
Hematopoietic Stem Cell Gene-Addition/Editing Therapy in Sickle Cell Disease

 ,
,
Abstract
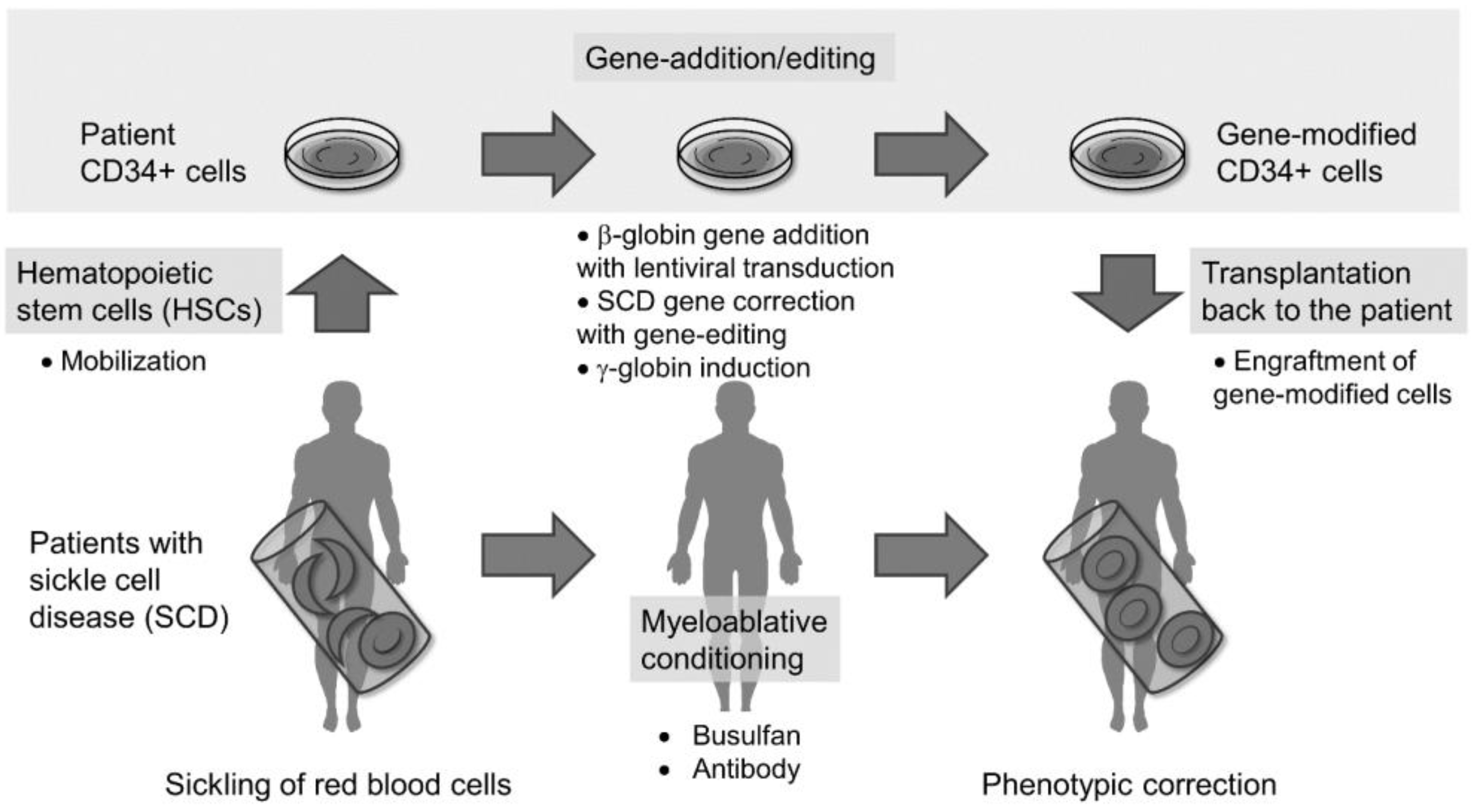
1. Introduction
2. HSC-Targeted Gene Therapy with Lentiviral Gene Addition in SCD
2.1. HSC Collection from Patients with SCD
2.2. HSC Culture Conditions for Gene Addition and Engraftment
2.3. Myeloablative Conditioning for Engraftment of Gene-Modified Cells
2.4. Potential Insertional Mutagenesis in Gene-Addition Therapy
2.5. Clinical Trials of Gene-Addition Therapy in SCD and β-Thalassemia
3. HSC-Targeted Gene-Editing Therapy in SCD
3.1. Endogenous DNA Repair Mechanisms
3.2. Engineered Endonucleases for Site-Specific DNA Break
3.3. Gene-Editing Delivery Methods
3.4. Gene Correction of the SCD Mutation with Gene Editing
3.5. Fetal Hemoglobin (HbF) Induction with Gene Editing
3.6. Base-Editing in SCD
3.7. Off-Target Effects with Gene Editing
3.8. Clinical Trials of Gene-Editing Therapy in SCD and β-Thalassemia
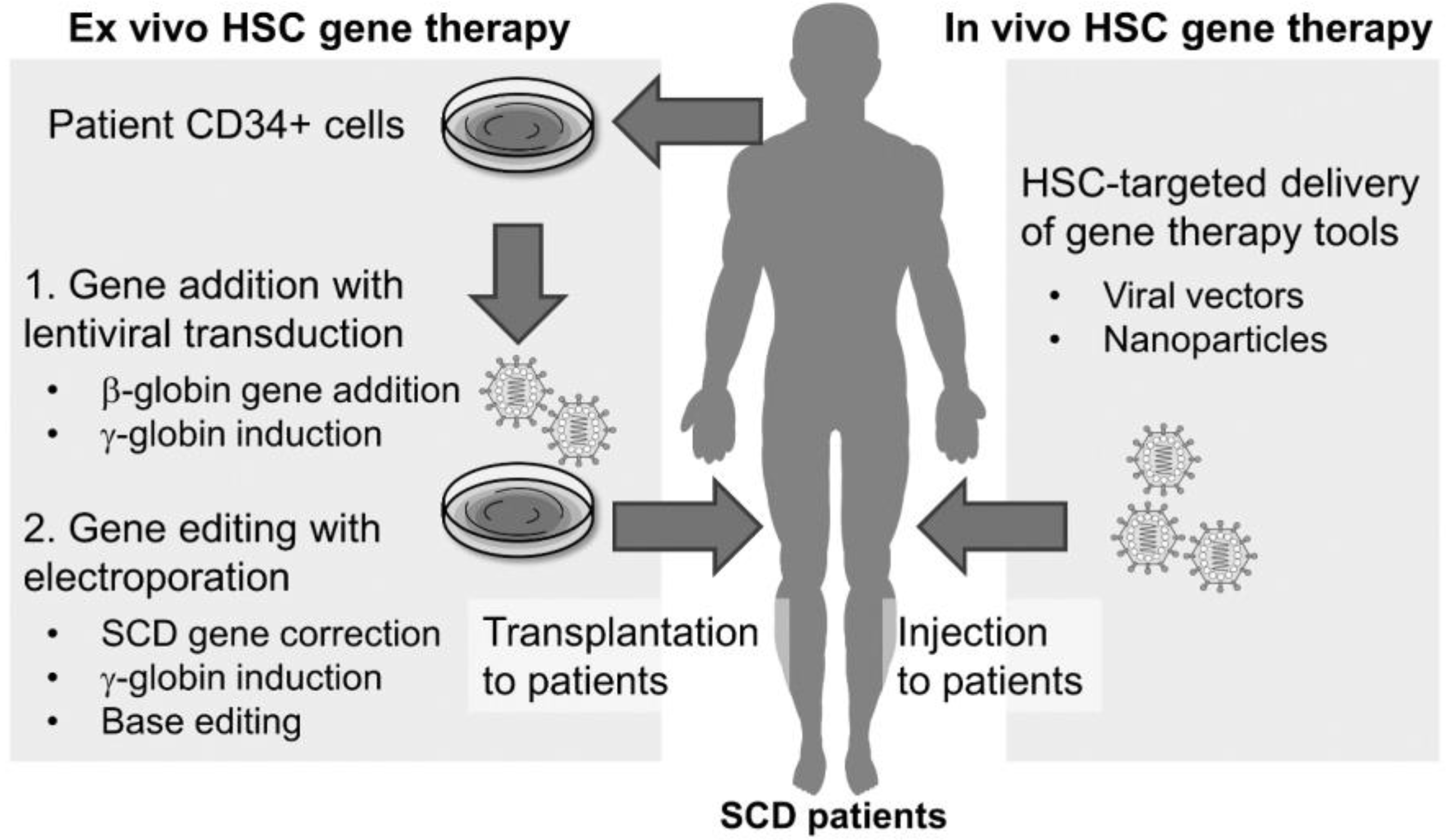
4. In Vivo Gene Editing for SCD
4.1. In Vivo Delivery with AAV Vectors
4.2. In Vivo Delivery with Ad Vectors
4.3. In Vivo Delivery with LVs
4.4. In vivo Delivery with Virus-Like Particles (VLPs)
4.5. In Vivo Delivery with LNPs
4.6. In Vivo Delivery with Polymer Nanoparticles (PNPs)
4.7. In Vivo Delivery with AuNPs
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Herrick, J.B. Peculiar elongated and sickle-shaped red blood corpuscles in a case of severe anemia. 1910. Yale J. Biol Med. 2001, 74, 179–184. [Google Scholar] [PubMed]
- Chakravorty, S.; Williams, T.N. Sickle cell disease: A neglected chronic disease of increasing global health importance. Arch. Dis. Child. 2015, 100, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Kato, G.J.; Piel, F.B.; Reid, C.D.; Gaston, M.H.; Ohene-Frempong, K.; Krishnamurti, L.; Smith, W.R.; Panepinto, J.A.; Weatherall, D.J.; Costa, F.F.; et al. Sickle cell disease. Nat. Rev. Dis Primers 2018, 4, 18010. [Google Scholar] [CrossRef] [PubMed]
- Eaton, W.A.; Hofrichter, J. Hemoglobin S gelation and sickle cell disease. Blood 1987, 70, 1245–1266. [Google Scholar] [CrossRef] [PubMed]
- Platt, O.S.; Brambilla, D.J.; Rosse, W.F.; Milner, P.F.; Castro, O.; Steinberg, M.H.; Klug, P.P. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N. Engl. J. Med. 1994, 330, 1639–1644. [Google Scholar] [CrossRef]
- Rund, D.; Rachmilewitz, E. Beta-thalassemia. N. Engl. J. Med. 2005, 353, 1135–1146. [Google Scholar] [CrossRef] [PubMed]
- Eaton, W.A.; Bunn, H.F. Treating sickle cell disease by targeting HbS polymerization. Blood 2017, 129, 2719–2726. [Google Scholar] [CrossRef]
- Ataga, K.I.; Kutlar, A.; Kanter, J.; Liles, D.; Cancado, R.; Friedrisch, J.; Guthrie, T.H.; Knight-Madden, J.; Alvarez, O.A.; Gordeuk, V.R.; et al. Crizanlizumab for the Prevention of Pain Crises in Sickle Cell Disease. N. Engl. J. Med. 2017, 376, 429–439. [Google Scholar] [CrossRef]
- Rai, P.; Ataga, K.I. Drug Therapies for the Management of Sickle Cell Disease. F1000Res 2020, 9. [Google Scholar] [CrossRef]
- Johnson, F.L.; Look, A.T.; Gockerman, J.; Ruggiero, M.R.; Dalla-Pozza, L.; Billings, F.T. 3rd. Bone-marrow transplantation in a patient with sickle-cell anemia. N. Engl. J. Med. 1984, 311, 780–783. [Google Scholar] [CrossRef]
- Gluckman, E.; Cappelli, B.; Bernaudin, F.; Labopin, M.; Volt, F.; Carreras, J.; Pinto Simões, B.; Ferster, A.; Dupont, S.; de la Fuente, J.; et al. Sickle cell disease: An international survey of results of HLA-identical sibling hematopoietic stem cell transplantation. Blood 2017, 129, 1548–1556. [Google Scholar] [CrossRef] [PubMed]
- Walters, M.C.; De Castro, L.M.; Sullivan, K.M.; Krishnamurti, L.; Kamani, N.; Bredeson, C.; Neuberg, D.; Hassell, K.L.; Farnia, S.; Campbell, A.; et al. Indications and Results of HLA-Identical Sibling Hematopoietic Cell Transplantation for Sickle Cell Disease. Biol. Blood Marrow Transplant. 2016, 22, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, M.M.; Kang, E.M.; Fitzhugh, C.D.; Link, M.B.; Bolan, C.D.; Kurlander, R.; Childs, R.W.; Rodgers, G.P.; Powell, J.D.; Tisdale, J.F. Allogeneic hematopoietic stem-cell transplantation for sickle cell disease. N. Engl. J. Med. 2009, 361, 2309–2317. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, M.M.; Fitzhugh, C.D.; Weitzel, R.P.; Link, M.E.; Coles, W.A.; Zhao, X.; Rodgers, G.P.; Powell, J.D.; Tisdale, J.F. Nonmyeloablative HLA-matched sibling allogeneic hematopoietic stem cell transplantation for severe sickle cell phenotype. Jama 2014, 312, 48–56. [Google Scholar] [CrossRef]
- Bernaudin, F.; Dalle, J.H.; Bories, D.; de Latour, R.P.; Robin, M.; Bertrand, Y.; Pondarre, C.; Vannier, J.P.; Neven, B.; Kuentz, M.; et al. Long-term event-free survival, chimerism and fertility outcomes in 234 patients with sickle-cell anemia younger than 30 years after myeloablative conditioning and matched-sibling transplantation in France. Haematologica 2020, 105, 91–101. [Google Scholar] [CrossRef]
- Abraham, A.; Cluster, A.; Jacobsohn, D.; Delgado, D.; Hulbert, M.L.; Kukadiya, D.; Murray, L.; Shenoy, S. Unrelated Umbilical Cord Blood Transplantation for Sickle Cell Disease Following Reduced-Intensity Conditioning: Results of a Phase I Trial. Biol Blood Marrow Transplant. 2017, 23, 1587–1592. [Google Scholar] [CrossRef]
- Booth, C.; Romano, R.; Roncarolo, M.G.; Thrasher, A.J. Gene therapy for primary immunodeficiency. Hum. Mol. Genet. 2019, 28. [Google Scholar] [CrossRef]
- Sii-Felice, K.; Giorgi, M.; Leboulch, P.; Payen, E. Hemoglobin disorders: Lentiviral gene therapy in the starting blocks to enter clinical practice. Exp. Hematol 2018, 64, 12–32. [Google Scholar] [CrossRef]
- Calado, R.T.; Clé, D.V. Treatment of inherited bone marrow failure syndromes beyond transplantation. Hematology Am. Soc. Hematol. Educ. Program. 2017, 2017, 96–101. [Google Scholar] [CrossRef]
- Chandler, R.J.; Venditti, C.P. Gene Therapy for Metabolic Diseases. Transl. Sci. Rare Dis. 2016, 1, 73–89. [Google Scholar] [CrossRef]
- Uchida, N.; Hsieh, M.M.; Raines, L.; Haro-Mora, J.J.; Demirci, S.; Bonifacino, A.C.; Krouse, A.E.; Metzger, M.E.; Donahue, R.E.; Tisdale, J.F. Development of a forward-oriented therapeutic lentiviral vector for hemoglobin disorders. Nat. Commun. 2019, 10, 4479. [Google Scholar] [CrossRef] [PubMed]
- Uchida, N.; Li, L.; Nassehi, T.; Drysdale, C.M.; Yapundich, M.; Gamer, J.; Haro-Mora, J.J.; Demirci, S.; Leonard, A.; Bonifacino, A.C.; et al. Preclinical evaluation for engraftment of CD34(+) cells gene-edited at the sickle cell disease locus in xenograft mouse and non-human primate models. Cell Rep. Med. 2021, 2, 100247. [Google Scholar] [CrossRef] [PubMed]
- Dever, D.P.; Bak, R.O.; Reinisch, A.; Camarena, J.; Washington, G.; Nicolas, C.E.; Pavel-Dinu, M.; Saxena, N.; Wilkens, A.B.; Mantri, S.; et al. CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nature 2016, 539, 384–389. [Google Scholar] [CrossRef] [PubMed]
- Uchida, N.; Fujita, A.; Hsieh, M.M.; Bonifacino, A.C.; Krouse, A.E.; Metzger, M.E.; Donahue, R.E.; Tisdale, J.F. Bone Marrow as a Hematopoietic Stem Cell Source for Gene Therapy in Sickle Cell Disease: Evidence from Rhesus and SCD Patients. Hum. Gene Ther. Clin. Dev. 2017, 28, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Leonard, A.; Bonifacino, A.; Dominical, V.M.; Luo, M.; Haro-Mora, J.J.; Demirci, S.; Uchida, N.; Pierciey, F.J., Jr.; Tisdale, J.F. Bone marrow characterization in sickle cell disease: Inflammation and stress erythropoiesis lead to suboptimal CD34 recovery. Br. J. Haematol. 2019, 186, 286–299. [Google Scholar] [CrossRef]
- Devine, S.M.; Vij, R.; Rettig, M.; Todt, L.; McGlauchlen, K.; Fisher, N.; Devine, H.; Link, D.C.; Calandra, G.; Bridger, G.; et al. Rapid mobilization of functional donor hematopoietic cells without G-CSF using AMD3100, an antagonist of the CXCR4/SDF-1 interaction. Blood 2008, 112, 990–998. [Google Scholar] [CrossRef] [PubMed]
- Ishii, A.; Jo, T.; Arai, Y.; Oshima, S.; Kanda, J.; Kitawaki, T.; Matsui, K.; Niwa, N.; Nakagawa, Y.; Takaori-Kondo, A.; et al. Development of a quantitative prediction model for peripheral blood stem cell collection yield in the plerixafor era. Cytotherapy 2022, 24, 49–58. [Google Scholar] [CrossRef]
