
Human Blood Bacteriome: Eubiotic and Dysbiotic States in Health and Diseases


Abstract
:1. Introduction
2. Profiles of the Eubiotic Blood Bacteriome
3. Profiles of Dysbiotic Blood Bacteriome
3.1. Infection-Related Diseases and Profiles of Blood Dysbiosis
3.2. Age-Related Metabolic Diseases and Profiles of Blood Dysbiosis
3.3. Oral, Gastrointestinal, and Hepatobiliary Diseases and Profile of Blood Dysbiosis
3.4. Neurological Disorders and Profiles of Blood Dysbiosis
3.5. Immune-Mediated Diseases and Profiles of Blood Dysbiosis
4. Limitations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 16s rRNA | 16s ribosomal RNA |
| ALS | amyotrophic lateral sclerosis |
| AMI | acute myocardial infarction |
| cART | combined antiviral therapy |
| CHD | congenital heart disease |
| CLABSI | central line bloodstream infection |
| CLF | compensated liver function |
| CRP | C-reactive protein |
| CT | chemotherapy |
| BMI | body mass index |
| BP | bipolar disorder |
| DC-CIK | dendritic cell/cytokine-induced killer cell |
| DLF | decompensated liver function |
| EVs | extracellular vesicles |
| GA | gestational age |
| GC | giant cell arteritis |
| HBV | chronic hepatitis B virus infection |
| HC | healthy controls |
| HCC | hepatocellular carcinoma |
| hCV | high cardiovascular risk |
| HIV | human immunodeficiency virus |
| HMP | Human Microbiome Project |
| ICS | inhaled corticosteroids |
| IHD | ischemic heart disease |
| IL | interleukin |
| INI | integrase-inhibitor-based regimen |
| LBP | LPS-binding protein |
| LPS | lipopolysaccharide |
| MDE | major depressive episode |
| MSA | multiple system atrophy |
| NAFLD | non-alcoholic fatty liver disease |
| NCBI | NCBI RefSeq database |
| NNRTI | non-nucleotide-reverse-transcriptase-inhibitor-based regimen |
| OCS | oral corticosteroids |
| PBMC | peripheral blood mononuclear cells |
| PI | protease-inhibitor-based regimen |
| PICC | peripherally inserted central catheter |
| RDP | Ribosomal Database Project |
| RNA-seq | RNA sequencing |
| SCZ | Schizophrenia |
| SLE | systemic lupus erythematosus |
| TAK | Takayasu’s arteritis |
| T2DM | type 2 diabetes mellitus |
| TNF | tumor necrosis factor |
| V | hypervariable region |
| VHD | valvular heart disease |
References
- Ursell, L.K.; Metcalf, J.L.; Parfrey, L.W.; Knight, R. Defining the human microbiome. Nutr. Rev. 2012, 70 (Suppl. 1), S38–S44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiffany, C.R.; Baumler, A.J. Dysbiosis: From fiction to function. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 317, G602–G608. [Google Scholar] [CrossRef] [PubMed]
- Petersen, C.; Round, J.L. Defining dysbiosis and its influence on host immunity and disease. Cell Microbiol. 2014, 16, 1024–1033. [Google Scholar] [CrossRef] [PubMed]
- Berg, G.; Rybakova, D.; Fischer, D.; Cernava, T.; Verges, M.C.; Charles, T.; Chen, X.; Cocolin, L.; Eversole, K.; Corral, G.H.; et al. Microbiome definition re-visited: Old concepts and new challenges. Microbiome 2020, 8, 103. [Google Scholar] [CrossRef]
- Potgieter, M.; Bester, J.; Kell, D.B.; Pretorius, E. The dormant blood microbiome in chronic, inflammatory diseases. FEMS Microbiol. Rev. 2015, 39, 567–591. [Google Scholar] [CrossRef] [Green Version]
- Markova, N.D. Eubiotic vs. dysbiotic human blood microbiota: The phenomenon of cell wall deficiency and disease-trigger potential of bacterial and fungal L-forms. Discov. Med. 2020, 29, 17–26. [Google Scholar]
- Markova, N. Dysbiotic microbiota in autistic children and their mothers: Persistence of fungal and bacterial wall-deficient L-form variants in blood. Sci. Rep. 2019, 9, 13401. [Google Scholar] [CrossRef] [Green Version]
- Errington, J.; Mickiewicz, K.; Kawai, Y.; Wu, L.J. L-form bacteria, chronic diseases and the origins of life. Philos. Trans. R Soc. B Biol. Sci. 2016, 371, 20150494. [Google Scholar] [CrossRef]
- Dimova, T.; Terzieva, A.; Djerov, L.; Dimitrova, V.; Nikolov, A.; Grozdanov, P.; Markova, N. Mother-to-newborn transmission of mycobacterial L-forms and Vdelta2 T-cell response in placentobiome of BCG-vaccinated pregnant women. Sci. Rep. 2017, 7, 17366. [Google Scholar] [CrossRef] [Green Version]
- Markova, N.; Slavchev, G.; Michailova, L. Presence of mycobacterial L-forms in human blood: Challenge of BCG vaccination. Hum. Vaccin. Immunother. 2015, 11, 1192–1200. [Google Scholar] [CrossRef]
- Edgar, R.C. Accuracy of taxonomy prediction for 16S rRNA and fungal ITS sequences. PeerJ 2018, 6, e4652. [Google Scholar] [CrossRef]
- Cangelosi, G.A.; Meschke, J.S. Dead or alive: Molecular assessment of microbial viability. Appl. Environ. Microbiol. 2014, 80, 5884–5891. [Google Scholar] [CrossRef] [Green Version]
- Panaiotov, S.; Filevski, G.; Equestre, M.; Nikolova, E.; Kalfin, R. Cultural Isolation and Characteristics of the Blood Microbiome of Healthy Individuals. Adv. Microbiol. 2018, 8, 16. [Google Scholar] [CrossRef] [Green Version]
- Panaiotov, S.; Hodzhev, Y.; Tsafarova, B.; Tolchkov, V.; Kalfin, R. Culturable and Non-Culturable Blood Microbiota of Healthy Individuals. Microorganisms 2021, 9, 1464. [Google Scholar] [CrossRef]
- Dinakaran, V.; Rathinavel, A.; Pushpanathan, M.; Sivakumar, R.; Gunasekaran, P.; Rajendhran, J. Elevated levels of circulating DNA in cardiovascular disease patients: Metagenomic profiling of microbiome in the circulation. PLoS ONE 2014, 9, e105221. [Google Scholar] [CrossRef]
- Castillo, D.J.; Rifkin, R.F.; Cowan, D.A.; Potgieter, M. The Healthy Human Blood Microbiome: Fact or Fiction? Front. Cell. Infect. Microbiol. 2019, 9, 148. [Google Scholar] [CrossRef] [Green Version]
- Kajihara, M.; Koido, S.; Kanai, T.; Ito, Z.; Matsumoto, Y.; Takakura, K.; Saruta, M.; Kato, K.; Odamaki, T.; Xiao, J.Z.; et al. Characterisation of blood microbiota in patients with liver cirrhosis. Eur. J. Gastroenterol. Hepatol. 2019, 31, 1577–1583. [Google Scholar] [CrossRef]
- Qiu, J.; Zhou, H.; Jing, Y.; Dong, C. Association between blood microbiome and type 2 diabetes mellitus: A nested case-control study. J. Clin. Lab. Anal. 2019, 33, e22842. [Google Scholar] [CrossRef] [Green Version]
- Gosiewski, T.; Ludwig-Galezowska, A.H.; Huminska, K.; Sroka-Oleksiak, A.; Radkowski, P.; Salamon, D.; Wojciechowicz, J.; Kus-Slowinska, M.; Bulanda, M.; Wolkow, P.P. Comprehensive detection and identification of bacterial DNA in the blood of patients with sepsis and healthy volunteers using next-generation sequencing method-the observation of DNAemia. Eur. J. Clin. Microbiol. Infect. Dis. 2017, 36, 329–336. [Google Scholar] [CrossRef] [Green Version]
- Whittle, E.; Leonard, M.O.; Harrison, R.; Gant, T.W.; Tonge, D.P. Multi-Method Characterization of the Human Circulating Microbiome. Front. Microbiol. 2018, 9, 3266. [Google Scholar] [CrossRef] [Green Version]
- Emery, D.C.; Cerajewska, T.L.; Seong, J.; Davies, M.; Paterson, A.; Allen-Birt, S.J.; West, N.X. Comparison of Blood Bacterial Communities in Periodontal Health and Periodontal Disease. Front. Cell. Infect. Microbiol. 2020, 10, 577485. [Google Scholar] [CrossRef]
