
Human Mesenchymal Stromal Cells Resolve Lipid Load in High Fat Diet-Induced Non-Alcoholic Steatohepatitis in Mice by Mitochondria Donation

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Isolation of MSC from Human Bone Marrow
2.2. Animals and MSC Application
2.3. Immunohistochemical Procedures
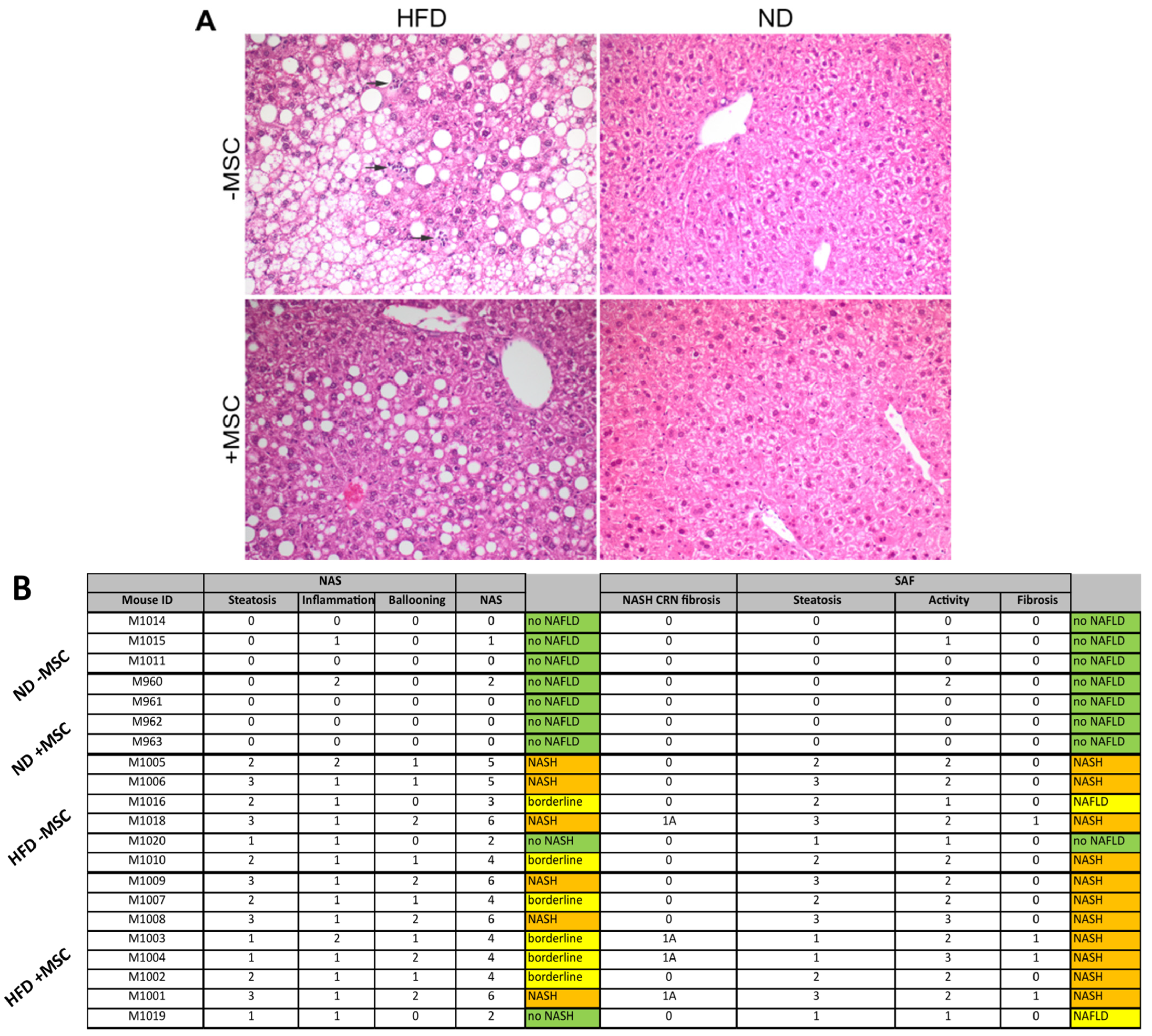
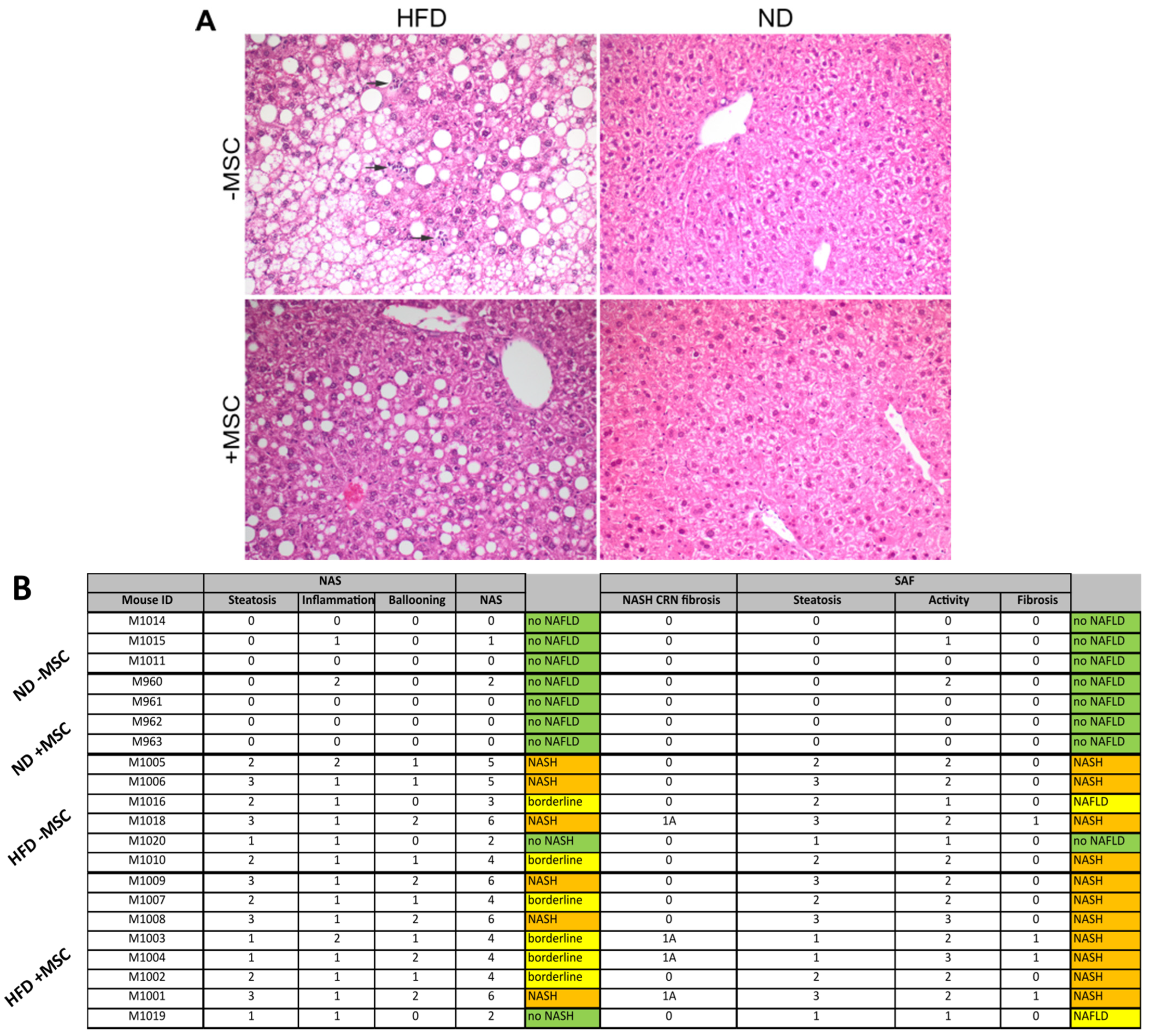
2.4. Histopathological Scoring
2.5. Semiquantitative PCR
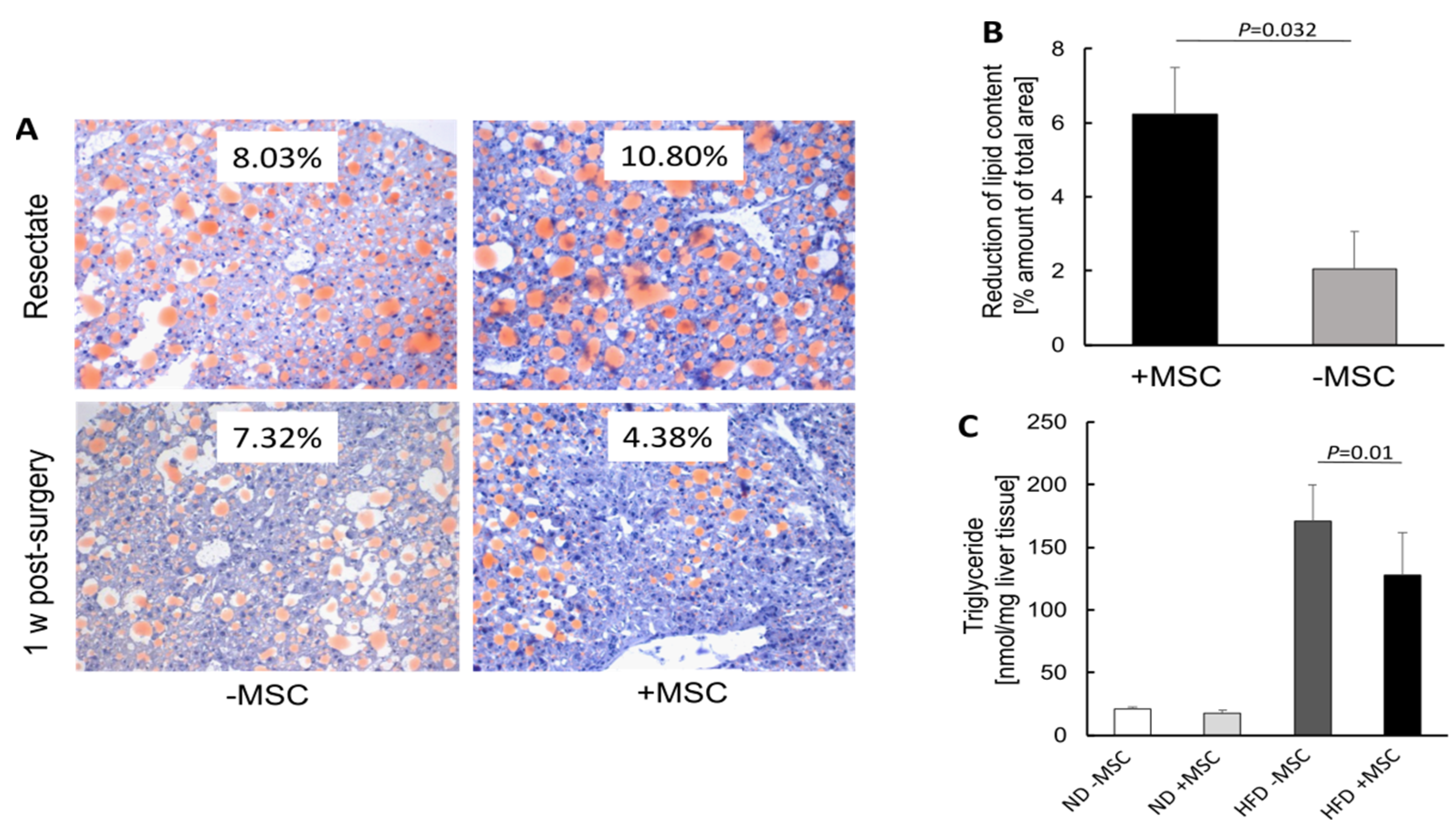
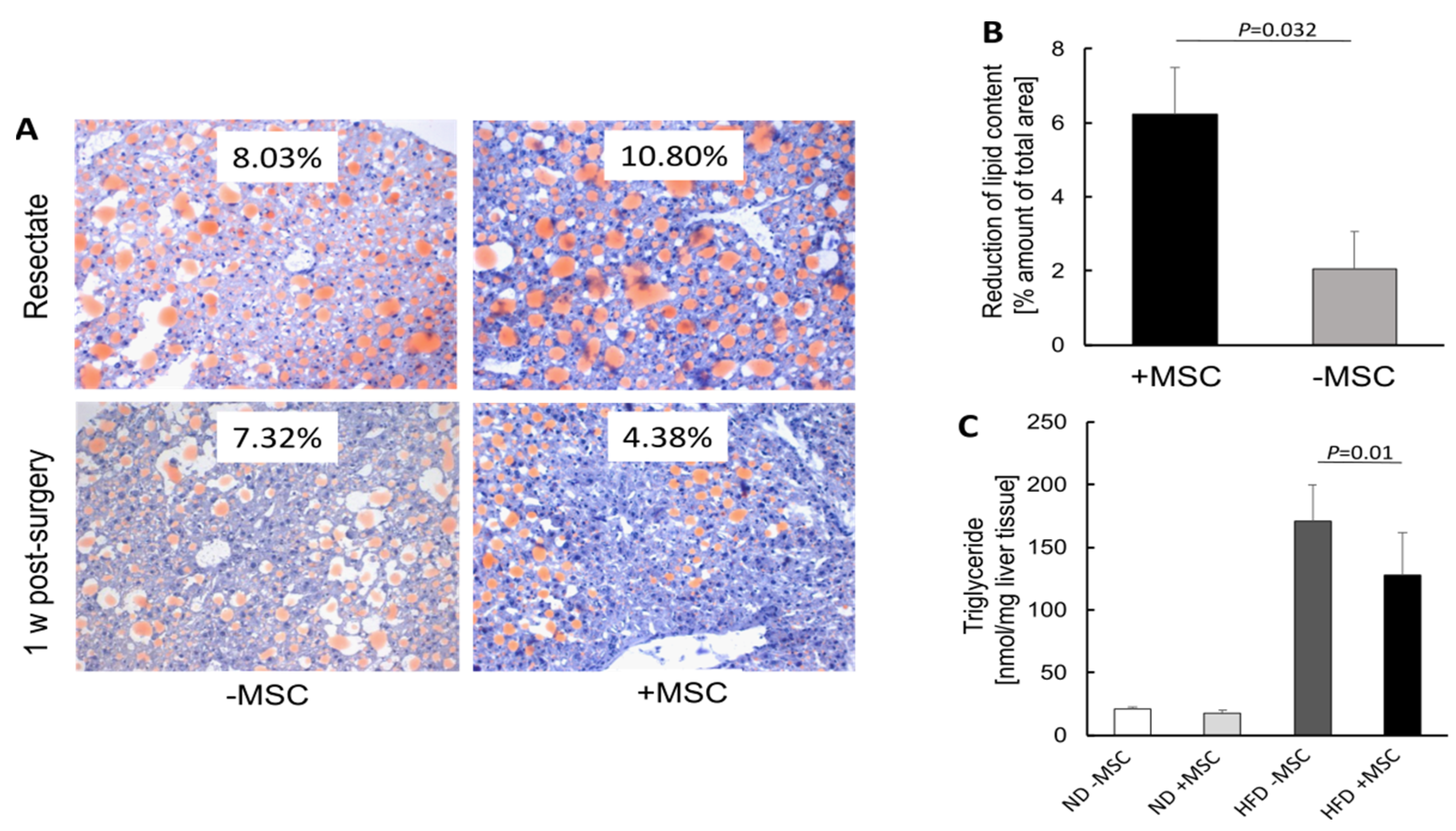
2.6. Quantification of Liver Triglyceride Content
2.7. Multiplex Assay
2.8. Statistics
3. Results
3.1. Hepatic Transplantation of MSC Decreases NASH-Associated Hepatic Lipid Storage
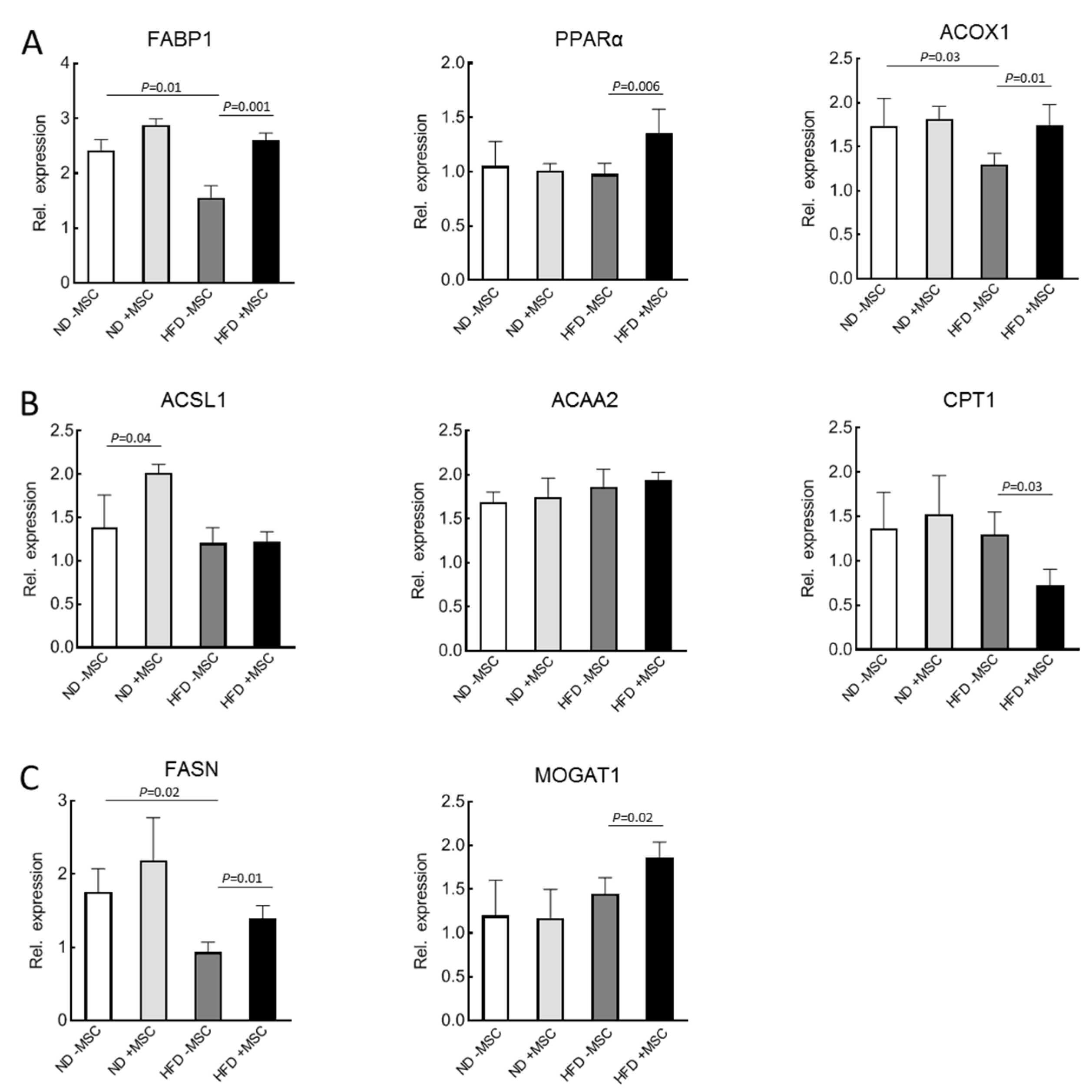
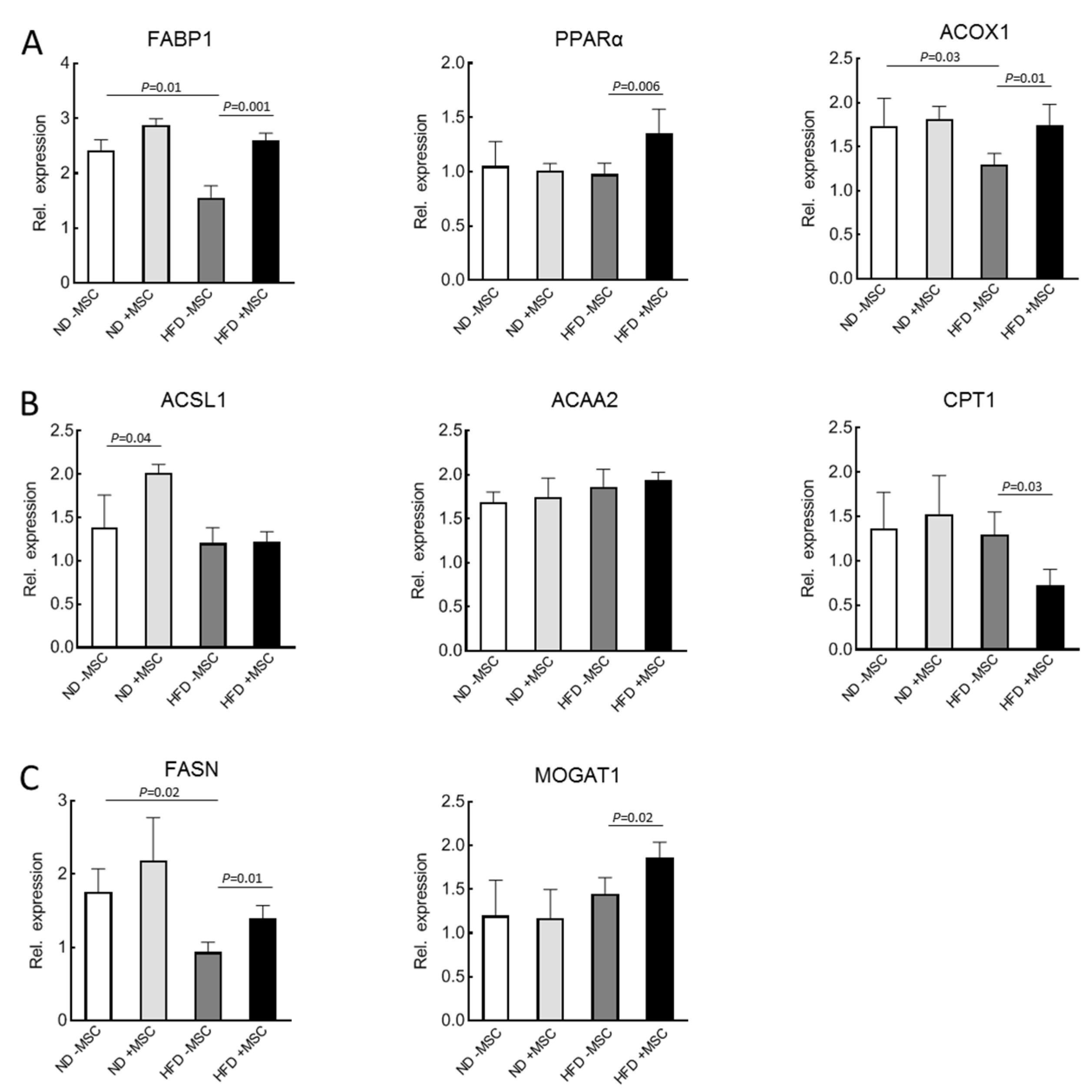
3.2. MSC-Treatment Augments Expression of Lipid Utilization Genes
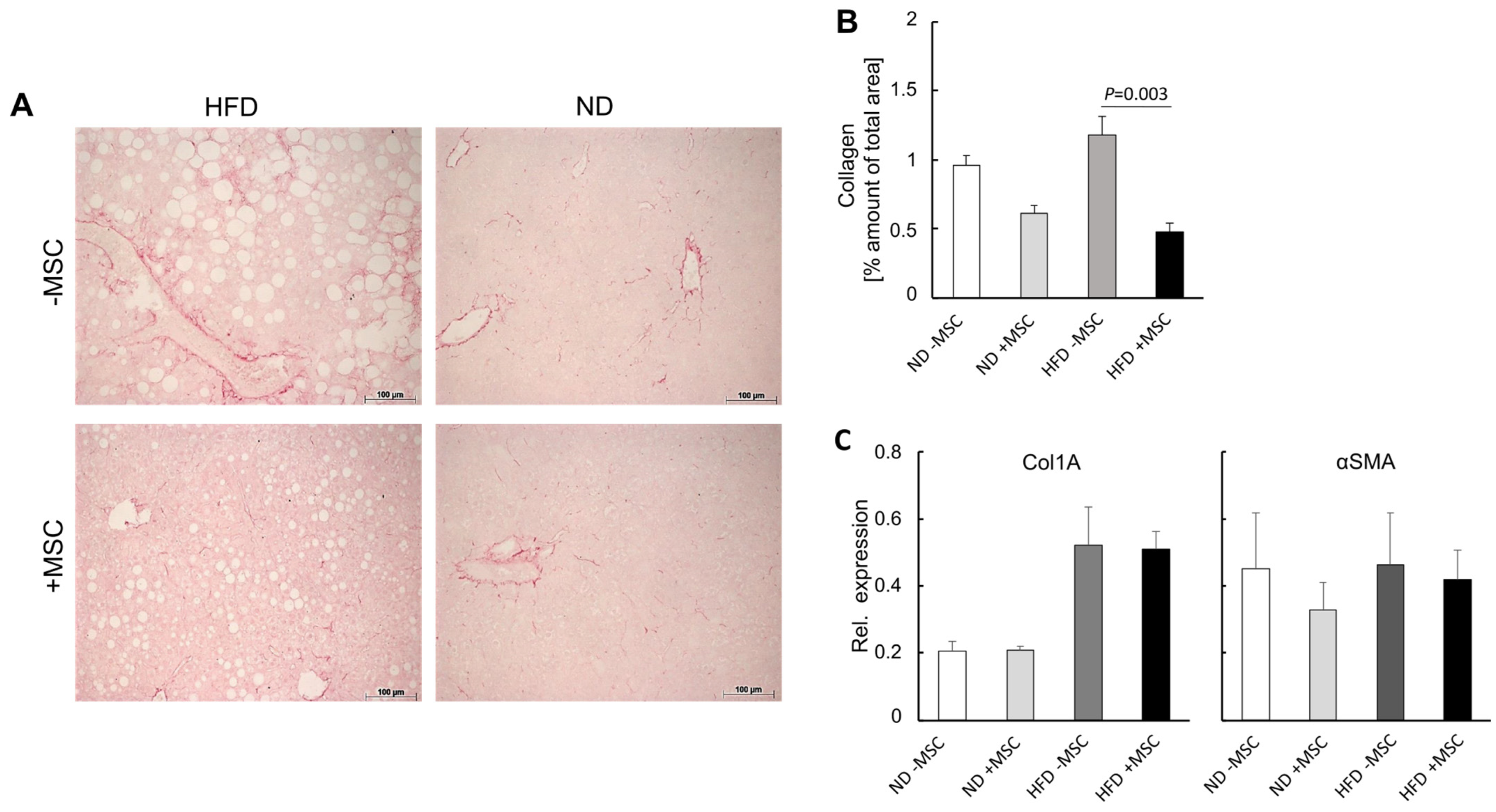
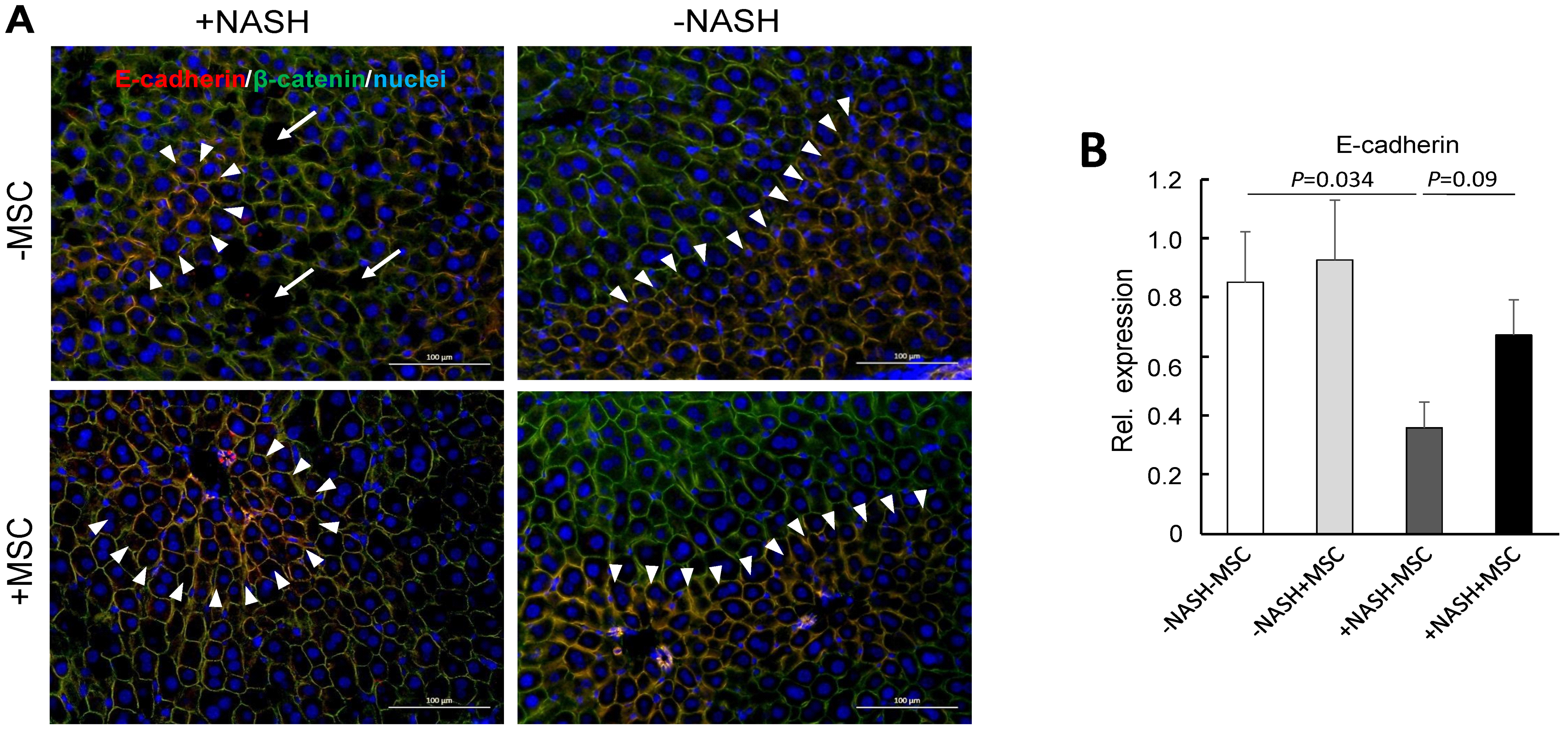
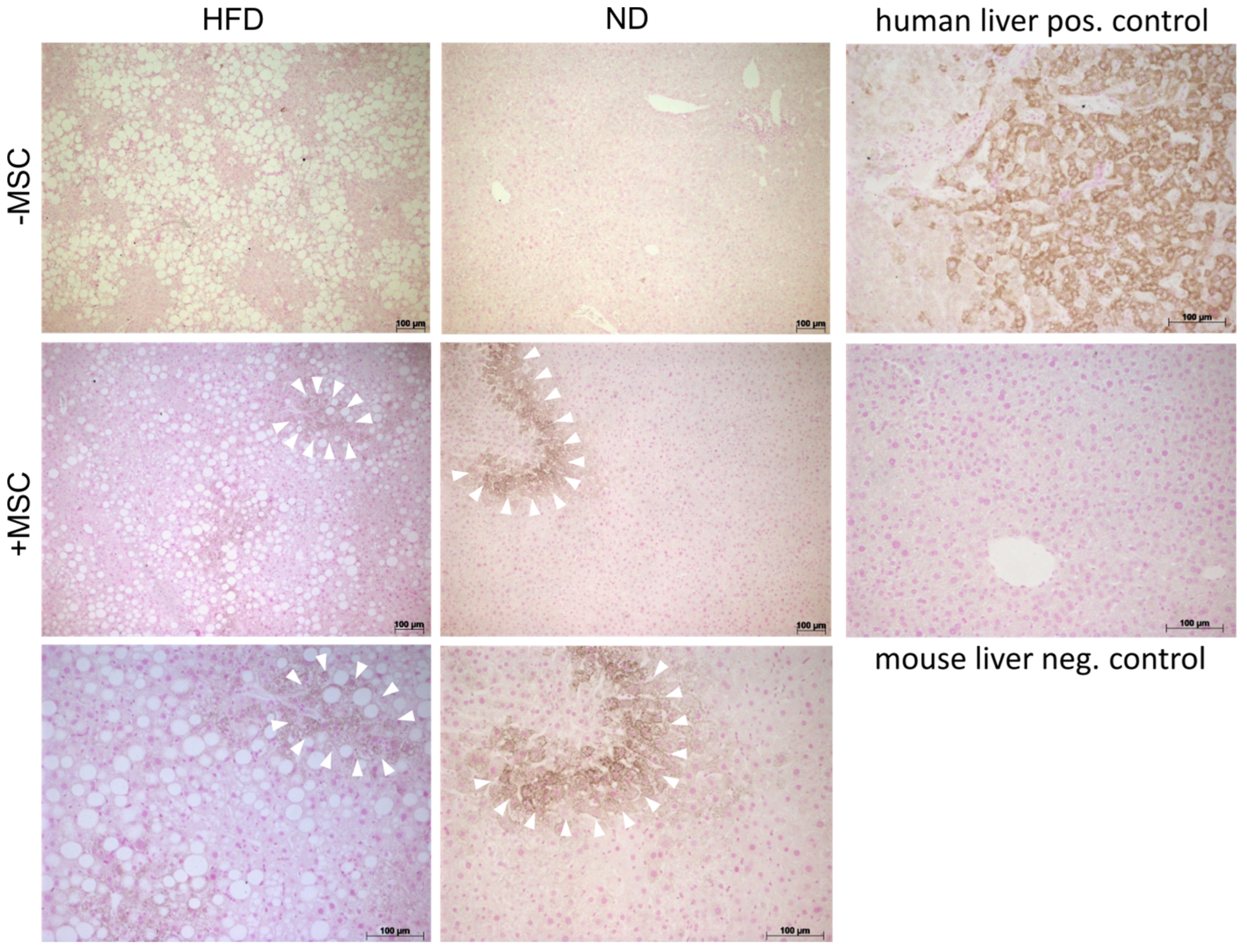
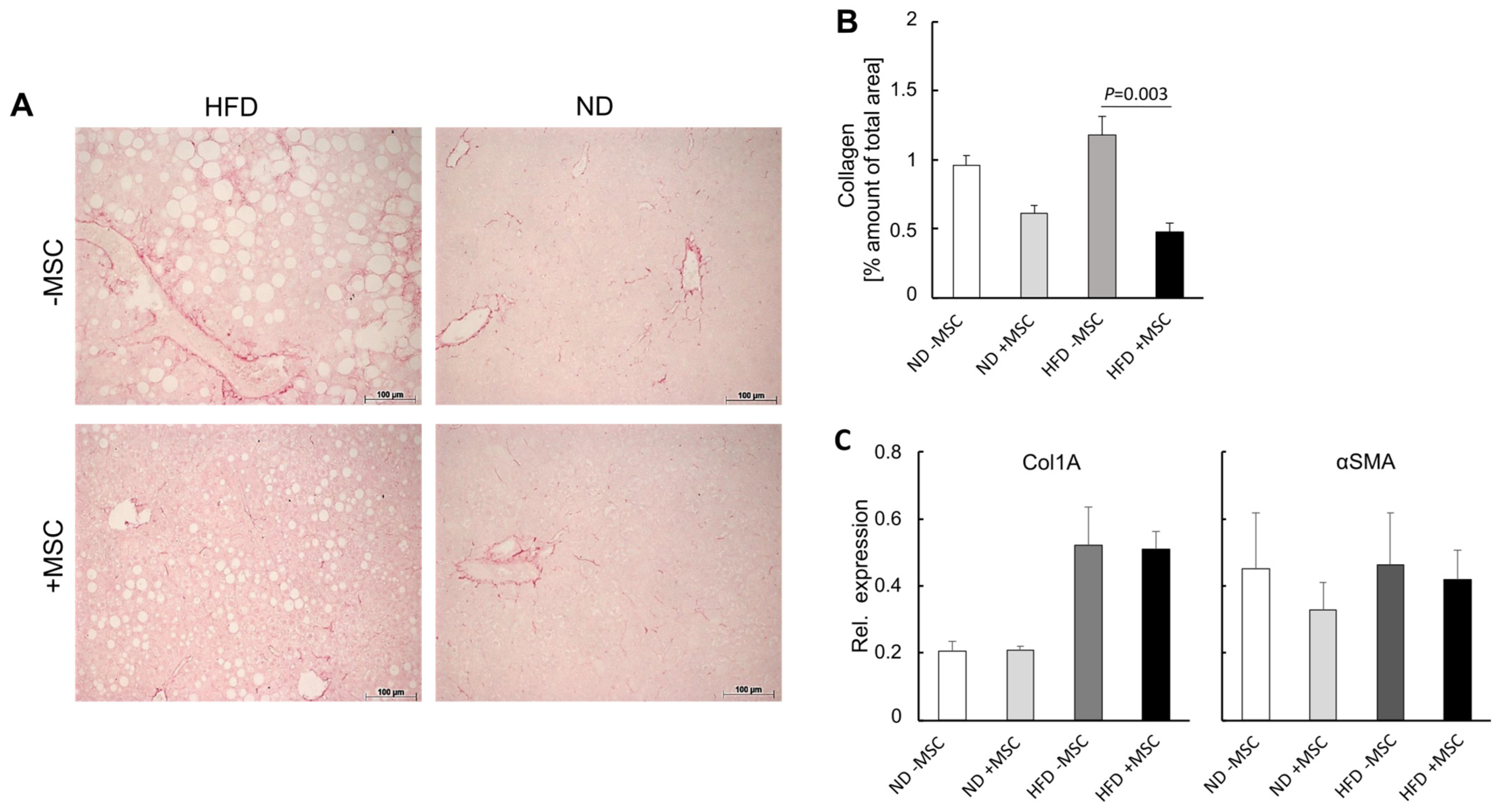
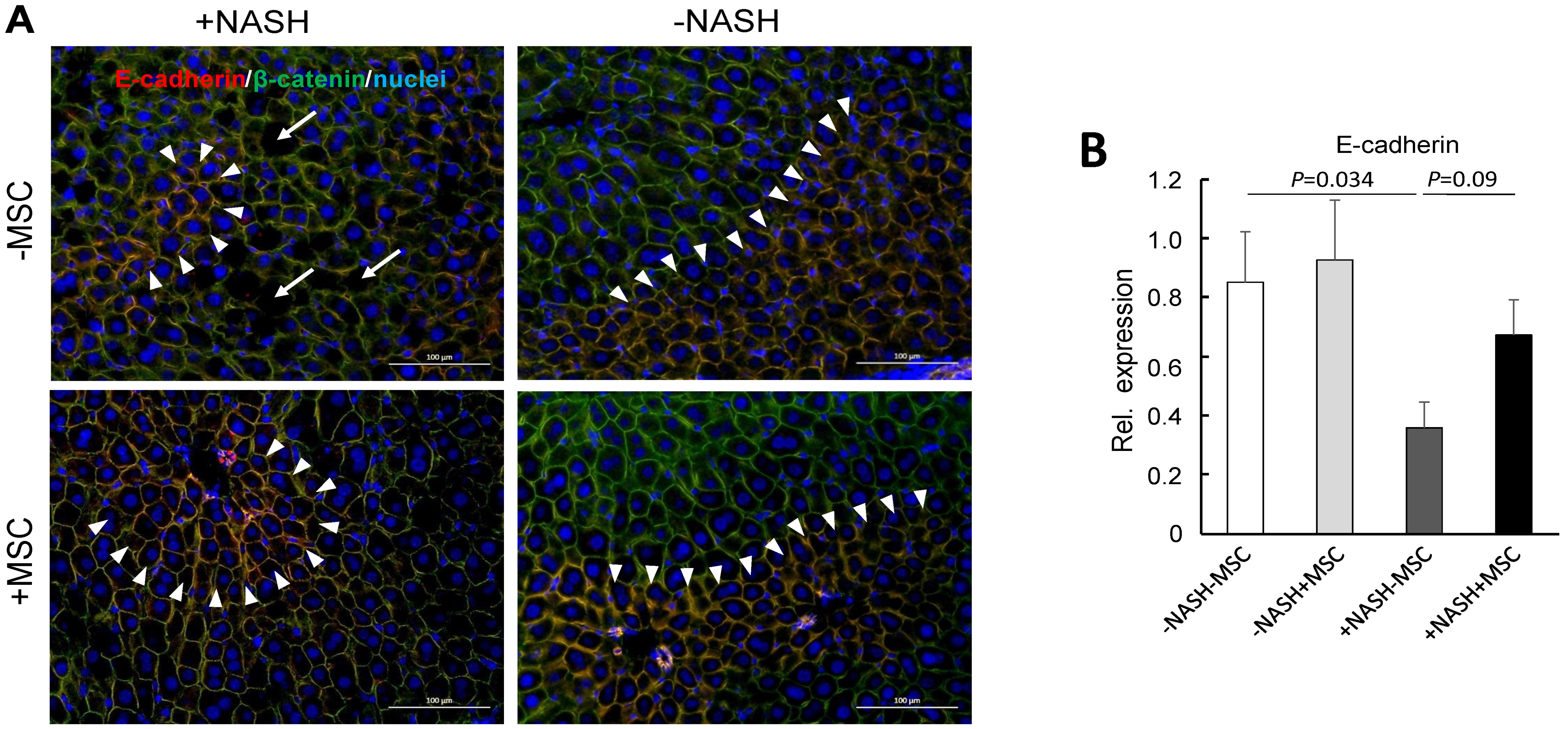
3.3. MSC Treatment Improves Tissue Homeostasis in NASH Livers
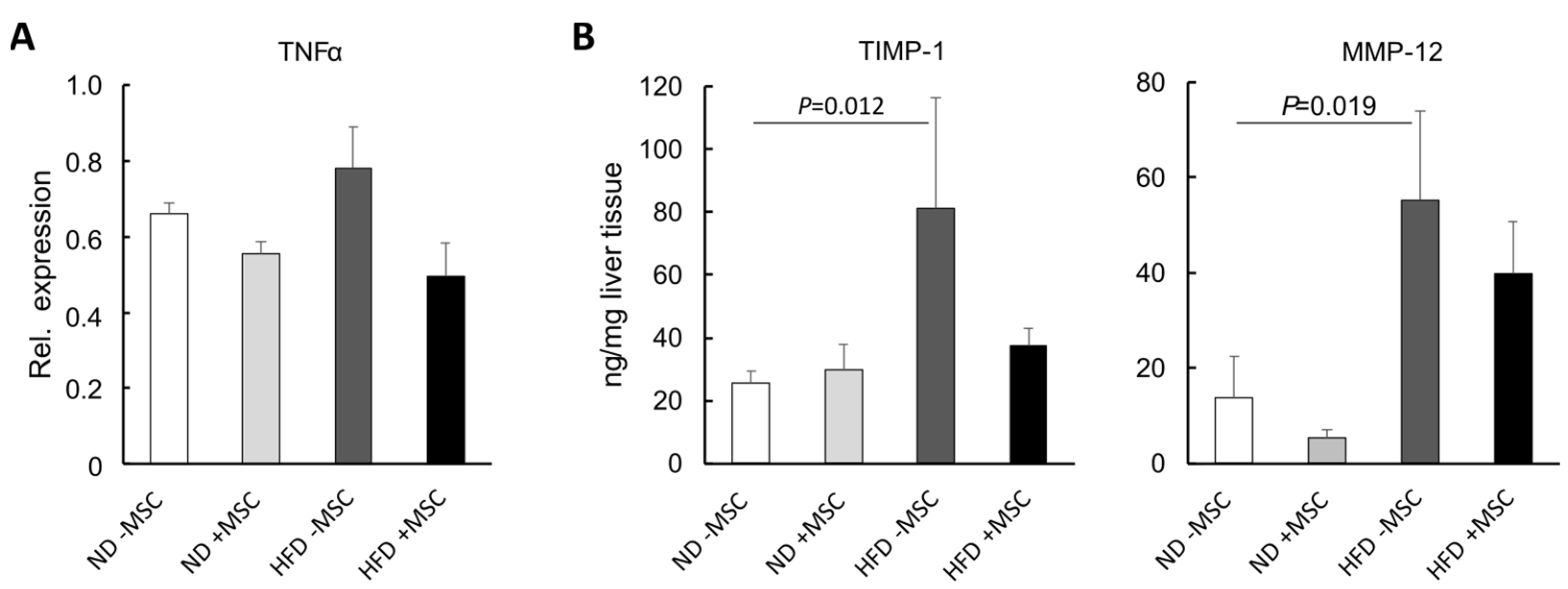
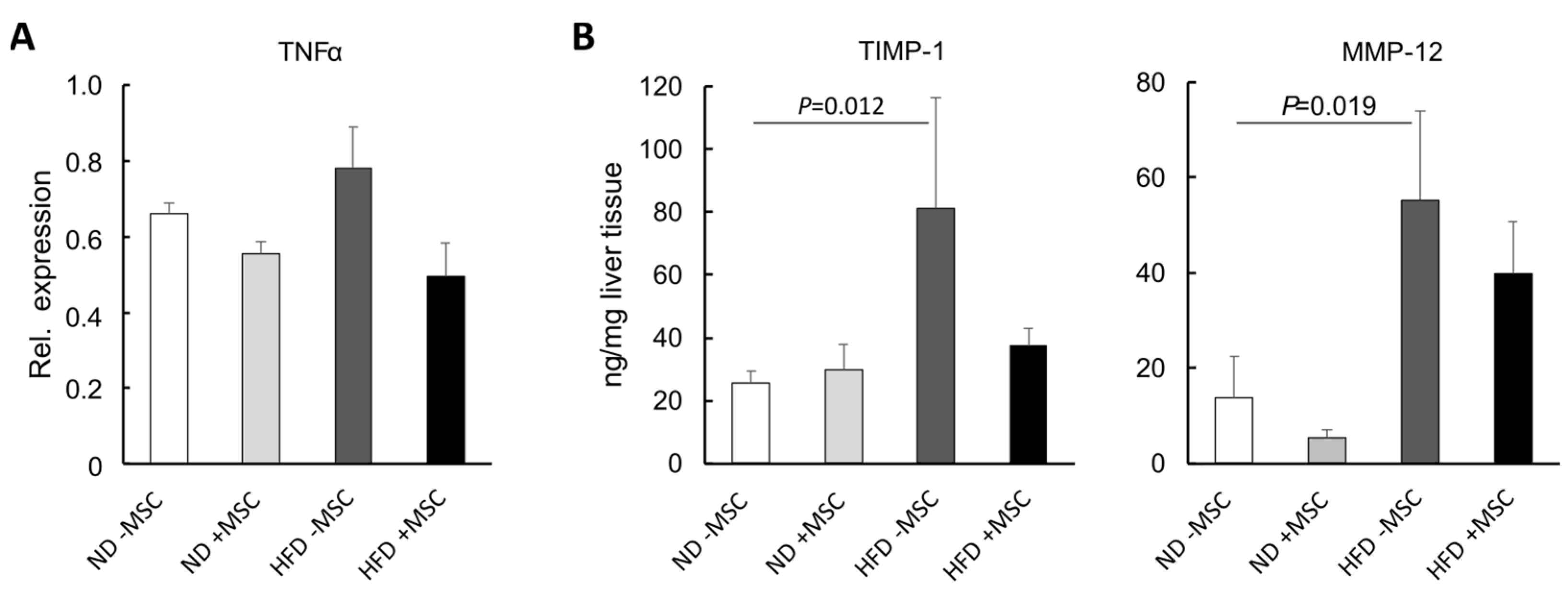
3.4. MSC Ameliorate NASH-Associated Inflammation
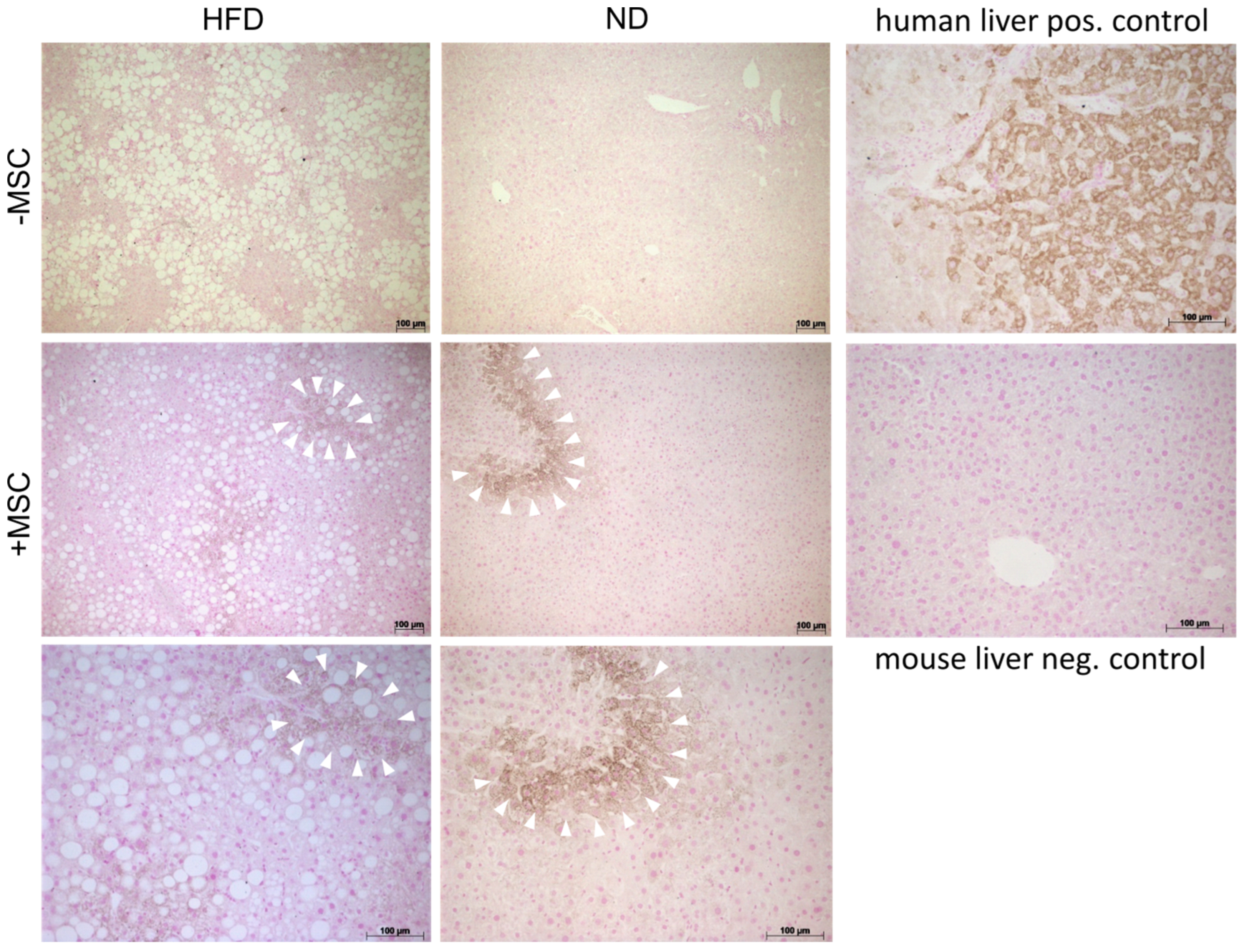
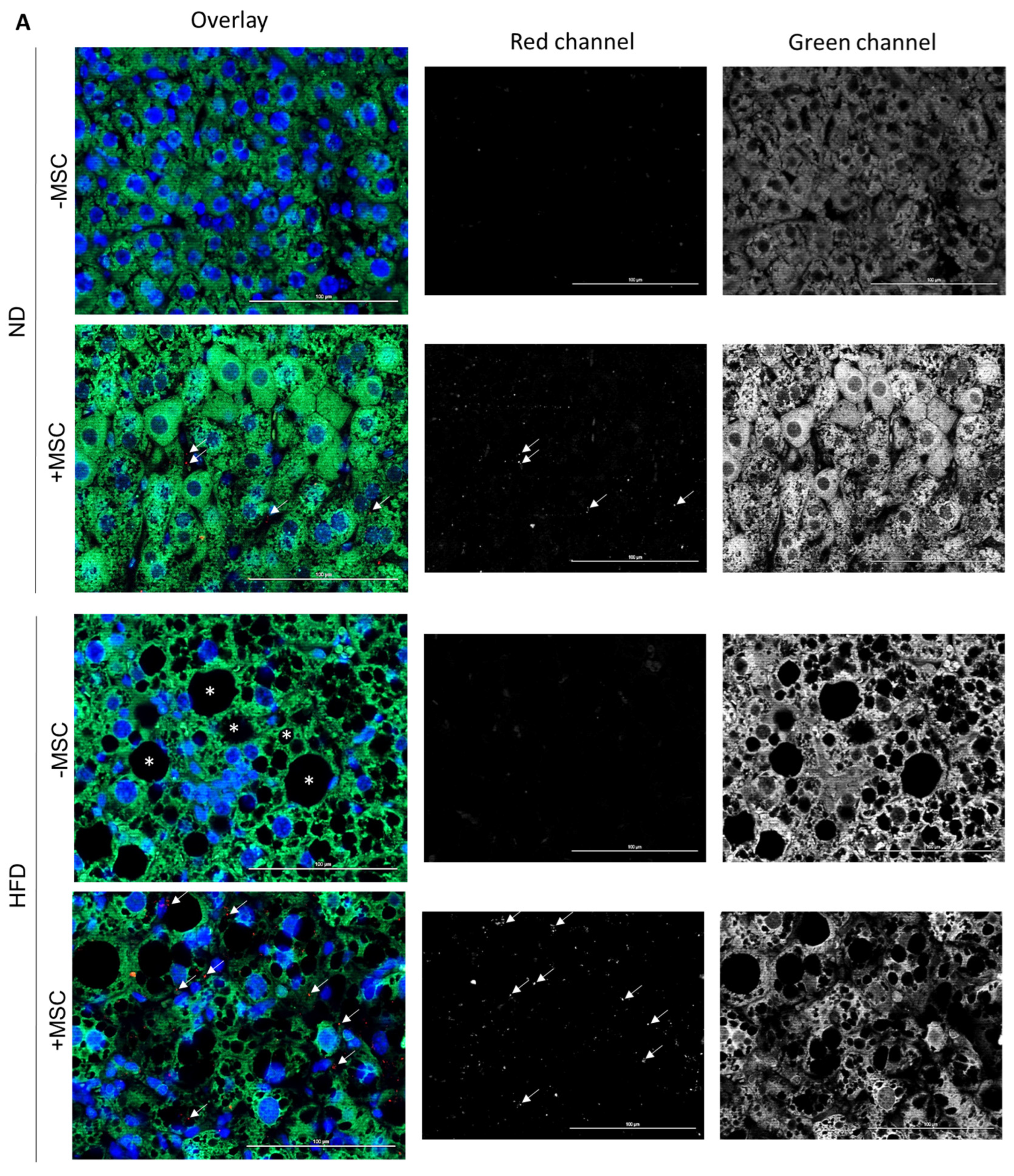
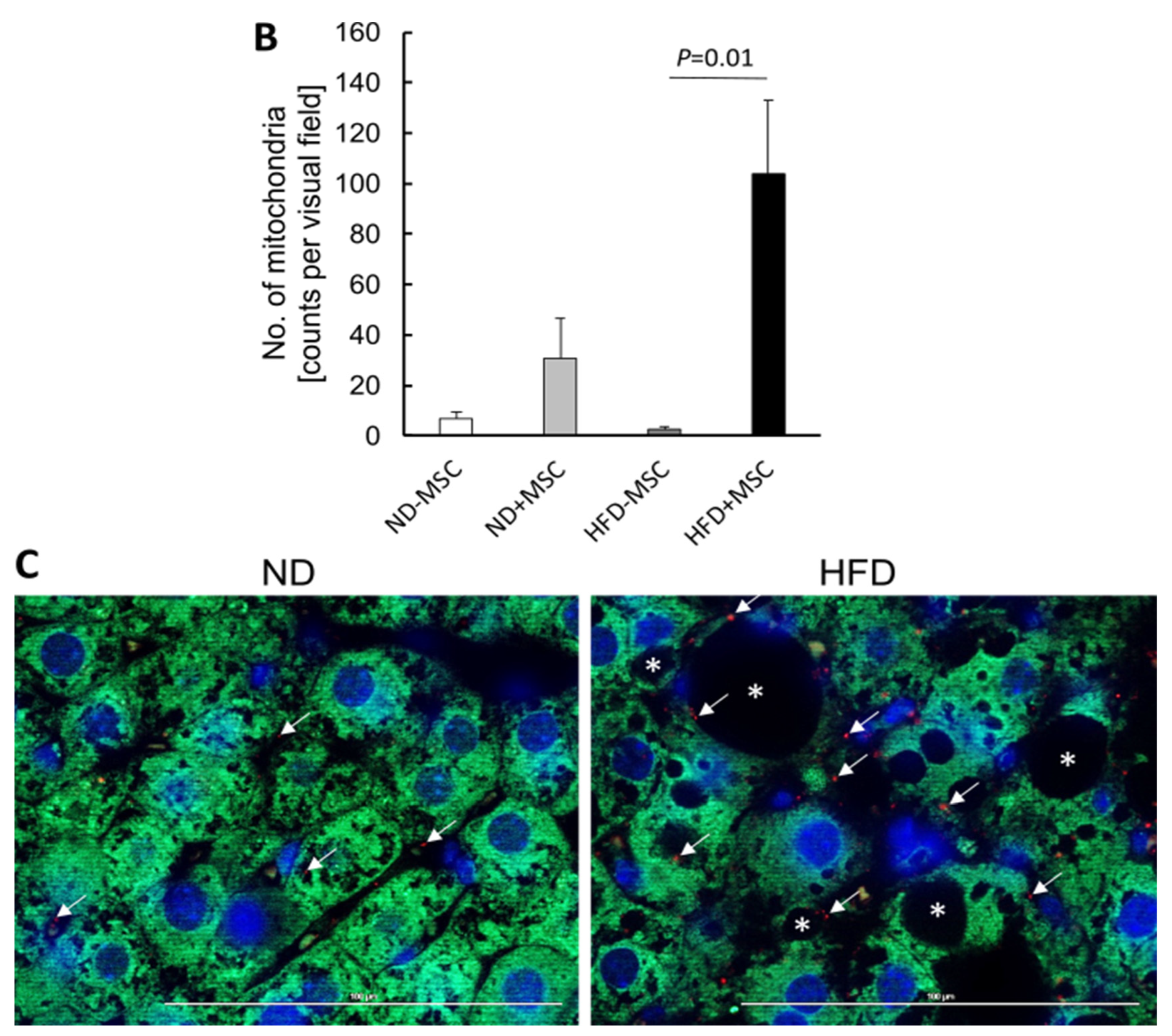
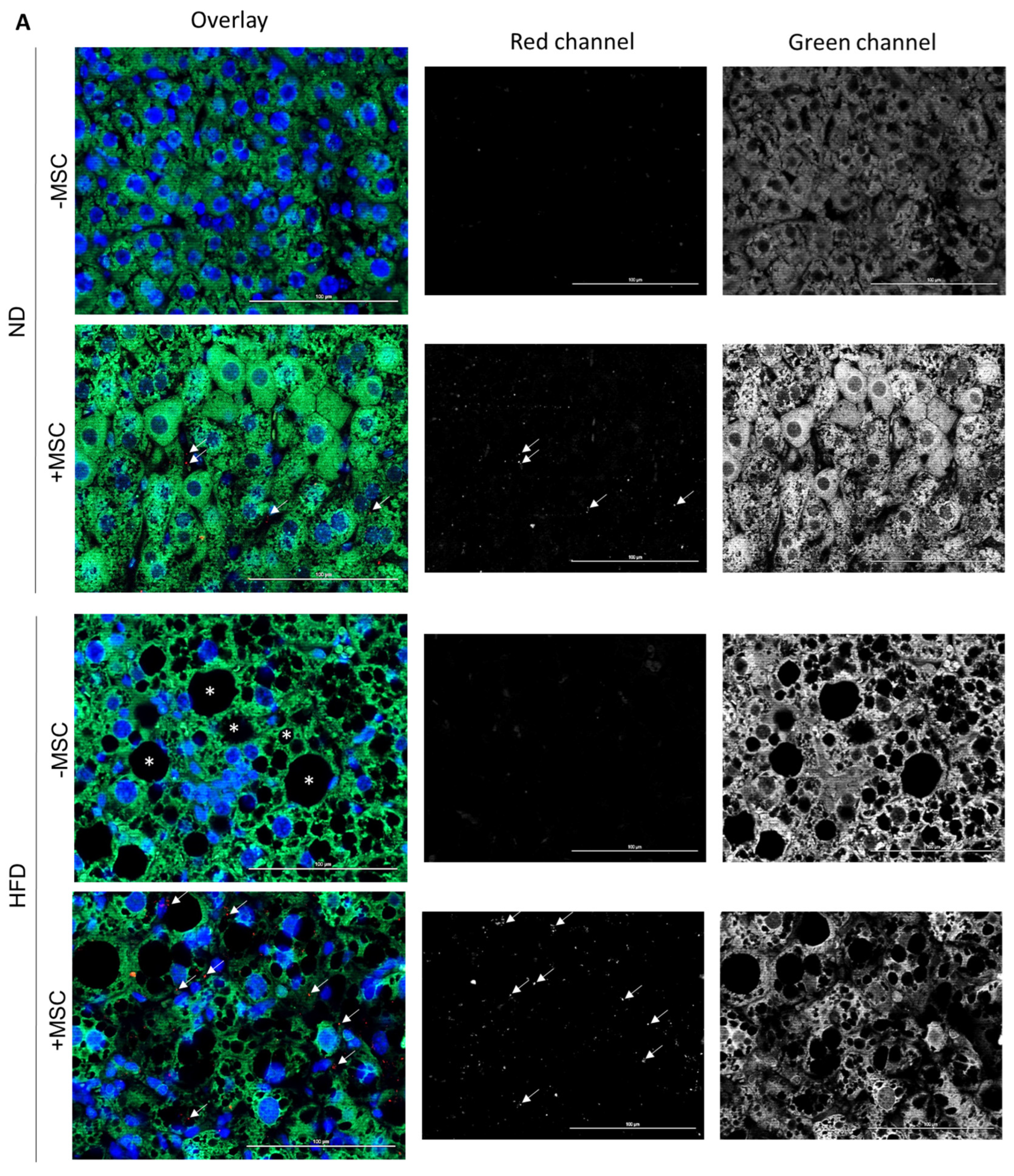
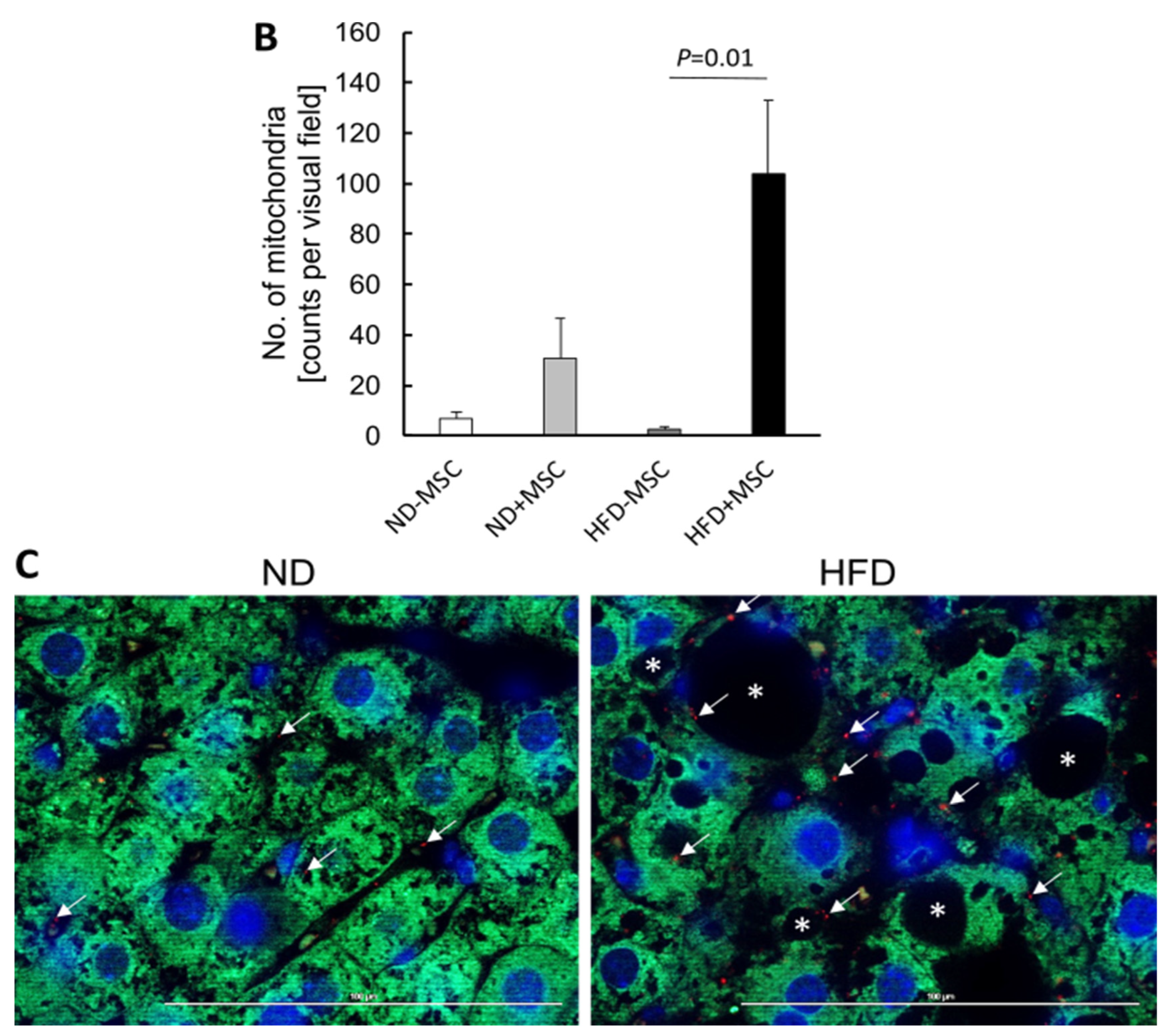
3.5. Human MSC Transplants Donate Mitochondria to Mouse Host Hepatocytes
4. Discussion
4.1. Potential Mechanisms of MSC-Mediated Improvement of Hepatocyte Functions
4.2. Mitochondria Transfer from Human MSC to Mouse Hepatocytes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dietrich, P.; Hellerbrand, C. Non-alcoholic fatty liver disease, obesity and the metabolic syndrome. Best Pract. Research. Clin. Gastroenterol. 2014, 28, 637–653. [Google Scholar] [CrossRef] [PubMed]
- Tacke, F.; Weiskirchen, R. Non-alcoholic fatty liver disease (nafld)/non-alcoholic steatohepatitis (nash)-related liver fibrosis: Mechanisms, treatment and prevention. Ann. Transl. Med. 2021, 9, 729. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, E.; Yatsuji, S.; Tobari, M.; Taniai, M.; Torii, N.; Tokushige, K.; Shiratori, K. Hepatocellular carcinoma in patients with nonalcoholic steatohepatitis. J. Gastroenterol. 2009, 44, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Henry, L. Epidemiology of non-alcoholic fatty liver disease and hepatocellular carcinoma. JHEP Rep. Innov. Hepatol. 2021, 3, 100305. [Google Scholar] [CrossRef] [PubMed]
- Roeb, E. Non-alcoholic fatty liver diseases: Current challenges and future directions. Ann. Transl. Med. 2021, 9, 726. [Google Scholar] [CrossRef]