- Malinowska, I.; Romiszewski, M.; Smalisz, K.; Stelmaszczyk-Emmel, A.; Nasilowska-Adamska, B.; Krol, M.; Urbanowska, E.; Brozyna, A.; Baginska-Dembowska, B. Plerixafor combined with G-CSF for stem cell mobilization in children qualified for autologous transplantation- single center experience. Transfus Apher. Sci. 2021, 60, 103077. [Google Scholar] [CrossRef]
- Leonard, A.; Sharma, A.; Uchida, N.; Stroncek, D.; Panch, S.R.; West, K.; Molloy, E.; Hughes, T.E.; Hauffe, S.; Taylor, T.; et al. Disease severity impacts plerixafor-mobilized stem cell collection in patients with sickle cell disease. Blood Adv. 2021, 5, 2403–2411. [Google Scholar] [CrossRef]
- Karpova, D.; Bräuninger, S.; Wiercinska, E.; Krämer, A.; Stock, B.; Graff, J.; Martin, H.; Wach, A.; Escot, C.; Douglas, G.; et al. Mobilization of hematopoietic stem cells with the novel CXCR4 antagonist POL6326 (balixafortide) in healthy volunteers-results of a dose escalation trial. J. Transl. Med. 2017, 15, 2. [Google Scholar] [CrossRef]
- Schmitt, S.; Weinhold, N.; Dembowsky, K.; Neben, K.; Witzens-Harig, M.; Braun, M.; Klemmer, J.; Wuchter, P.; Ludin, C.; Ho, A.D.; et al. First Results of a Phase-II Study with the New CXCR4 Antagonist POL6326 to Mobilize Hematopoietic Stem Cells (HSC) In Multiple Myeloma (MM). Blood 2010, 116, 824. [Google Scholar] [CrossRef]
- Abraham, M.; Biyder, K.; Begin, M.; Wald, H.; Weiss, I.D.; Galun, E.; Nagler, A.; Peled, A. Enhanced unique pattern of hematopoietic cell mobilization induced by the CXCR4 antagonist 4F-benzoyl-TN14003. Stem Cells 2007, 25, 2158–2166. [Google Scholar] [CrossRef] [PubMed]
- Domingues, M.J.; Nilsson, S.K.; Cao, B. New agents in HSC mobilization. Int. J. Hematol 2017, 105, 141–152. [Google Scholar] [CrossRef]
- Uchida, N.; Hsieh, M.M.; Hayakawa, J.; Madison, C.; Washington, K.N.; Tisdale, J.F. Optimal conditions for lentiviral transduction of engrafting human CD34+ cells. Gene Ther 2011, 18, 1078–1086. [Google Scholar] [CrossRef] [PubMed]
- Kluge, K.A.; Bonifacino, A.C.; Sellers, S.; Agricola, B.A.; Donahue, R.E.; Dunbar, C.E. Retroviral transduction and engraftment ability of primate hematopoietic progenitor and stem cells transduced under serum-free versus serum-containing conditions. Mol. Ther. 2002, 5, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, A.C.; Ishida, R.; Kikuchi, M.; Sudo, K.; Morita, M.; Crisostomo, R.V.; Yamamoto, R.; Loh, K.M.; Nakamura, Y.; Watanabe, M.; et al. Long-term ex vivo haematopoietic-stem-cell expansion allows nonconditioned transplantation. Nature 2019, 571, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Uchida, N.; Nassehi, T.; Drysdale, C.M.; Gamer, J.; Yapundich, M.; Demirci, S.; Haro-Mora, J.J.; Leonard, A.; Hsieh, M.M.; Tisdale, J.F. High-Efficiency Lentiviral Transduction of Human CD34(+) Cells in High-Density Culture with Poloxamer and Prostaglandin E2. Mol. Ther. Methods Clin. Dev. 2019, 13, 187–196. [Google Scholar] [CrossRef]
- Kanter, J.; Walters, M.C.; Krishnamurti, L.; Mapara, M.Y.; Kwiatkowski, J.L.; Rifkin-Zenenberg, S.; Aygun, B.; Kasow, K.A.; Pierciey, F.J., Jr.; Bonner, M.; et al. Biologic and Clinical Efficacy of LentiGlobin for Sickle Cell Disease. N. Engl. J. Med. 2022, 386, 617–628. [Google Scholar] [CrossRef]
- Copelan, E.A. Hematopoietic stem-cell transplantation. N. Engl. J. Med. 2006, 354, 1813–1826. [Google Scholar] [CrossRef]
- Bacigalupo, A.; Ballen, K.; Rizzo, D.; Giralt, S.; Lazarus, H.; Ho, V.; Apperley, J.; Slavin, S.; Pasquini, M.; Sandmaier, B.M.; et al. Defining the intensity of conditioning regimens: Working definitions. Biol. Blood Marrow Transplant. 2009, 15, 1628–1633. [Google Scholar] [CrossRef]
- Uchida, N.; Weitzel, R.P.; Shvygin, A.; Skala, L.P.; Raines, L.; Bonifacino, A.C.; Krouse, A.E.; Metzger, M.E.; Donahue, R.E.; Tisdale, J.F. Total body irradiation must be delivered at high dose for efficient engraftment and tolerance in a rhesus stem cell gene therapy model. Mol. Ther. Methods Clin. Dev. 2016, 3, 16059. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Uchida, N.; Nassehi, T.; Drysdale, C.M.; Gamer, J.; Yapundich, M.; Bonifacino, A.C.; Krouse, A.E.; Linde, N.; Hsieh, M.M.; Donahue, R.E.; et al. Busulfan Combined with Immunosuppression Allows Efficient Engraftment of Gene-Modified Cells in a Rhesus Macaque Model. Mol. Ther. 2019, 27, 1586–1596. [Google Scholar] [CrossRef] [PubMed]
- Drysdale, C.M.; Tisdale, J.F.; Uchida, N. Immunoresponse to Gene-Modified Hematopoietic Stem Cells. Mol. Ther Methods Clin. Dev. 2020, 16, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, M.E.; Aiuti, A. The Role of Conditioning in Hematopoietic Stem-Cell Gene Therapy. Hum. Gene Ther. 2016, 27, 741–748. [Google Scholar] [CrossRef]
- Mansilla-Soto, J.; Riviere, I.; Boulad, F.; Sadelain, M. Cell and Gene Therapy for the Beta-Thalassemias: Advances and Prospects. Hum. Gene Ther. 2016, 27, 295–304. [Google Scholar] [CrossRef]
- Czechowicz, A.; Palchaudhuri, R.; Scheck, A.; Hu, Y.; Hoggatt, J.; Saez, B.; Pang, W.W.; Mansour, M.K.; Tate, T.A.; Chan, Y.Y.; et al. Selective hematopoietic stem cell ablation using CD117-antibody-drug-conjugates enables safe and effective transplantation with immunity preservation. Nat. Commun 2019, 10, 617. [Google Scholar] [CrossRef]
- Kwon, H.S.; Logan, A.C.; Chhabra, A.; Pang, W.W.; Czechowicz, A.; Tate, K.; Le, A.; Poyser, J.; Hollis, R.; Kelly, B.V.; et al. Anti-human CD117 antibody-mediated bone marrow niche clearance in nonhuman primates and humanized NSG mice. Blood 2019, 133, 2104–2108. [Google Scholar] [CrossRef]
- Uchida, N.; Stasula, U.; Hinds, M.; Germino-Watnick, P.; Krouse, A.E.; Linde, N.S.; Bonifacino, A.C.; Latimer, K.; Bhattarai, P.; Yoder, N.C.; et al. CD117 Antibody Drug Conjugate-Based Conditioning Allows for Efficient Engraftment of Gene-Modified CD34+ Cells in a Rhesus Gene Therapy Model. Blood 2021, 138, 560. [Google Scholar] [CrossRef]
- Bankova, A.K.; Pang, W.W.; Velasco, B.J.; Long-Boyle, J.R.; Shizuru, J.A. 5-Azacytidine depletes HSCs and synergizes with an anti-CD117 antibody to augment donor engraftment in immunocompetent mice. Blood Adv. 2021, 5, 3900–3912. [Google Scholar] [CrossRef]
- Cavazzana, M.; Bushman, F.D.; Miccio, A.; André-Schmutz, I.; Six, E. Gene therapy targeting haematopoietic stem cells for inherited diseases: Progress and challenges. Nat. Rev. Drug Discov. 2019, 18, 447–462. [Google Scholar] [CrossRef]
- Habib, O.; Habib, G.; Hwang, G.H.; Bae, S. Comprehensive analysis of prime editing outcomes in human embryonic stem cells. Nucleic Acids Res. 2022, 50, 1187–1197. [Google Scholar] [CrossRef] [PubMed]
- Hacein-Bey-Abina, S.; von Kalle, C.; Schmidt, M.; Le Deist, F.; Wulffraat, N.; McIntyre, E.; Radford, I.; Villeval, J.L.; Fraser, C.C.; Cavazzana-Calvo, M.; et al. A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. N. Engl. J. Med. 2003, 348, 255–256. [Google Scholar] [CrossRef] [PubMed]
- Howe, S.J.; Mansour, M.R.; Schwarzwaelder, K.; Bartholomae, C.; Hubank, M.; Kempski, H.; Brugman, M.H.; Pike-Overzet, K.; Chatters, S.J.; de Ridder, D.; et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J. Clin. Investig. 2008, 118, 3143–3150. [Google Scholar] [CrossRef]
- Dull, T.; Zufferey, R.; Kelly, M.; Mandel, R.J.; Nguyen, M.; Trono, D.; Naldini, L. A third-generation lentivirus vector with a conditional packaging system. J. Virol. 1998, 72, 8463–8471. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, M.M.; Bonner, M.; Pierciey, F.J.; Uchida, N.; Rottman, J.; Demopoulos, L.; Schmidt, M.; Kanter, J.; Walters, M.C.; Thompson, A.A.; et al. Myelodysplastic syndrome unrelated to lentiviral vector in a patient treated with gene therapy for sickle cell disease. Blood Adv. 2020, 4, 2058–2063. [Google Scholar] [CrossRef]
- Goyal, S.; Tisdale, J.; Schmidt, M.; Kanter, J.; Jaroscak, J.; Whitney, D.; Bitter, H.; Gregory, P.D.; Parsons, G.; Foos, M.; et al. Acute Myeloid Leukemia Case after Gene Therapy for Sickle Cell Disease. N. Engl. J. Med. 2022, 386, 138–147. [Google Scholar] [CrossRef]
- Brunson, A.; Keegan, T.H.M.; Bang, H.; Mahajan, A.; Paulukonis, S.; Wun, T. Increased risk of leukemia among sickle cell disease patients in California. Blood 2017, 130, 1597–1599. [Google Scholar] [CrossRef]