- Amar, J.; Serino, M.; Lange, C.; Chabo, C.; Iacovoni, J.; Mondot, S.; Lepage, P.; Klopp, C.; Mariette, J.; Bouchez, O.; et al. Involvement of tissue bacteria in the onset of diabetes in humans: Evidence for a concept. Diabetologia 2011, 54, 3055–3061. [Google Scholar] [CrossRef] [Green Version]
- Olde Loohuis, L.M.; Mangul, S.; Ori, A.P.S.; Jospin, G.; Koslicki, D.; Yang, H.T.; Wu, T.; Boks, M.P.; Lomen-Hoerth, C.; Wiedau-Pazos, M.; et al. Transcriptome analysis in whole blood reveals increased microbial diversity in schizophrenia. Transl. Psychiatry 2018, 8, 96. [Google Scholar] [CrossRef] [Green Version]
- Rajendhran, J.; Shankar, M.; Dinakaran, V.; Rathinavel, A.; Gunasekaran, P. Contrasting circulating microbiome in cardiovascular disease patients and healthy individuals. Int. J. Cardiol. 2013, 168, 5118–5120. [Google Scholar] [CrossRef]
- Li, Q.; Wang, C.; Tang, C.; Zhao, X.; He, Q.; Li, J. Identification and Characterization of Blood and Neutrophil-Associated Microbiomes in Patients with Severe Acute Pancreatitis Using Next-Generation Sequencing. Front. Cell. Infect. Microbiol. 2018, 8, 5. [Google Scholar] [CrossRef] [Green Version]
- Lelouvier, B.; Servant, F.; Paisse, S.; Brunet, A.C.; Benyahya, S.; Serino, M.; Valle, C.; Ortiz, M.R.; Puig, J.; Courtney, M.; et al. Changes in blood microbiota profiles associated with liver fibrosis in obese patients: A pilot analysis. Hepatology 2016, 64, 2015–2027. [Google Scholar] [CrossRef]
- Wang, C.; Li, Q.; Tang, C.; Zhao, X.; He, Q.; Tang, X.; Ren, J. Characterization of the blood and neutrophil-specific microbiomes and exploration of potential bacterial biomarkers for sepsis in surgical patients. Immun. Inflamm. Dis. 2021, 9, 1343–1357. [Google Scholar] [CrossRef]
- Paisse, S.; Valle, C.; Servant, F.; Courtney, M.; Burcelin, R.; Amar, J.; Lelouvier, B. Comprehensive description of blood microbiome from healthy donors assessed by 16S targeted metagenomic sequencing. Transfusion 2016, 56, 1138–1147. [Google Scholar] [CrossRef]
- Suppli, M.P.; Bagger, J.I.; Lelouvier, B.; Broha, A.; Demant, M.; Konig, M.J.; Strandberg, C.; Lund, A.; Vilsboll, T.; Knop, F.K. Hepatic microbiome in healthy lean and obese humans. JHEP Rep. 2021, 3, 100299. [Google Scholar] [CrossRef]
- Shah, N.B.; Allegretti, A.S.; Nigwekar, S.U.; Kalim, S.; Zhao, S.; Lelouvier, B.; Servant, F.; Serena, G.; Thadhani, R.I.; Raj, D.S.; et al. Blood Microbiome Profile in CKD: A Pilot Study. Clin. J. Am. Soc. Nephrol. 2019, 14, 692–701. [Google Scholar] [CrossRef]
- Mo, X.B.; Dong, C.Y.; He, P.; Wu, L.F.; Lu, X.; Zhang, Y.H.; Deng, H.W.; Deng, F.Y.; Lei, S.F. Alteration of circulating microbiome and its associated regulation role in rheumatoid arthritis: Evidence from integration of multiomics data. Clin. Transl. Med. 2020, 10, e229. [Google Scholar] [CrossRef] [PubMed]
- Han, D.S.C.; Lo, Y.M.D. The Nexus of cfDNA and Nuclease Biology. Trends Genet. 2021, 37, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Rai, A.; Chen, M.; Suwakulsiri, W.; Greening, D.W.; Simpson, R.J. Extracellular vesicles in cancer-implications for future improvements in cancer care. Nat. Rev. Clin. Oncol. 2018, 15, 617–638. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Choi, J.P.; Yang, J.; Won, H.K.; Park, C.S.; Song, W.J.; Kwon, H.S.; Kim, T.B.; Kim, Y.K.; Park, H.S.; et al. Metagenome analysis using serum extracellular vesicles identified distinct microbiota in asthmatics. Sci. Rep. 2020, 10, 15125. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Woo, H.G.; Jeong, J.H.; Kim, G.H.; Park, K.D.; Song, T.J. Microbiota dysbiosis and functional outcome in acute ischemic stroke patients. Sci. Rep. 2021, 11, 10977. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.J.; Zhang, J.; Tsai, Y.L.; Chen, C.B.; Lu, C.W.; Huo, Y.P.; Liou, H.M.; Ji, C.; Chung, W.H. Compositional Features of Distinct Microbiota Base on Serum Extracellular Vesicle Metagenomics Analysis in Moderate to Severe Psoriasis Patients. Cells 2021, 10, 2349. [Google Scholar] [CrossRef] [PubMed]
- Jones, E.; Stentz, R.; Telatin, A.; Savva, G.M.; Booth, C.; Baker, D.; Rudder, S.; Knight, S.C.; Noble, A.; Carding, S.R. The Origin of Plasma-Derived Bacterial Extracellular Vesicles in Healthy Individuals and Patients with Inflammatory Bowel Disease: A Pilot Study. Genes 2021, 12, 1636. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Lee, H.K.; Min, S.K.; Lee, W.H. 16S rDNA microbiome composition pattern analysis as a diagnostic biomarker for biliary tract cancer. World J. Surg. Oncol. 2020, 18, 19. [Google Scholar] [CrossRef] [Green Version]
- Byndloss, M.X.; Baumler, A.J. The germ-organ theory of non-communicable diseases. Nat. Rev. Microbiol. 2018, 16, 103–110. [Google Scholar] [CrossRef]
- Libertucci, J.; Young, V.B. The role of the microbiota in infectious diseases. Nat. Microbiol. 2019, 4, 35–45. [Google Scholar] [CrossRef]
- Dabke, K.; Hendrick, G.; Devkota, S. The gut microbiome and metabolic syndrome. J. Clin. Investig. 2019, 129, 4050–4057. [Google Scholar] [CrossRef]
- Puri, P.; Liangpunsakul, S.; Christensen, J.E.; Shah, V.H.; Kamath, P.S.; Gores, G.J.; Walker, S.; Comerford, M.; Katz, B.; Borst, A.; et al. The circulating microbiome signature and inferred functional metagenomics in alcoholic hepatitis. Hepatology 2018, 67, 1284–1302. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Zheng, H.; Tan, K.; Sun, X.; Ye, J.; Zhang, Y. Circulating metabolomics profiling reveals novel pathways associated with cognitive decline in patients with hypertension. Ther. Adv. Neurol. Disord. 2020, 13, 1756286420947973. [Google Scholar] [CrossRef]
- Buford, T.W.; Carter, C.S.; VanDerPol, W.J.; Chen, D.; Lefkowitz, E.J.; Eipers, P.; Morrow, C.D.; Bamman, M.M. Composition and richness of the serum microbiome differ by age and link to systemic inflammation. Geroscience 2018, 40, 257–268. [Google Scholar] [CrossRef] [Green Version]
- Cho, E.J.; Leem, S.; Kim, S.A.; Yang, J.; Lee, Y.B.; Kim, S.S.; Cheong, J.Y.; Cho, S.W.; Kim, J.W.; Kim, S.M.; et al. Circulating Microbiota-Based Metagenomic Signature for Detection of Hepatocellular Carcinoma. Sci. Rep. 2019, 9, 7536. [Google Scholar] [CrossRef]
- Dong, Z.; Chen, B.; Pan, H.; Wang, D.; Liu, M.; Yang, Y.; Zou, M.; Yang, J.; Xiao, K.; Zhao, R.; et al. Detection of Microbial 16S rRNA Gene in the Serum of Patients With Gastric Cancer. Front. Oncol. 2019, 9, 608. [Google Scholar] [CrossRef]
- Hammad, D.B.M.; Hider, S.L.; Liyanapathirana, V.C.; Tonge, D.P. Molecular Characterization of Circulating Microbiome Signatures in Rheumatoid Arthritis. Front. Cell. Infect. Microbiol. 2019, 9, 440. [Google Scholar] [CrossRef]
- Desbois, A.C.; Ciocan, D.; Saadoun, D.; Perlemuter, G.; Cacoub, P. Specific microbiome profile in Takayasu’s arteritis and giant cell arteritis. Sci. Rep. 2021, 11, 5926. [Google Scholar] [CrossRef]
- Ancona, G.; Merlini, E.; Tincati, C.; Barassi, A.; Calcagno, A.; Augello, M.; Bono, V.; Bai, F.; Cannizzo, E.S.; d’Arminio Monforte, A.; et al. Long-Term Suppressive cART Is Not Sufficient to Restore Intestinal Permeability and Gut Microbiota Compositional Changes. Front. Immunol. 2021, 12, 639291. [Google Scholar] [CrossRef]
