
Golgi Complex: A Signaling Hub in Cancer



Abstract
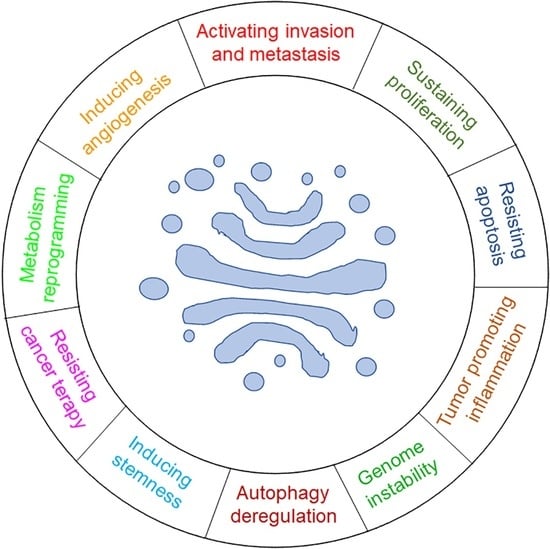
1. Introduction
2. GC-Centered Signaling Pathways Regulate Cancer Hallmarks
2.1. GC-Centered Signaling Pathways that Regulate Cancer Migration, Invasion and Metastasis Formation
2.1.1. ADP-Ribosylation Factor 1 (ARF1)
2.1.2. GM130
2.1.3. Golgi Membrane Protein 1 (GOLM1)/Golgi Protein 73 (GP73)/Golgi Phosphoprotein 2 (GOLPH2)
2.1.4. Similar Expression to FGF (Sef) (also Known as Interleukin-17 Receptor D (IL-17RD))
2.1.5. Golgin-97
2.1.6. TMED Family of p24 Proteins
2.1.7. Secretory Carrier-Associated Membrane Protein 1 (SCAMP1)
2.1.8. RKTG (Raf Kinase Trapping to Golgi)/PAQR3
2.1.9. Protein Kinase D (PKD) Family
2.2. GC-Centered Signaling Pathways that Regulate Cancer Proliferation
2.2.1. ADP-Ribosylation Factor 1 (ARF1)
2.2.2. Golgi Membrane Protein 1 (GOLM1)/Golgi Protein 73 (GP73)/Golgi Phosphoprotein 2 (GOLPH2)
2.2.3. Vesicle Transport Factor (USO1) (also Known as Vesicle Docking Protein, 115-KD (p115))
2.2.4. RKTG (Raf Kinase Trapping to Golgi)/PAQR3
2.2.5. TMED Family of p24 Proteins
2.2.6. Similar Expression to FGF (Sef) (also Known as Interleukin-17 Receptor D (IL-17RD))
2.2.7. UbiA Prenyltransferase Domain-Containing Protein 1 (UBIAD1)
2.2.8. Secretory Carrier-Associated Membrane Protein 3 (SCAMP3)
2.2.9. Golgi Calcium Pump Secretory Pathway Calcium ATPase 1 (SPCA1)
2.2.10. Protein Kinase D (PKD) Family
2.3. GC-Centered Signaling Pathways that Regulate Survival and Apoptosis
2.3.1. CLIPR-59 (Cytoplasmic Linker Protein 170-Related 59 kDa Protein)
2.3.2. Ras
2.3.3. TMED Family of p24 Proteins
2.3.4. Protein Kinase D (PKD) Family
2.4. GC-Centered Signaling Pathways that Regulate Autophagy
2.4.1. TMED Family of p24 Proteins
2.4.2. VPS53
2.5. GC-Centered Signaling Pathways that Regulate Angiogenesis
2.5.1. RKTG (Raf Kinase Trapping to Golgi)/PAQR3
2.5.2. Protein Kinase D (PKD) Family
2.6. GC-Centered Signaling Pathways that Regulate Cancer Stemness
2.6.1. TMED Family of p24 Proteins
2.6.2. Protein Kinase D (PKD) Family
2.7. GC-Centered Signaling Pathways that Regulate Cancer Resistance to Therapies
2.7.1. TMED Family of p24 Proteins
2.7.2. Protein Kinase D (PKD) Family
2.8. GC-Centered Signaling Pathways that Reprogram Cancer Metabolism
Protein Kinase D (PKD) Family
2.9. GC-Centered Signaling Pathways that Regulate Chronic Inflammation
2.9.1. Golgi Membrane Protein 1 (GOLM1)/Golgi Protein 73 (GP73)/Golgi Phosphoprotein 2 (GOLPH2)
2.9.2. Similar Expression to FGF (Sef) (also Known as INTERLEUKIN-17 receptor D (IL-17RD))
2.10. GC-Centered Signaling Pathways that Regulate Cancer Genomic Instability
Similar Expression to FGF (Sef) (also Known as Interleukin-17 Receptor D (IL-17RD))
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Makhoul, C.; Gosavi, P.; Gleeson, P.A. The Golgi Architecture and Cell Sensing. Biochem. Soc. Trans. 2018, 46, 1063–1072. [Google Scholar] [CrossRef]
- Farhan, H.; Rabouille, C. Signalling to and from the Secretory Pathway. J. Cell Sci. 2011, 124, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Luini, A.; Parashuraman, S. Signaling at the Golgi: Sensing and Controlling the Membrane Fluxes. Curr. Opin. Cell Biol. 2016, 39, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Mayinger, P. Signaling at the Golgi. Cold Spring Harb. Perspect. Biol. 2011, 3, a005314. [Google Scholar] [CrossRef] [PubMed]
- Cancino, J.; Luini, A. Signaling Circuits on the Golgi Complex. Traffic 2013, 14, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Petrosyan, A. Onco-Golgi: Is Fragmentation a Gate to Cancer Progression? Biochem. Mol. Biol. J. 2015, 1, 16. [Google Scholar] [CrossRef]
- Zhang, X. Alterations of Golgi Structural Proteins and Glycosylation Defects in Cancer. Front. Cell Dev. Biol. 2021, 9, 665289. [Google Scholar] [CrossRef]
- Huang, H.; Jiang, Y.; Wang, Y.; Chen, T.; Yang, L.; He, H.; Lin, Z.; Liu, T.; Yang, T.; Kamp, D.W.; et al. MiR-5100 Promotes Tumor Growth in Lung Cancer by Targeting Rab6. Cancer Lett. 2015, 362, 15–24. [Google Scholar] [CrossRef]
- Shimada, K.; Uzawa, K.; Kato, M.; Endo, Y.; Shiiba, M.; Bukawa, H.; Yokoe, H.; Seki, N.; Tanzawa, H. Aberrant Expression of RAB1A in Human Tongue Cancer. Br. J. Cancer 2005, 92, 1915–1921. [Google Scholar] [CrossRef]
- Bravo-Cordero, J.J.; Marrero-Diaz, R.; Megías, D.; Genís, L.; García-Grande, A.; García, M.A.; Arroyo, A.G.; Montoya, M.C. MT1-MMP Proinvasive Activity Is Regulated by a Novel Rab8-Dependent Exocytic Pathway. EMBO J. 2007, 26, 1499–1510. [Google Scholar] [CrossRef]
- Waugh, M.G. The Great Escape: How Phosphatidylinositol 4-Kinases and PI4P Promote Vesicle Exit from the Golgi (and Drive Cancer). Biochem. J. 2019, 476, 2321–2346. [Google Scholar] [CrossRef] [PubMed]
- Lan, L.; Han, H.; Zuo, H.; Chen, Z.; Du, Y.; Zhao, W.; Gu, J.; Zhang, Z. Upregulation of Myosin Va by Snail Is Involved in Cancer Cell Migration and Metastasis. Int. J. Cancer 2010, 126, 53–64. [Google Scholar] [CrossRef]
- Bhide, G.P.; Colley, K.J. Sialylation of N-Glycans: Mechanism, Cellular Compartmentalization and Function. Histochem. Cell Biol. 2017, 147, 149–174. [Google Scholar] [CrossRef] [PubMed]
- Donizy, P.; Marczuk, J. Selected Golgi-Localized Proteins and Carcinogenesis: What Do We Know? In The Golgi Apparatus and Centriole; Kloc, M., Ed.; Results and Problems in Cell Differentiation; Springer International Publishing: Cham, Switzerland, 2019; Volume 67, pp. 487–529. ISBN 978-3-030-23172-9. [Google Scholar]
- Bui, S.; Mejia, I.; Díaz, B.; Wang, Y. Adaptation of the Golgi Apparatus in Cancer Cell Invasion and Metastasis. Front. Cell Dev. Biol. 2021, 9, 806482. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Sechi, S.; Frappaolo, A.; Karimpour-Ghahnavieh, A.; Piergentili, R.; Giansanti, M.G. Oncogenic Roles of GOLPH3 in the Physiopathology of Cancer. Int. J. Mol. Sci. 2020, 21, 933. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, R.; Russo, D.; Kurokawa, K.; Sahu, P.; Lombardi, B.; Supino, D.; Zhukovsky, M.A.; Vocat, A.; Pothukuchi, P.; Kunnathully, V.; et al. Golgi Maturation-Dependent Glycoenzyme Recycling Controls Glycosphingolipid Biosynthesis and Cell Growth via GOLPH3. EMBO J. 2021, 40, e107238. [Google Scholar] [CrossRef]