- Li, Y.; Maule, J.; Neff, J.L.; McCall, C.M.; Rapisardo, S.; Lagoo, A.S.; Yang, L.H.; Crawford, R.D.; Zhao, Y.; Wang, E. Myeloid neoplasms in the setting of sickle cell disease: An intrinsic association with the underlying condition rather than a coincidence; report of 4 cases and review of the literature. Mod. Pathol. 2019, 32, 1712–1726. [Google Scholar] [CrossRef]
- Ghannam, J.Y.; Xu, X.; Maric, I.; Dillon, L.; Li, Y.; Hsieh, M.M.; Hourigan, C.S.; Fitzhugh, C.D. Baseline TP53 mutations in adults with SCD developing myeloid malignancy following hematopoietic cell transplantation. Blood 2020, 135, 1185–1188. [Google Scholar] [CrossRef]
- Cavazzana-Calvo, M.; Payen, E.; Negre, O.; Wang, G.; Hehir, K.; Fusil, F.; Down, J.; Denaro, M.; Brady, T.; Westerman, K.; et al. Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia. Nature 2010, 467, 318–322. [Google Scholar] [CrossRef]
- Uchida, N.; Washington, K.N.; Lap, C.J.; Hsieh, M.M.; Tisdale, J.F. Chicken HS4 insulators have minimal barrier function among progeny of human hematopoietic cells transduced with an HIV1-based lentiviral vector. Mol. Ther. 2011, 19, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Magrin, E.; Semeraro, M.; Hebert, N.; Joseph, L.; Magnani, A.; Chalumeau, A.; Gabrion, A.; Roudaut, C.; Marouene, J.; Lefrere, F.; et al. Long-term outcomes of lentiviral gene therapy for the β-hemoglobinopathies: The HGB-205 trial. Nat. Med. 2022, 28, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, J.L.; Thompson, A.A.; Rasko, J.E.J.; Hongeng, S.; Schiller, G.J.; Anurathapan, U.; Cavazzana, M.; Ho, P.J.; Schmidt, M.; Kletzel, M.; et al. Long-Term Clinical Outcomes of Lentiglobin Gene Therapy for Transfusion-Dependent β-Thalassemia in the Northstar (HGB-204) Study. Blood 2019, 134, 4628. [Google Scholar] [CrossRef]
- Thompson, A.A.; Walters, M.C.; Kwiatkowski, J.; Rasko, J.E.J.; Ribeil, J.A.; Hongeng, S.; Magrin, E.; Schiller, G.J.; Payen, E.; Semeraro, M.; et al. Gene Therapy in Patients with Transfusion-Dependent β-Thalassemia. N. Engl. J. Med. 2018, 378, 1479–1493. [Google Scholar] [CrossRef]
- Thompson, A.A.; Kwiatkowski, J.L.; Porter, J.B.; Hongeng, S.; Yannaki, E.; Kulozik, A.E.; Sauer, M.G.; Thrasher, A.J.; Thuret, I.; Lal, A.; et al. Favorable Outcomes in Pediatric Patients in the Phase 3 Hgb-207 (Northstar-2) and Hgb-212 (Northstar-3) Studies of Betibeglogene Autotemcel Gene Therapy for the Treatment of Transfusion-Dependent β-Thalassemia. Blood 2020, 136, 52–54. [Google Scholar] [CrossRef]
- Leonard, A.; Tisdale, J.F. A pause in gene therapy: Reflecting on the unique challenges of sickle cell disease. Mol. Ther 2021, 29, 1355–1356. [Google Scholar] [CrossRef]
- Boulad, F.; Riviere, I.; Wang, X.; Bartido, S.; Prockop, S.E.; Barone, R.; Moi, P.; Maggio, A.; Sadelain, M. First US Phase I Clinical Trial Of Globin Gene Transfer For The Treatment Of Beta-Thalassemia Major. Blood 2013, 122, 716. [Google Scholar] [CrossRef]
- Marktel, S.; Scaramuzza, S.; Cicalese, M.P.; Giglio, F.; Galimberti, S.; Lidonnici, M.R.; Calbi, V.; Assanelli, A.; Bernardo, M.E.; Rossi, C.; et al. Intrabone hematopoietic stem cell gene therapy for adult and pediatric patients affected by transfusion-dependent ß-thalassemia. Nat. Med. 2019, 25, 234–241. [Google Scholar] [CrossRef]
- Miccio, A.; Cesari, R.; Lotti, F.; Rossi, C.; Sanvito, F.; Ponzoni, M.; Routledge, S.J.; Chow, C.M.; Antoniou, M.N.; Ferrari, G. In vivo selection of genetically modified erythroblastic progenitors leads to long-term correction of beta-thalassemia. Proc. Natl. Acad. Sci. USA 2008, 105, 10547–10552. [Google Scholar] [CrossRef]
- Kanter, J.; Tisdale, J.F.; Mapara, M.Y.; Kwiatkowski, J.L.; Krishnamurti, L.; Schmidt, M.; Miller, A.L.; Pierciey, F.J., Jr.; Huang, W.; Ribeil, J.-A.; et al. Resolution of Sickle Cell Disease Manifestations in Patients Treated with Lentiglobin Gene Therapy: Updated Results from the Phase 1/2 Hgb-206 Group C Study. Blood 2019, 134, 990. [Google Scholar] [CrossRef]
- Tisdale, J.F.; Thompson, A.A.; Mapara, M.Y.; Kwiatkowski, J.L.; Krishnamurti, L.; Aygun, B.; Kasow, K.A.; Rifkin-Zenenberg, S.; Schmidt, M.; Pierciey, F.J., Jr.; et al. Polyclonality Strongly Correlates with Biological Outcomes and Is Significantly Increased Following Improvements to the Phase 1/2 HGB-206 Protocol and Manufacturing of LentiGlobin for Sickle Cell Disease (SCD; bb1111) Gene Therapy (GT). Blood 2021, 138, 561. [Google Scholar] [CrossRef]
- Grimley, M.; Asnani, M.; Shrestha, A.; Felker, S.; Lutzko, C.; Arumugam, P.I.; Witting, S.; Knight-Madden, J.; Niss, O.; Quinn, C.T.; et al. Early Results from a Phase 1/2 Study of Aru-1801 Gene Therapy for Sickle Cell Disease (SCD): Manufacturing Process Enhancements Improve Efficacy of a Modified Gamma Globin Lentivirus Vector and Reduced Intensity Conditioning Transplant. Blood 2020, 136, 20–21. [Google Scholar] [CrossRef]
- Magrin, E.; Magnani, A.; Semeraro, M.; Hebert, N.; Joseph, L.; Leblanc, O.; Gabrion, A.; Roudaut, C.; Maulet, A.; Chalumeau, A.; et al. Clinical Results of the Drepaglobe Trial for Sickle Cell Disease Patients. Blood 2021, 138, 1854. [Google Scholar] [CrossRef]
- Esrick, E.B.; Lehmann, L.E.; Biffi, A.; Achebe, M.; Brendel, C.; Ciuculescu, M.F.; Daley, H.; MacKinnon, B.; Morris, E.; Federico, A.; et al. Post-Transcriptional Genetic Silencing of BCL11A to Treat Sickle Cell Disease. N. Engl. J. Med. 2021, 384, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Bibikova, M.; Golic, M.; Golic, K.G.; Carroll, D. Targeted chromosomal cleavage and mutagenesis in Drosophila using zinc-finger nucleases. Genetics 2002, 161, 1169–1175. [Google Scholar] [CrossRef] [PubMed]
- Sung, P.; Klein, H. Mechanism of homologous recombination: Mediators and helicases take on regulatory functions. Nat. Rev. Mol. Cell Biol. 2006, 7, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kim, J.S. A guide to genome engineering with programmable nucleases. Nat. Rev. Genet. 2014, 15, 321–334. [Google Scholar] [CrossRef]
- Hoban, M.D.; Lumaquin, D.; Kuo, C.Y.; Romero, Z.; Long, J.; Ho, M.; Young, C.S.; Mojadidi, M.; Fitz-Gibbon, S.; Cooper, A.R.; et al. CRISPR/Cas9-Mediated Correction of the Sickle Mutation in Human CD34+ cells. Mol. Ther 2016, 24, 1561–1569. [Google Scholar] [CrossRef]
- Zhang, M.; Yang, C.; Tasan, I.; Zhao, H. Expanding the Potential of Mammalian Genome Engineering via Targeted DNA Integration. ACS Synth. Biol. 2021, 10, 429–446. [Google Scholar] [CrossRef]
- Hsu, P.D.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef]
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reyon, D.; Joung, J.K.; Sander, J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Unti, M.J.; Aleshe, B.; Brown, D.; Osborne, K.S.; Koziol, C.; Ayoub, P.G.; Smith, O.B.; O’Brien, R.; Tam, C.; et al. Improved Titer and Gene Transfer by Lentiviral Vectors Using Novel, Small β-Globin Locus Control Region Elements. Mol. Ther. 2020, 28, 328–340. [Google Scholar] [CrossRef] [PubMed]
- Simhadri, V.L.; McGill, J.; McMahon, S.; Wang, J.; Jiang, H.; Sauna, Z.E. Prevalence of Pre-existing Antibodies to CRISPR-Associated Nuclease Cas9 in the USA Population. Mol. Ther. Methods Clin. Dev. 2018, 10, 105–112. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, L.; Liu, H.; Cheng, K. Delivery strategies of the CRISPR-Cas9 gene-editing system for therapeutic applications. J. Control Release 2017, 266, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Flotte, T.R. Gene therapy progress and prospects: Recombinant adeno-associated virus (rAAV) vectors. Gene Ther. 2004, 11, 805–810. [Google Scholar] [CrossRef] [PubMed]
- DeWitt, M.A.; Magis, W.; Bray, N.L.; Wang, T.; Berman, J.R.; Urbinati, F.; Heo, S.J.; Mitros, T.; Muñoz, D.P.; Boffelli, D.; et al. Selection-free genome editing of the sickle mutation in human adult hematopoietic stem/progenitor cells. Sci. Transl. Med. 2016, 8, 360ra134. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.M.; Ikeda, K.; Cromer, M.K.; Uchida, N.; Nishimura, T.; Romano, R.; Tong, A.J.; Lemgart, V.T.; Camarena, J.; Pavel-Dinu, M.; et al. Highly Efficient and Marker-free Genome Editing of Human Pluripotent Stem Cells by CRISPR-Cas9 RNP and AAV6 Donor-Mediated Homologous Recombination. Cell Stem Cell 2019, 24, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Vakulskas, C.A.; Dever, D.P.; Rettig, G.R.; Turk, R.; Jacobi, A.M.; Collingwood, M.A.; Bode, N.M.; McNeill, M.S.; Yan, S.; Camarena, J.; et al. A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nat. Med. 2018, 24, 1216–1224. [Google Scholar] [CrossRef]