- Ogunrinde, E.; Zhou, Z.; Luo, Z.; Alekseyenko, A.; Li, Q.Z.; Macedo, D.; Kamen, D.L.; Oates, J.C.; Gilkeson, G.S.; Jiang, W. A Link Between Plasma Microbial Translocation, Microbiome, and Autoantibody Development in First-Degree Relatives of Systemic Lupus Erythematosus Patients. Arthritis Rheumatol. 2019, 71, 1858–1868. [Google Scholar] [CrossRef]
- Luo, Z.; Li, M.; Wu, Y.; Meng, Z.; Martin, L.; Zhang, L.; Ogunrinde, E.; Zhou, Z.; Qin, S.; Wan, Z.; et al. Systemic translocation of Staphylococcus drives autoantibody production in HIV disease. Microbiome 2019, 7, 25. [Google Scholar] [CrossRef]
- Jing, Y.; Zhou, H.; Lu, H.; Chen, X.; Zhou, L.; Zhang, J.; Wu, J.; Dong, C. Associations between peripheral blood microbiome and the risk of hypertension. Am. J. Hypertens. 2021, 34, 1064–1070. [Google Scholar] [CrossRef]
- Luo, Z.; Alekseyenko, A.V.; Ogunrinde, E.; Li, M.; Li, Q.Z.; Huang, L.; Tsao, B.P.; Kamen, D.L.; Oates, J.C.; Li, Z.; et al. Rigorous Plasma Microbiome Analysis Method Enables Disease Association Discovery in Clinic. Front. Microbiol. 2020, 11, 613268. [Google Scholar] [CrossRef]
- Somsouk, M.; Estes, J.D.; Deleage, C.; Dunham, R.M.; Albright, R.; Inadomi, J.M.; Martin, J.N.; Deeks, S.G.; McCune, J.M.; Hunt, P.W. Gut epithelial barrier and systemic inflammation during chronic HIV infection. AIDS 2015, 29, 43–51. [Google Scholar] [CrossRef] [Green Version]
- Marsland, B.J.; Trompette, A.; Gollwitzer, E.S. The Gut-Lung Axis in Respiratory Disease. Ann. Am. Thorac. Soc. 2015, 12 (Suppl. S2), S150–S156. [Google Scholar] [CrossRef]
- Sze, M.A.; Tsuruta, M.; Yang, S.W.; Oh, Y.; Man, S.F.; Hogg, J.C.; Sin, D.D. Changes in the bacterial microbiota in gut, blood, and lungs following acute LPS instillation into mice lungs. PLoS ONE 2014, 9, e111228. [Google Scholar] [CrossRef] [Green Version]
- Pammi, M.; Thapa, S.; Balderas, M.; Runge, J.K.; Venkatachalam, A.; Luna, R.A. Microbiome signatures in neonatal central line associated bloodstream infections. PLoS ONE 2020, 15, e0227967. [Google Scholar] [CrossRef] [Green Version]
- Subramaniam, A.; Van Der Pol, W.J.; Ptacek, T.; Lobashevsky, E.; Neely, C.; Biggio, J.R., Jr.; Lefkowitz, E.J.; Morrow, C.D.; Edwards, R.K. Midtrimester microbial DNA variations in maternal serum of women who experience spontaneous preterm birth. J. Matern. Fetal. Neonatal Med. 2020, 33, 359–367. [Google Scholar] [CrossRef]
- You, Y.A.; Yoo, J.Y.; Kwon, E.J.; Kim, Y.J. Blood Microbial Communities During Pregnancy Are Associated With Preterm Birth. Front. Microbiol. 2019, 10, 1122. [Google Scholar] [CrossRef]
- Edwards, S.M.; Cunningham, S.A.; Dunlop, A.L.; Corwin, E.J. The Maternal Gut Microbiome During Pregnancy. MCN Am. J. Matern. Child Nurs. 2017, 42, 310–317. [Google Scholar] [CrossRef]
- Bankoski, A.; Harris, T.B.; McClain, J.J.; Brychta, R.J.; Caserotti, P.; Chen, K.Y.; Berrigan, D.; Troiano, R.P.; Koster, A. Sedentary activity associated with metabolic syndrome independent of physical activity. Diabetes Care 2011, 34, 497–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saad, M.J.; Santos, A.; Prada, P.O. Linking Gut Microbiota and Inflammation to Obesity and Insulin Resistance. Physiology 2016, 31, 283–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Aquila, P.; Giacconi, R.; Malavolta, M.; Piacenza, F.; Burkle, A.; Villanueva, M.M.; Dolle, M.E.T.; Jansen, E.; Grune, T.; Gonos, E.S.; et al. Microbiome in Blood Samples From the General Population Recruited in the MARK-AGE Project: A Pilot Study. Front. Microbiol. 2021, 12, 707515. [Google Scholar] [CrossRef] [PubMed]
- Anhe, F.F.; Jensen, B.A.H.; Varin, T.V.; Servant, F.; Van Blerk, S.; Richard, D.; Marceau, S.; Surette, M.; Biertho, L.; Lelouvier, B.; et al. Type 2 diabetes influences bacterial tissue compartmentalisation in human obesity. Nat. Metab. 2020, 2, 233–242. [Google Scholar] [CrossRef] [Green Version]
- Massier, L.; Chakaroun, R.; Tabei, S.; Crane, A.; Didt, K.D.; Fallmann, J.; von Bergen, M.; Haange, S.B.; Heyne, H.; Stumvoll, M.; et al. Adipose tissue derived bacteria are associated with inflammation in obesity and type 2 diabetes. Gut 2020, 69, 1796–1806. [Google Scholar] [CrossRef]
- Crovesy, L.; Masterson, D.; Rosado, E.L. Profile of the gut microbiota of adults with obesity: A systematic review. Eur. J. Clin. Nutr. 2020, 74, 1251–1262. [Google Scholar] [CrossRef]
- Amar, J.; Lelouvier, B.; Servant, F.; Lluch, J.; Burcelin, R.; Bongard, V.; Elbaz, M. Blood Microbiota Modification After Myocardial Infarction Depends Upon Low-Density Lipoprotein Cholesterol Levels. J. Am. Heart Assoc. 2019, 8, e011797. [Google Scholar] [CrossRef]
- Koliarakis, I.; Athanasakis, E.; Sgantzos, M.; Mariolis-Sapsakos, T.; Xynos, E.; Chrysos, E.; Souglakos, J.; Tsiaoussis, J. Intestinal Microbiota in Colorectal Cancer Surgery. Cancers 2020, 12, 3011. [Google Scholar] [CrossRef]
- Soby, J.H.; Watt, S.K.; Vogelsang, R.P.; Servant, F.; Lelouvier, B.; Raskov, H.; Knop, F.K.; Gogenur, I. Alterations in blood microbiota after colonic cancer surgery. BJS Open 2020, 4, 1227–1237. [Google Scholar] [CrossRef]
- Yang, D.; Wang, X.; Zhou, X.; Zhao, J.; Yang, H.; Wang, S.; Morse, M.A.; Wu, J.; Yuan, Y.; Li, S.; et al. Blood microbiota diversity determines response of advanced colorectal cancer to chemotherapy combined with adoptive T cell immunotherapy. Oncoimmunology 2021, 10, 1976953. [Google Scholar] [CrossRef]
- Nishida, A.; Inoue, R.; Inatomi, O.; Bamba, S.; Naito, Y.; Andoh, A. Gut microbiota in the pathogenesis of inflammatory bowel disease. Clin. J. Gastroenterol. 2018, 11, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Yan, Q.; Luo, F.; Shang, D.; Wu, D.; Zhang, H.; Shang, X.; Kang, X.; Abdo, M.; Liu, B.; et al. Acute cholecystitis associated with infection of Enterobacteriaceae from gut microbiota. Clin. Microbiol. Infect. 2015, 21, e851–e859. [Google Scholar] [CrossRef] [Green Version]
- Pagliari, D.; Saviano, A.; Newton, E.E.; Serricchio, M.L.; Dal Lago, A.A.; Gasbarrini, A.; Cianci, R. Gut Microbiota-Immune System Crosstalk and Pancreatic Disorders. Mediat. Inflamm. 2018, 2018, 7946431. [Google Scholar] [CrossRef] [Green Version]
- Kummen, M.; Hov, J.R. The gut microbial influence on cholestatic liver disease. Liver Int. 2019, 39, 1186–1196. [Google Scholar] [CrossRef] [Green Version]
- Yun, Y.; Kim, H.N.; Lee, E.J.; Ryu, S.; Chang, Y.; Shin, H.; Kim, H.L.; Kim, T.H.; Yoo, K.; Kim, H.Y. Fecal and blood microbiota profiles and presence of nonalcoholic fatty liver disease in obese versus lean subjects. PLoS ONE 2019, 14, e0213692. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zhao, R.; Shi, D.; Sun, S.; Ren, H.; Zhao, H.; Wu, W.; Jin, L.; Sheng, J.; Shi, Y. Characterization of the circulating microbiome in acute-on-chronic liver failure associated with hepatitis B. Liver Int. 2019, 39, 1207–1216. [Google Scholar] [CrossRef]
- Zhu, S.; Jiang, Y.; Xu, K.; Cui, M.; Ye, W.; Zhao, G.; Jin, L.; Chen, X. The progress of gut microbiome research related to brain disorders. J. Neuroinflamm. 2020, 17, 25. [Google Scholar] [CrossRef] [Green Version]