- Boulay, P.-L.; Cotton, M.; Melançon, P.; Claing, A. ADP-Ribosylation Factor 1 Controls the Activation of the Phosphatidylinositol 3-Kinase Pathway to Regulate Epidermal Growth Factor-Dependent Growth and Migration of Breast Cancer Cells. J. Biol. Chem. 2008, 283, 36425–36434. [Google Scholar] [CrossRef]
- Gu, G.; Chen, Y.; Duan, C.; Zhou, L.; Chen, C.; Chen, J.; Cheng, J.; Shi, N.; Jin, Y.; Xi, Q.; et al. Overexpression of ARF1 Is Associated with Cell Proliferation and Migration through PI3K Signal Pathway in Ovarian Cancer. Oncol. Rep. 2017, 37, 1511–1520. [Google Scholar] [CrossRef]
- Lewis-Saravalli, S.; Campbell, S.; Claing, A. ARF1 Controls Rac1 Signaling to Regulate Migration of MDA-MB-231 Invasive Breast Cancer Cells. Cell. Signal. 2013, 25, 1813–1819. [Google Scholar] [CrossRef] [PubMed]
- Schlienger, S.; Ramirez, R.A.M.; Claing, A. ARF1 Regulates Adhesion of MDA-MB-231 Invasive Breast Cancer Cells through Formation of Focal Adhesions. Cell. Signal. 2015, 27, 403–415. [Google Scholar] [CrossRef] [PubMed]
- Schlienger, S.; Campbell, S.; Claing, A. ARF1 Regulates the Rho/MLC Pathway to Control EGF-Dependent Breast Cancer Cell Invasion. MBoC 2014, 25, 17–29. [Google Scholar] [CrossRef]
- Khater, M.; Bryant, C.N.; Wu, G. Gβγ Translocation to the Golgi Apparatus Activates ARF1 to Spatiotemporally Regulate G Protein–Coupled Receptor Signaling to MAPK. J. Biol. Chem. 2021, 296, 100805. [Google Scholar] [CrossRef]
- Boulay, P.-L.; Schlienger, S.; Lewis-Saravalli, S.; Vitale, N.; Ferbeyre, G.; Claing, A. ARF1 Controls Proliferation of Breast Cancer Cells by Regulating the Retinoblastoma Protein. Oncogene 2011, 30, 3846–3861. [Google Scholar] [CrossRef]
- Zhao, J.; Yang, C.; Guo, S.; Wu, Y. GM130 Regulates Epithelial-to-Mesenchymal Transition and Invasion of Gastric Cancer Cells via Snail. Int. J. Clin. Exp. Pathol. 2015, 8, 10784–10791. [Google Scholar] [PubMed]
- Baschieri, F.; Confalonieri, S.; Bertalot, G.; Di Fiore, P.P.; Dietmaier, W.; Leist, M.; Crespo, P.; Macara, I.G.; Farhan, H. Spatial Control of Cdc42 Signalling by a GM130–RasGRF Complex Regulates Polarity and Tumorigenesis. Nat. Commun. 2014, 5, 4839. [Google Scholar] [CrossRef] [PubMed]
- Baschieri, F.; Uetz-von Allmen, E.; Legler, D.F.; Farhan, H. Loss of GM130 in Breast Cancer Cells and Its Effects on Cell Migration, Invasion and Polarity. Cell Cycle 2015, 14, 1139–1147. [Google Scholar] [CrossRef] [PubMed]
- Jin, D.; Tao, J.; Li, D.; Wang, Y.; Li, L.; Hu, Z.; Zhou, Z.; Chang, X.; Qu, C.; Zhang, H. Golgi Protein 73 Activation of MMP-13 Promotes Hepatocellular Carcinoma Cell Invasion. Oncotarget 2015, 6, 33523–33533. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, X.; Zhou, S.; Shi, J.; Xu, Y.; He, J.; Lin, F.; Wei, A.; Zhou, L.; Chen, Z. Knockdown of Golgi Phosphoprotein 73 Blocks the Trafficking of Matrix Metalloproteinase-2 in Hepatocellular Carcinoma Cells and Inhibits Cell Invasion. J. Cell Mol. Med. 2019, 23, 2399–2409. [Google Scholar] [CrossRef]
- Ye, Q.-H.; Zhu, W.-W.; Zhang, J.-B.; Qin, Y.; Lu, M.; Lin, G.-L.; Guo, L.; Zhang, B.; Lin, Z.-H.; Roessler, S.; et al. GOLM1 Modulates EGFR/RTK Cell-Surface Recycling to Drive Hepatocellular Carcinoma Metastasis. Cancer Cell 2016, 30, 444–458. [Google Scholar] [CrossRef]
- Gai, X.; Tang, B.; Liu, F.; Wu, Y.; Wang, F.; Jing, Y.; Huang, F.; Jin, D.; Wang, L.; Zhang, H. MTOR/MiR-145-Regulated Exosomal GOLM1 Promotes Hepatocellular Carcinoma through Augmented GSK-3β/MMPs. J. Genet. Genom. 2019, 46, 235–245. [Google Scholar] [CrossRef]
- Yan, G.; Ru, Y.; Wu, K.; Yan, F.; Wang, Q.; Wang, J.; Pan, T.; Zhang, M.; Han, H.; Li, X.; et al. GOLM1 Promotes Prostate Cancer Progression through Activating PI3K-AKT-mTOR Signaling. Prostate 2018, 78, 166–177. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Ji, J.; Zhang, X.; Han, M.; Zhang, C.; Xu, Y.; Wei, Y.; Wang, S.; Huang, B.; Chen, A.; et al. PDGFA/PDGFRα-Regulated GOLM1 Promotes Human Glioma Progression through Activation of AKT. J. Exp. Clin. Cancer Res. 2017, 36, 193. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liu, Q.; Li, Z.; Zhang, R.; Jia, C.; Yang, Z.; Zhao, H.; Ya, S.; Mao, R.; Ailijiang, T.; et al. GP73 Promotes Epithelial–Mesenchymal Transition and Invasion Partly by Activating TGF-Β1/Smad2 Signaling in Hepatocellular Carcinoma. Carcinogenesis 2018, 39, 900–910. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.-J.; Liu, G.-L.; Liu, B.; Liu, T. GP73 Promotes Invasion and Metastasis of Bladder Cancer by Regulating the Epithelial-Mesenchymal Transition through the TGF-Β1/Smad2 Signalling Pathway. J. Cell. Mol. Med. 2018, 22, 1650–1665. [Google Scholar] [CrossRef]
- Ding, X.; Deng, G.; Liu, J.; Liu, B.; Yuan, F.; Yang, X.; Chen, Q. GOLM1 Silencing Inhibits the Proliferation and Motility of Human Glioblastoma Cells via the Wnt/β-Catenin Signaling Pathway. Brain Res. 2019, 1717, 117–126. [Google Scholar] [CrossRef]
- Song, Q.; He, X.; Xiong, Y.; Wang, J.; Zhang, L.; Leung, E.L.-H.; Li, G. The Functional Landscape of Golgi Membrane Protein 1 (GOLM1) Phosphoproteome Reveal GOLM1 Regulating P53 That Promotes Malignancy. Cell Death Discov. 2021, 7, 42. [Google Scholar] [CrossRef]
- Chen, X.; Wang, Y.; Tao, J.; Shi, Y.; Gai, X.; Huang, F.; Ma, Q.; Zhou, Z.; Chen, H.; Zhang, H.; et al. MTORC1 Up-Regulates GP73 to Promote Proliferation and Migration of Hepatocellular Carcinoma Cells and Growth of Xenograft Tumors in Mice. Gastroenterology 2015, 149, 741–752.e14. [Google Scholar] [CrossRef]
- Pu, Y.; Song, Y.; Zhang, M.; Long, C.; Li, J.; Wang, Y.; Xu, Y.; Pan, F.; Zhao, N.; Zhang, X.; et al. GOLM1 Restricts Colitis and Colon Tumorigenesis by Ensuring Notch Signaling Equilibrium in Intestinal Homeostasis. Signal Transduct. Target 2021, 6, 148. [Google Scholar] [CrossRef] [PubMed]
- Darby, S.; Sahadevan, K.; Khan, M.M.; Robson, C.N.; Leung, H.Y.; Gnanapragasam, V.J. Loss of Sef (Similar Expression to FGF) Expression Is Associated with High Grade and Metastatic Prostate Cancer. Oncogene 2006, 25, 4122–4127. [Google Scholar] [CrossRef][Green Version]
- Darby, S.; Murphy, T.; Thomas, H.; Robson, C.N.; Leung, H.Y.; Mathers, M.E.; Gnanapragasam, V.J. Similar Expression to FGF (Sef) Inhibits Fibroblast Growth Factor-Induced Tumourigenic Behaviour in Prostate Cancer Cells and Is Downregulated in Aggressive Clinical Disease. Br. J. Cancer 2009, 101, 1891–1899. [Google Scholar] [CrossRef]
