirata, N.; Yamada, S.D.; Yanagida, S.; Ono, A.; Yasuhiko, Y.; Nishida, M.; Kanada, Y. Lysophosphatidic acid promotes the expansion of cancer stem cells via TRPC3 channels in triple-negative breast cancer. Int. J. Mol. Sci. 2022, 23, 1967. [Google Scholar] [CrossRef]
- Klein, R.M.; Zheng, M.; Ambesi, A.; van de Water, L.; McKeown-Longo, P.J. Stimulation of extracellular matrix remodeling by the first type III repeat in fibronectin. J. Cell Sci. 2003, 116, 4663–4674. [Google Scholar] [CrossRef]
- Kelsh-Lasher, R.; Ambesi, A.; Bertram, C.; McKeown-Longo, P.J. Integrin a4b1 and TLR4 cooperate to induce fibrotic gene expression in response to fibronectin’s EDA domain. J. Investig. Dermatol. 2017, 137, 2505–2512. [Google Scholar] [CrossRef]
- Valenty, L.M.; Longo, C.M.; Horzempa, C.; Ambesi, A.; McKeown-Longo, P.J. TLR4 ligands selectively synergize to induce expression of IL-8. Adv. Wound Care 2017, 6, 309–319. [Google Scholar] [CrossRef]
- Okamura, Y.; Watari, M.; Jerud, E.S.; Young, D.W.; Ishizaka, S.T.; Rose, J.; Chow, J.C.; Strauss, J.R., 3rd. The extra domain A of fibronectin activates Toll-like receptor 4. J. Biol. Chem. 2001, 276, 10229–10233. [Google Scholar] [CrossRef] [PubMed]
- Gondokaryono, S.P.; Ushio, H.; Niyonsaba, F.; Hara, M.; Takenaka, H.; Jayawardana, S.T.; Ikeda, S.; Okumura, K.; Ogawa, H. The extra domain A of fibronectin stimulates murine mast cells via toll-like receptor 4. J. Leukoc. Biol. 2007, 82, 657–665. [Google Scholar] [CrossRef] [PubMed]
- McFadden, J.P.; Basketter, D.A.; Dearman, R.J.; Kimber, I.R. Extra domain A-positive fibronectin-positive feedback loops and their association with cutaneous inflammatory disease. Clin. Dermatol. 2011, 29, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Tamaki, Z.; Wang, W.; Hinchcliff, M.; Hoover, P.; Getsios, S.; White, E.S.; Varga, J. Fibronectin EDA promotes chronic cutaneous fibrosis through toll-like receptor signaling. Sci. Transl. Med. 2014, 6, 232–250. [Google Scholar] [CrossRef] [PubMed]
- Doddapattar, P.; Gandhi, C.; Prakash, P.; Dhanesha, N.; Grumbach, I.M.; Dailey, M.E.; Lentz, S.R.; Chauhan, A.K. Fibronectin splicing variants containing extra domain A promote atherosclerosis in mice through Toll-like Receptor 4. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2391–2400. [Google Scholar] [CrossRef]
- Prakash, P.; Kulkarni, P.P.; Lentz, S.R.; Chauhan, A.K. Cellular fibronectin containing extra domain A promotes arterial thrombosis in mice through platelet Toll-like receptor 4. Blood 2015, 125, 3164–3172. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Varga, J. Endogenous ligands of TLR4 promote unresolving tissue fibrosis: Implications for systemic sclerosis and its targeted therapy. Immunol. Lett. 2018, 195, 9–17. [Google Scholar] [CrossRef]
- Roberts, A.L.; Mavlyutov, T.A.; Perlmutter, T.E.; Curry, S.M.; Larris, S.L.; Chauhan, A.K.; McDowell, C.M. Fibronectin extra domain A (FN-EDA) elevates intraocular pressure through Toll-like receptor 4 signaling. Sci. Rep. 2020, 10, 9815. [Google Scholar] [CrossRef]