- Ciocan, D.; Cassard, A.M.; Becquemont, L.; Verstuyft, C.; Voican, C.S.; El Asmar, K.; Colle, R.; David, D.; Trabado, S.; Feve, B.; et al. Blood microbiota and metabolomic signature of major depression before and after antidepressant treatment: A prospective case-control study. J. Psychiatry Neurosci. 2021, 46, E358–E368. [Google Scholar] [CrossRef]
- Qian, Y.; Yang, X.; Xu, S.; Wu, C.; Qin, N.; Chen, S.D.; Xiao, Q. Detection of Microbial 16S rRNA Gene in the Blood of Patients With Parkinson’s Disease. Front. Aging Neurosci. 2018, 10, 156. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Huang, P.; Qian, Y.; Yang, X.; Cui, S.; Lin, Y.; Gao, C.; Zhang, P.; He, Y.; Xiao, Q.; et al. Fecal and Blood Microbial 16s rRNA Gene Alterations in Chinese Patients with Multiple System Atrophy and Its Subtypes. J. Park. Dis. 2019, 9, 711–721. [Google Scholar] [CrossRef] [Green Version]
- Yun, Y.; Kim, H.N.; Chang, Y.; Lee, Y.; Ryu, S.; Shin, H.; Kim, W.S.; Kim, H.L.; Nam, J.H. Characterization of the Blood Microbiota in Korean Females with Rosacea. Dermatology 2019, 235, 255–259. [Google Scholar] [CrossRef]
- Whittle, E.; Leonard, M.O.; Gant, T.W.; Tonge, D.P. Multi-Method Molecular Characterisation of Human Dust-Mite-associated Allergic Asthma. Sci. Rep. 2019, 9, 8912. [Google Scholar] [CrossRef] [Green Version]
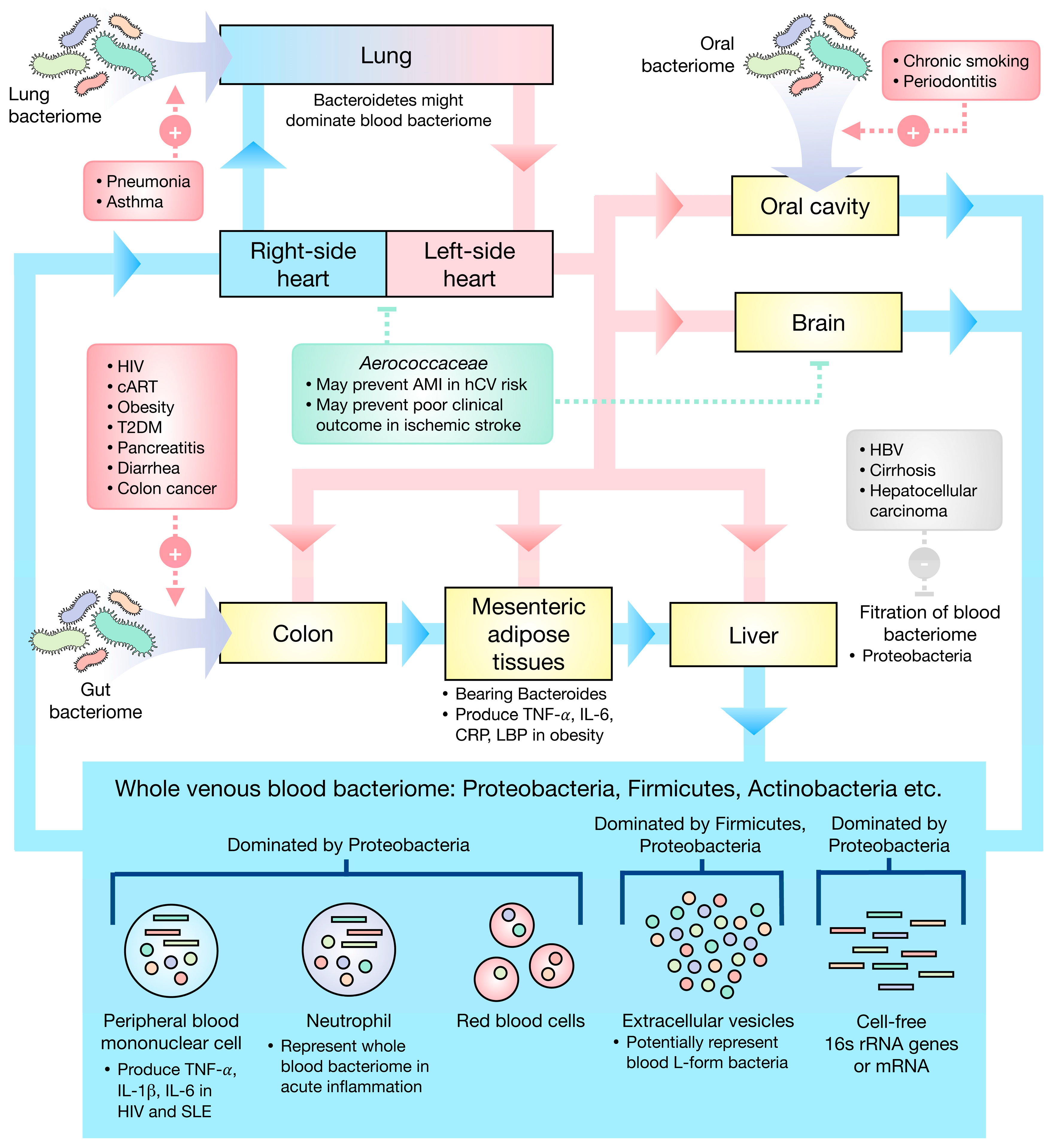

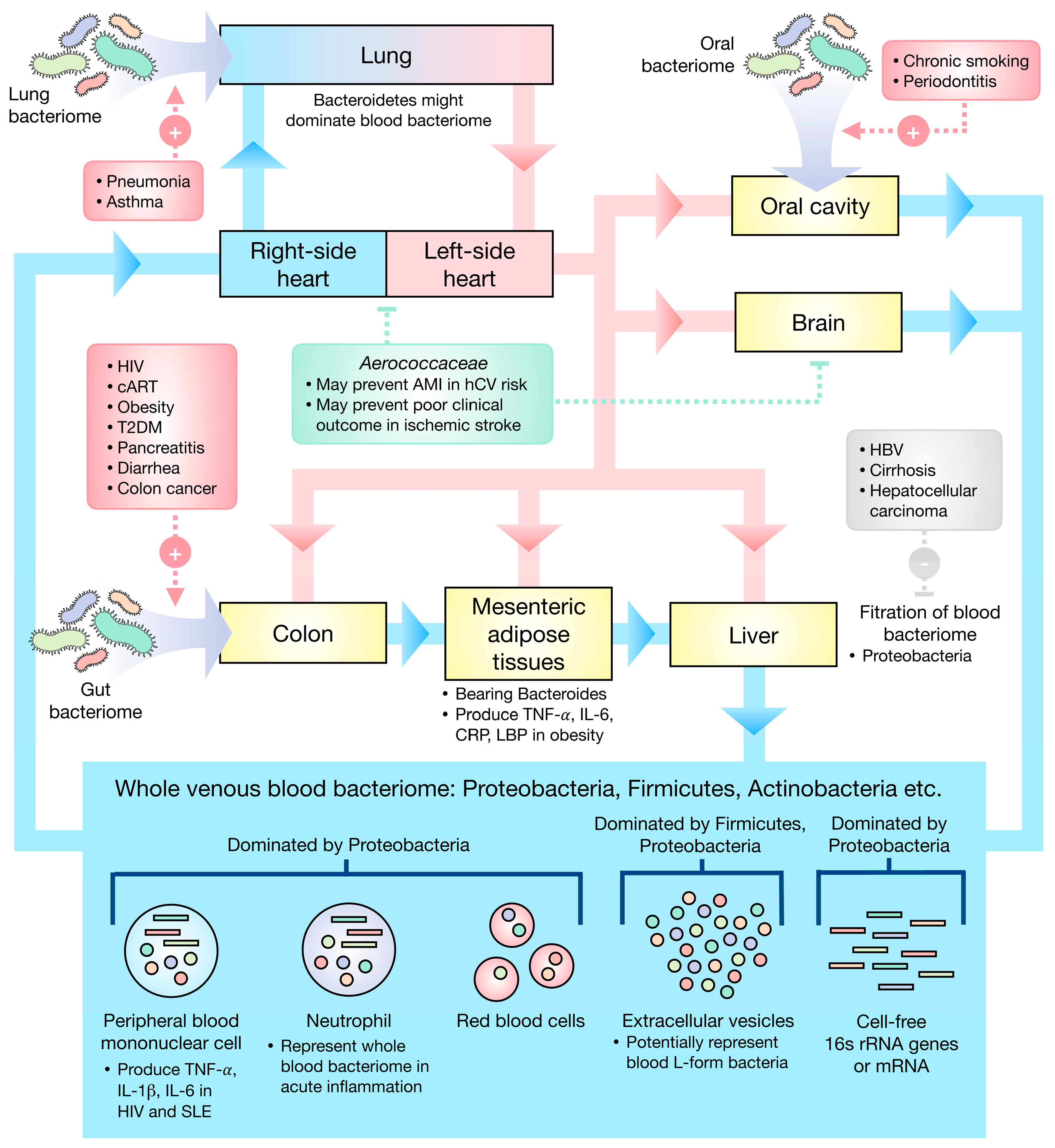{kind=link}
| Blood Specimen | Subjects n (M/F) | Age # | Country | Hypervariable Region (V) | Taxonomic Database | Order of Relative Abundance at Phylum Level | Ref. | |||
|---|---|---|---|---|---|---|---|---|---|---|
| First | Second | Third | Other | |||||||
| DNA | ||||||||||
| Whole blood | 10 (9/1) | 29.2 ± 11.26 | India | V3 | Greengenes | Proteobacteria | Firmicutes | Actinobacteria | NA | [23] |
| 12 (10/2) | 29.2 ± 3.8 | China | V3 | RDP | Proteobacteria | Actinobacteria | Firmicutes | Bacteroidetes | [24] | |
| 3 (2/1) | 38.33 ± 20.98 | UK | V3–V4 | SILVA | Proteobacteria | Actinobacteria | Firmicutes | Bacteroidetes, Fusobacteria | [21] | |
| 60 (18/42) | 39.8 ± 9.5 | Italian | V3–V4 | NCBI | Proteobacteria | Actinobacteria | Firmicutes | Bacteroidetes | [25] | |
| 19 (4/15) | 39.89 ± 13.69 | UK | V3–V4 | SILVA | Firmicutes | Proteobacteria | Actinobacteria | Bacteroidetes, Fusobacteria | [21] † | |
| 20 (5/15) | 41.9 ± 10.7 | USA | V3–V4 | Greengenes | Firmicutes | Proteobacteria | Actinobacteria | Bacteroidetes | [26] | |
| 28 (14/14) | 45 ± 12 | Bulgaria | V3–V4 | Greengenes | Proteobacteria | Firmicutes | Actinobacteria | Planctomycetes | [14] † | |
| 28 (14/14) | 45 ± 12 | Bulgaria | V3–V4 | Greengenes | Proteobacteria | Firmicutes | Actinobacteria | Bacteroidetes, Cyanobacteria | [14] †,‡ | |
| 23 (10/13) | 59 | Poland | V3–V4 | RDP, Greengenes | Actinobacteria | Proteobacteria | Firmicutes | Bacteroidetes, Cyanobacteria | [19] | |
| 28 (NA) | NA | China | V3, V4, V3–V4, V4–V5 | Greengenes | Firmicutes | Bacteroidetes | Proteobacteria | Actinobacteria, Cyanobacteria | [27] | |
| 5 (NA) | NA | China | V3 | RDP | Proteobacteria | Actinobacteria | Firmicutes | Bacteroidetes | [28] | |
| 28 (NA) | NA | Bulgaria | V3–V4 | SILVA | Proteobacteria | Firmicutes | Actinobacteria | Planctomycetes, Armatimonadetes | [13] † | |
| 28 (NA) | NA | Bulgaria | V3–V4 | SILVA | Proteobacteria | Firmicutes | Actinobacteria | Bacteroidetes, Fusobacteria | [13] †,‡ | |
| Buffy coat | 30 (9/21) | 21 (18–53) | France | V3–V4 | NCBI | Proteobacteria | Actinobacteria | Firmicutes | Bacteroidetes | [29] |
| 15 (15/0) | 40 (25–68) | Denmark | V3–V4 | NCBI, SILVA | Proteobacteria | Actinobacteria | Firmicutes | Acidobacteria, Bacteroidetes | [30] | |
| 20 (7/13) | 44 (39–53) | USA | V3–V4 | SILVA | Proteobacteria | Bacteroidetes | Actinobacteria | Firmicutes | [31] | |
| 26 (5/21) | 46.2 ± 8.9 | Spain | V3–V4 | NCBI | Proteobacteria | Actinobacteria | Firmicutes | Bacteroidetes | [25] | |
| 28 (NA) | 47 ± 10 | France | V1–V2 | SILVA | Proteobacteria | Bacteroidetes | Actinobacteria | Firmicutes, Acidobacteria | [22] | |
| Neutrophil | 12 (10/2) | 29.2 ± 3.8 | China | V3 | RDP | Proteobacteria | Actinobacteria | Firmicutes | Bacteroidetes | [24] |
| 5 (NA) | NA | China | V3 | RDP | Proteobacteria | Actinobacteria | Firmicutes | Bacteroidetes | [28] | |
| PBMC | 14 (0/15) | 50.48 ± 14.05 | China | V3–V4 | SILVA | Proteobacteria | Actinobacteria | Bacteroidetes | Deinococcus–Thermus, Firmicutes | [32] |
| Red blood cell | 30 (9/21) | 21 (18–53) | France | V3–V4 | NCBI | Proteobacteria | Actinobacteria | Firmicutes | Bacteroidetes, Fusobacteria | [29] |
| Serum | 24 (10/14) | 27.8 ± 4.0 | USA | V4 | RDP | Firmicutes | Bacteroidetes | Proteobacteria | Fusobacteria, Actinobacteria | [33] |
| 201 (119/82) | 57.6 ± 10.4 | Korea | V3–V4 | Greengenes | Firmicutes | Proteobacteria | Actinobacteria | Bacteroidetes, Verrucomicrobia | [34] | |
| 24 (10/14) | 63.9 ± 3.2 | USA | V4 | RDP | Firmicutes | Bacteroidetes | Proteobacteria | Actinobacteria, Fusobacteria | [33] | |
| 13 (NA) | NA | China | V1–V2 | RDP, Greengenes | Proteobacteria | Actinobacteria | Firmicutes | Deinococcus–Thermus, Bacteroidetes | [35] | |
| 4 (NA) | NA | UK | V4 | SILVA | Proteobacteria | Firmicutes | Bacteroidetes | Actinobacteria, Fusobacteria | [36] | |
| 15 (NA) | NA | France | V3–V4 | Greengenes | Proteobacteria | Bacteroidetes | Actinobacteria | Firmicutes, Gemmatimonadetes | [37] | |
| Plasma | 30 (9/21) | 21 (18–53) | France | V3–V4 | NCBI | Proteobacteria | Actinobacteria | Firmicutes | Bacteroidetes | [29] |
| 3 (2/1) | 27 ± 3.46 | India | Shotgun | MG-RAST/ SEED | Proteobacteria | Actinobacteria | Firmicutes | NA | [15] | |
| 15 (15/0) | 29 (24–33) | Italy | V3–V4 | NCBI, SILVA | Proteobacteria | Actinobacteria | Firmicutes | Bacteroidetes | [38] | |
| 19 (0/19) | 34.2 ± 9.4 | USA | V4 | Greengenes | Proteobacteria | Fusobacteria | Actinobacteria | Firmicutes, Bacteroidetes | [39] | |
| 16 (5/11) | 38 (33–55) | USA | V4 | NCBI, RDP | Proteobacteria | Firmicutes | Actinobacteria | Bacteroidetes | [40] | |
| 18 (3/15) | 38.6 ± 12.4 | USA | V4 | Greengenes | Proteobacteria | Actinobacteria | Firmicutes | Bacteroidetes, Cyanobacteria | [39] | |
| 5 (0/5) | 39.4 ± 10.3 | UK | V4 | SILVA | Proteobacteria | Actinobacteria | Firmicutes | Bacteroidetes | [20] | |
| 150 (66/84) | 48.13 ± 6.22 | China | V6–V7 | NA | Proteobacteria | Firmicutes | Actinobacteria | Bacteroidetes | [41] | |
| 100 (64/36) | 51.98 ± 8.05 | China | V5–V6 | NA | Proteobacteria | Bacteroidetes | Firmicutes | Actinobacteria | [18] | |
| EVs | 8 (5/3) | 49.63 ± 15.16 | Taiwan | V1–V9 | NCBI | Proteobacteria | Firmicutes | Actinobacteria | Bacteroidetes, Fusobacteria | [42] |
| 88 (37/51) | 54.4 ± 12.8 | Korea | V3–V4 | Greengenes | Proteobacteria | Firmicutes | Actinobacteria | Bacteroidetes, Cyanobacteria | [43] | |
| 260 (105/155) | 56 | Korea | V3–V4 | SILVA | Firmicutes | Proteobacteria | Actinobacteria | Bacteroidetes, Verrucomicrobia | [44] | |
| 200 (117/83) | 63.5 ± 12.5 | Korea | V3–V4 | Greengenes | Firmicutes | Bacteroidetes | Proteobacteria | Verrucomicrobia, Actinobacteria | [45] | |
| 5 (NA) | NA | UK | V3–V4 | SILVA | Proteobacteria | Firmicutes | Actinobacteria | Bacteroidetes, Fusobacteria | [46] † | |