- Lomova, A.; Clark, D.N.; Campo-Fernandez, B.; Flores-Bjurström, C.; Kaufman, M.L.; Fitz-Gibbon, S.; Wang, X.; Miyahira, E.Y.; Brown, D.; DeWitt, M.A.; et al. Improving Gene Editing Outcomes in Human Hematopoietic Stem and Progenitor Cells by Temporal Control of DNA Repair. Stem Cells 2019, 37, 284–294. [Google Scholar] [CrossRef]
- Fitzhugh, C.D.; Cordes, S.; Taylor, T.; Coles, W.; Roskom, K.; Link, M.; Hsieh, M.M.; Tisdale, J.F. At least 20% donor myeloid chimerism is necessary to reverse the sickle phenotype after allogeneic HSCT. Blood 2017, 130, 1946–1948. [Google Scholar] [CrossRef]
- Drysdale, C.M.; Nassehi, T.; Gamer, J.; Yapundich, M.; Tisdale, J.F.; Uchida, N. Hematopoietic-Stem-Cell-Targeted Gene-Addition and Gene-Editing Strategies for β-hemoglobinopathies. Cell Stem Cell 2021, 28, 191–208. [Google Scholar] [CrossRef] [PubMed]
- Demirci, S.; Uchida, N.; Tisdale, J.F. Gene therapy for sickle cell disease: An update. Cytotherapy 2018, 20, 899–910. [Google Scholar] [CrossRef] [PubMed]
- Charache, S.; Terrin, M.L.; Moore, R.D.; Dover, G.J.; Barton, F.B.; Eckert, S.V.; McMahon, R.P.; Bonds, D.R. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. N. Engl. J. Med. 1995, 332, 1317–1322. [Google Scholar] [CrossRef] [PubMed]
- Yahouédéhou, S.; Adorno, E.V.; da Guarda, C.C.; Ndidi, U.S.; Carvalho, S.P.; Santiago, R.P.; Aleluia, M.M.; de Oliveira, R.M.; Gonçalves, M.S. Hydroxyurea in the management of sickle cell disease: Pharmacogenomics and enzymatic metabolism. Pharmacogenomics J. 2018, 18, 730–739. [Google Scholar] [CrossRef]
- Antoniani, C.; Meneghini, V.; Lattanzi, A.; Felix, T.; Romano, O.; Magrin, E.; Weber, L.; Pavani, G.; El Hoss, S.; Kurita, R.; et al. Induction of fetal hemoglobin synthesis by CRISPR/Cas9-mediated editing of the human β-globin locus. Blood 2018, 131, 1960–1973. [Google Scholar] [CrossRef]
- Humbert, O.; Peterson, C.W.; Norgaard, Z.K.; Radtke, S.; Kiem, H.P. A Nonhuman Primate Transplantation Model to Evaluate Hematopoietic Stem Cell Gene Editing Strategies for β-Hemoglobinopathies. Mol. Ther. Methods Clin. Dev. 2018, 8, 75–86. [Google Scholar] [CrossRef]
- Liu, N.; Hargreaves, V.V.; Zhu, Q.; Kurland, J.V.; Hong, J.; Kim, W.; Sher, F.; Macias-Trevino, C.; Rogers, J.M.; Kurita, R.; et al. Direct Promoter Repression by BCL11A Controls the Fetal to Adult Hemoglobin Switch. Cell 2018, 173, 430–442. [Google Scholar] [CrossRef]
- Sankaran, V.G.; Menne, T.F.; Xu, J.; Akie, T.E.; Lettre, G.; Van Handel, B.; Mikkola, H.K.; Hirschhorn, J.N.; Cantor, A.B.; Orkin, S.H. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science 2008, 322, 1839–1842. [Google Scholar] [CrossRef]
- Xu, J.; Peng, C.; Sankaran, V.G.; Shao, Z.; Esrick, E.B.; Chong, B.G.; Ippolito, G.C.; Fujiwara, Y.; Ebert, B.L.; Tucker, P.W.; et al. Correction of sickle cell disease in adult mice by interference with fetal hemoglobin silencing. Science 2011, 334, 993–996. [Google Scholar] [CrossRef]
- Canver, M.C.; Smith, E.C.; Sher, F.; Pinello, L.; Sanjana, N.E.; Shalem, O.; Chen, D.D.; Schupp, P.G.; Vinjamur, D.S.; Garcia, S.P.; et al. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature 2015, 527, 192–197. [Google Scholar] [CrossRef]
- Vierstra, J.; Reik, A.; Chang, K.H.; Stehling-Sun, S.; Zhou, Y.; Hinkley, S.J.; Paschon, D.E.; Zhang, L.; Psatha, N.; Bendana, Y.R.; et al. Functional footprinting of regulatory DNA. Nat. Methods 2015, 12, 927–930. [Google Scholar] [CrossRef] [PubMed]
- Bauer, D.E.; Kamran, S.C.; Lessard, S.; Xu, J.; Fujiwara, Y.; Lin, C.; Shao, Z.; Canver, M.C.; Smith, E.C.; Pinello, L.; et al. An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science 2013, 342, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Psatha, N.; Reik, A.; Phelps, S.; Zhou, Y.; Dalas, D.; Yannaki, E.; Levasseur, D.N.; Urnov, F.D.; Holmes, M.C.; Papayannopoulou, T. Disruption of the BCL11A Erythroid Enhancer Reactivates Fetal Hemoglobin in Erythroid Cells of Patients with β-Thalassemia Major. Mol. Ther. Methods Clin. Dev. 2018, 10, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zeng, J.; Roscoe, B.P.; Liu, P.; Yao, Q.; Lazzarotto, C.R.; Clement, K.; Cole, M.A.; Luk, K.; Baricordi, C.; et al. Highly efficient therapeutic gene editing of human hematopoietic stem cells. Nat. Med. 2019, 25, 776–783. [Google Scholar] [CrossRef] [PubMed]
- Demirci, S.; Zeng, J.; Wu, Y.; Uchida, N.; Shen, A.H.; Pellin, D.; Gamer, J.; Yapundich, M.; Drysdale, C.; Bonanno, J.; et al. BCL11A enhancer-edited hematopoietic stem cells persist in rhesus monkeys without toxicity. J. Clin. Investig. 2020, 130, 6677–6687. [Google Scholar] [CrossRef] [PubMed]
- Martyn, G.E.; Wienert, B.; Yang, L.; Shah, M.; Norton, L.J.; Burdach, J.; Kurita, R.; Nakamura, Y.; Pearson, R.C.M.; Funnell, A.P.W.; et al. Natural regulatory mutations elevate the fetal globin gene via disruption of BCL11A or ZBTB7A binding. Nat. Genet. 2018, 50, 498–503. [Google Scholar] [CrossRef]
- Weber, L.; Frati, G.; Felix, T.; Hardouin, G.; Casini, A.; Wollenschlaeger, C.; Meneghini, V.; Masson, C.; De Cian, A.; Chalumeau, A.; et al. Editing a γ-globin repressor binding site restores fetal hemoglobin synthesis and corrects the sickle cell disease phenotype. Sci Adv. 2020, 6. [Google Scholar] [CrossRef]
- Gillinder, K.R.; Reed, C.L.; Malelang, S.; Mitchell, H.L.; Hoskin, E.; Magor, G.W.; Kaplan, Z.; Perkins, A.C. Gene Editing of KLF1 to Cure Sickle Cell Disease. Blood 2020, 136, 30–31. [Google Scholar] [CrossRef]
- Métais, J.Y.; Doerfler, P.A.; Mayuranathan, T.; Bauer, D.E.; Fowler, S.C.; Hsieh, M.M.; Katta, V.; Keriwala, S.; Lazzarotto, C.R.; Luk, K.; et al. Genome editing of HBG1 and HBG2 to induce fetal hemoglobin. Blood Adv. 2019, 3, 3379–3392. [Google Scholar] [CrossRef]
- Lakin, N.D.; Jackson, S.P. Regulation of p53 in response to DNA damage. Oncogene 1999, 18, 7644–7655. [Google Scholar] [CrossRef]
- Kosicki, M.; Tomberg, K.; Bradley, A. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018, 36, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Doerfler, P.A.; Feng, R.; Li, Y.; Palmer, L.E.; Porter, S.N.; Bell, H.W.; Crossley, M.; Pruett-Miller, S.M.; Cheng, Y.; Weiss, M.J. Activation of γ-globin gene expression by GATA1 and NF-Y in hereditary persistence of fetal hemoglobin. Nat. Genet. 2021, 53, 1177–1186. [Google Scholar] [CrossRef] [PubMed]
- Bauer, D.E.; Orkin, S.H. Update on fetal hemoglobin gene regulation in hemoglobinopathies. Curr. Opin. Pediatr. 2011, 23, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ravi, N.S.; Wienert, B.; Wyman, S.K.; Bell, H.W.; George, A.; Mahalingam, G.; Vu, J.T.; Prasad, K.; Bandlamudi, B.P.; Devaraju, N.; et al. Identification of novel HPFH-like mutations by CRISPR base editing that elevate the expression of fetal hemoglobin. Elife 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [PubMed]
- Komor, A.C.; Zhao, K.T.; Packer, M.S.; Gaudelli, N.M.; Waterbury, A.L.; Koblan, L.W.; Kim, Y.B.; Badran, A.H.; Liu, D.R. Improved base excision repair inhibition and bacteriophage Mu Gam protein yields C:G-to-T:A base editors with higher efficiency and product purity. Sci. Adv. 2017, 3, eaao4774. [Google Scholar] [CrossRef]
- Zeng, J.; Wu, Y.; Ren, C.; Bonanno, J.; Shen, A.H.; Shea, D.; Gehrke, J.M.; Clement, K.; Luk, K.; Yao, Q.; et al. Therapeutic base editing of human hematopoietic stem cells. Nat. Med. 2020, 26, 535–541. [Google Scholar] [CrossRef]
- Liang, P.; Ding, C.; Sun, H.; Xie, X.; Xu, Y.; Zhang, X.; Sun, Y.; Xiong, Y.; Ma, W.; Liu, Y.; et al. Correction of β-thalassemia mutant by base editor in human embryos. Protein Cell 2017, 8, 811–822. [Google Scholar] [CrossRef]
- Gehrke, J.M.; Cervantes, O.; Clement, M.K.; Wu, Y.; Zeng, J.; Bauer, D.E.; Pinello, L.; Joung, J.K. An APOBEC3A-Cas9 base editor with minimized bystander and off-target activities. Nat. Biotechnol. 2018, 36, 977–982. [Google Scholar] [CrossRef]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef]