- Tannapfel, A.; Denk, H.; Dienes, H.P.; Langner, C.; Schirmacher, P.; Trauner, M.; Flott-Rahmel, B. Histopathological diagnosis of non-alcoholic and alcoholic fatty liver disease. Virchows Arch. Int. J. Pathol. 2011, 458, 511–523. [Google Scholar] [CrossRef] [Green Version]
- Day, C.P.; Saksena, S. Non-alcoholic steatohepatitis: Definitions and pathogenesis. J. Gastroenterol. Hepatol. 2002, 17, S377–S384. [Google Scholar] [CrossRef]
- Pascale, A.; Pais, R.; Ratziu, V. An overview of nonalcoholic steatohepatitis: Past, present and future directions. J. Gastrointest. Liver Dis. JGLD 2010, 19, 415–423. [Google Scholar]
- Bessone, F.; Razori, M.V.; Roma, M.G. Molecular pathways of nonalcoholic fatty liver disease development and progression. Cell. Mol. Life Sci. CMLS 2019, 76, 99–128. [Google Scholar] [CrossRef]
- Kim, J.Y.; He, F.; Karin, M. From liver fat to cancer: Perils of the western diet. Cancers 2021, 13, 1095. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Friedman, S.L.; Shulman, G.I. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell 2021, 184, 2537–2564. [Google Scholar] [CrossRef] [PubMed]
- Pittenger, M.F.; Discher, D.E.; Peault, B.M.; Phinney, D.G.; Hare, J.M.; Caplan, A.I. Mesenchymal stem cell perspective: Cell biology to clinical progress. NPJ Regen. Med. 2019, 4, 22. [Google Scholar] [CrossRef] [Green Version]
- Le Blanc, K.; Davies, L.C. Mesenchymal stromal cells and the innate immune response. Immunol. Lett. 2015, 168, 140–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, R.E.; Yoo, D.; LeRoux, M.A.; Danilkovitch-Miagkova, A. Treatment of inflammatory diseases with mesenchymal stem cells. Inflamm. Allergy Drug Targets 2009, 8, 110–123. [Google Scholar] [CrossRef]
- Salomone, F.; Barbagallo, I.; Puzzo, L.; Piazza, C.; Li Volti, G. Efficacy of adipose tissue-mesenchymal stem cell transplantation in rats with acetaminophen liver injury. Stem Cell Res. 2013, 11, 1037–1044. [Google Scholar] [CrossRef] [Green Version]
- Stock, P.; Bruckner, S.; Winkler, S.; Dollinger, M.M.; Christ, B. Human bone marrow mesenchymal stem cell-derived hepatocytes improve the mouse liver after acute acetaminophen intoxication by preventing progress of injury. Int. J. Mol. Sci. 2014, 15, 7004–7028. [Google Scholar] [CrossRef] [Green Version]
- Tanimoto, H.; Terai, S.; Taro, T.; Murata, Y.; Fujisawa, K.; Yamamoto, N.; Sakaida, I. Improvement of liver fibrosis by infusion of cultured cells derived from human bone marrow. Cell Tissue Res. 2013, 354, 717–728. [Google Scholar] [CrossRef]
- Bruckner, S.; Zipprich, A.; Hempel, M.; Thonig, A.; Schwill, F.; Roderfeld, M.; Roeb, E.; Christ, B. Improvement of portal venous pressure in cirrhotic rat livers by systemic treatment with adipose tissue-derived mesenchymal stromal cells. Cytotherapy 2017, 19, 1462–1473. [Google Scholar] [CrossRef]