- Hori, S.; Wadhwa, K.; Pisupati, V.; Zecchini, V.; Ramos-Montoya, A.; Warren, A.Y.; Neal, D.E.; Gnanapragasam, V.J. Loss of HSef Promotes Metastasis through Upregulation of EMT in Prostate Cancer. Int. J. Cancer 2017, 140, 1881–1887. [Google Scholar] [CrossRef]
- He, Q.; Gong, Y.; Gower, L.; Yang, X.; Friesel, R.E. Sef Regulates Epithelial-Mesenchymal Transition in Breast Cancer Cells. J. Cell Biochem. 2016, 117, 2346–2356. [Google Scholar] [CrossRef]
- Zisman-Rozen, S.; Fink, D.; Ben-Izhak, O.; Fuchs, Y.; Brodski, A.; Kraus, M.H.; Bejar, J.; Ron, D. Downregulation of Sef, an Inhibitor of Receptor Tyrosine Kinase Signaling, Is Common to a Variety of Human Carcinomas. Oncogene 2007, 26, 6093–6098. [Google Scholar] [CrossRef][Green Version]
- Zhang, H.; Zhao, X.; Yan, L.; Li, M. Similar Expression to FGF (Sef) Reduces Endometrial Adenocarcinoma Cells Proliferation via Inhibiting Fibroblast Growth Factor 2-Mediated MAPK/ERK Signaling Pathway. Gynecol. Oncol. 2011, 122, 669–674. [Google Scholar] [CrossRef]
- Fuchs, Y.; Brunwasser, M.; Haif, S.; Haddad, J.; Shneyer, B.; Goldshmidt-Tran, O.; Korsensky, L.; Abed, M.; Zisman-Rozen, S.; Koren, L.; et al. Sef Is an Inhibitor of Proinflammatory Cytokine Signaling, Acting by Cytoplasmic Sequestration of NF-ΚB. Dev. Cell 2012, 23, 611–623. [Google Scholar] [CrossRef]
- Mellett, M.; Atzei, P.; Bergin, R.; Horgan, A.; Floss, T.; Wurst, W.; Callanan, J.J.; Moynagh, P.N. Orphan Receptor IL-17RD Regulates Toll-like Receptor Signalling via SEFIR/TIR Interactions. Nat. Commun. 2015, 6, 6669. [Google Scholar] [CrossRef]
- Girondel, C.; Lévesque, K.; Langlois, M.-J.; Pasquin, S.; Saba-El-Leil, M.K.; Rivard, N.; Friesel, R.; Servant, M.J.; Gauchat, J.-F.; Lesage, S.; et al. Loss of Interleukin-17 Receptor D Promotes Chronic Inflammation-Associated Tumorigenesis. Oncogene 2021, 40, 452–464. [Google Scholar] [CrossRef]
- Duhamel, S.; Hébert, J.; Gaboury, L.; Bouchard, A.; Simon, R.; Sauter, G.; Basik, M.; Meloche, S. Sef Downregulation by Ras Causes MEK1/2 to Become Aberrantly Nuclear Localized Leading to Polyploidy and Neoplastic Transformation. Cancer Res. 2012, 72, 626–635. [Google Scholar] [CrossRef]
- Hsu, R.-M.; Zhong, C.-Y.; Wang, C.-L.; Liao, W.-C.; Yang, C.; Lin, S.-Y.; Lin, J.-W.; Cheng, H.-Y.; Li, P.-Y.; Yu, C.-J. Golgi Tethering Factor Golgin-97 Suppresses Breast Cancer Cell Invasiveness by Modulating NF-ΚB Activity. Cell Commun. Signal. 2018, 16, 19. [Google Scholar] [CrossRef]
- Shi-Peng, G.; Chun-Lin, C.; Huan, W.; Fan-Liang, M.; Yong-Ning, C.; Ya-Di, Z.; Guang-Ping, Z.; Ye-Ping, C. TMED2 Promotes Epithelial Ovarian Cancer Growth. Oncotarget 2017, 8, 94151–94165. [Google Scholar] [CrossRef]
- Duquet, A.; Melotti, A.; Mishra, S.; Malerba, M.; Seth, C.; Conod, A.; Ruiz i Altaba, A. A Novel Genome-Wide in Vivo Screen for Metastatic Suppressors in Human Colon Cancer Identifies the Positive WNT-TCF Pathway Modulators TMED3 and SOX12. EMBO Mol. Med. 2014, 6, 882–901. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Yang, Y.; Han, J.; Jiang, W.; Chen, C.; Wang, M.; Gao, R.; Li, S.; Tian, T.; Wang, J.; et al. TMED3 Promotes Hepatocellular Carcinoma Progression via IL-11/STAT3 Signaling. Sci. Rep. 2016, 6, 37070. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Luo, Y.; Li, Q. TMED3 Promotes Proliferation and Migration in Breast Cancer Cells by Activating Wnt/β-Catenin Signaling. Onco Targets Ther. 2020, 13, 5819–5830. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Sun, L.; Zhang, J. TMED3 Exerts a Protumor Function in Non-Small Cell Lung Cancer by Enhancing the Wnt/β-Catenin Pathway via Regulation of AKT. Toxicol. Appl. Pharm. 2021, 433, 115793. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Huang, H.; Xiao, D.; Duan, Y.; Zheng, Y.; Chen, Z. Knockdown of TMED3 Inhibits Cell Viability and Migration and Increases Apoptosis in Human Chordoma Cells. Int. J. Oncol. 2021, 58, 15. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Bernal, C.; Silvano, M.; Anand, S.; Ruiz I Altaba, A. The Protein Secretion Modulator TMED9 Drives CNIH4/TGFα/GLI Signaling Opposing TMED3-WNT-TCF to Promote Colon Cancer Metastases. Oncogene 2019, 38, 5817–5837. [Google Scholar] [CrossRef] [PubMed]
- Nakano, N.; Tsuchiya, Y.; Kako, K.; Umezaki, K.; Sano, K.; Ikeno, S.; Otsuka, E.; Shigeta, M.; Nakagawa, A.; Sakata, N.; et al. TMED10 Protein Interferes with Transforming Growth Factor (TGF)-β Signaling by Disrupting TGF-β Receptor Complex Formation. J. Biol. Chem. 2017, 292, 4099–4112. [Google Scholar] [CrossRef]
- Wang, H.; Xiao, L.; Kazanietz, M.G. P23/Tmp21 Associates with Protein Kinase Cδ (PKCδ) and Modulates Its Apoptotic Function. J. Biol. Chem. 2011, 286, 15821–15831. [Google Scholar] [CrossRef]
- Xu, X.; Gao, H.; Qin, J.; He, L.; Liu, W. TMP21 Modulates Cell Growth in Papillary Thyroid Cancer Cells by Inducing Autophagy through Activation of the AMPK/MTOR Pathway. Int. J. Clin. Exp. Pathol. 2015, 8, 10824–10831. [Google Scholar]
- Vadakekolathu, J.; Al-Juboori, S.I.K.; Johnson, C.; Schneider, A.; Buczek, M.E.; Di Biase, A.; Pockley, A.G.; Ball, G.R.; Powe, D.G.; Regad, T. MTSS1 and SCAMP1 Cooperate to Prevent Invasion in Breast Cancer. Cell Death Dis 2018, 9, 344. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Wang, S.; Wang, J. PAQR3 Inhibits the Proliferation and Tumorigenesis in Esophageal Cancer Cells. Oncol Res 2017, 25, 663–671. [Google Scholar] [CrossRef]
- Bai, G.; Chu, J.; Eli, M.; Bao, Y.; Wen, H. PAQR3 Overexpression Suppresses the Aggressive Phenotype of Esophageal Squamous Cell Carcinoma Cells via Inhibition of ERK Signaling. Biomed. Pharm. 2017, 94, 813–819. [Google Scholar] [CrossRef]
- Bai, G.; Yang, M.; Zheng, C.; Zhang, L.; Eli, M. Suppressor PAQR3 Associated with the Clinical Significance and Prognosis in Esophageal Squamous Cell Carcinoma. Oncol. Lett. 2018, 15, 5703–5711. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Zhuang, K.; Li, H. PAQR3 Plays a Suppressive Role in Laryngeal Squamous Cell Carcinoma. Tumour Biol 2016, 37, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Ling, Z.Q.; Guo, W.; Lu, X.X.; Zhu, X.; Hong, L.L.; Wang, Z.; Wang, Z.; Chen, Y. A Golgi-Specific Protein PAQR3 Is Closely Associated with the Progression, Metastasis and Prognosis of Human Gastric Cancers. Ann. Oncol. 2014, 25, 1363–1372. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Guo, W.; You, X.; Pan, Y.; Dong, Z.; Jia, G.; Yang, C.; Chen, Y. PAQR3 Suppresses the Proliferation, Migration and Tumorigenicity of Human Prostate Cancer Cells. Oncotarget 2017, 8, 53948–53958. [Google Scholar] [CrossRef]