- Gaudelli, N.M.; Lam, D.K.; Rees, H.A.; Solá-Esteves, N.M.; Barrera, L.A.; Born, D.A.; Edwards, A.; Gehrke, J.M.; Lee, S.J.; Liquori, A.J.; et al. Directed evolution of adenine base editors with increased activity and therapeutic application. Nat. Biotechnol. 2020, 38, 892–900. [Google Scholar] [CrossRef] [PubMed]
- Chu, S.H.; Packer, M.S.; Olins, J.R.; Lung, G.; Cheng, L.-I.; Liquori, A.; Lee, C.; Marshall, J.; Yan, B.; Decker, J.; et al. A novel base editing approach to directly edit the causative mutation in sickle cell disease. Mol. Ther. 2020, 28, 808. [Google Scholar] [CrossRef]
- Newby, G.A.; Yen, J.S.; Woodard, K.J.; Mayuranathan, T.; Lazzarotto, C.R.; Li, Y.; Sheppard-Tillman, H.; Porter, S.N.; Yao, Y.; Mayberry, K.; et al. Base editing of haematopoietic stem cells rescues sickle cell disease in mice. Nature 2021, 595, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Rybak, A.P.; Akrawi, E.Z.; Rinaldi, C.; Haskett, S.J.; Lin, L.; Marshall, J.; Liquori, A.; Barrera, L.; Olins, J.; Chu, S.H.; et al. Base Editing of Gamma Globin Gene Promoters Generates Durable Expression of Fetal Hemoglobin for the Treatment of Sickle Cell Disease. Mol. Ther. 2020, 28, 547. [Google Scholar] [CrossRef]
- Jeong, Y.K.; Song, B.; Bae, S. Current Status and Challenges of DNA Base Editing Tools. Mol. Ther. 2020, 28, 1938–1952. [Google Scholar] [CrossRef]
- Hu, J.H.; Miller, S.M.; Geurts, M.H.; Tang, W.; Chen, L.; Sun, N.; Zeina, C.M.; Gao, X.; Rees, H.A.; Lin, Z.; et al. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature 2018, 556, 57–63. [Google Scholar] [CrossRef]
- Chu, S.H.; Packer, M.; Rees, H.; Lam, D.; Yu, Y.; Marshall, J.; Cheng, L.I.; Lam, D.; Olins, J.; Ran, F.A.; et al. Rationally Designed Base Editors for Precise Editing of the Sickle Cell Disease Mutation. Crispr J. 2021, 4, 169–177. [Google Scholar] [CrossRef]
- Walton, R.T.; Christie, K.A.; Whittaker, M.N.; Kleinstiver, B.P. Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants. Science 2020, 368, 290–296. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef]
- Hoban, M.D.; Cost, G.J.; Mendel, M.C.; Romero, Z.; Kaufman, M.L.; Joglekar, A.V.; Ho, M.; Lumaquin, D.; Gray, D.; Lill, G.R.; et al. Correction of the sickle cell disease mutation in human hematopoietic stem/progenitor cells. Blood 2015, 125, 2597–2604. [Google Scholar] [CrossRef]
- Long, J.; Hoban, M.D.; Cooper, A.R.; Kaufman, M.L.; Kuo, C.Y.; Campo-Fernandez, B.; Lumaquin, D.; Hollis, R.P.; Wang, X.; Kohn, D.B.; et al. Characterization of Gene Alterations following Editing of the β-Globin Gene Locus in Hematopoietic Stem/Progenitor Cells. Mol. Ther. 2018, 26, 468–479. [Google Scholar] [CrossRef] [PubMed]
- Kleinstiver, B.P.; Pattanayak, V.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Zheng, Z.; Joung, J.K. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 2016, 529, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Sander, J.D.; Reyon, D.; Cascio, V.M.; Joung, J.K. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat. Biotechnol. 2014, 32, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.Q.; Zheng, Z.; Nguyen, N.T.; Liebers, M.; Topkar, V.V.; Thapar, V.; Wyvekens, N.; Khayter, C.; Iafrate, A.J.; Le, L.P.; et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 2015, 33, 187–197. [Google Scholar] [CrossRef]
- Zhang, X.H.; Tee, L.Y.; Wang, X.G.; Huang, Q.S.; Yang, S.H. Off-target Effects in CRISPR/Cas9-mediated Genome Engineering. Mol. Ther. Nucleic Acids 2015, 4, e264. [Google Scholar] [CrossRef]
- Tsai, S.Q.; Nguyen, N.T.; Malagon-Lopez, J.; Topkar, V.V.; Aryee, M.J.; Joung, J.K. CIRCLE-seq: A highly sensitive in vitro screen for genome-wide CRISPR-Cas9 nuclease off-targets. Nat. Methods 2017, 14, 607–614. [Google Scholar] [CrossRef]
- Yan, W.X.; Mirzazadeh, R.; Garnerone, S.; Scott, D.; Schneider, M.W.; Kallas, T.; Custodio, J.; Wernersson, E.; Li, Y.; Gao, L.; et al. BLISS is a versatile and quantitative method for genome-wide profiling of DNA double-strand breaks. Nat. Commun. 2017, 8, 15058. [Google Scholar] [CrossRef]
- Wienert, B.; Wyman, S.K.; Yeh, C.D.; Conklin, B.R.; Corn, J.E. CRISPR off-target detection with DISCOVER-seq. Nat. Protoc 2020, 15, 1775–1799. [Google Scholar] [CrossRef]
- Frangoul, H.; Bobruff, Y.; Cappellini, M.D.; Corbacioglu, S.; Fernandez, C.M.; de la Fuente, J.; Grupp, S.A.; Handgretinger, R.; Ho, T.W.; Imren, S.; et al. Safety and Efficacy of CTX001 in Patients with Transfusion-Dependent β-Thalassemia and Sickle Cell Disease: Early Results from the Climb THAL-111 and Climb SCD-121 Studies of Autologous CRISPR-CAS9-Modified CD34+ Hematopoietic Stem and Progenitor Cells. Blood 2020, 136, 3–4. [Google Scholar] [CrossRef]
- Grupp, S.; Bloberger, N.; Campbell, C.; Carroll, C.; Hankins, J.S.; Ho, T.W.; Hobbs, W.; Imren, S.; Lu, Y.; Mapara, M.; et al. CTX001 FOR SICKLE CELL DISEASE: SAFETY AND EFFICACY RESULTS FROM THE ONGOING CLIMB SCD-121 STUDY OF AUTOLOGOUS CRISPR-CAS9-MODIFIED CD34+ HEMATOPOIETIC STEM AND PROGENITOR CELLS. HemaSphere 2021, 5, 365. [Google Scholar]
- Walters, M.C.; Smith, A.R.; Schiller, G.J.; Esrick, E.B.; Williams, D.A.; Gogoleva, T.; Rouy, D.; Cockroft, B.M.; Vercellotti, G.M. Updated Results of a Phase 1/2 Clinical Study of Zinc Finger Nuclease-Mediated Editing of BCL11A in Autologous Hematopoietic Stem Cells for Transfusion-Dependent Beta Thalassemia. Blood 2021, 138, 3974. [Google Scholar] [CrossRef]
- Alavi, A.; Krishnamurti, L.; Abedi, M.; Galeon, I.; Reiner, D.; Smith, S.E.; Wang, L.; Ramezi, A.; Rendo, P.; Walters, M.C. Preliminary Safety and Efficacy Results from Precizn-1: An Ongoing Phase 1/2 Study on Zinc Finger Nuclease-Modified Autologous CD34+ HSPCs for Sickle Cell Disease (SCD). Blood 2021, 138, 2930. [Google Scholar] [CrossRef]
- Kanter, J.; DiPersio, J.F.; Leavey, P.; Shyr, D.C.; Thompson, A.A.; Porteus, M.H.; Intondi, A.; Lahiri, P.; Dever, D.P.; Petrusich, A.; et al. Cedar Trial in Progress: A First in Human, Phase 1/2 Study of the Correction of a Single Nucleotide Mutation in Autologous HSCs (GPH101) to Convert HbS to HbA for Treating Severe SCD. Blood 2021, 138, 1864. [Google Scholar] [CrossRef]
- Frangoul, H.; Altshuler, D.; Cappellini, M.D.; Chen, Y.S.; Domm, J.; Eustace, B.K.; Foell, J.; de la Fuente, J.; Grupp, S.; Handgretinger, R.; et al. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. N. Engl. J. Med. 2021, 384, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Shen, J.; Li, D.; Cheng, Y. Strategies in the delivery of Cas9 ribonucleoprotein for CRISPR/Cas9 genome editing. Theranostics 2021, 11, 614–648. [Google Scholar] [CrossRef] [PubMed]
- Sung, Y.K.; Kim, S.W. Recent advances in the development of gene delivery systems. Biomater. Res. 2019, 23, 8. [Google Scholar] [CrossRef]
- Aghamiri, S.; Talaei, S.; Ghavidel, A.A.; Zandsalimi, F.; Masoumi, S.; Hafshejani, N.H.; Jajarmi, V. Nanoparticles-mediated CRISPR/Cas9 delivery: Recent advances in cancer treatment. J. Drug Deliv. Sci. Technol. 2020, 56, 101533. [Google Scholar] [CrossRef]
- Taha, E.A.; Lee, J.; Hotta, A. Delivery of CRISPR-Cas tools for in vivo genome editing therapy: Trends and challenges. J. Control Release 2022, 342, 345–361. [Google Scholar] [CrossRef]
- Richards, D.Y.; Winn, S.R.; Dudley, S.; Nygaard, S.; Mighell, T.L.; Grompe, M.; Harding, C.O. AAV-Mediated CRISPR/Cas9 Gene Editing in Murine Phenylketonuria. Mol. Ther. Methods Clin. Dev. 2020, 17, 234–245. [Google Scholar] [CrossRef]
- Zhao, H.; Li, Y.; He, L.; Pu, W.; Yu, W.; Li, Y.; Wu, Y.T.; Xu, C.; Wei, Y.; Ding, Q.; et al. In Vivo AAV-CRISPR/Cas9-Mediated Gene Editing Ameliorates Atherosclerosis in Familial Hypercholesterolemia. Circulation 2020, 141, 67–79. [Google Scholar] [CrossRef]
- Gao, J.; Bergmann, T.; Zhang, W.; Schiwon, M.; Ehrke-Schulz, E.; Ehrhardt, A. Viral Vector-Based Delivery of CRISPR/Cas9 and Donor DNA for Homology-Directed Repair in an In Vitro Model for Canine Hemophilia B. Mol. Ther. Nucleic Acids 2019, 14, 364–376. [Google Scholar] [CrossRef] [PubMed]