- Wang, H.; Wang, D.; Yang, L.; Wang, Y.; Jia, J.; Na, D.; Chen, H.; Luo, Y.; Liu, C. Compact bone-derived mesenchymal stem cells attenuate nonalcoholic steatohepatitis in a mouse model by modulation of cd4 cells differentiation. Int. Immunopharmacol. 2017, 42, 67–73. [Google Scholar] [CrossRef] [Green Version]
- Winkler, S.; Borkham-Kamphorst, E.; Stock, P.; Bruckner, S.; Dollinger, M.; Weiskirchen, R.; Christ, B. Human mesenchymal stem cells towards non-alcoholic steatohepatitis in an immunodeficient mouse model. Exp. Cell Res. 2014, 326, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Ezquer, M.; Ezquer, F.; Ricca, M.; Allers, C.; Conget, P. Intravenous administration of multipotent stromal cells prevents the onset of non-alcoholic steatohepatitis in obese mice with metabolic syndrome. J. Hepatol. 2011, 55, 1112–1120. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.W.; Hsiao, W.T.; Lee, O.K. Mesenchymal stromal cell-based therapies reduce obesity and metabolic syndromes induced by a high-fat diet. Transl. Res. J. Lab. Clin. Med. 2017, 182, 61–74 e68. [Google Scholar] [CrossRef] [PubMed]
- Domingues, C.C.; Kundu, N.; Kropotova, Y.; Ahmadi, N.; Sen, S. Antioxidant-upregulated mesenchymal stem cells reduce inflammation and improve fatty liver disease in diet-induced obesity. Stem Cell Res. Ther. 2019, 10, 280. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Zeng, X.; Liu, Z.; Chen, X.; Li, L.; Luo, R.; Liu, X.; Zhang, J.; Liu, J.; Lu, Y.; et al. Mesenchymal stromal cells protect hepatocytes from lipotoxicity through alleviation of endoplasmic reticulum stress by restoring serca activity. J. Cell. Mol. Med. 2021, 25, 2976–2993. [Google Scholar] [CrossRef]
- Yang, M.; Cui, Y.; Song, J.; Cui, C.; Wang, L.; Liang, K.; Wang, C.; Sha, S.; He, Q.; Hu, H.; et al. Mesenchymal stem cell-conditioned medium improved mitochondrial function and alleviated inflammation and apoptosis in non-alcoholic fatty liver disease by regulating sirt1. Biochem. Biophys. Res. Commun. 2021, 546, 74–82. [Google Scholar] [CrossRef]
- Nassir, F.; Ibdah, J.A. Role of mitochondria in nonalcoholic fatty liver disease. Int. J. Mol. Sci. 2014, 15, 8713–8742. [Google Scholar] [CrossRef] [Green Version]
- Stankova, P.; Kucera, O.; Peterova, E.; Elkalaf, M.; Rychtrmoc, D.; Melek, J.; Podhola, M.; Zubanova, V.; Cervinkova, Z. Western diet decreases the liver mitochondrial oxidative flux of succinate: Insight from a murine nafld model. Int. J. Mol. Sci. 2021, 22, 6908. [Google Scholar] [CrossRef]
- Takeichi, Y.; Miyazawa, T.; Sakamoto, S.; Hanada, Y.; Wang, L.; Gotoh, K.; Uchida, K.; Katsuhara, S.; Sakamoto, R.; Ishihara, T.; et al. Non-alcoholic fatty liver disease in mice with hepatocyte-specific deletion of mitochondrial fission factor. Diabetologia 2021, 64, 2092–2107. [Google Scholar] [CrossRef]
- Hsu, M.J.; Karkossa, I.; Schafer, I.; Christ, M.; Kuhne, H.; Schubert, K.; Rolle-Kampczyk, U.E.; Kalkhof, S.; Nickel, S.; Seibel, P.; et al. Mitochondrial transfer by human mesenchymal stromal cells ameliorates hepatocyte lipid load in a mouse model of nash. Biomedicines 2020, 8, 350. [Google Scholar] [CrossRef]