| RNA | ||||||||||
| Whole blood | 14 (12/1) | 37.4 ± 10 | Japan | V3–V4 | Greengenes | Firmicutes | Bacteroidetes | Fusobacteria | Proteobacteria, Actinobacteria | [17] |
| 49 (38/11) | 41.1 ± 10.7 | USA | RNA-Seq | PhyloSift | Proteobacteria | Firmicutes | Cyanobacteria | Bacteroidetes, Thermotogae | [47] | |
| Plasma | 5 (0/5) | 39.4 ± 10.3 | UK | RNA-Seq | Kraken/ NCBI | Proteobacteria | Firmicutes | Bacteroidetes | Actinobacteria | [20] |
| Subjects (n; Mean Age) | Samples | Dysbiotic Blood Bacteriome of Patients vs. Controls | Other | Interpretation | Ref. | |||
|---|---|---|---|---|---|---|---|---|
| Diversity | Differential Abundance | |||||||
| α-R | α-E | β | ||||||
| HIV Infection | ||||||||
The Italian study (56):
| Plasma DNA | ↑ | ↑ | NA | Family: ↑Prevotellaceae, ↑Lactobacillaceae, ↓Ruminococcaceae, and ↓Bacteroidaceae | NA | Blood dysbiosis in HIV infection might be characterized by an increase in Prevotellaceae and Lactobacillaceae but a decrease in Ruminococcaceae and Bacteroidaceae | [38] |
The Italian study (41; age = 42 (31.5–50.5)):
| Plasma DNA | ↕ | ↕ | NS | Family: ↑Staphylococcaceae, ↑Sphingomonadaceae, and ↓Pseudomonadaceae | NA | cART could modify blood bacteriome with an increase in Staphylococcaceae and Sphingomonadaceae but a decrease in Pseudomonadaceae | [38] |
| Subgroup Analysis | ||||||||
| NNRTI vs. PI and INI | ||||||||
| NA | NA | NA | Family: ↑Veillonellaceae, ↓Coriobacteriaceae, and ↓Peptococcaceae | After treated with NNRTI
| HIV infection treated with either NNRTI or PI may lead to an increase in disruption of the gut epithelial barrier, and NNRTI could distinctly modify blood bacteriome by an increase in Veillonellaceae but a decrease in Coriobacteriaceae and Peptococcaceae compared with PI and INI | |||
The American study (91):
| Plasma DNA | NA | NA | S | Genus: ↑Massilia, ↑Haemophilus, ↑Veillonella, ↑Arthrobacter, ↑Fusobacterium, ↓Altererythrobacter, ↓Cryobacterium, and ↓Anaerococcus | Validated by in vitro study
| An increase in Massilia and Haemophilus in blood bacteriome of HIV infection could lead to chronic systemic inflammation | [53] |
The American study (42):
| Plasma DNA (before vaccinated) | NA | ↕ | NS | Phylum: ↑Proteobacteria, ↓Actinobacteria, and ↓Firmicutes Genus: ↑Pseudomonas |
| Blood dysbiosis in HIV infection could initiate production of autoantibody, which may be characterized by an increase in Proteobacteria, Pseudomonas in particular, and Actinobacteria but a decrease in Firmicutes | [40] |
| Subgroup Analysis | ||||||||
| HIV with high anti-nuclear antibody vs. HIV with low anti-nuclear antibody after vaccinated | ||||||||
| NA | ↕ | NS | Phylum: ↑Firmicutes Genus: ↑Staphylococcus Species: ↑Staphylococcus epidermidis and ↑Staphylococcus haemolyticus | Validated by in vivo study
| An increased proportion of Staphylococcus in blood bacteriome in HIV infection may be involved in the pathophysiology of the autoantibody production after receiving influenza vaccine | |||
| Sepsis | ||||||||
The Polish study (85):
| Whole blood DNA | ↑ | NA | S | Phylum: ↑Proteobacteria and ↓Actinobacteria Order: ↑Rhizobiales, ↑Aeromonadales, ↑Sphingomonadales, ↑Actinomycetales and ↓Bifidobacteriales | NA | Blood dysbiosis in sepsis might be characterized by an increase in Proteobacteria but a decrease in Actinobacteria, Bifidobacteriales in particular | [19] |
The Chinese study (51):
| Whole blood DNA | ↓ | NA | S | NA |
| Blood dysbiosis in post-operative patients with infection may originate from the gut microbiome, and Agrococcus may play a role in septic progression | [28] |
| Subgroup Analysis | ||||||||
| Septic shock vs. HC | ||||||||
| NA | NA | NA | Phylum: ↑Bacteroidetes and ↓Actinobacteria Class: ↑Flavobacteria, ↑Bacteroidia, ↑Clostridia, ↑Betaproteobacteria, and ↓Gammaproteobacteria Subclass: ↓Actinobacteridae | NA | Blood dysbiosis in septic shock may be characterized by an increase in Bacteroidetes but a decrease in Actinobacteria | |||
The Chinese study (34):
| Neutrophil DNA | ↑ (SS) but ↕ (S) | NA | S | Phylum: ↑Proteobacteria and ↓Actinobacteria Class: ↑Betaproteobacteria, ↑Alphaproteobacteria, and ↓Gammaproteobacteria Subclass: ↓Actinobacteridae |
| Neutrophil bacteriome in post-operative patients with sepsis may originate from the gut microbiome and be characterized by an increase in Proteobacteria but a decrease in Actinobacteria | [28] |
The American study (30):
| Whole blood DNA | ↕ | ↕ | S | CLABSI (n = 3) vs. non-CLABSI (n = 3) Family: ↑Enterobacteriaceae Genus: ↑Proteus and ↑Staphylococcus | Bacteriome of catheter biofilm in CLABSI (n = 15) vs. non-CLABSI (n = 27)
| Blood dysbiosis of CLABSI might be associated with ascending infection from catheter biofilm | [54] |
| Pregnancy with Pre-Term Delivery | ||||||||
The Korean study (41):
| Plasma-separated blood cell DNA(at labor stage) | ↑ | NA | S | Phylum: ↑Firmicutes, ↑Bacteroidetes, and ↓Proteobacteria Family: ↑Ruminococcaceae, ↑Saccharibacteria, and ↑Lachnospiraceae Genus: ↑Bacteroides, ↑Lactobacillus, ↑Sphingomonas, ↑Fastidiosipila, ↑Butyricicoccus, ↑Methanobrevibacter, ↓Delftia, ↓Pseudomonas, ↓Massilia, and ↓Stenotrophomonas | NA | Blood dysbiosis in pregnant women who had pre-term delivery might be characterized by an increase in Firmicutes and Bacteroidetes but a decrease in Proteobacteria | [55] |
The American study (40):
| Serum DNA (at GA 15–20 weeks) | ↑ | ↑ | S | Phylum: ↑Proteobacteria, ↑Actinobacteria, ↓Firmicutes, and ↓Bacteroidetes | NA | Blood dysbiosis in mid-trimester pregnant women who had pre-term delivery might be characterized by an increase in Proteobacteria and Actinobacteria but a decrease in Firmicutes and Bacteroidetes | [56] |
| Subjects (n; Mean Age) | Samples | Dysbiotic Blood Bacteriome Patients vs. Controls | Other | Interpretation | Ref. | |||
|---|---|---|---|---|---|---|---|---|
| Diversity | Differential Abundance | |||||||
| α-R | α-E | β | ||||||
| Type 2 Diabetes Mellitus (T2DM) and Obesity | ||||||||
The French study (42):
| Buffy coat DNA | NA | NA | NA | Phylum: ↑Proteobacteria and ↓Actinobacteria |
| Blood dysbiosis characterized by an increase in Proteobacteria and a decrease in Actinobacteria as well as an upsurge in baseline 16s rRNA gene concentration may be involved in the development of T2DM in healthy subjects | [22] |
The Chinese study (150):
| Plasma DNA | ↕ | ↕ | NA | Order: ↓Rhodospirillales and ↓Myxococcales Genus: ↑Actinotalea, ↑Alishewanella, ↑Sediminibacterium, ↑Pseudoclavibacter, ↓Aquabacterium, ↓Xanthomonas, and ↓Pseudonocardia |
| Blood dysbiosis in T2DM might be characterized by a decrease in Rhodospirillales together with Myxococcales, and Bacteroides might be a protective factor for T2DM, while Sediminibacterium might be a risk factor for T2DM | [18] |
The Canadian study (40):
| Plasma DNA | ↕ | ↕ | NS | Family: ↑Enterobacteriaceae and ↑Neisseriaceae Genus: ↑Escherichia-Shigella and ↑Serratia |
| Blood dysbiosis in morbid obesity with T2DM might be characterized by an increase in Enterobacteriaceae and Neisseriaceae; in addition, liver might filter microbes in blood derived from gut bacterial translocation | [57] |