- Tang, S.-L.; Gao, Y.-L.; Hu, W.-Z. PAQR3 Inhibits the Proliferation, Migration and Invasion in Human Glioma Cells. Biomed. Pharm. 2017, 92, 24–32. [Google Scholar] [CrossRef]
- Guo, W.; You, X.; Xu, D.; Zhang, Y.; Wang, Z.; Man, K.; Wang, Z.; Chen, Y. PAQR3 Enhances Twist1 Degradation to Suppress Epithelial-Mesenchymal Transition and Metastasis of Gastric Cancer Cells. Carcinogenesis 2016, 37, 397–407. [Google Scholar] [CrossRef]
- Fan, F.; Feng, L.; He, J.; Wang, X.; Jiang, X.; Zhang, Y.; Wang, Z.; Chen, Y. RKTG Sequesters B-Raf to the Golgi Apparatus and Inhibits the Proliferation and Tumorigenicity of Human Malignant Melanoma Cells. Carcinogenesis 2008, 29, 1157–1163. [Google Scholar] [CrossRef]
- Xie, X.; Zhang, Y.; Jiang, Y.; Liu, W.; Ma, H.; Wang, Z.; Chen, Y. Suppressive Function of RKTG on Chemical Carcinogen-Induced Skin Carcinogenesis in Mouse. Carcinogenesis 2008, 29, 1632–1638. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Deng, N.; Wang, X.; Chen, Y.; Li, G.; Fan, H. RKTG Overexpression Inhibits Proliferation and Induces Apoptosis of Human Leukemia Cells via Suppression of the ERK and PI3K/AKT Signaling Pathways. Oncol. Lett. 2017, 14, 965–970. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, M.; Chen, D.; Shi, G.; Zhao, H. PAQR3 Inhibits Proliferation via Suppressing PI3K/AKT Signaling Pathway in Non-Small Cell Lung Cancer. Arch. Med. Sci. 2018, 14, 1289–1297. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, X.; Fan, F.; Jiao, S.; Wang, L.; Zhu, L.; Pan, Y.; Wu, G.; Ling, Z.-Q.; Fang, J.; et al. PAQR3 Plays a Suppressive Role in the Tumorigenesis of Colorectal Cancers. Carcinogenesis 2012, 33, 2228–2235. [Google Scholar] [CrossRef][Green Version]
- Zhang, Y.; Jiang, X.; Qin, X.; Ye, D.; Yi, Z.; Liu, M.; Bai, O.; Liu, W.; Xie, X.; Wang, Z.; et al. RKTG Inhibits Angiogenesis by Suppressing MAPK-Mediated Autocrine VEGF Signaling and Is Downregulated in Clear-Cell Renal Cell Carcinoma. Oncogene 2010, 29, 5404–5415. [Google Scholar] [CrossRef][Green Version]
- Jin, Y.; Dai, Z. USO1 Promotes Tumor Progression via Activating Erk Pathway in Multiple Myeloma Cells. Biomed. Pharm. 2016, 78, 264–271. [Google Scholar] [CrossRef]
- Xia, Y.; Wei, X.; Wu, S.; Wang, B.; Wang, X.; Hong, L. Down-Regulation of TERE1/UBIAD1 Activated Ras-MAPK Signalling and Induced Cell Proliferation. Cell Biol. Int. Rep. 2010, 17, e00005. [Google Scholar] [CrossRef]
- McGarvey, T.W.; Nguyen, T.; Puthiyaveettil, R.; Tomaszewski, J.E.; Malkowicz, S.B. TERE1, a Novel Gene Affecting Growth Regulation in Prostate Carcinoma. Prostate 2003, 54, 144–155. [Google Scholar] [CrossRef]
- Xu, Z.; Duan, F.; Lu, H.; Abdulkadhim Dragh, M.; Xia, Y.; Liang, H.; Hong, L. UBIAD1 Suppresses the Proliferation of Bladder Carcinoma Cells by Regulating H-Ras Intracellular Trafficking via Interaction with the C-Terminal Domain of H-Ras. Cell Death Dis. 2018, 9, 1170. [Google Scholar] [CrossRef]
- Aoh, Q.L.; Castle, A.M.; Hubbard, C.H.; Katsumata, O.; Castle, J.D. SCAMP3 Negatively Regulates Epidermal Growth Factor Receptor Degradation and Promotes Receptor Recycling. MBoC 2009, 20, 1816–1832. [Google Scholar] [CrossRef]
- Li, C.; Zhang, Z.; Lv, P.; Zhan, Y.; Zhong, Q. SCAMP3 Promotes Glioma Proliferation and Indicates Unfavorable Prognosis via Multiple Pathways. Onco Targets 2020, 13, 3677–3687. [Google Scholar] [CrossRef] [PubMed]
- Beaumatin, F.; O’Prey, J.; Barthet, V.J.A.; Zunino, B.; Parvy, J.-P.; Bachmann, A.M.; O’Prey, M.; Kania, E.; Gonzalez, P.S.; Macintosh, R.; et al. MTORC1 Activation Requires DRAM-1 by Facilitating Lysosomal Amino Acid Efflux. Mol. Cell 2019, 76, 163–176.e8. [Google Scholar] [CrossRef] [PubMed]
- Grice, D.M.; Vetter, I.; Faddy, H.M.; Kenny, P.A.; Roberts-Thomson, S.J.; Monteith, G.R. Golgi Calcium Pump Secretory Pathway Calcium ATPase 1 (SPCA1) Is a Key Regulator of Insulin-like Growth Factor Receptor (IGF1R) Processing in the Basal-like Breast Cancer Cell Line MDA-MB-231. J. Biol. Chem. 2010, 285, 37458–37466. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Liu, Y.; Yao, L.; Wang, D.; Zhang, J.; Cui, G.; Yang, X.; Huang, X.; Liu, F.; Shen, A. Spy1 Induces De-Ubiquitinating of RIP1 Arrest and Confers Glioblastoma’s Resistance to Tumor Necrosis Factor (TNF-α)-Induced Apoptosis through Suppressing the Association of CLIPR-59 and CYLD. Cell Cycle 2015, 14, 2149–2159. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Casar, B.; Badrock, A.P.; Jiménez, I.; Arozarena, I.; Colón-Bolea, P.; Lorenzo-Martín, L.F.; Barinaga-Rementería, I.; Barriuso, J.; Cappitelli, V.; Donoghue, D.J.; et al. RAS at the Golgi Antagonizes Malignant Transformation through PTPRκ-Mediated Inhibition of ERK Activation. Nat. Commun. 2018, 9, 3595. [Google Scholar] [CrossRef]
- Peng, H.; Zheng, J.; Su, Q.; Feng, X.; Peng, M.; Gong, L.; Wu, H.; Pan, X. VPS53 Suppresses Malignant Properties in Colorectal Cancer by Inducing the Autophagy Signaling Pathway. OTT 2020, 13, 10667–10675. [Google Scholar] [CrossRef]
- Du, C.; Zhang, C.; Li, Z.; Biswas, M.H.U.; Balaji, K.C. Beta-Catenin Phosphorylated at Threonine 120 Antagonizes Generation of Active Beta-Catenin by Spatial Localization in Trans-Golgi Network. PLoS ONE 2012, 7, e33830. [Google Scholar] [CrossRef]
- Du, C.; Zhang, C.; Hassan, S.; Biswas, M.H.U.; Balaji, K.C. Protein Kinase D1 Suppresses Epithelial-to-Mesenchymal Transition through Phosphorylation of Snail. Cancer Res. 2010, 70, 7810–7819. [Google Scholar] [CrossRef]
- Jaggi, M.; Rao, P.S.; Smith, D.J.; Wheelock, M.J.; Johnson, K.R.; Hemstreet, G.P.; Balaji, K.C. E-Cadherin Phosphorylation by Protein Kinase D1/Protein Kinase C{mu} Is Associated with Altered Cellular Aggregation and Motility in Prostate Cancer. Cancer Res. 2005, 65, 483–492. [Google Scholar] [CrossRef]
- Biswas, M.H.U.; Du, C.; Zhang, C.; Straubhaar, J.; Languino, L.R.; Balaji, K.C. Protein Kinase D1 Inhibits Cell Proliferation through Matrix Metalloproteinase-2 and Matrix Metalloproteinase-9 Secretion in Prostate Cancer. Cancer Res. 2010, 70, 2095–2104. [Google Scholar] [CrossRef][Green Version]