- Stock, P.; Bruckner, S.; Ebensing, S.; Hempel, M.; Dollinger, M.M.; Christ, B. The generation of hepatocytes from mesenchymal stem cells and engraftment into murine liver. Nat. Protoc. 2010, 5, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Aurich, I.; Mueller, L.P.; Aurich, H.; Luetzkendorf, J.; Tisljar, K.; Dollinger, M.M.; Schormann, W.; Walldorf, J.; Hengstler, J.G.; Fleig, W.E.; et al. Functional integration of hepatocytes derived from human mesenchymal stem cells into mouse livers. Gut 2007, 56, 405–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkler, S.; Hempel, M.; Bruckner, S.; Tautenhahn, H.M.; Kaufmann, R.; Christ, B. Identification of pathways in liver repair potentially targeted by secretory proteins from human mesenchymal stem cells. Int. J. Mol. Sci. 2016, 17, 1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- Bedossa, P.; Poitou, C.; Veyrie, N.; Bouillot, J.L.; Basdevant, A.; Paradis, V.; Tordjman, J.; Clement, K. Histopathological algorithm and scoring system for evaluation of liver lesions in morbidly obese patients. Hepatology 2012, 56, 1751–1759. [Google Scholar] [CrossRef]
- Filali-Mouncef, Y.; Hunter, C.; Roccio, F.; Zagkou, S.; Dupont, N.; Primard, C.; Proikas-Cezanne, T.; Reggiori, F. The menage a trois of autophagy, lipid droplets and liver disease. Autophagy 2021, 18, 50–72. [Google Scholar] [CrossRef]
- Irungbam, K.; Churin, Y.; Matono, T.; Weglage, J.; Ocker, M.; Glebe, D.; Hardt, M.; Koeppel, A.; Roderfeld, M.; Roeb, E. Cannabinoid receptor 1 knockout alleviates hepatic steatosis by downregulating perilipin 2. Lab. Investig. 2020, 100, 454–465. [Google Scholar] [CrossRef]
- Wang, Y.; Nakajima, T.; Gonzalez, F.J.; Tanaka, N. Ppars as metabolic regulators in the liver: Lessons from liver-specific ppar-null mice. Int. J. Mol. Sci. 2020, 21, 2061. [Google Scholar] [CrossRef] [Green Version]
- Shah, S.S.; Chandan, V.S. Hepatocyte antigen expression in barrett esophagus and associated neoplasia. Appl. Immunohistochem. Mol. Morphol. AIMM 2018, 26, 557–561. [Google Scholar] [CrossRef]
- Hagstrom, H.; Nasr, P.; Ekstedt, M.; Hammar, U.; Stal, P.; Hultcrantz, R.; Kechagias, S. Fibrosis stage but not nash predicts mortality and time to development of severe liver disease in biopsy-proven nafld. J. Hepatol. 2017, 67, 1265–1273. [Google Scholar] [CrossRef]