- Tabebordbar, M.; Lagerborg, K.A.; Stanton, A.; King, E.M.; Ye, S.; Tellez, L.; Krunnfusz, A.; Tavakoli, S.; Widrick, J.J.; Messemer, K.A.; et al. Directed evolution of a family of AAV capsid variants enabling potent muscle-directed gene delivery across species. Cell 2021, 184, 4919–4938. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yang, Y.; Breton, C.; Bell, P.; Li, M.; Zhang, J.; Che, Y.; Saveliev, A.; He, Z.; White, J.; et al. A mutation-independent CRISPR-Cas9-mediated gene targeting approach to treat a murine model of ornithine transcarbamylase deficiency. Sci. Adv. 2020, 6, eaax5701. [Google Scholar] [CrossRef] [PubMed]
- Min, Y.L.; Chemello, F.; Li, H.; Rodriguez-Caycedo, C.; Sanchez-Ortiz, E.; Mireault, A.A.; McAnally, J.R.; Shelton, J.M.; Zhang, Y.; Bassel-Duby, R.; et al. Correction of Three Prominent Mutations in Mouse and Human Models of Duchenne Muscular Dystrophy by Single-Cut Genome Editing. Mol. Ther. 2020, 28, 2044–2055. [Google Scholar] [CrossRef]
- Duan, W.; Guo, M.; Yi, L.; Liu, Y.; Li, Z.; Ma, Y.; Zhang, G.; Liu, Y.; Bu, H.; Song, X.; et al. The deletion of mutant SOD1 via CRISPR/Cas9/sgRNA prolongs survival in an amyotrophic lateral sclerosis mouse model. Gene Ther. 2020, 27, 157–169. [Google Scholar] [CrossRef]
- Monteys, A.M.; Ebanks, S.A.; Keiser, M.S.; Davidson, B.L. CRISPR/Cas9 Editing of the Mutant Huntingtin Allele In Vitro and In Vivo. Mol. Ther. 2017, 25, 12–23. [Google Scholar] [CrossRef]
- Yu, W.; Mookherjee, S.; Chaitankar, V.; Hiriyanna, S.; Kim, J.W.; Brooks, M.; Ataeijannati, Y.; Sun, X.; Dong, L.; Li, T.; et al. Nrl knockdown by AAV-delivered CRISPR/Cas9 prevents retinal degeneration in mice. Nat. Commun. 2017, 8, 14716. [Google Scholar] [CrossRef]
- Truong, D.J.; Kühner, K.; Kühn, R.; Werfel, S.; Engelhardt, S.; Wurst, W.; Ortiz, O. Development of an intein-mediated split-Cas9 system for gene therapy. Nucleic Acids Res. 2015, 43, 6450–6458. [Google Scholar] [CrossRef]
- Zetsche, B.; Volz, S.E.; Zhang, F. A split-Cas9 architecture for inducible genome editing and transcription modulation. Nat. Biotechnol. 2015, 33, 139–142. [Google Scholar] [CrossRef]
- Ran, F.A.; Cong, L.; Yan, W.X.; Scott, D.A.; Gootenberg, J.S.; Kriz, A.J.; Zetsche, B.; Shalem, O.; Wu, X.; Makarova, K.S.; et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 2015, 520, 186–191. [Google Scholar] [CrossRef]
- Kim, E.; Koo, T.; Park, S.W.; Kim, D.; Kim, K.; Cho, H.Y.; Song, D.W.; Lee, K.J.; Jung, M.H.; Kim, S.; et al. In vivo genome editing with a small Cas9 orthologue derived from Campylobacter jejuni. Nat. Commun. 2017, 8, 14500. [Google Scholar] [CrossRef] [PubMed]
- Koo, T.; Park, S.W.; Jo, D.H.; Kim, D.; Kim, J.H.; Cho, H.Y.; Kim, J.; Kim, J.H.; Kim, J.S. CRISPR-LbCpf1 prevents choroidal neovascularization in a mouse model of age-related macular degeneration. Nat. Commun. 2018, 9, 1855. [Google Scholar] [CrossRef] [PubMed]
- Strecker, J.; Jones, S.; Koopal, B.; Schmid-Burgk, J.; Zetsche, B.; Gao, L.; Makarova, K.S.; Koonin, E.V.; Zhang, F. Engineering of CRISPR-Cas12b for human genome editing. Nat. Commun. 2019, 10, 212. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Tanner, M.R.; Lee, C.M.; Hurley, A.E.; De Giorgi, M.; Jarrett, K.E.; Davis, T.H.; Doerfler, A.M.; Bao, G.; Beeton, C.; et al. AAV-CRISPR Gene Editing Is Negated by Pre-existing Immunity to Cas9. Mol. Ther. 2020, 28, 1432–1441. [Google Scholar] [CrossRef] [PubMed]
- Erles, K.; Sebökovà, P.; Schlehofer, J.R. Update on the prevalence of serum antibodies (IgG and IgM) to adeno-associated virus (AAV). J. Med. Virol. 1999, 59, 406–411. [Google Scholar] [CrossRef]
- Bartel, M.A.; Weinstein, J.R.; Schaffer, D.V. Directed evolution of novel adeno-associated viruses for therapeutic gene delivery. Gene Ther. 2012, 19, 694–700. [Google Scholar] [CrossRef]
- Ates, I.; Rathbone, T.; Stuart, C.; Bridges, P.H.; Cottle, R.N. Delivery Approaches for Therapeutic Genome Editing and Challenges. Genes (Basel) 2020, 11, 1113. [Google Scholar] [CrossRef]
- Hinderer, C.; Katz, N.; Buza, E.L.; Dyer, C.; Goode, T.; Bell, P.; Richman, L.K.; Wilson, J.M. Severe Toxicity in Nonhuman Primates and Piglets Following High-Dose Intravenous Administration of an Adeno-Associated Virus Vector Expressing Human SMN. Hum. Gene Ther. 2018, 29, 285–298. [Google Scholar] [CrossRef]
- Nelson, C.E.; Wu, Y.; Gemberling, M.P.; Oliver, M.L.; Waller, M.A.; Bohning, J.D.; Robinson-Hamm, J.N.; Bulaklak, K.; Castellanos Rivera, R.M.; Collier, J.H.; et al. Long-term evaluation of AAV-CRISPR genome editing for Duchenne muscular dystrophy. Nat. Med. 2019, 25, 427–432. [Google Scholar] [CrossRef]
- Hanlon, K.S.; Kleinstiver, B.P.; Garcia, S.P.; Zaborowski, M.P.; Volak, A.; Spirig, S.E.; Muller, A.; Sousa, A.A.; Tsai, S.Q.; Bengtsson, N.E.; et al. High levels of AAV vector integration into CRISPR-induced DNA breaks. Nat. Commun. 2019, 10, 4439. [Google Scholar] [CrossRef]
- Buchlis, G.; Podsakoff, G.M.; Radu, A.; Hawk, S.M.; Flake, A.W.; Mingozzi, F.; High, K.A. Factor IX expression in skeletal muscle of a severe hemophilia B patient 10 years after AAV-mediated gene transfer. Blood 2012, 119, 3038–3041. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Lee, C.M.; Hurley, A.E.; Jarrett, K.E.; De Giorgi, M.; Lu, W.; Balderrama, K.S.; Doerfler, A.M.; Deshmukh, H.; Ray, A.; et al. A Self-Deleting AAV-CRISPR System for In Vivo Genome Editing. Mol. Ther. Methods Clin. Dev. 2019, 12, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.M.; Flotte, T.R. Moving Forward After Two Deaths in a Gene Therapy Trial of Myotubular Myopathy. Hum. Gene Ther. 2020, 31, 695–696. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, M.A.; Siegfried, W.; Yoshimura, K.; Yoneyama, K.; Fukayama, M.; Stier, L.E.; Pääkkö, P.K.; Gilardi, P.; Stratford-Perricaudet, L.D.; Perricaudet, M.; et al. Adenovirus-mediated transfer of a recombinant alpha 1-antitrypsin gene to the lung epithelium in vivo. Science 1991, 252, 431–434. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, H.A.; Danel, C.; Longenecker, G.; Metzger, M.; Setoguchi, Y.; Rosenfeld, M.A.; Gant, T.W.; Thorgeirsson, S.S.; Stratford-Perricaudet, L.D.; Perricaudet, M.; et al. Adenovirus-mediated in vivo gene transfer and expression in normal rat liver. Nat. Genet. 1992, 1, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Zabner, J.; Couture, L.A.; Gregory, R.J.; Graham, S.M.; Smith, A.E.; Welsh, M.J. Adenovirus-mediated gene transfer transiently corrects the chloride transport defect in nasal epithelia of patients with cystic fibrosis. Cell 1993, 75, 207–216. [Google Scholar] [CrossRef]
- Rosengart, T.K.; Lee, L.Y.; Patel, S.R.; Sanborn, T.A.; Parikh, M.; Bergman, G.W.; Hachamovitch, R.; Szulc, M.; Kligfield, P.D.; Okin, P.M.; et al. Angiogenesis gene therapy: Phase I assessment of direct intramyocardial administration of an adenovirus vector expressing VEGF121 cDNA to individuals with clinically significant severe coronary artery disease. Circulation 1999, 100, 468–474. [Google Scholar] [CrossRef]
- Crystal, R.G. Adenovirus: The first effective in vivo gene delivery vector. Hum. Gene Ther. 2014, 25, 3–11. [Google Scholar] [CrossRef]
- Raper, S.E.; Chirmule, N.; Lee, F.S.; Wivel, N.A.; Bagg, A.; Gao, G.P.; Wilson, J.M.; Batshaw, M.L. Fatal systemic inflammatory response syndrome in a ornithine transcarbamylase deficient patient following adenoviral gene transfer. Mol. Genet. Metab. 2003, 80, 148–158. [Google Scholar] [CrossRef]
- Schnell, M.A.; Zhang, Y.; Tazelaar, J.; Gao, G.P.; Yu, Q.C.; Qian, R.; Chen, S.J.; Varnavski, A.N.; LeClair, C.; Raper, S.E.; et al. Activation of innate immunity in nonhuman primates following intraportal administration of adenoviral vectors. Mol. Ther. 2001, 3, 708–722. [Google Scholar] [CrossRef]