- Karam, M.; Legay, C.; Auclair, C.; Ricort, J.-M. Protein Kinase D1 Stimulates Proliferation and Enhances Tumorigenesis of MCF-7 Human Breast Cancer Cells through a MEK/ERK-Dependent Signaling Pathway. Exp. Cell Res. 2012, 318, 558–569. [Google Scholar] [CrossRef]
- Liou, G.-Y.; Döppler, H.; DelGiorno, K.E.; Zhang, L.; Leitges, M.; Crawford, H.C.; Murphy, M.P.; Storz, P. Mutant KRas-Induced Mitochondrial Oxidative Stress in Acinar Cells Upregulates EGFR Signaling to Drive Formation of Pancreatic Precancerous Lesions. Cell Rep 2016, 14, 2325–2336. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.-Y.; Döppler, H.; Braun, U.B.; Panayiotou, R.; Scotti Buzhardt, M.; Radisky, D.C.; Crawford, H.C.; Fields, A.P.; Murray, N.R.; Wang, Q.J.; et al. Protein Kinase D1 Drives Pancreatic Acinar Cell Reprogramming and Progression to Intraepithelial Neoplasia. Nat. Commun. 2015, 6, 6200. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.Y.; Park, E.Y.; Chang, E.; Kang, H.-G.; Koo, Y.; Lee, E.J.; Ko, J.Y.; Kong, H.K.; Chun, K.-H.; Park, J.H. A Novel MiR-34a Target, Protein Kinase D1, Stimulates Cancer Stemness and Drug Resistance through GSK3/β-Catenin Signaling in Breast Cancer. Oncotarget 2016, 7, 14791–14802. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Guo, Y.; Hao, J.; Guenter, R.; Lathia, J.; Beck, A.W.; Hattaway, R.; Hurst, D.; Wang, Q.J.; Liu, Y.; et al. Development of an Arteriolar Niche and Self-Renewal of Breast Cancer Stem Cells by Lysophosphatidic Acid/Protein Kinase D Signaling. Commun. Biol. 2021, 4, 780. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Cui, B.; Fan, Y.; Li, X.; Li, Q.; Du, Y.; Feng, Y.; Zhang, P. Protein Kinase D1 Regulates Hypoxic Metabolism through HIF-1α and Glycolytic Enzymes Incancer Cells. Oncol. Rep. 2018, 40, 1073–1082. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Khan, S.; Sekhri, R.; Mandil, H.; Behrman, S.; Yallapu, M.M.; Chauhan, S.C.; Jaggi, M. Protein Kinase D1 Regulates Metabolic Switch in Pancreatic Cancer via Modulation of MTORC1. Br. J. Cancer 2020, 122, 121–131. [Google Scholar] [CrossRef]
- Zhu, Y.; Cheng, Y.; Guo, Y.; Chen, J.; Chen, F.; Luo, R.; Li, A. Protein Kinase D2 Contributes to TNF-α-Induced Epithelial Mesenchymal Transition and Invasion via the PI3K/GSK-3β/β-Catenin Pathway in Hepatocellular Carcinoma. Oncotarget 2016, 7, 5327–5341. [Google Scholar] [CrossRef]
- Zou, Z.; Zeng, F.; Xu, W.; Wang, C.; Ke, Z.; Wang, Q.J.; Deng, F. PKD2 and PKD3 Promote Prostate Cancer Cell Invasion by Modulating NF-ΚB- and HDAC1-Mediated Expression and Activation of UPA. J. Cell Sci. 2012, 125, 4800–4811. [Google Scholar] [CrossRef]
- Zhou, X.; Xue, P.; Yang, M.; Shi, H.; Lu, D.; Wang, Z.; Shi, Q.; Hu, J.; Xie, S.; Zhan, W.; et al. Protein Kinase D2 Promotes the Proliferation of Glioma Cells by Regulating Golgi Phosphoprotein 3. Cancer Lett. 2014, 355, 121–129. [Google Scholar] [CrossRef]
- Wei, N.; Chu, E.; Wipf, P.; Schmitz, J.C. Protein Kinase d as a Potential Chemotherapeutic Target for Colorectal Cancer. Mol. Cancer 2014, 13, 1130–1141. [Google Scholar] [CrossRef] [PubMed]
- Azoitei, N.; Diepold, K.; Brunner, C.; Rouhi, A.; Genze, F.; Becher, A.; Kestler, H.; van Lint, J.; Chiosis, G.; Koren, J.; et al. HSP90 Supports Tumor Growth and Angiogenesis through PRKD2 Protein Stabilization. Cancer Res. 2014, 74, 7125–7136. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Giridhar, K.V.; Zhang, L.; Xu, S.; Wang, Q.J. A Protein Kinase C/Protein Kinase D Pathway Protects LNCaP Prostate Cancer Cells from Phorbol Ester-Induced Apoptosis by Promoting ERK1/2 and NF-{kappa}B Activities. Carcinogenesis 2011, 32, 1198–1206. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Deng, F.; Singh, S.V.; Wang, Q.J. Protein Kinase D3 (PKD3) Contributes to Prostate Cancer Cell Growth and Survival through a PKCepsilon/PKD3 Pathway Downstream of Akt and ERK 1/2. Cancer Res 2008, 68, 3844–3853. [Google Scholar] [CrossRef]
- Huck, B.; Duss, S.; Hausser, A.; Olayioye, M.A. Elevated Protein Kinase D3 (PKD3) Expression Supports Proliferation of Triple-Negative Breast Cancer Cells and Contributes to MTORC1-S6K1 Pathway Activation. J Biol Chem 2014, 289, 3138–3147. [Google Scholar] [CrossRef]
- Xu, W.; Qian, J.; Zeng, F.; Li, S.; Guo, W.; Chen, L.; Li, G.; Zhang, Z.; Wang, Q.J.; Deng, F. Protein Kinase Ds Promote Tumor Angiogenesis through Mast Cell Recruitment and Expression of Angiogenic Factors in Prostate Cancer Microenvironment. J Exp. Clin. Cancer Res. 2019, 38, 114. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.L. Activators and Effectors of the Small G Protein Arf1 in Regulation of Golgi Dynamics During the Cell Division Cycle. Front. Cell Dev. Biol. 2018, 6, 29. [Google Scholar] [CrossRef]
- Cohen, L.A.; Honda, A.; Varnai, P.; Brown, F.D.; Balla, T.; Donaldson, J.G. Active Arf6 Recruits ARNO/Cytohesin GEFs to the PM by Binding Their PH Domains. MBoC 2007, 18, 2244–2253. [Google Scholar] [CrossRef]
- Li, H.-S.; Shome, K.; Rojas, R.; Rizzo, M.A.; Vasudevan, C.; Fluharty, E.; Santy, L.C.; Casanova, J.E.; Romero, G. The Guanine Nucleotide Exchange Factor ARNO Mediates the Activation of ARF and Phospholipase D by Insulin. BMC Cell Biol. 2003, 4, 13. [Google Scholar] [CrossRef]
- Wei, Z.; Xu, X.; Fang, Y.; Khater, M.; Naughton, S.X.; Hu, G.; Terry, A.V.; Wu, G. Rab43 GTPase Directs Postsynaptic Trafficking and Neuron-Specific Sorting of G Protein–Coupled Receptors. J. Biol. Chem. 2021, 296, 100517. [Google Scholar] [CrossRef]
- Nakamura, N. Emerging New Roles of GM130, a Cis-Golgi Matrix Protein, in Higher Order Cell Functions. J Pharm. Sci 2010, 112, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Mascanzoni, F.; Iannitti, R.; Colanzi, A. Functional Coordination among the Golgi Complex, the Centrosome and the Microtubule Cytoskeleton during the Cell Cycle. Cells 2022, 11, 354. [Google Scholar] [CrossRef] [PubMed]
- Calvo, F.; Sanz-Moreno, V.; Agudo-Ibáñez, L.; Wallberg, F.; Sahai, E.; Marshall, C.J.; Crespo, P. RasGRF Suppresses Cdc42-Mediated Tumour Cell Movement, Cytoskeletal Dynamics and Transformation. Nat. Cell Biol. 2011, 13, 819–826. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A. Epithelial Plasticity: A Common Theme in Embryonic and Cancer Cells. Science 2013, 342, 1234850. [Google Scholar] [CrossRef]