- Popov, Y.; Sverdlov, D.Y.; Sharma, A.K.; Bhaskar, K.R.; Li, S.; Freitag, T.L.; Lee, J.; Dieterich, W.; Melino, G.; Schuppan, D. Tissue transglutaminase does not affect fibrotic matrix stability or regression of liver fibrosis in mice. Gastroenterology 2011, 140, 1642–1652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hempel, M.; Schmitz, A.; Winkler, S.; Kucukoglu, O.; Bruckner, S.; Niessen, C.; Christ, B. Pathological implications of cadherin zonation in mouse liver. Cell. Mol. Life Sci. CMLS 2015, 72, 2599–2612. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Diehl, A.M. Evidence for and against epithelial-to-mesenchymal transition in the liver. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G881–G890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roeb, E.; Graeve, L.; Mullberg, J.; Matern, S.; Rose-John, S. Timp-1 protein expression is stimulated by il-1 beta and il-6 in primary rat hepatocytes. FEBS Lett. 1994, 349, 45–49. [Google Scholar] [CrossRef] [Green Version]
- El-Youssef, M.; Mu, Y.; Huang, L.; Stellmach, V.; Crawford, S.E. Increased expression of transforming growth factor-beta1 and thrombospondin-1 in congenital hepatic fibrosis: Possible role of the hepatic stellate cell. J. Pediatric Gastroenterol. Nutr. 1999, 28, 386–392. [Google Scholar] [CrossRef]
- Min-DeBartolo, J.; Schlerman, F.; Akare, S.; Wang, J.; McMahon, J.; Zhan, Y.; Syed, J.; He, W.; Zhang, B.; Martinez, R.V. Thrombospondin-i is a critical modulator in non-alcoholic steatohepatitis (nash). PLoS ONE 2019, 14, e0226854. [Google Scholar] [CrossRef] [Green Version]
- Gwag, T.; Reddy Mooli, R.G.; Li, D.; Lee, S.; Lee, E.Y.; Wang, S. Macrophage-derived thrombospondin 1 promotes obesity-associated non-alcoholic fatty liver disease. JHEP Rep. Innov. Hepatol. 2021, 3, 100193. [Google Scholar] [CrossRef]
- Nickel, S.; Vlaic, S.; Christ, M.; Schubert, K.; Henschler, R.; Tautenhahn, F.; Burger, C.; Kuhne, H.; Erler, S.; Roth, A.; et al. Mesenchymal stromal cells mitigate liver damage after extended resection in the pig by modulating thrombospondin-1/tgf-beta. NPJ Regen. Med. 2021, 6, 84. [Google Scholar] [CrossRef]
- Luukkonen, P.K.; Qadri, S.; Ahlholm, N.; Porthan, K.; Mannisto, V.; Sammalkorpi, H.; Penttila, A.K.; Hakkarainen, A.; Lehtimaki, T.E.; Gaggini, M.; et al. Distinct contributions of metabolic dysfunction and genetic risk factors in the pathogenesis of non-alcoholic fatty liver disease. J. Hepatol. 2022, 76, 526–535. [Google Scholar] [CrossRef]
- Al-Sulaiti, H.; Diboun, I.; Agha, M.V.; Mohamed, F.F.S.; Atkin, S.; Domling, A.S.; Elrayess, M.A.; Mazloum, N.A. Metabolic signature of obesity-associated insulin resistance and type 2 diabetes. J. Transl. Med. 2019, 17, 348. [Google Scholar] [CrossRef] [Green Version]
- Sliz, E.; Sebert, S.; Wurtz, P.; Kangas, A.J.; Soininen, P.; Lehtimaki, T.; Kahonen, M.; Viikari, J.; Mannikko, M.; Ala-Korpela, M.; et al. Nafld risk alleles in pnpla3, tm6sf2, gckr and lyplal1 show divergent metabolic effects. Hum. Mol. Genet. 2018, 27, 2214–2223. [Google Scholar] [CrossRef] [Green Version]
- Speliotes, E.K.; George, J. Metabolic and genetic contributions to nafld: Really distinct and homogeneous? J. Hepatol. 2022, 76, 498–500. [Google Scholar] [CrossRef] [PubMed]