- Amin, A.; Mokhdomi, T.A.; Rukhari, S.; Wani, Z.; Chikan, N.A.; Shah, B.A.; Koul, A.M.; Majeed, U.; Farooq, F.; Qadri, A.; et al. Lung cancer cell-derived EDA-containing fibronectin induces an inflammatory response from monocytes and promotes metastatic tumor microenvironment. J. Cell Biochem. 2021, 122, 562–576. [Google Scholar] [CrossRef]
- Schaefer, L. Complexity of danger: The diverse nature of damage-associated molecular patterns. J. Biol. Chem. 2014, 289, 35237–35245. [Google Scholar] [CrossRef]
- Yan, L.; Liang, J.; Yao, C.; Wu, P.; Zeng, X.; Cheng, K.; Yin, H. Pyrimidine triazole thioether derivatives as Toll-like receptor 5 (TLR5)/flagellin complex inhibitors. Chem. Med. Chem. 2016, 11, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Mistry, P.; Laird, M.H.; Schwarz, R.S.; Greene, S.; Dyson, T.; Snyder, G.A.; Xiao, T.S.; Chauhan, J.; Fletcher, S.; Toshchakov, V.Y.; et al. Inhibition of TLR2 signaling by small molecule inhibitors targeting a pocket within the TLR2 TIR domain. Proc. Natl. Acad. Sci. USA 2015, 112, 5455–5460. [Google Scholar] [CrossRef] [PubMed]
- Dalton, C.J.; Lemmon, C.A. Fibronectin: Molecular structure, fibrillar structure, and mechanochemical signaling. Cells 2021, 10, 2433. [Google Scholar] [CrossRef] [PubMed]
- Pratt, S.J.; Lee, R.M.; Martin, S.D. The mechanical microenvironment in breast cancer. Cancers 2020, 12, 1452. [Google Scholar] [CrossRef]
- Wang, K.; Wu, F.; Seo, B.R.; Fischbach, C.; Chen, W.; Hsu, L.; Gourdon, D. Breast cancer cells alter the dynamics of stromal fibronectin-collagen interactions. Matrix Biol. 2017, 60, 86–95. [Google Scholar] [CrossRef]
- Efthymiou, G.; Sant, A.; Ruff, M.; Rekad, Z.; Ciais, D.; Van Obberghen-Schilling, E. Shaping up the tumor microenvironment with cellular fibronectin. Front. Oncol. 2020, 10, 641. [Google Scholar] [CrossRef]
- Ffrench-Constant, C.; Hynes, R.O. Alternative splicing of fibronectin is temporally and spatially regulated in the chicken embryo. Development 1989, 106, 375–388. [Google Scholar] [CrossRef]
- Oyama, F.; Murata, Y.; Suganuma, N.; Kimura, T.; Titani, K.; Sekiguchi, K. Patterns of alternative splicing of fibronectin pre-mRNA in human adult and fetal tissues. Biochemistry 1989, 28, 1428–1434. [Google Scholar] [CrossRef]
- Insua-Rodriguez, J.; Oskarsson, T. The extracellular matrix in breast cancer. Adv. Drug Deliv. Rev. 2016, 97, 41–55. [Google Scholar] [CrossRef]
- Han, Z.; Lu, Z.-R. Targeting fibronectin for cancer imaging and therapy. J. Mater. Chem. B Mater. Biol. Med. 2017, 5, 639–654. [Google Scholar] [CrossRef]
- Gao, M.; Craig, D.; Lequin, O.; Campbell, I.D.; Vogel, V.; Schulten, K. Structure and functional significance of mechanically unfolded fibronectin type III1 intermediates. Proc. Natl. Acad. Sci. USA 2003, 100, 14784–14789. [Google Scholar] [CrossRef] [PubMed]
- Cho, C.; Kelsh-Lasher, R.; Ambesi, A.; McKeown-Longo, P.J. Cryptic activity within the Type III 1 domain of fibronectin regulates tissue inflammation and angiogenesis. Curr. Top. Pept. Protein Res. 2015, 16, 37–47. [Google Scholar] [PubMed]