- Fausther-Bovendo, H.; Kobinger, G.P. Pre-existing immunity against Ad vectors: Humoral, cellular, and innate response, what’s important? Hum. Vaccin Immunother. 2014, 10, 2875–2884. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.; Singh, N.; Vemula, S.V.; Couëtil, L.; Katz, J.M.; Donis, R.; Sambhara, S.; Mittal, S.K. Impact of preexisting adenovirus vector immunity on immunogenicity and protection conferred with an adenovirus-based H5N1 influenza vaccine. PLoS ONE 2012, 7, e33428. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Psatha, N.; Wang, H.; Singh, M.; Samal, H.B.; Zhang, W.; Ehrhardt, A.; Izsvák, Z.; Papayannopoulou, T.; Lieber, A. Integrating HDAd5/35++ Vectors as a New Platform for HSC Gene Therapy of Hemoglobinopathies. Mol. Ther. Methods Clin. Dev. 2018, 9, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, H.; Gil, S.; Germond, A.; Fountain, C.; Baldessari, A.; Kim, J.; Liu, Z.; Georgakopoulou, A.; Radtke, S.; et al. Safe and efficient in vivo hematopoietic stem cell transduction in nonhuman primates using HDAd5/35++ vectors. Mol. Ther. Methods Clin. Dev. 2022, 24, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Mishra, A.S.; Gil, S.; Wang, M.; Georgakopoulou, A.; Papayannopoulou, T.; Hawkins, R.D.; Lieber, A. Targeted Integration and High-Level Transgene Expression in AAVS1 Transgenic Mice after In Vivo HSC Transduction with HDAd5/35++ Vectors. Mol. Ther. 2019, 27, 2195–2212. [Google Scholar] [CrossRef] [PubMed]
- Voysey, M.; Clemens, S.A.C.; Madhi, S.A.; Weckx, L.Y.; Folegatti, P.M.; Aley, P.K.; Angus, B.; Baillie, V.L.; Barnabas, S.L.; Bhorat, Q.E.; et al. Safety and efficacy of the ChAdOx1 nCoV-19 vaccine (AZD1222) against SARS-CoV-2: An interim analysis of four randomised controlled trials in Brazil, South Africa, and the UK. Lancet 2021, 397, 99–111. [Google Scholar] [CrossRef]
- Sadoff, J.; Le Gars, M.; Shukarev, G.; Heerwegh, D.; Truyers, C.; de Groot, A.M.; Stoop, J.; Tete, S.; Van Damme, W.; Leroux-Roels, I.; et al. Interim Results of a Phase 1-2a Trial of Ad26.COV2.S Covid-19 Vaccine. N. Engl. J. Med. 2021, 384, 1824–1835. [Google Scholar] [CrossRef]
- Schucht, R.; Coroadinha, A.S.; Zanta-Boussif, M.A.; Verhoeyen, E.; Carrondo, M.J.; Hauser, H.; Wirth, D. A new generation of retroviral producer cells: Predictable and stable virus production by Flp-mediated site-specific integration of retroviral vectors. Mol. Ther. 2006, 14, 285–292. [Google Scholar] [CrossRef]
- Follenzi, A.; Battaglia, M.; Lombardo, A.; Annoni, A.; Roncarolo, M.G.; Naldini, L. Targeting lentiviral vector expression to hepatocytes limits transgene-specific immune response and establishes long-term expression of human antihemophilic factor IX in mice. Blood 2004, 103, 3700–3709. [Google Scholar] [CrossRef]
- Wanisch, K.; Yáñez-Muñoz, R.J. Integration-deficient lentiviral vectors: A slow coming of age. Mol. Ther. 2009, 17, 1316–1332. [Google Scholar] [CrossRef]
- Lombardo, A.; Genovese, P.; Beausejour, C.M.; Colleoni, S.; Lee, Y.L.; Kim, K.A.; Ando, D.; Urnov, F.D.; Galli, C.; Gregory, P.D.; et al. Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nat. Biotechnol. 2007, 25, 1298–1306. [Google Scholar] [CrossRef] [PubMed]
- Uchida, N.; Drysdale, C.M.; Nassehi, T.; Gamer, J.; Yapundich, M.; DiNicola, J.; Shibata, Y.; Hinds, M.; Gudmundsdottir, B.; Haro-Mora, J.J.; et al. Cas9 protein delivery non-integrating lentiviral vectors for gene correction in sickle cell disease. Mol. Ther. Methods Clin. Dev. 2021, 21, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Blasco, R.B.; Karaca, E.; Ambrogio, C.; Cheong, T.C.; Karayol, E.; Minero, V.G.; Voena, C.; Chiarle, R. Simple and rapid in vivo generation of chromosomal rearrangements using CRISPR/Cas9 technology. Cell Rep. 2014, 9, 1219–1227. [Google Scholar] [CrossRef] [PubMed]
- Holmgaard, A.; Askou, A.L.; Benckendorff, J.N.E.; Thomsen, E.A.; Cai, Y.; Bek, T.; Mikkelsen, J.G.; Corydon, T.J. In Vivo Knockout of the Vegfa Gene by Lentiviral Delivery of CRISPR/Cas9 in Mouse Retinal Pigment Epithelium Cells. Mol. Ther. Nucleic Acids 2017, 9, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.H.; Cai, H.; Steinmetz, N.F. Viral nanoparticles for drug delivery, imaging, immunotherapy, and theranostic applications. Adv. Drug Deliv. Rev. 2020, 156, 214–235. [Google Scholar] [CrossRef] [PubMed]
- Nooraei, S.; Bahrulolum, H.; Hoseini, Z.S.; Katalani, C.; Hajizade, A.; Easton, A.J.; Ahmadian, G. Virus-like particles: Preparation, immunogenicity and their roles as nanovaccines and drug nanocarriers. J. Nanobiotechnol. 2021, 19, 59. [Google Scholar] [CrossRef] [PubMed]
- Rohovie, M.J.; Nagasawa, M.; Swartz, J.R. Virus-like particles: Next-generation nanoparticles for targeted therapeutic delivery. Bioeng Transl. Med. 2017, 2, 43–57. [Google Scholar] [CrossRef]
- Choi, J.G.; Dang, Y.; Abraham, S.; Ma, H.; Zhang, J.; Guo, H.; Cai, Y.; Mikkelsen, J.G.; Wu, H.; Shankar, P.; et al. Lentivirus pre-packed with Cas9 protein for safer gene editing. Gene Ther. 2016, 23, 627–633. [Google Scholar] [CrossRef]
- Montagna, C.; Petris, G.; Casini, A.; Maule, G.; Franceschini, G.M.; Zanella, I.; Conti, L.; Arnoldi, F.; Burrone, O.R.; Zentilin, L.; et al. VSV-G-Enveloped Vesicles for Traceless Delivery of CRISPR-Cas9. Mol. Ther. Nucleic Acids 2018, 12, 453–462. [Google Scholar] [CrossRef]
- Mangeot, P.E.; Risson, V.; Fusil, F.; Marnef, A.; Laurent, E.; Blin, J.; Mournetas, V.; Massouridès, E.; Sohier, T.J.M.; Corbin, A.; et al. Genome editing in primary cells and in vivo using viral-derived Nanoblades loaded with Cas9-sgRNA ribonucleoproteins. Nat. Commun. 2019, 10, 45. [Google Scholar] [CrossRef]
- Campbell, L.A.; Coke, L.M.; Richie, C.T.; Fortuno, L.V.; Park, A.Y.; Harvey, B.K. Gesicle-Mediated Delivery of CRISPR/Cas9 Ribonucleoprotein Complex for Inactivating the HIV Provirus. Mol. Ther. 2019, 27, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Cullis, P.R.; Hope, M.J. Lipid Nanoparticle Systems for Enabling Gene Therapies. Mol. Ther. 2017, 25, 1467–1475. [Google Scholar] [CrossRef] [PubMed]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Pérez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef] [PubMed]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef]
- Wei, T.; Cheng, Q.; Min, Y.L.; Olson, E.N.; Siegwart, D.J. Systemic nanoparticle delivery of CRISPR-Cas9 ribonucleoproteins for effective tissue specific genome editing. Nat. Commun. 2020, 11, 3232. [Google Scholar] [CrossRef]
- Glass, Z.; Lee, M.; Li, Y.; Xu, Q. Engineering the Delivery System for CRISPR-Based Genome Editing. Trends. Biotechnol. 2018, 36, 173–185. [Google Scholar] [CrossRef]
- Jayaraman, M.; Ansell, S.M.; Mui, B.L.; Tam, Y.K.; Chen, J.; Du, X.; Butler, D.; Eltepu, L.; Matsuda, S.; Narayanannair, J.K.; et al. Maximizing the potency of siRNA lipid nanoparticles for hepatic gene silencing in vivo. Angew. Chem. Int. Ed. Engl. 2012, 51, 8529–8533. [Google Scholar] [CrossRef]
- Hou, X.; Zaks, T.; Langer, R.; Dong, Y. Lipid nanoparticles for mRNA delivery. Nat. Rev. Mater. 2021, 6, 1078–1094. [Google Scholar] [CrossRef]
- Jokerst, J.V.; Lobovkina, T.; Zare, R.N.; Gambhir, S.S. Nanoparticle PEGylation for imaging and therapy. Nanomedicine (Lond) 2011, 6, 715–728. [Google Scholar] [CrossRef]
- Miller, J.B.; Zhang, S.; Kos, P.; Xiong, H.; Zhou, K.; Perelman, S.S.; Zhu, H.; Siegwart, D.J. Non-Viral CRISPR/Cas Gene Editing In Vitro and In Vivo Enabled by Synthetic Nanoparticle Co-Delivery of Cas9 mRNA and sgRNA. Angew. Chem. Int. Ed. Engl. 2017, 56, 1059–1063. [Google Scholar] [CrossRef]
- Finn, J.D.; Smith, A.R.; Patel, M.C.; Shaw, L.; Youniss, M.R.; van Heteren, J.; Dirstine, T.; Ciullo, C.; Lescarbeau, R.; Seitzer, J.; et al. A Single Administration of CRISPR/Cas9 Lipid Nanoparticles Achieves Robust and Persistent In Vivo Genome Editing. Cell Rep. 2018, 22, 2227–2235. [Google Scholar] [CrossRef] [PubMed]
- Gillmore, J.D.; Gane, E.; Taubel, J.; Kao, J.; Fontana, M.; Maitland, M.L.; Seitzer, J.; O’Connell, D.; Walsh, K.R.; Wood, K.; et al. CRISPR-Cas9 In Vivo Gene Editing for Transthyretin Amyloidosis. N. Engl. J. Med. 2021, 385, 493–502. [Google Scholar] [CrossRef]
- Liu, Q.; Zhao, K.; Wang, C.; Zhang, Z.; Zheng, C.; Zhao, Y.; Zheng, Y.; Liu, C.; An, Y.; Shi, L.; et al. Multistage Delivery Nanoparticle Facilitates Efficient CRISPR/dCas9 Activation and Tumor Growth Suppression In Vivo. Adv. Sci. (Weinh) 2019, 6, 1801423. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.L.; Xu, C.F.; Li, H.J.; Cao, Z.T.; Liu, J.; Wang, J.L.; Du, X.J.; Yang, X.Z.; Gu, Z.; Wang, J. Macrophage-Specific in Vivo Gene Editing Using Cationic Lipid-Assisted Polymeric Nanoparticles. ACS Nano 2018, 12, 994–1005. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.X.; Song, Z.; Lao, Y.H.; Xu, X.; Gong, J.; Cheng, D.; Chakraborty, S.; Park, J.S.; Li, M.; Huang, D.; et al. Nonviral gene editing via CRISPR/Cas9 delivery by membrane-disruptive and endosomolytic helical polypeptide. Proc. Natl. Acad. Sci. USA 2018, 115, 4903–4908. [Google Scholar] [CrossRef]