- Byrne, A.-M.; Bekiaris, S.; Duggan, G.; Prichard, D.; Kirca, M.; Finn, S.; Reynolds, J.V.; Kelleher, D.; Long, A. Golgi Phosphoprotein 2 (GOLPH2) Is a Novel Bile Acid-Responsive Modulator of Oesophageal Cell Migration and Invasion. Br. J. Cancer 2015, 113, 1332–1342. [Google Scholar] [CrossRef][Green Version]
- Katayama, H.; Sasai, K.; Kawai, H.; Yuan, Z.-M.; Bondaruk, J.; Suzuki, F.; Fujii, S.; Arlinghaus, R.B.; Czerniak, B.A.; Sen, S. Phosphorylation by Aurora Kinase A Induces Mdm2-Mediated Destabilization and Inhibition of P53. Nat. Genet. 2004, 36, 55–62. [Google Scholar] [CrossRef]
- Murphy, T.; Darby, S.; Mathers, M.E.; Gnanapragasam, V.J. Evidence for Distinct Alterations in the FGF Axis in Prostate Cancer Progression to an Aggressive Clinical Phenotype. J. Pathol. 2010, 220, 452–460. [Google Scholar] [CrossRef]
- Grimaldi, G.; Filograna, A.; Schembri, L.; Lo Monte, M.; Di Martino, R.; Pirozzi, M.; Spano, D.; Beccari, A.R.; Parashuraman, S.; Luini, A.; et al. PKD-Dependent PARP12-Catalyzed Mono-ADP-Ribosylation of Golgin-97 Is Required for E-Cadherin Transport from Golgi to Plasma Membrane. Proc. Natl. Acad. Sci. USA 2022, 119, e2026494119. [Google Scholar] [CrossRef]
- Strating, J.R.P.M.; Martens, G.J.M. The P24 Family and Selective Transport Processes at the ER-Golgi Interface. Biol Cell 2009, 101, 495–509. [Google Scholar] [CrossRef]
- Xu, W.; Li, Y.; Ye, X.; Ji, Y.; Chen, Y.; Zhang, X.; Li, Z. TMED3/RPS15A Axis Promotes the Development and Progression of Osteosarcoma. Cancer Cell Int. 2021, 21, 630. [Google Scholar] [CrossRef]
- Buechling, T.; Chaudhary, V.; Spirohn, K.; Weiss, M.; Boutros, M. P24 Proteins Are Required for Secretion of Wnt Ligands. EMBO Rep. 2011, 12, 1265–1272. [Google Scholar] [CrossRef] [PubMed]
- Port, F.; Hausmann, G.; Basler, K. A Genome-Wide RNA Interference Screen Uncovers Two P24 Proteins as Regulators of Wingless Secretion. EMBO Rep. 2011, 12, 1144–1152. [Google Scholar] [CrossRef] [PubMed]
- Varnat, F.; Siegl-Cachedenier, I.; Malerba, M.; Gervaz, P.; Ruiz i Altaba, A. Loss of WNT-TCF Addiction and Enhancement of HH-GLI1 Signalling Define the Metastatic Transition of Human Colon Carcinomas. EMBO Mol. Med. 2010, 2, 440–457. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Liu, L.; Lin, X.; Wang, Y.; Li, Y.; Guo, Q.; Li, S.; Sun, Y.; Tao, X.; Zhang, D.; et al. A Translocation Pathway for Vesicle-Mediated Unconventional Protein Secretion. Cell 2020, 181, 637–652.e15. [Google Scholar] [CrossRef]
- Zhang, Y.E. Non-Smad Signaling Pathways of the TGF-β Family. Cold Spring Harb. Perspect. Biol. 2017, 9, a022129. [Google Scholar] [CrossRef]
- Hewavitharana, T.; Wedegaertner, P.B. PAQR3 Regulates Golgi Vesicle Fission and Transport via the Gβγ-PKD Signaling Pathway. Cell Signal 2015, 27, 2444–2451. [Google Scholar] [CrossRef]
- Lei, L.; Ling, Z.-N.; Chen, X.-L.; Hong, L.-L.; Ling, Z.-Q. Characterization of the Golgi Scaffold Protein PAQR3, and Its Role in Tumor Suppression and Metabolic Pathway Compartmentalization. Cancer Manag. Res. 2020, 12, 353–362. [Google Scholar] [CrossRef]
- Lounglaithong, K.; Bychkov, A.; Sampatanukul, P. Aberrant Promoter Methylation of the PAQR3 Gene Is Associated with Prostate Cancer. Pathol. Res. Pr. 2018, 214, 126–129. [Google Scholar] [CrossRef]
- Zhao, C.; Li, Y.; Chen, G.; Wang, F.; Shen, Z.; Zhou, R. Overexpression of MiR-15b-5p Promotes Gastric Cancer Metastasis by Regulating PAQR3. Oncol. Rep. 2017, 38, 352–358. [Google Scholar] [CrossRef]
- Qiao, S.; Guo, W.; Liao, L.; Wang, L.; Wang, Z.; Zhang, R.; Xu, D.; Zhang, Y.; Pan, Y.; Wang, Z.; et al. DDB2 Is Involved in Ubiquitination and Degradation of PAQR3 and Regulates Tumorigenesis of Gastric Cancer Cells. Biochem. J. 2015, 469, 469–480. [Google Scholar] [CrossRef]
- Feng, L.; Xie, X.; Ding, Q.; Luo, X.; He, J.; Fan, F.; Liu, W.; Wang, Z.; Chen, Y. Spatial Regulation of Raf Kinase Signaling by RKTG. Proc. Natl. Acad. Sci. USA 2007, 104, 14348–14353. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Xie, X.; Zhang, Y.; Luo, X.; Wang, X.; Fan, F.; Zheng, D.; Wang, Z.; Chen, Y. Regulation of G-Protein Signaling by RKTG via Sequestration of the G Betagamma Subunit to the Golgi Apparatus. Mol. Cell Biol. 2010, 30, 78–90. [Google Scholar] [CrossRef]
- Wang, X.; Wang, L.; Zhu, L.; Pan, Y.; Xiao, F.; Liu, W.; Wang, Z.; Guo, F.; Liu, Y.; Thomas, W.G.; et al. PAQR3 Modulates Insulin Signaling by Shunting Phosphoinositide 3-Kinase P110α to the Golgi Apparatus. Diabetes 2013, 62, 444–456. [Google Scholar] [CrossRef][Green Version]
- Van Lint, J.; Rykx, A.; Maeda, Y.; Vantus, T.; Sturany, S.; Malhotra, V.; Vandenheede, J.R.; Seufferlein, T. Protein Kinase D: An Intracellular Traffic Regulator on the Move. Trends Cell Biol. 2002, 12, 193–200. [Google Scholar] [CrossRef]
- Olayioye, M.A.; Barisic, S.; Hausser, A. Multi-Level Control of Actin Dynamics by Protein Kinase D. Cell Signal 2013, 25, 1739–1747. [Google Scholar] [CrossRef]
- Zhang, X.; Connelly, J.; Chao, Y.; Wang, Q.J. Multifaceted Functions of Protein Kinase D in Pathological Processes and Human Diseases. Biomolecules 2021, 11, 483. [Google Scholar] [CrossRef] [PubMed]
- Jaggi, M.; Rao, P.S.; Smith, D.J.; Hemstreet, G.P.; Balaji, K.C. Protein Kinase C μ Is Down-Regulated in Androgen-Independent Prostate Cancer. Biochem. Biophys. Res. Commun. 2003, 307, 254–260. [Google Scholar] [CrossRef]
- Kim, M.; Jang, H.-R.; Kim, J.-H.; Noh, S.-M.; Song, K.-S.; Cho, J.-S.; Jeong, H.-Y.; Norman, J.C.; Caswell, P.T.; Kang, G.H.; et al. Epigenetic Inactivation of Protein Kinase D1 in Gastric Cancer and Its Role in Gastric Cancer Cell Migration and Invasion. Carcinogenesis 2007, 29, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Eiseler, T.; Döppler, H.; Yan, I.K.; Goodison, S.; Storz, P. Protein Kinase D1 Regulates Matrix Metalloproteinase Expression and Inhibits Breast Cancer Cell Invasion. Breast Cancer Res. 2009, 11, R13. [Google Scholar] [CrossRef] [PubMed]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt Signaling in Cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef]
- Sui, J.; Li, X.; Xing, J.; Cao, F.; Wang, H.; Gong, H.; Zhang, W. Lentivirus-Mediated Silencing of USO1 Inhibits Cell Proliferation and Migration of Human Colon Cancer Cells. Med. Oncol. 2015, 32, 218. [Google Scholar] [CrossRef] [PubMed]