- Kohrmann, A.; Kammerer, U.; Kapp, M.; Dietl, J.; Anacker, J. Expression of matrix metalloproteinases (MMPs) in primary human breast cancer and breast cancer cell lines: New findings and review of the literature. BMC Cancer 2009, 9, 188. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Rhee, S. Matrix metalloproteinase-2 regulates MDA-NBA-231 breast cancer cell invasion induced by active mammalian diaphanous-related form 1. Mol. Med. Rep. 2016, 14, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lee, J.; Jeon, M.; Lee, J.E.; Nam, S.J. MEK-dependent IL-8 induction regulates the invasiveness of triple-negative breast cancer cells. Tumour Biol. 2016, 37, 4991–4999. [Google Scholar] [CrossRef]
- Tang, C.P.; Zhou, H.J.; Qin, J.; Luo, Y.; Zhang, T. MicroRNA-520c-3p negatively regulates EMT by targeting IL-8 to suppress the invasion and migration of breast cancer. Oncol. Rep. 2017, 38, 3144–3152. [Google Scholar] [CrossRef]
- Dominguez, C.; McCampbell, K.K.; David, J.M.; Palena, C. Neutralizaation of IL-8 decreases tumor PMN-MDSCs and reduces mesenchymalization of claudin-low triple-negative breast cancer. J. Clin. Investig. Insight 2017, 2, e94296. [Google Scholar] [CrossRef]
- Ignacio, R.M.C.; Gibbs, C.R.; Kim, S.; Lee, E.S.; Adunyah, S.E.; Son, D.S. Serum amyloid A predisposes inflammatory tumor microenvironment in triple negative breast cancer. Oncotarget 2019, 10, 511–526. [Google Scholar] [CrossRef]
- Singh, J.K.; Simoes, B.M.; Howell, S.J.; Farnie, G.; Clark, R.B. Recent advances reveal IL-8 signaling as a potential key to targeting breast cancer stem cells. Breast Cancer Res. 2013, 15, 210. [Google Scholar] [CrossRef]
- Alraouji, N.N.; Aboussekhra, A. Tocilizumab inhibits IL-8 and the proangiogenic potential of triple negative breast cancer cells. Mol. Carcinog. 2021, 60, 51–59. [Google Scholar] [CrossRef]
- David, J.M.; Dominguez, C.; Hamilton, D.H.; Pajena, C. The IL-8/IL-8R axis: A double agent in tumor immune resistance. Vaccines 2016, 4, 22. [Google Scholar] [CrossRef]
- Fang, Q.I.; Wang, X.; Luo, G.; Yu, M.; Zhang, X.; Xu, N. Increased CXCL8 expression is negatively correlated with the overall survival of patients with ER-negative breast cancer. Anticancer Res. 2017, 37, 4845–4852. [Google Scholar] [PubMed]
- Milovanovic, J.; Todorovic-Rakovic, N.; Radulovic, M. Interleukin-6 and interleukin-8 serum levels in prognosis of hormone-dependent breast cancer. Cytokine 2018, 118, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Khazali, A.S.; Clark, A.M.; Wells, A. Inflammatory cytokine IL-8/CXCL8 promotes tumour escape from hepatocyte-induced dormancy. Brit. J. Cancer 2018, 118, 566–576. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Xu, C.; Fang, X.; Zhang, Y.; Li, H.; Wen, W.; Yang, G. Expression profile of Toll-like receptors in human breast cancer. Mol. Med. Rep. 2020, 21, 784–794. [Google Scholar] [CrossRef]
- Cai, Z.M.; Sanchez, A.; Shi, Z.; Zhang, T.; Liu, M.; Zhang, D. Activtion of Toll-like receptor 5 on breast cancer cells by flagellin suppresses cell proliferation and tumor growth. Cancer Res. 2011, 71, 2466–2475. [Google Scholar] [CrossRef]