The German study (75):
| Plasma DNA | ↕ | ↕ | NA | Genus: ↑Tahibacter, ↓Delftia, ↓Lactobacillus, and ↓Lactococcus |
| Blood dysbiosis in T2DM might be characterized by an increase in Tahibacter but a decrease in Delftia, Lactobacillus, and Lactococcus; furthermore, adipose tissues which were exposed to bacteria might initiate chronic systemic inflammation leading to obesity | [58] |
The Danish study (29):
| Buffy coat DNA | ↕ | NA | NA | Order: ↑Propionibactereles, ↑Sphingomonadales, and ↑Norcardioides Family: ↑Comamonodaceae Genus: ↓Enterobacter |
| Blood dysbiosis in obesity might be characterized by an increase in Propionibactereles, Sphingomonadales, and Norcardioides; moreover, liver might filter microbes in blood, especially Proteobacteria, and an increase in 16s rRNA gene concentration in liver might play a role in pathogenesis of fatty liver | [30] |
| Hypertension | ||||||||
The Chinese study (69):
| Whole blood DNA | ↕ | ↕ | S | Genus: ↑Streptococcus, ↑Lactobacillus, ↑Parabacteroides, ↑Helicobacter, ↓Stenotrophomonas, and ↓Turicibacter |
| Blood dysbiosis in hypertension might be characterized by an increase in Streptococcus, Lactobacillus, Parabacteroides, and Helicobacter but a decrease in Stenotrophomonas and Turicibacter; additionally, blood bacteriome might originate from inflammatory state of gut and lung | [27] |
The Chinese study (300):
| Plasma DNA | ↓ | ↕ | NA | Phylum: ↑Proteobacteria, ↓Firmicutes, and ↓Bacteroidetes Genus: ↑Sphingomonas, ↑Acinetobacter, and ↓Staphylococcus |
| Blood dysbiosis in hypertension might be characterized by an increase in Proteobacteria but a decrease in Firmicutes and Bacteroidetes; furthermore, Staphylococcus might be a protective factor for hypertension while either Acinetobacter or Sphingomonas might be a risk factor for hypertension | [41] |
| Cardiac Diseases | ||||||||
The Indian study (41):
| Whole blood DNA | NA | NA | NA | Phylum: ↑Proteobacteria and ↓Firmicutes Family: ↓Staphylococcaceae | NA | Blood dysbiosis in cardiac diseases might be characterized by an increase in Proteobacteria but a decrease in Firmicutes | [23] |
The Indian study (6):
| Whole blood DNA | NA | NA | NA | Phylum: ↑Actinobacteria and ↓Proteobacteria Family: ↑Propionibacteriaceae and ↓Pseudomonadaceae | ↑16s rRNA gene concentration in all patients vs. HC | Blood dysbiosis in cardiac disease might be characterized by an increase in Actinobacteria but a decrease in Proteobacteria as well as an upsurge in 16s rRNA gene concentration | [15] |
The French study (202):
| Whole blood DNA | ↓ | NA | NS | Family: ↓Caulobacteraceae, ↓Norcardiaceae *, and ↓Aerococcaceae * Genus: ↓Gordonia *, ↓Propionibacterium *, and ↓Chryseobacterium * * Cholesterol-degrading microbes |
| Blood dysbiosis in patients with myocardial infarction compared with controls with high cardiovascular risk may be characterized by a decrease in Cholesterol-degrading microbes, including Norcardiaceae and Aerococcaceae, as well as an increase in 16s rRNA gene concentration | [59] |
| Cerebrovascular Accidents | ||||||||
The Korean study (398):
| EVs DNA (at the onset of stroke) | NA | NA | S | Phylum: ↑Proteobacteria and ↓Firmicutes Order: ↓Clostridiales, Family: ↑Aerococcaceae Genus: ↑Flavobacterium, ↓Stenotrophomonas, ↓Lactobacillus, ↓Akkermansia, and ↓Mucispirillum | NA | Blood dysbiosis in acute ischemic stroke might be characterized by an increase in Proteobacteria but a decrease in Firmicutes | [45] |
The Korean study (398):
| EVs DNA (at the onset of stroke) | Subgroup Analysis | [45] | |||||
| Good vs. poor clinical outcomes | ||||||||
| NA | NA | NS | Family: ↑Aerococcaceae, ↑Microbacteriaceae, and ↓Ruminococcaceae Genus: ↑Anaerococcus, ↑Blautia, ↑Dialister, ↑Propionibacterium, ↑Rothia, and ↓Prevotella | NA | An increase in Aerococcaceae together with Microbacteriaceae and a reduction in Ruminococcaceae in blood of patients with acute ischemic stroke might lead to good clinical outcomes | |||
| Chronic Kidney Disease | ||||||||
The American study (40):
| Buffy coat DNA | ↓ | NA | NS | Phylum: ↑Proteobacteria Family: ↑Enterobacteriaceae and ↑Pseudomonadaceae |
| Blood dysbiosis in chronic kidney disease might be characterized by an increase in Proteobacteria, which may play a role in progression of chronic kidney disease | [31] |
| Subjects (n; Mean Age) | Samples | Dysbiotic Blood Bacteriome of Patients vs. Controls | Other | Interpretation | Ref. | |||
|---|---|---|---|---|---|---|---|---|
| Diversity | Differential Abundance | |||||||
| α-R | α-E | β | ||||||
| Oral Diseases | ||||||||
The American study (41):
| Plasma DNA | NA | ↑ | S | Genus: ↑Streptococcus Species: ↑Streptococcus parasanguinis, ↑Streptococcus australis, and ↑Streptococcus oligofermentans | NA | Blood dysbiosis in association with tobacco smoking might be characterized by an increase in Streptococcus | [53] |
The British study (40):
| Whole blood DNA | ↕ | ↕ | S | Phylum: ↓Candidatus Saccharibacteria Order: ↓ Myxococcales |
| Blood dysbiosis in periodontitis might be characterized by a decrease in Candidatus Saccharibacteria; in addition, blood bacteriome might originate from oral bacteriome | [21] |
| Stomach Diseases | ||||||||
The Chinese study (84):
| Serum DNA | NA | ↓ | S | Genus: ↑Haemophilus, ↑Acinetobacter, ↑Bacteroides, ↓Comamonas, ↓Sphingomonas, and ↓Pseudomonas |
| Blood dysbiosis in gastric cancer might be characterized by an increase in Haemophilus, Acinetobacter, Bacteroides, and Comamonas but a decrease in Sphingomonas and Pseudomonas; furthermore, Enterococcus might play a role in progression of gastric cancer | [35] |
| Bowel Diseases | ||||||||
The Danish study with colon cancer (30; age = 67.6 ± 8.8):
| Whole blood DNA | ↓ | NA | NS | Post-operative vs. Pre-operative Phylum: ↑Proteobacteria and ↓Actinobacteria Order: ↑Pseudomonadales and ↑Enterobacteriales |
| Blood dysbiosis in post-operative patients with colon cancer might be characterized by an increase in Proteobacteria but a decrease in Actinobacteria as well as a decline in 16s rRNA gene concentration | [60] |
The Chinese study with colon cancer (19; age = 64 (36–81)):
| Plasma DNA | ↕ | ↕ | NA | Post-treatment vs. Pre-treatment Phylum: ↑Verrucomicrobia |
| CT could modify blood bacteriome in colon cancer as an increase in Verrucomicrobia while 16s rRNA gene concentration was decreased | [61] |
| Subgroup Analysis | ||||||||
| Patients who later became drug responders vs. drug non-responders | ||||||||
| ↕ | ↕ | S | Phylum: ↑Firmicutes and ↑Fusobacteria | NA | An increase in Firmicutes and Fusobacteria in baseline blood bacteriome of colon cancer could predict the responsiveness of CT | |||
The Chinese study with colon cancer (20; age = 60 (36–86)):
| Plasma DNA | ↑ | ↓ | NA | Post-treatment vs. Pre-treatment Phylum: ↑Bacteroidetes |
| CT together with DC-CIK could modify blood bacteriome in colon cancer as an increase in Bacteroidetes while 16s rRNA gene concentration was decreased | [61] |
| Subgroup Analysis | ||||||||
| Patients who later became drug responders vs. drug non-responders | ||||||||