- Arrese, M.; Arab, J.P.; Barrera, F.; Kaufmann, B.; Valenti, L.; Feldstein, A.E. Insights into nonalcoholic fatty-liver disease heterogeneity. Semin. Liver Dis. 2021, 41, 421–434. [Google Scholar] [CrossRef] [PubMed]
- Guilherme, A.; Virbasius, J.V.; Puri, V.; Czech, M.P. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat. Reviews. Mol. Cell Biol. 2008, 9, 367–377. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, K.; Yang, L.; McCall, S.; Huang, J.; Yu, X.X.; Pandey, S.K.; Bhanot, S.; Monia, B.P.; Li, Y.X.; Diehl, A.M. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology 2007, 45, 1366–1374. [Google Scholar] [CrossRef]
- Zhang, Q.; Liu, X.; Piao, C.; Jiao, Z.; Ma, Y.; Wang, Y.; Liu, T.; Xu, J.; Wang, H. Effect of conditioned medium from adipose derived mesenchymal stem cells on endoplasmic reticulum stress and lipid metabolism after hepatic ischemia reperfusion injury and hepatectomy in swine. Life Sci. 2022, 289, 120212. [Google Scholar] [CrossRef] [PubMed]
- Schwabe, R.F.; Tabas, I.; Pajvani, U.B. Mechanisms of fibrosis development in nonalcoholic steatohepatitis. Gastroenterology 2020, 158, 1913–1928. [Google Scholar] [CrossRef]
- El-Derany, M.O.; AbdelHamid, S.G. Upregulation of mir-96-5p by bone marrow mesenchymal stem cells and their exosomes alleviate non-alcoholic steatohepatitis: Emphasis on caspase-2 signaling inhibition. Biochem. Pharmacol. 2021, 190, 114624. [Google Scholar] [CrossRef]
- Ohara, M.; Ohnishi, S.; Hosono, H.; Yamamoto, K.; Yuyama, K.; Nakamura, H.; Fu, Q.; Maehara, O.; Suda, G.; Sakamoto, N. Extracellular vesicles from amnion-derived mesenchymal stem cells ameliorate hepatic inflammation and fibrosis in rats. Stem Cells Int. 2018, 2018, 3212643. [Google Scholar] [CrossRef] [Green Version]
- Pellicoro, A.; Aucott, R.L.; Ramachandran, P.; Robson, A.J.; Fallowfield, J.A.; Snowdon, V.K.; Hartland, S.N.; Vernon, M.; Duffield, J.S.; Benyon, R.C.; et al. Elastin accumulation is regulated at the level of degradation by macrophage metalloelastase (mmp-12) during experimental liver fibrosis. Hepatology 2012, 55, 1965–1975. [Google Scholar] [CrossRef]
- Oh, S.H.; Swiderska-Syn, M.; Jewell, M.L.; Premont, R.T.; Diehl, A.M. Liver regeneration requires yap1-tgfbeta-dependent epithelial-mesenchymal transition in hepatocytes. J. Hepatol. 2018, 69, 359–367. [Google Scholar] [CrossRef] [Green Version]
- Di Ciaula, A.; Passarella, S.; Shanmugam, H.; Noviello, M.; Bonfrate, L.; Wang, D.Q.; Portincasa, P. Nonalcoholic fatty liver disease (nafld). Mitochondria as players and targets of therapies? Int. J. Mol. Sci. 2021, 22, 5375. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.J.; Campbell-Sargent, C.; Mirshahi, F.; Rizzo, W.B.; Contos, M.J.; Sterling, R.K.; Luketic, V.A.; Shiffman, M.L.; Clore, J.N. Nonalcoholic steatohepatitis: Association of insulin resistance and mitochondrial abnormalities. Gastroenterology 2001, 120, 1183–1192. [Google Scholar] [CrossRef]