- Chen, S.; Yuan, W.; Fu, Z.; Huang, Y.; Yang, J.; Xue, J.; Liu, X.; Li, Y.; Li, D. Toll-like receptor 5 gene polymorphism is associated with breast cancer susceptibility. Oncotarget 2017, 8, 88622–88629. [Google Scholar]
- Shi, M.; Yao, Y.; Han, F.; Li, Y.; Li, Y. MAP1S controls breast cancer cell TLR5 signaling pathway and promotes TLR5 signaling-baased tumor suppression. PLoS ONE 2014, 9, e86839. [Google Scholar]
- Burdelya, L.G.; Brackett, C.M.; Kojouharov, B.; Gitlin, I.I.; Lenova, K.I.; Bleiberman, A.S.; Aygun-Sunar, S.; Veith, J.; Johnson, C.; Haderski, G.J.; et al. Central role of liver in anticancer and radioprotective activities of Toll-like receptor 5 agonist. Proc. Natl. Acad. Sci. USA 2013, 110, E1857–E1866. [Google Scholar] [CrossRef]
- Yang, H.; Brackett, C.M.; Morales-Tirado, V.M.; Li, Z.; Zhang, Q.; Wilson, M.W.; Benjamin, C.; Harris, W.; Waller, E.K.; Gudkov, A.V.; et al. The Toll-like receptor 5 agonist entolimod suppresses hepatic metastasis in a murine model of ocular melanoma via an NK cell-dependent mechanism. Oncotarget 2016, 7, 2936–2950. [Google Scholar] [CrossRef]
- Brackett, C.M.; Kojouharov, B.; Veith, J.; Greene, K.F.; Burdelya, L.; Gollnick, S.O.; Abrams, S.I.; Gudkov, A.V. Toll-like receptor-5 agonist, entolimod, suppresses metastasis and induced immunity by stimiulating an NK-dendritic-CD8+ T-cell axis. Proc. Natl. Acad. Sci. USA 2016, 113, E874–E883. [Google Scholar] [CrossRef] [PubMed]
- Das, N.; Dewan, V.; Grace, P.M.; Gunn, R.J.; Tamura, R.; Tzarum, N.; Watkins, L.R.; Wilson, I.A.; Yin, H. HMGB1 activates proinflammatory signaling via TLR5 leading to allodynia. Cell Rep. 2016, 17, 1128–1140. [Google Scholar] [CrossRef] [PubMed]
- Trofimova, O.; Kiorotkaja, K.; Skrastina, D.; Jansons, J.; Spunde, K.; Isaguliants, M.; Zajakina, A. Alphavirus-driven interferon gamma (IFNg) expression inhibits tumor growth in orthotopic 4T1 breast cancer model. Vaccines 2021, 9, 1247. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, C.; Li, L.; Jin, X.; Zhang, Z.; Zheng, H.; Pan, J.; Shi, L.; Jiang, Z.; Su, K.; et al. Tumor-derived HMGB1 induces CD62Ldim neutrophil polarization and promotes lung metastasis in triple negative breast cancer. Oncogenesis 2020, 9, 82. [Google Scholar] [CrossRef] [PubMed]
- El-Kharashy, G.; Gowily, A.; Okda, T.; Houssen, M. Association between serum soluble Toll-like receptor 2 and 4 and the risk of breast cancer. Mol. Clin. Oncol. 2021, 14, 38. [Google Scholar] [CrossRef]
- Bezhaeva, T.; Karper, J.C.; Quax, P.H.A.; de Vries, M.R. The intriguing role of TLR accessory molecules in cardiovascular health and disease. Front. Cardiovasc. Med. 2022, 9, 820962. [Google Scholar] [CrossRef]
- Roedig, H.; Damiescu, R.; Zeng-Brouwers, J.; Kutija, I.; Trebicka, J.; Wygrecka, M.; Schaefer, L. Danger matrix molecules orchestrate CD14/CD44 signaling in cancer development. Semin. Cancer Biol. 2020, 62, 31–47. [Google Scholar] [CrossRef]
- De Nardo, D.; De Nardo, C.M.; Nguyen, T.; Hamilton, J.A.; Scholz, G.M. Signaling crosstalk during sequential TLR4 and TLR9 activation amplifies the inflammatory response of mouse macrophages. J. Immunol. 2009, 183, 8110–8118. [Google Scholar] [CrossRef]