| ↓ | ↕ | NA | Genus: ↑Lactobacillus, ↑Bifidobacterium, ↑Enterococcus, and ↑Pseudomonas |
| An increase in Lactobacillus, Bifidobacterium, and Enterococcus but a decrease in Pseudomonas in baseline blood bacteriome of colon cancer could predict the responsiveness of CT with DC-CIK | |||
The British study (18):
| EVs DNA | ↕ | NA | NS | NA | NA | Blood bacteriome in treated inflammatory bowel diseases might not be different from healthy controls | [46] |
| Pancreatobiliary Diseases | ||||||||
The Chinese study (62):
| Whole blood DNA | ↑ | NA | S | Phylum: ↑Bacteroidetes and ↓Actinobacteria |
| Blood dysbiosis in acute pancreatitis might be characterized by an increase in Bacteroidetes but a decrease in Actinobacteria; moreover, blood bacteriome might originate from gut | [24] |
The Chinese study (62):
| Neutrophil DNA | ↑ | NA | S | Phylum: ↑Bacteroidetes, ↑Firmicutes, ↓Actinobacteria, and ↓Proteobacteria |
| Neutrophil dysbiosis in acute pancreatitis might be characterized by an increase in Bacteroidetes and Firmicutes but a decrease in Actinobacteria and Proteobacteria; additionally, blood bacteriome might originate from gut | [24] |
The Korean study (155):
| EVs DNA | ↕ | NA | S | Class: ↑Clostridia and ↓Gammaproteobacteria | NA | Blood dysbiosis in biliary diseases might be characterized by an increase in Clostridia but a decrease in Gammaproteobacteria | [43] |
| Subgroup Analysis | ||||||||
| Patients with biliary tract cancers vs. HC | ||||||||
| ↕ | NA | S | Family: ↑Bifidobacteriaceae and ↓Pseudomonadaceae Genus: ↑Ralstonia, ↓Corynebacterium, and ↓Comamonas | NA | Blood dysbiosis in biliary tract cancers might be characterized by an increase in Bifidobacteriaceae but a decrease in Pseudomonadaceae | |||
| Liver Diseases | ||||||||
The American study (76):
| Whole blood DNA | ↕ | NA | NS | Phylum: ↓Bacteroidetes |
| Blood dysbiosis in association with alcoholic hepatitis might be characterized by a decrease in Bacteroidetes as well as an increase in 16s rRNA gene concentration | [26] |
The morbid obese in Spanish study (37):
| Buffy coat DNA | ↓ | NA | NA | Phylum: ↑Proteobacteria and ↓Actinobacteria Class: ↑Alphaproteobacteria Family: ↑Bradyrhizobiaceae and ↑Sphigomonadaceae |
| Blood dysbiosis in morbidly obese patients with cirrhosis compared with morbidly obese patients without cirrhosis might be characterized by an increase in Proteobacteria but a decrease in Actinobacteria as well as an increase in 16s rRNA gene concentration | [25] |
The Korean study with NAFLD (76):
| Buffy coat DNA | NA | NA | NS | Family: ↑Succinivibrionaceae and ↓Leukonostocaceae | NA | Blood dysbiosis in obese patients with NAFLD might be characterized by an increase in Succinivibrionaceae but a decrease in Leukonostocaceae compared with lean patients with NAFLD | [62] |
The Korean study (363):
| Serum DNA | ↓ | NA | S | HCC and cirrhosis vs. HC Phylum: ↑Proteobacteria and ↓Firmicutes | NA | Blood dysbiosis in liver diseases (HCC and cirrhosis) might be characterized by an increase in Proteobacteria but a decrease in Firmicutes | [34] |
| Subgroup Analysis | ||||||||
| HCC vs. HC | ||||||||
| ↓ | NA | S | Genus: ↑Staphylococcus, ↑Acinetobacter, ↑Klebsiella, ↑Trabusiella, ↓Pseudomonas, ↓Streptococcus, and ↓Bifidobacterium | NA | Blood dysbiosis in HCC might be characterized by an increase in Staphylococcus, Acinetobacter, Klebsiella, and Trabusiella but a decrease in Streptococcus and Bifidobacterium | |||
The Japanese study (80):
| Whole blood RNA | ↕ | ↕ | NS | Order: ↓ Erysipelotrichales Family: ↑Enterobacteriaceae and ↓Rikenellaceae Genus: ↓Akkermansia | NA | Blood dysbiosis in cirrhosis might be characterized by a decrease in Erysipelotrichales but an increase in Enterobacteriaceae regardless of age | [17] |
| Subgroup Analysis | ||||||||
| HCC vs. HC | ||||||||
| NA | NA | NA | Family: ↑Enterobacteriaceae Genus: ↑Bacteroides and ↓Bifidobacterium | NA | Blood dysbiosis in HCC might be characterized by an increase in Enterobacteriaceae regardless of age | |||
The Chinese study (98):
| Plasma DNA | ↕ | ↓ | NS | Phylum: ↓Actinobacteria and ↓Deinococcus-Thermus Order: ↓Enterobacteriales Family: ↑Moraxellaceae and ↓Enterobacteriaceae Genus: ↑Sulfurovum and ↓Meiothermus |
| Blood dysbiosis in HBV-DLF compared with HBV-CLF and HC might be characterized by a decrease in Actinobacteria and Deinococcus-Thermus; in addition, the liver may filter bacteriome in blood, and its efficacy might depend on liver function | [63] |
| Subgroup Analysis | ||||||||
| HBV-DLF vs. HBV-CLF | ||||||||
| ↕ | ↓ | NS | Order: ↑Campylobacterales and ↓Xanthomonadales Family: ↓Xanthomonadaceae | NA | Blood dysbiosis in HBV-DLF compared with HBV-CLF might be characterized by an increase in Campylobacterales but a decrease in Xanthomonadales | |||
| HBV-DLF vs. HC | ||||||||
| ↕ | ↓ | NS | Class: ↓Alphaproteobacteria Family: ↑Burkholderiaceae and ↑Moraxellaceae Genus: ↑Acinetobacter and ↑Comamonas | NA | Blood dysbiosis in HBV-DLF might be characterized by a decrease in Alphaproteobacteria | |||
| Patients who died within 28 days after diagnosis vs. who survived for 28 days after diagnosis | ||||||||
| NA | NA | NA | Family: ↑Enterobacteriaceae and ↓Prevotellaceae | NA | Blood dysbiosis in HBV-DLF with poor prognosis might be characterized by an increase in Enterobacteriaceae but a decrease in Prevotellaceae | |||
| Subjects (n; Mean Age) | Samples | Dysbiotic Blood Bacteriome of Patients vs. Controls | Other | Interpretation | Ref. | |||
|---|---|---|---|---|---|---|---|---|
| Diversity | Differential Abundance | |||||||
| α-R | α-E | β | ||||||
| Psychiatric Disorders | ||||||||
The France study (112):
| Plasma DNA | ↕ | ↕ | S | Phylum: ↓Fusobacteria and ↓Candidatus Saccharibacteria Genus: ↑Janthinobacterium and ↓Neisseria | NA | Blood dysbiosis in MDE might be characterized by a decrease in Fusobacteria and Candidatus Saccharibacteria | [64] |
The France study (56; 41.9 ± 11.6):
| Plasma DNA | NA | NA | NA | Genus: ↑Neisseria and ↓Janthinobacterium | NA | Blood dysbiosis in MDE might be reversed by anti-depressive drugs as an increase in Neisseria and a decrease in Janthinobacterium | [64] |
| Subgroup Analysis | ||||||||
| Responder vs. non-responder before receiving anti-depressants | ||||||||
| NA | NA | NA | Phylum: ↑Firmicutes, ↓Proteobacteria, and ↓Actinobacteria | NA | MDE patients whose baseline blood has increased Firmicutes and a reduction in Proteobacteria and Actinobacteria may respond to anti-depressive drugs | |||
The American study (192):
| Whole blood RNA | NA | ↑ | S | Phylum: ↑Planctomycetes and ↑Thermotogae |
| Human blood bacteriome may originate from gut as well as oral bacteriome, and a reduction in diversity of T cell population in SCZ might relate to blood dysbiosis, which was characterized by an increase in Planctomycetes and Thermotogae | [47] |
| Neurodegenerative Diseases | ||||||||
The Chinese study (90):
| Buffy coat DNA | ↕ | ↕ | NS | Genus: ↑Myroides, ↑Isoptericola, ↑Microbacterium, ↑Cloacibacterium, ↑Enhydrobacter, and ↓Limnobacter | NA | Blood dysbiosis in Parkinson’s disease might be characterized by an increase in Myroides, Isoptericola, Microbacterium, Cloacibacterium, and Enhydrobacter, as well as a decrease in Limnobacter | [65] |