- Pessayre, D.; Fromenty, B. Nash: A mitochondrial disease. J. Hepatol. 2005, 42, 928–940. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Qian, H.; Chen, A.; Ni, H.M.; Ding, W.X. Perspectives on mitochondria-er and mitochondria-lipid droplet contact in hepatocytes and hepatic lipid metabolism. Cells 2021, 10, 2273. [Google Scholar] [CrossRef] [PubMed]
- Benador, I.Y.; Veliova, M.; Liesa, M.; Shirihai, O.S. Mitochondria bound to lipid droplets: Where mitochondrial dynamics regulate lipid storage and utilization. Cell Metab. 2019, 29, 827–835. [Google Scholar] [CrossRef] [Green Version]
- Hsu, Y.C.; Wu, Y.T.; Yu, T.H.; Wei, Y.H. Mitochondria in mesenchymal stem cell biology and cell therapy: From cellular differentiation to mitochondrial transfer. Semin. Cell Dev. Biol. 2016, 52, 119–131. [Google Scholar] [CrossRef]
- Velarde, F.; Ezquerra, S.; Delbruyere, X.; Caicedo, A.; Hidalgo, Y.; Khoury, M. Mesenchymal stem cell-mediated transfer of mitochondria: Mechanisms and functional impact. Cell. Mol. Life Sci. CMLS 2022, 79, 177. [Google Scholar] [CrossRef]
- Gomzikova, M.O.; James, V.; Rizvanov, A.A. Mitochondria donation by mesenchymal stem cells: Current understanding and mitochondria transplantation strategies. Front. Cell Dev. Biol. 2021, 9, 653322. [Google Scholar] [CrossRef]
- Kubat, G.B.; Ozler, M.; Ulger, O.; Ekinci, O.; Atalay, O.; Celik, E.; Safali, M.; Budak, M.T. The effects of mesenchymal stem cell mitochondrial transplantation on doxorubicin-mediated nephrotoxicity in rats. J. Biochem. Mol. Toxicol. 2021, 35, e22612. [Google Scholar] [CrossRef]
- McCully, J.D.; Cowan, D.B.; Pacak, C.A.; Toumpoulis, I.K.; Dayalan, H.; Levitsky, S. Injection of isolated mitochondria during early reperfusion for cardioprotection. Am. J. Physiology. Heart Circ. Physiol. 2009, 296, H94–H105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.C.; Liu, S.Y.; Lai, H.S.; Lai, I.R. Isolated mitochondria infusion mitigates ischemia-reperfusion injury of the liver in rats. Shock 2013, 39, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Emani, S.M.; Piekarski, B.L.; Harrild, D.; Del Nido, P.J.; McCully, J.D. Autologous mitochondrial transplantation for dysfunction after ischemia-reperfusion injury. J. Thorac. Cardiovasc. Surg. 2017, 154, 286–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antigen | Cat. No., Supplier | Dilution |
|---|---|---|
| HepPar1 [OCH1E5] | MAB7927, Abnova, Taipeh, Taiwan | 1:100 |
| Perilipin 1 | ab3526, Abcam, Cambridge, England | 1:600 |
| E-cadherin | 610182, BD Biosciences, Heidelberg, Germany | 1:200 |
| β-catenin | 610154, BD Biosciences, Heidelberg, Germany | 1:200 |
| THBS1 | ab1823, Abcam, Cambridge, England | 1:100 |
| CD42b | ab183345, Abcam, Cambridge, England | 1:200 |
| N-cadherin | 610921, BD Biosciences, Heidelberg, Germany | 1:200 |