- Falguières, T.; Castle, D.; Gruenberg, J. Regulation of the MVB Pathway by SCAMP3. Traffic 2012, 13, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Suárez-Arroyo, I.J.; Feliz-Mosquea, Y.R.; Pérez-Laspiur, J.; Arju, R.; Giashuddin, S.; Maldonado-Martínez, G.; Cubano, L.A.; Schneider, R.J.; Martínez-Montemayor, M.M. The Proteome Signature of the Inflammatory Breast Cancer Plasma Membrane Identifies Novel Molecular Markers of Disease. Am. J. Cancer Res. 2016, 6, 1720–1740. [Google Scholar] [PubMed]
- Zhang, X.; Sheng, J.; Zhang, Y.; Tian, Y.; Zhu, J.; Luo, N.; Xiao, C.; Li, R. Overexpression of SCAMP3 Is an Indicator of Poor Prognosis in Hepatocellular Carcinoma. Oncotarget 2017, 8, 109247–109257. [Google Scholar] [CrossRef]
- Lissandron, V.; Podini, P.; Pizzo, P.; Pozzan, T. Unique Characteristics of Ca2+ Homeostasis of the Trans-Golgi Compartment. Proc. Natl. Acad. Sci. USA 2010, 107, 9198–9203. [Google Scholar] [CrossRef]
- Rigiracciolo, D.C.; Nohata, N.; Lappano, R.; Cirillo, F.; Talia, M.; Scordamaglia, D.; Gutkind, J.S.; Maggiolini, M. IGF-1/IGF-1R/FAK/YAP Transduction Signaling Prompts Growth Effects in Triple-Negative Breast Cancer (TNBC) Cells. Cells 2020, 9, 1010. [Google Scholar] [CrossRef]
- Steinhusen, U.; Weiske, J.; Badock, V.; Tauber, R.; Bommert, K.; Huber, O. Cleavage and Shedding of E-Cadherin after Induction of Apoptosis. J. Biol. Chem. 2001, 276, 4972–4980. [Google Scholar] [CrossRef]
- Fouquet, S.; Lugo-Martínez, V.-H.; Faussat, A.-M.; Renaud, F.; Cardot, P.; Chambaz, J.; Pinçon-Raymond, M.; Thenet, S. Early Loss of E-Cadherin from Cell-Cell Contacts Is Involved in the Onset of Anoikis in Enterocytes. J. Biol. Chem. 2004, 279, 43061–43069. [Google Scholar] [CrossRef]
- Azoitei, N.; Kleger, A.; Schoo, N.; Thal, D.R.; Brunner, C.; Pusapati, G.V.; Filatova, A.; Genze, F.; Möller, P.; Acker, T.; et al. Protein Kinase D2 Is a Novel Regulator of Glioblastoma Growth and Tumor Formation. Neuro-Oncol. 2011, 13, 710–724. [Google Scholar] [CrossRef]
- Wille, C.; Köhler, C.; Armacki, M.; Jamali, A.; Gössele, U.; Pfizenmaier, K.; Seufferlein, T.; Eiseler, T. Protein Kinase D2 Induces Invasion of Pancreatic Cancer Cells by Regulating Matrix Metalloproteinases. Mol. Biol. Cell 2014, 25, 324–336. [Google Scholar] [CrossRef]
- Hao, Q.; McKenzie, R.; Gan, H.; Tang, H. Protein Kinases D2 and D3 Are Novel Growth Regulators in HCC1806 Triple-Negative Breast Cancer Cells. Anticancer Res. 2013, 33, 393–399. [Google Scholar]
- Scott, K.L.; Kabbarah, O.; Liang, M.-C.; Ivanova, E.; Anagnostou, V.; Wu, J.; Dhakal, S.; Wu, M.; Chen, S.; Feinberg, T.; et al. GOLPH3 Modulates MTOR Signalling and Rapamycin Sensitivity in Cancer. Nature 2009, 459, 1085–1090. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, J.; Ma, Z.; Zhang, J.; Wang, Y.; Yu, Z.; Lin, X.; Xu, Z.; Su, Q.; An, L.; et al. Oncogenic Functions of Protein Kinase D2 and D3 in Regulating Multiple Cancer-Related Pathways in Breast Cancer. Cancer Med. 2019, 8, 729–741. [Google Scholar] [CrossRef] [PubMed]
- Fujikura, D.; Ito, M.; Chiba, S.; Harada, T.; Perez, F.; Reed, J.C.; Uede, T.; Miyazaki, T. CLIPR-59 Regulates TNF-α-Induced Apoptosis by Controlling Ubiquitination of RIP1. Cell Death Dis. 2012, 3, e264. [Google Scholar] [CrossRef][Green Version]
- McAndrew, C.W.; Gastwirt, R.F.; Donoghue, D.J. The Atypical CDK Activator Spy1 Regulates the Intrinsic DNA Damage Response and Is Dependent upon P53 to Inhibit Apoptosis. Cell Cycle 2009, 8, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Fehrenbacher, N.; Bar-Sagi, D.; Philips, M. Ras/MAPK Signaling from Endomembranes. Mol. Oncol. 2009, 3, 297–307. [Google Scholar] [CrossRef]
- de Ruiter, N.D.; Wolthuis, R.M.; van Dam, H.; Burgering, B.M.; Bos, J.L. Ras-Dependent Regulation of c-Jun Phosphorylation Is Mediated by the Ral Guanine Nucleotide Exchange Factor-Ral Pathway. Mol. Cell Biol. 2000, 20, 8480–8488. [Google Scholar] [CrossRef][Green Version]
- Ouwens, D.M.; de Ruiter, N.D.; van der Zon, G.C.M.; Carter, A.P.; Schouten, J.; van der Burgt, C.; Kooistra, K.; Bos, J.L.; Maassen, J.A.; van Dam, H. Growth Factors Can Activate ATF2 via a Two-Step Mechanism: Phosphorylation of Thr71 through the Ras-MEK-ERK Pathway and of Thr69 through RalGDS-Src-P38. EMBO J. 2002, 21, 3782–3793. [Google Scholar] [CrossRef]
- Fan, S.; Meng, Q.; Laterra, J.J.; Rosen, E.M. Ras Effector Pathways Modulate Scatter Factor-Stimulated NF-KappaB Signaling and Protection against DNA Damage. Oncogene 2007, 26, 4774–4796. [Google Scholar] [CrossRef]
- Huang, T.; Song, X.; Yang, Y.; Wan, X.; Alvarez, A.A.; Sastry, N.; Feng, H.; Hu, B.; Cheng, S.-Y. Autophagy and Hallmarks of Cancer. Crit. Rev. Oncol. 2018, 23, 247–267. [Google Scholar] [CrossRef]
- Fröhlich, F.; Petit, C.; Kory, N.; Christiano, R.; Hannibal-Bach, H.-K.; Graham, M.; Liu, X.; Ejsing, C.S.; Farese, R.V.; Walther, T.C. The GARP Complex Is Required for Cellular Sphingolipid Homeostasis. Elife 2015, 4, e08712. [Google Scholar] [CrossRef] [PubMed]
- Khakurel, A.; Kudlyk, T.; Bonifacino, J.S.; Lupashin, V.V. The Golgi-Associated Retrograde Protein (GARP) Complex Plays an Essential Role in the Maintenance of the Golgi Glycosylation Machinery. Mol. Biol. Cell 2021, 32, 1594–1610. [Google Scholar] [CrossRef] [PubMed]
- Luchsinger, C.; Aguilar, M.; Burgos, P.V.; Ehrenfeld, P.; Mardones, G.A. Functional Disruption of the Golgi Apparatus Protein ARF1 Sensitizes MDA-MB-231 Breast Cancer Cells to the Antitumor Drugs Actinomycin D and Vinblastine through ERK and AKT Signaling. PLoS ONE 2018, 13, e0195401. [Google Scholar] [CrossRef] [PubMed]
- Haines, E.; Schlienger, S.; Claing, A. The Small GTPase ADP-Ribosylation Factor 1 Mediates the Sensitivity of Triple Negative Breast Cancer Cells to EGFR Tyrosine Kinase Inhibitors. Cancer Biol. 2015, 16, 1535–1547. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Yi, J.; Song, Y.; Zhao, M.; Fan, L.; Zhao, L. Suppression of GOLM1 by EGCG through HGF/HGFR/AKT/GSK-3β/β-Catenin/c-Myc Signaling Pathway Inhibits Cell Migration of MDA-MB-231. Food Chem. Toxicol. 2021, 157, 112574. [Google Scholar] [CrossRef] [PubMed]
- Mishel, S.; Shneyer, B.; Korsensky, L.; Goldshmidt-Tran, O.; Haber, T.; Machluf, M.; Ron, D. Delivery of the Gene Encoding the Tumor Suppressor Sef into Prostate Tumors by Therapeutic-Ultrasound Inhibits Both Tumor Angiogenesis and Growth. Sci. Rep. 2017, 7, 15060. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Protein | Expression Change in Cancer | Function in Cancer | Molecular and Signaling Pathways Regulated | Cancer Hallmarks | References |