- McNeer, N.A.; Schleifman, E.B.; Cuthbert, A.; Brehm, M.; Jackson, A.; Cheng, C.; Anandalingam, K.; Kumar, P.; Shultz, L.D.; Greiner, D.L.; et al. Systemic delivery of triplex-forming PNA and donor DNA by nanoparticles mediates site-specific genome editing of human hematopoietic cells in vivo. Gene Ther. 2013, 20, 658–669. [Google Scholar] [CrossRef]
- Roca, M.; Haes, A.J. Probing cells with noble metal nanoparticle aggregates. Nanomedicine 2008, 3, 555–565. [Google Scholar] [CrossRef]
- Hong, R.; Han, G.; Fernández, J.M.; Kim, B.J.; Forbes, N.S.; Rotello, V.M. Glutathione-mediated delivery and release using monolayer protected nanoparticle carriers. J. Am. Chem. Soc. 2006, 128, 1078–1079. [Google Scholar] [CrossRef]
- Sun, M.; Peng, D.; Hao, H.; Hu, J.; Wang, D.; Wang, K.; Liu, J.; Guo, X.; Wei, Y.; Gao, W. Thermally Triggered in Situ Assembly of Gold Nanoparticles for Cancer Multimodal Imaging and Photothermal Therapy. ACS Appl. Mater. Interfaces 2017, 9, 10453–10460. [Google Scholar] [CrossRef]
- Amina, S.J.; Guo, B. A Review on the Synthesis and Functionalization of Gold Nanoparticles as a Drug Delivery Vehicle. Int J. Nanomed. 2020, 15, 9823–9857. [Google Scholar] [CrossRef]
- Ngobili, T.A.; Daniele, M.A. Nanoparticles and direct immunosuppression. Exp. Biol. Med. 2016, 241, 1064–1073. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Jiang, Z.; Saha, K.; Kim, C.S.; Kim, S.T.; Landis, R.F.; Rotello, V.M. Gold nanoparticles for nucleic acid delivery. Mol. Ther. 2014, 22, 1075–1083. [Google Scholar] [CrossRef] [PubMed]
- Shahbazi, R.; Sghia-Hughes, G.; Reid, J.L.; Kubek, S.; Haworth, K.G.; Humbert, O.; Kiem, H.P.; Adair, J.E. Targeted homology-directed repair in blood stem and progenitor cells with CRISPR nanoformulations. Nat. Mater. 2019, 18, 1124–1132. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Zhang, L.; Zheng, W.; Cong, L.; Guo, Z.; Xie, Y.; Wang, L.; Tang, R.; Feng, Q.; Hamada, Y.; et al. Thermo-triggered Release of CRISPR-Cas9 System by Lipid-Encapsulated Gold Nanoparticles for Tumor Therapy. Angew. Chem. Int. Ed. Engl. 2018, 57, 1491–1496. [Google Scholar] [CrossRef]
- Lee, K.; Conboy, M.; Park, H.M.; Jiang, F.; Kim, H.J.; Dewitt, M.A.; Mackley, V.A.; Chang, K.; Rao, A.; Skinner, C.; et al. Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nat. Biomed. Eng. 2017, 1, 889–901. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Trial | Phase | Year | Study Drug | Target Gene | HSC Source | Conditioning | Sponsor | Location | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Sickle cell disease | |||||||||
| NCT04628585 | N/A | 2020 | BB305 | βT87Q-globin | N/A | N/A | bluebird bio | USA, France | [62] |
| NCT04293185 | 3 | 2020 | BB305 | βT87Q-globin | Plerixafor mobilization | Myeloablative busulfan | bluebird bio | USA | N/A |
| NCT04091737 | 1 | 2019 | CSL200 | γG16D-globin, shRNA-HPRT | Plerixafor mobilization | RIC melphalan | CSL Behring | USA | N/A |
| NCT03964792 | 1/2 | 2019 | GLOBE1 | βAS3-globin | Plerixafor mobilization | Myeloablative busulfan | APHP | France | [68,69] |
| NCT03282656 | 1 | 2018 | BCH-BB694 | shmiR-BCL11A | BM | Myeloablative busulfan | Boston Children’s Hospital | USA | [74] |
| NCT02247843 | 1/2 | 2014 | βAS3-FB | βAS3-globin | Plerixafor mobilization | Myeloablative busulfan | University of California, Los Angeles | USA | N/A |
| NCT02186418 | 1/2 | 2014 | ARU-1801 | γ-globin | BM/Plerixafor mobilization | RIC melphalan | Aruvant Sciences | USA, Canada, Jamaica | [72] |
| NCT02140554 | 1/2 | 2014 | BB305 | βT87Q-globin | BM (Group A/B), Plerixafor mobilization (Group C) | Myeloablative busulfan | bluebird bio | USA | [38] |
| NCT02151526 | 1/2 | 2013 | BB305 | βT87Q-globin | BM | Myeloablative busulfan | bluebird bio | France | [62,68] |
| β-Thalassemia | |||||||||
| NCT05015920 | 1 | 2021 | BD211 | βT87Q-globin | BM | Myeloablative busulfan | Shanghai Bdgene | China | N/A |
| NCT04592458 | 1 | 2020 | LentiHBBT87Q | β-globin | BM | N/A | Shenzhen Children’s Hospital, BGI-Research | China | N/A |
| NCT03207009 | 3 | 2017 | BB305 | βT87Q-globin | G-CSF and plerixafor mobilization | Myeloablative busulfan | bluebird bio | USA, France, Germany, Greece, Italy, UK | [65] |
| NCT02906202 | 3 | 2016 | BB305 | βT87Q-globin | G-CSF and plerixafor mobilization | Myeloablative busulfan | bluebird bio | USA, France, Germany, Italy, Thailand, UK | [65] |
| NCT02453477 | 1/2 | 2015 | GLOBE | β-globin | G-CSF and plerixafor mobilization | Myeloablative treosulfan/thiotepa | TIGET | Italy | [68] |
| NCT01745120 | 1/2 | 2013 | BB305 | βT87Q-globin | BM | Myeloablative busulfan | bluebird bio | USA, Australia, Thailand | [64] |
| NCT02151526 | 1/2 | 2013 | BB305 | βT87Q-globin | BM | Myeloablative busulfan | bluebird bio | France | [64] |
| NCT02633943 | N/A | 2013 | BB305 | BM/G-CSF and plerixafor mobilization | Myeloablative busulfan | bluebird bio | USA, Australia, France, Germany, Italy, Thailand, UK | [62] | |
| NCT01639690 | 1 | 2012 | TNS9.3.55 | β-globin | G-CSF mobilization | RIC busulfan | Memorial Sloan Kettering | USA | [45,67] |
| LG001 | 1/2 | 2007 | BM | Myeloablative busulfan | bluebird bio | France | [50,60] |
| Trial | Phase | Year | Study Drug | Target Gene | HSC Source | Conditioning | Sponsor | Location | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Sickle cell disease | |||||||||
| NCT04774536 | 1/2 | 2021 | CRISPR/Cas9: CRISPR_SCD001 | β-globin | BM | Myeloablative Busulfan | University of California, San Francisco | USA | N/A |
| NCT04853576 | 1/2 | 2021 | CRISPR/Cas12: EDIT-301 | BCL11A ESE | Plerixafor mobilization | Myeloablative busulfan | Editas Medicine | USA | N/A |
| NCT04819841 | 1/2 | 2021 | CRISPR/Cas9: GPH101 | β-globin | BM | Myeloablative busulfan | Graphite Bio | USA | N/A |
| NCT05145062 | N/A | 2021 | ZFN: BIVV003 | BCL11A ESE | Plerixafor mobilization | Myeloablative busulfan | Bioverativ | USA | [142] |
| NCT04443907 | 1/2 | 2020 | CRISPR/Cas9: OTQ923 / HIX763 | BCL11A ESE | N/A | N/A | Novartis Pharmaceuticals | USA | N/A |
| NCT03653247 | 1/2 | 2019 | ZFN: BIVV003 | BCL11A ESE | Plerixafor mobilization | Myeloablative busulfan | Bioverativ | USA | [142] |
| NCT04208529 | N/A | 2019 | CRISPR/Cas9: CTX001 | BCL11A ESE | BM | Myeloablative busulfan | Vertex Pharmaceuticals | USA | [144] |
| NCT03745287 | 2/3 | 2018 | CRISPR/Cas9: CTX001 | BCL11A ESE | BM | Myeloablative busulfan | Vertex Pharmaceuticals | USA | [144] |
| β-Thalassemia | |||||||||
| NCT04205435 | 1/2 | 2021 | CRISPR/Cas9 | β-globin | N/A | N/A | Biorary Laboratories | China | N/A |
| NCT04208529 | N/A | 2021 | CRISPR/Cas9: CTX001 | BCL11A ESE | BM | Myeloablative busulfan | Vertex Pharmaceuticals | USA | [144] |
| NCT04211480 | 1/2 | 2020 | CRISPR/Cas9 | BCL11A ESE | N/A | N/A | Biorary Laboratories | China | N/A |
| NCT03655678 | 2/3 | 2018 | CRISPR/Cas9: CTX001 | BCL11A ESE | BM | Myeloablative busulfan | Vertex Pharmaceuticals | USA, Canada, Germany, Italy, UK | [144] |
| NCT03432364 | 1/2 | 2018 | ZFN: ST-400 | BCL11A ESE | G-CSF & plerixafor mobilization | Myeloablative busulfan | Sangamo Therapeutics | USA | [141] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Germino-Watnick, P.; Hinds, M.; Le, A.; Chu, R.; Liu, X.; Uchida, N. Hematopoietic Stem Cell Gene-Addition/Editing Therapy in Sickle Cell Disease. Cells 2022, 11, 1843. https://doi.org/10.3390/cells11111843
Germino-Watnick P, Hinds M, Le A, Chu R, Liu X, Uchida N. Hematopoietic Stem Cell Gene-Addition/Editing Therapy in Sickle Cell Disease. Cells. 2022; 11(11):1843. https://doi.org/10.3390/cells11111843
Chicago/Turabian StyleGermino-Watnick, Paula, Malikiya Hinds, Anh Le, Rebecca Chu, Xiong Liu, and Naoya Uchida. 2022. "Hematopoietic Stem Cell Gene-Addition/Editing Therapy in Sickle Cell Disease" Cells 11, no. 11: 1843. https://doi.org/10.3390/cells11111843
APA StyleGermino-Watnick, P., Hinds, M., Le, A., Chu, R., Liu, X., & Uchida, N. (2022). Hematopoietic Stem Cell Gene-Addition/Editing Therapy in Sickle Cell Disease. Cells, 11(11), 1843. https://doi.org/10.3390/cells11111843