| ZO-1 | 44-2200, InVitrogen, Carlsbad, CA, USA | 1:200 |
| Anti-human mitochondria | MAB1273, Millipore, Darmstadt, Germany | 1:200 |
| Cyclophilin | 2175, Cell Signaling, Cambridge, England | 1:100 |
| HRP goat anti-mouse Ig | 554002, BD Biosciences, Heidelberg, Germany | 1:200 |
| Goat anti-mouse Cy3 | 115-165-003, Dianova, Hamburg, Germany | 1:200 |
| Goat anti-rabbit AlexaFluor 488 | A11008, Life Technologies, Ober-Olm, Germany | 1:300 |
| Goat anti-mouse AlexaFluor 488 | 115-545-003, Dianova, Hamburg, Germany | 1:200 |
| Target Transcript | F (5′→3′) | R (5′→3′) | bp | h/m |
|---|---|---|---|---|
| ACAA2 | GGGCACTGAAGAAAGCAGGA | CGTGAACCAGGTGTGCAGTA | 198 | h |
| ACATAACTTCACGCCCCTGG | GCAAAAGCTTCGTTCACGTCT | 162 | m | |
| ACOX1 | GCCGCCGAGAGATCGAGAAC | CGCCCTCGGTGCACAAAA | 193 | h |
| CGTCGAGAAATCGAGAACTTG | GGTTCCACAAAATTGACCATATGTA | 200 | m | |
| ACSL1 | GTGGAACTACAGGCAACCCC | ATCATCTGGGCAAGGATTGAC | 113 | h |
| AAGTGGAACTACAGGCAACCC | AGCTCCATGACACAGCATTACA | 190 | m | |
| CPT1 | AGTTCTCTTGCCCTGAGACG | GGGGCTTTGGACCGGTTG | 139 | h |
| CATTCCGCCGCCGCC | ACCAGTGATGATGCCATTCTTG | 200 | m | |
| FABP1 | GCTGGGTCCAAAGTGATCCA | TATGTCGCCGTTGAGTTCGG | 165 | h |
| GGGAAAAAGTCAAGGCAGTCG | GACAATGTCGCCCAATGTCA | 131 | m | |
| FASN | TATGAAGCCATCGTGGACGG | GAAGAAGGAGAGCCGGTTGG | 183 | h |
| TGGATTACCCAAGCGGTCTG | AGTGTTCGTTCCTCGGAGTG | 181 | m | |
| MOGAT1 | GGTAGAGTTTGCACCGCTCA | CTGGGGTATGCCAGTCAAAGT | 191 | h |
| GGCTCACCCAGGAACATTCA | GTGGCAAGGCTACTCCCATT | 200 | m | |
| PPAR-α | GCCTCCTTCGGCGTTCG | GAGCTCCAAGCTACTGTGGT | 148 | h |
| GGGAACTTAGAGGAGAGCCAAG | CAGTGGGGAGAGAGGACAGA | 183 | m | |
| β2-microglobulin | TGTCTTTCAGCAAGGACTGGT | TTCAAACCTCCATGATGCTGC | 161 | h |
| TCTACTGGGATCGAGACATGTGA | ATTGCTATTTCTTTCTGCGTGCAT | 124 | m |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nickel, S.; Christ, M.; Schmidt, S.; Kosacka, J.; Kühne, H.; Roderfeld, M.; Longerich, T.; Tietze, L.; Bosse, I.; Hsu, M.-J.; et al. Human Mesenchymal Stromal Cells Resolve Lipid Load in High Fat Diet-Induced Non-Alcoholic Steatohepatitis in Mice by Mitochondria Donation. Cells 2022, 11, 1829. https://doi.org/10.3390/cells11111829
Nickel S, Christ M, Schmidt S, Kosacka J, Kühne H, Roderfeld M, Longerich T, Tietze L, Bosse I, Hsu M-J, et al. Human Mesenchymal Stromal Cells Resolve Lipid Load in High Fat Diet-Induced Non-Alcoholic Steatohepatitis in Mice by Mitochondria Donation. Cells. 2022; 11(11):1829. https://doi.org/10.3390/cells11111829
Chicago/Turabian StyleNickel, Sandra, Madlen Christ, Sandra Schmidt, Joanna Kosacka, Hagen Kühne, Martin Roderfeld, Thomas Longerich, Lysann Tietze, Ina Bosse, Mei-Ju Hsu, and et al. 2022. "Human Mesenchymal Stromal Cells Resolve Lipid Load in High Fat Diet-Induced Non-Alcoholic Steatohepatitis in Mice by Mitochondria Donation" Cells 11, no. 11: 1829. https://doi.org/10.3390/cells11111829