|---|---|---|---|---|---|
| ARF1 | Increased | Oncogene | ↑PI3K/AKT | ↑Migration | [19,20] |
| ↑Rac1 | ↑Invasion | [21] | |||
| ↑FAK | [22] | ||||
| ↑RhoA, RhoC | [23] | ||||
| ↑GPCR-Gβγ-PI3Kγ-ARF1-ERK | [24] | ||||
| ↑PI3K/AKT | |||||
| ↑pRb phosphorylation | ↑Proliferation | [19,20] | |||
| [25] | |||||
| GM130 | Increased | Oncogene | ↑Snail transcription | ↑Migration ↑Invasion | [26] |
| ↓Migration ↓Invasion | |||||
| Decreased | Tumor suppressor | ↑GC-localized Cdc42 | [27,28] | ||
| GOLM1 | Increased | Oncogene | ↑CREB expression | ↑Migration ↑Invasion | [29] |
| ↑MMP2 trafficking and transcription | ↑Metastasis | [30] | |||
| ↑EGFR(RTK)/AKT/S6K | |||||
| ↑GSK3β | [31] | ||||
| ↑PI3K/AKT/mTOR | [32] | ||||
| ↑TGF-β1/Smad2/Smad3 | [33,34] | ||||
| ↑Wnt/β-catenin | [35,36] | ||||
| ↓p53 stability | [37] | ||||
| [38] | |||||
| ↑PI3K/AKT/mTOR | ↑Proliferation | ||||
| ↑Wnt/β-catenin | ↑Tumor growth | [31,32,33,34,39] | |||
| [37] | |||||
| ↓Notch2 | ↓ Cancer inflammation | ||||
| Decreased | Tumor suppressor | [40] | |||
| Sef | Decreased | Tumor suppressor | ↓ERK MAPK | ↓Migration ↓Invasion | [41,42] |
| ↓JNK MAPK | ↓Metastasis | [43] | |||
| ↓p38 MAPK | [43] | ||||
| ↓Wnt/β-catenin | [44] | ||||
| ↓Proliferation | |||||
| ↓ERK MAPK | ↓Tumor growth | [41,42,45,46] | |||
| ↓Cancer inflammation | |||||
| ↓NF-kB | [47] | ||||
| ↓IRF | [48] | ||||
| ↓TLR | ↓Polyploidization | [48,49] | |||
| ↓Genomic instability | |||||
| ↓ERK1/2 MAPK | [50] | ||||
| Golgin-97 | Decreased | Tumor suppressor | ↓NF-κB | ↓Migration | [51] |
| ↓Invasion | |||||
| TMED2 | Increased | Oncogene | ↑IGF2/IGF1R/PI3K/AKT | ↑Migration | [52] |
| ↑Invasion | |||||
| ↑Proliferation | |||||
| TMED3 | Increased | Metastasis suppressor | ↑WNT-TCF | ↓Metastasis ↓Embryonic-like metastatic CSCs population | [53] |
| ↓HH-GLI signaling | |||||
| ↑Migration | |||||
| ↑Invasion | |||||
| Metastasis promoter | ↑Metastasis | ||||
| ↑IL-11/STAT3 | [54] | ||||
| ↑Migration | |||||
| Oncogene | ↑Invasion | ||||
| ↑Proliferation | |||||
| ↑Wnt/β-catenin | ↑Tumor growth | [55] | |||
| ↑AKT/GSK3β/β-catenin | [56] | ||||
| ↑Survival | |||||
| Oncogene | ↑Proliferation | ||||
| ↑Tumor growth | |||||
| ↑PI3K/AKT | ↓Apoptosis | [57] | |||
| ↓MAPK9/JNK2 | |||||
| ↓Apoptosis signaling | ↑Chemoresistance | ||||
| ↑AKT/GSK3β/β-catenin axis | [56] | ||||
| TMED9 | Increased | Metastasis promoter | ↓WNT-TCF | ↑Migration | [58] |
| ↑CNIH4/TGFα/GLI | ↑Metastasis | ||||
| TMED10 | Increased | Tumor suppressor | ↓TGF-β/Smad | ↓Migration | [59] |
| ↓ TGF-β/JNK MAPK | ↓Tumor growth | ||||
| ↓ TGF-β/p38 MAPK | |||||
| Oncogene | ↓PKCδ | ↓Apoptosis | [60] | ||
| ↓AMPK/mTOR | ↑Proliferation | [61] | |||
| SCAMP1 | Tumor suppressor | ↑MTSS1/Rac1-GTP axis | ↓Migration ↓Invasion | [62] | |
| RKTG/PAQR3 | Decreased | Tumor suppressor | ↓Ras/Raf/MEK/ERK | ↓Migration ↓Invasion | [63,64,65,66] |
| ↓PI3K/AKT | ↓Metastasis | [67,68,69] | |||
| ↓Twist1 stability | [70] | ||||
| ↓Proliferation | |||||
| ↓Ras/Raf/MEK/ERK | ↓Tumor growth | [63,64,66,68,71,72,73] | |||
| [68,69,73,74] | |||||
| ↓PI3K/AKT | [75] | ||||
| ↓WNT/ β-catenin | |||||
| ↓Angiogenesis | [76] | ||||
| ↓Ras/Raf/MEK/ERK | ↓Endothelial cells proliferation, migration and tube formation | ||||
| ↓VEGF/ERK axis | |||||
| USO1/p115 | Increased | Oncogene | ↑ERK1/2 MAPK | ↑Proliferation | [77] |
| UBIAD1 | Decreased | Tumor suppressor | ↓Ras MAPK | ↓Proliferation | [78,79,80] |
| SCAMP3 | Increased | Oncogene | ↑EGFR signaling | ↑Proliferation | [81,82] |
| ↑mTORC1 signaling | [82,83] | ||||
| SPCA1 | Increased | Oncogene | ↑IGF-1/IGF-1R/FAK/YAP | ↑Proliferation | [84] |
| CLIPR-59 | Decreased | Tumor suppressor | ↑Caspase-8 activation | ↑TNFα-mediated apoptosis | [85] |
| GC-localized Ras | ↑JNK MAPK | ↑Apoptosis | [86] | ||
| ↑p38 MAPK | |||||
| ↓NF-κB | |||||
| ↓ERK | |||||
| VPS53 | Decreased | Tumor suppressor | ↑Autophagy signaling | ↑Apoptosis | [87] |
| ↓Proliferation | |||||
| ↓Migration | |||||
| ↓Invasion | |||||
| PKD1 | Decreased | Tumor suppressor | ↓Wnt/β-catenin | ↓Migration ↓Invasion | [88] |
| ↓Snail activity | [89] | ||||
| ↑Interaction of E-cadherin with catenins | [90] | ||||
| ↑MEK/ERK | ↓Proliferation | ||||
| Increased | Tumor suppressor | [91] | |||
| ↑MEK/ERK | ↑Proliferation | ||||
| Increased | Oncogene | ↑Oncogenic Kras/ROS/PKD1/NF-κB | ↑Tumor growth | [92] | |
| ↑Proliferation | [93] | ||||
| ↑Notch | |||||
| ↑Malignant trasformation | |||||
| [94] | |||||
| ↑GSK3/β-catenin | ↑CSCs population | ||||
| ↑LPA/PKD1/ERK | ↑Cancer stemness | ||||
| [95] | |||||
| ↑GSK3/β-catenin | ↑Chemotherapy resistance | [96] | |||
| ↑Metabolic reprogrammimng | [95] | ||||
| ↑p38 MAPK/HIF-1α | |||||
| ↑mTORC1/pS6K, 4EBP1 | |||||
| [97] | |||||
| [98] | |||||
| PKD2 | Increased | Oncogene | ↑PI3K/AKT/ GSK3β/β-catenin axis | ↑Migration ↑Invasion | [99] |
| ↑NF-κB | ↑Metastasis | ||||
| [100] | |||||
| ↑ PI4KIIIβ/GOLPH3/PI3K/AKT/mTOR axis | ↑Proliferation | ||||
| ↑AKT, ERK, NF-κB | [101] | ||||
| ↑HIF-1α accumulation | |||||
| ↑NF-κB | [102] | ||||
| ↑Angiogenesis | |||||
| [103] | |||||
| PKD1/PKD2 | ↑ERK1/2 | ↑Survival | [104] | ||
| ↑NF-kB | ↓Apoptosis | ||||
| ↓SAKP/JNK | |||||
| PKD3 | Increased | Oncogene | ↓HDAC1 expression | ↑Migration ↑Invasion | [100] |
| ↑Metastasis | |||||
| ↑Proliferation | |||||
| ↑PI3K, p38, ERK1/2 | ↑Tumor growth | [105] | |||
| ↑mTORC1- S6K1 | [106] | ||||
| PKD2/3 | Increased | Oncogene | ↑ERK1/2 | ↑Tumor micro-environment remodeling | [107] |
| ↑NF-κB | ↑Angiogenesis |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spano, D.; Colanzi, A. Golgi Complex: A Signaling Hub in Cancer. Cells 2022, 11, 1990. https://doi.org/10.3390/cells11131990
Spano D, Colanzi A. Golgi Complex: A Signaling Hub in Cancer. Cells. 2022; 11(13):1990. https://doi.org/10.3390/cells11131990
Chicago/Turabian StyleSpano, Daniela, and Antonino Colanzi. 2022. "Golgi Complex: A Signaling Hub in Cancer" Cells 11, no. 13: 1990. https://doi.org/10.3390/cells11131990
APA StyleSpano, D., & Colanzi, A. (2022). Golgi Complex: A Signaling Hub in Cancer. Cells, 11(13), 1990. https://doi.org/10.3390/cells11131990