The Chinese study (80):
| Buffy coat DNA | ↕ | ↕ | S | Genus: ↑Bacteroides and ↓Leucobacter | NA | Blood dysbiosis in MSA might be characterized by an increase in Bacteroides and a decrease in Leucobacter | [66] |
| Subgroup Analysis | ||||||||
| Cerebellar MSA vs. Parkinsonian MSA | ||||||||
| NA | NA | NA | Genus: ↑Acinetobacter, ↓Blastococcus and ↓Bacillus | NA | Blood dysbiosis in Cerebellar MSA might be characterized by an increase in Acinetobacter and a decrease in Blastococcus and Bacillus compared with Parkinsonian MSA | |||
| Subjects (n; Mean Age) | Samples | Dysbiotic Blood Bacteriome of Patients vs. Controls | Other | Interpretation | Ref. | |||
|---|---|---|---|---|---|---|---|---|
| Diversity | Differential Abundance | |||||||
| α-R | α-E | β | ||||||
| Autoimmune Diseases | ||||||||
The American (40):
| Plasma DNA | ↕ | ↕ | NS | Phylum: ↑Fusobacteria Genus: ↓Paenibacillus | NA | Blood dysbiosis in well-treated SLE might be characterized by an increase in Fusobacteria | [39] |
The American (36):
| Plasma DNA | ↓ | ↓ | S | Phylum: ↓Firmicutes Genus: ↓Paenibacillus Species: ↑Thermoanaerobacterium saccharolyticum and ↑Lactobacillus iners | NA | Blood dysbiosis in first-degree relatives of SLE patients might be characterized by a decrease in Firmicutes | [39] |
The American (49):
| Plasma DNA | NA | NA | NA | Genus: ↑Planococcus, ↑Desulfoconvexum, ↑Desulfofrigus, ↑Desulfovibrio, ↑Draconibacterium, ↑Planomicrobium, ↑Psychrilyobacter, ↑Corynebacterium, and ↑Ochrobactrum | Validated by in vitro study
| An increase in Planococcus in blood bacteriome of SLE could lead to chronic systemic inflammation | [53] |
The Chinese (42):
| PBMCDNA | ↕ | ↕ | S | Phylum: ↑Candidatus Saccharibacteria and ↓Bacteroidetes | Genus Pelagibacterium in family Hyphomicrobiaceae in order Rhizobiales correlated with PARP9 mRNA levels (r = 0.65, 0.66, and 0.60, respectively) | Blood dysbiosis in rheumatoid arthritis may be characterized by an increase in Candidatus Saccharibacteria, but a decrease in Bacteroidetes and an increase in Pelagibacterium, Hyphomicrobiaceae, and Rhizobiales, might play a role in pathophysiology of rheumatoid arthritis | [32] |
The British (30):
| Serum DNA | NA | NA | NA | Family: ↑Lachnospiraceae Genus: ↑Halomonas, ↑Shewanella, ↓Corynebacterium 1, and ↓Streptococcus | NA | Blood dysbiosis in rheumatoid arthritis might be characterized by an increase in Lachnospiraceae, Halomonas, and Shewanella but a decrease in Corynebacterium 1 and Streptococcus | [36] |
The British (20; age = NA):
| Serum DNA | NA | NA | NA | Family: ↑Lachnospiraceae Genus: ↑Corynebacterium 1, ↑Streptococcus, ↓Halomonas, and ↓Shewanella | NA | Anti-rheumatic drugs might cause a reversion of blood dysbiosis in rheumatoid arthritis by an increase in Corynebacterium 1 and Streptococcus but a decrease in Shewanella; in addition, the persistent increase in Lachnospiraceae after treatment might indicate that there might be compensatory effect for blood dysbiosis and could alleviate the disease | [36] |
The Taiwanese (28):
| EVsDNA | ↓ | ↓ | S | Phylum: ↓Firmicutes and ↓Fusobacteria Order: ↑Bacillales and ↓Lactobacillales Family: ↓Brucellaceae Genus: ↑Staphylococcus, ↑Sphingomonas, and ↓Streptococcus Species: ↑Ralstonia insidiosa, ↓Kingella oralis, and ↓Aquabacterium parvum | NA | Blood dysbiosis in psoriasis might be characterized by a decrease in Firmicutes and Fusobacteria | [42] |
The French (47):
| Serum DNA | ↕ | ↕ | NS | Class: ↑Cytophagia and ↑Clostridia Genus: ↓Zooloea and ↓Staphylococcus | NA | Blood dysbiosis in large vessel arteritis might be characterized by an increase in Cytophagia and Clostridia | [37] |
| Subgroup Analysis | ||||||||
| GCA vs. HC | Blood dysbiosis in both GCA and TAK characterized by an increase in Cytophagia and an upsurge in Staphylococcus in TAK might play a role in disease activity | |||||||
| NA | NA | NA | Class: ↑Cytophagia Genus: ↑Rhodococcus | NA | ||||
| TAK vs. HC | ||||||||
| NA | NA | NA | Class: ↑Cytophagia, ↑Clostridia, and ↑Deltaproteobacteria Genus: ↓Hyphomicrobium and ↓Staphylococcus | NA | ||||
| GCA vs. TAK | ||||||||
| NA | NA | NA | Family: ↑Hyphomonaceae Genus: ↑Rhodococcus, and ↓CloacibacteriumSpecies: ↓Candidatus aquiluna | NA | ||||
| Active TAK vs. inactive TAK | ||||||||
| NA | NA | NA | Genus: ↑Staphylococcus | NA | ||||
| Rosacea | ||||||||
The Korean (40):
| Whole blood DNA | ↕ | ↕ | S | Family: ↑Chromaticeae and ↑Fusobacteriaceae Genus: ↑Rheinheimera | NA | Blood dysbiosis in rosacea might be characterized by an increase in Chromaticeae, Rheinheimera in particular, and Fusobacteriaceae | [67] |
| Asthma | ||||||||
The British (10):
| Plasma DNA | ↕ | ↕ | NA | Phylum: ↑Firmicutes, ↑Bacteroidetes, and ↓Proteobacteria Order: ↓Bacteroidales Class: ↑Bacilli and ↓Bacteroidia Family: ↑Xanthomonadaceae Genus: ↑Kocuria and ↑Strenotrophomonas | NA | Blood dysbiosis in asthma might be characterized by an increase in Firmicutes and Bacteroidetes but a decrease in Proteobacteria | [68] |
The Korean (450):
| EVsDNA | ↑ | ↓ | S | Phylum: ↑Bacteroidetes, ↓Actinobacteria, ↓Verrucomicrobia, and ↓Cyanobacteria Genus: ↑Klebsiella, ↑Bacteroides, ↑Alistipes, ↑Subdoligranulum, ↑Bifidobacterium, ↓Akkermansia, ↓Citrobacter, ↓Staphylococcus, and ↓Micrococcus | NA | Blood dysbiosis in treated and untreated asthma might be characterized by an increase in Bacteroidetes and Actinobacteria but a decrease in Verrucomicrobia and Cyanobacteria | [44] |
| Subgroup Analysis | ||||||||
| Steroid use vs. steroid naïve | ||||||||
| NA | NA | NA | Genus: ↓Staphylococcus and ↓Rothia | NA | Asthma treated with steroids might affect blood bacteriome by a decrease in Staphylococcus and Rothia | |||
| Both ICS and OCS use vs. steroid naïve and ICS only | ||||||||
| NA | NA | NA | Genus: ↑Prevotella 9, ↑Intestinibacter, ↑Lactobacillus, and ↑Blautia | NA | Asthma treated with a combination of ICS and OCS compared with ICS only might affect blood bacteriome by an increase in Prevotella 9, Intestinibacter, Lactobacillus, and Blautia | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suparan, K.; Sriwichaiin, S.; Chattipakorn, N.; Chattipakorn, S.C. Human Blood Bacteriome: Eubiotic and Dysbiotic States in Health and Diseases. Cells 2022, 11, 2015. https://doi.org/10.3390/cells11132015
Suparan K, Sriwichaiin S, Chattipakorn N, Chattipakorn SC. Human Blood Bacteriome: Eubiotic and Dysbiotic States in Health and Diseases. Cells. 2022; 11(13):2015. https://doi.org/10.3390/cells11132015
Chicago/Turabian StyleSuparan, Kanokphong, Sirawit Sriwichaiin, Nipon Chattipakorn, and Siriporn C. Chattipakorn. 2022. "Human Blood Bacteriome: Eubiotic and Dysbiotic States in Health and Diseases" Cells 11, no. 13: 2015. https://doi.org/10.3390/cells11132015
APA StyleSuparan, K., Sriwichaiin, S., Chattipakorn, N., & Chattipakorn, S. C. (2022). Human Blood Bacteriome: Eubiotic and Dysbiotic States in Health and Diseases. Cells, 11(13), 2015. https://doi.org/10.3390/cells11132015