- Hsu, Y.-L.; Wang, M.-Y.; Ho, L.-J.; Lai, J.-H. Denguie virus infection induces interferon-l1 to facilitate cell migration. Sci. Rep. 2016, 6, 24530. [Google Scholar] [CrossRef]
- Prantner, D.; Nallar, S.; Vogel, S.N. The role of RAGE in host pathology and crosstalk between RAGE and TLR4 in innate immune signal transduction pathways. FASEB J. 2020, 34, 15659–15674. [Google Scholar] [CrossRef]
- Ryu, J.-K.; Kim, S.J.; Rah, S.-H.; Kang, J.I.; Jung, H.E.; Lee, D.; Lee, H.K.; Lee, J.-O.; Park, B.S.; Yoon, T.-Y.; et al. Reconstruction of LPS transfer cascade reveals structural determinants within LBP, CD14, and TLR4-MD2 for efficient LPS recognition and transfer. Immunity 2017, 46, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Ranoa, D.R.E.; Kelley, S.L.; Tapping, R.I. Human llipopolysaccharide-binding protein (LBP) and CD14 independently deliver triacylated lipoproteins to Toll-like receptor 1 (TLR1) and TLR2 and enhance formation of the ternary signaling complex. J. Biol. Chem. 2013, 288, 9729–9741. [Google Scholar] [CrossRef] [PubMed]
- Raby, A.-C.; Labeta, M.O. Therapeutic boosting of the immune response: Turning to CD14 for help. Curr. Pharm. Biotechnol. 2016, 17, 414–418. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hoang, T.X.; Jung, J.H.; Kim, J.Y. All-trans retinoic acid enhances bacterial flagellin-stimulated proinflammatory responses in human monocyte THP-1 cells by upregulating CD14. Biomed. Res. Intl. 2019, 2019, 8059312. [Google Scholar] [CrossRef]
- Di Gioia, M.; Zanoni, I. Toll-like receptor co-receptors as master regulators of the immune response. Mol. Immunol. 2015, 63, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Barratt-Due, A.; Pischke, S.E.; Hilsson, P.H.; Espevik, T.; Mollnes, T.E. Dual inhibition of complement and Toll-like receptors as a novel approach to treat inflammatory diseases-C3 or C5 emerge together with CD14 as promising targets. J. Leukoc. Biol. 2017, 101, 193–204. [Google Scholar] [CrossRef]
- Allen, M.; Jones, J.L. Jekyll and Hyde: The role of the microenvironment on the progression of cancer. J. Pathol. 2011, 223, 162–176. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ambesi, A.; Maddali, P.; McKeown-Longo, P.J. Fibronectin Functions as a Selective Agonist for Distinct Toll-like Receptors in Triple-Negative Breast Cancer. Cells 2022, 11, 2074. https://doi.org/10.3390/cells11132074
Ambesi A, Maddali P, McKeown-Longo PJ. Fibronectin Functions as a Selective Agonist for Distinct Toll-like Receptors in Triple-Negative Breast Cancer. Cells. 2022; 11(13):2074. https://doi.org/10.3390/cells11132074
Chicago/Turabian StyleAmbesi, Anthony, Pranav Maddali, and Paula J. McKeown-Longo. 2022. "Fibronectin Functions as a Selective Agonist for Distinct Toll-like Receptors in Triple-Negative Breast Cancer" Cells 11, no. 13: 2074. https://doi.org/10.3390/cells11132074
APA StyleAmbesi, A., Maddali, P., & McKeown-Longo, P. J. (2022). Fibronectin Functions as a Selective Agonist for Distinct Toll-like Receptors in Triple-Negative Breast Cancer. Cells, 11(13), 2074. https://doi.org/10.3390/cells11132074