
Pharmacological Inhibition of Spleen Tyrosine Kinase Suppressed Neuroinflammation and Cognitive Dysfunction in LPS-Induced Neurodegeneration Model

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal
- Control group (Saline for 9 days).
- LPS group (0.25 mg/kg for 9 days).
- LPS + BAY61-3606 (0.25 mg/kg + 10 mg/kg for 9 days).
- BAY61-3606 (10 mg/kg for 9 days).
2.2. Y-Maze and Passive Avoidance Task
2.3. Tissue Processing
2.4. Nissl Staining
2.5. Immunofluorescence
2.6. Cell Culture
2.7. Transwell Co-Culture Assay
2.8. Western Blot Analysis
2.9. Antibodies
2.10. RNA Isolation and Quantitative PCR
2.11. Statistical Analysis
3. Results
3.1. Pharmacological Inhibition of Syk Mitigated the Expression of Microglial Cells and Inflammatory Mediators in LPS-Induced Mouse
3.2. Pharmacological Inhibition of Syk Mitigated the Inflammatory Mediators in LPS-Induced Mouse
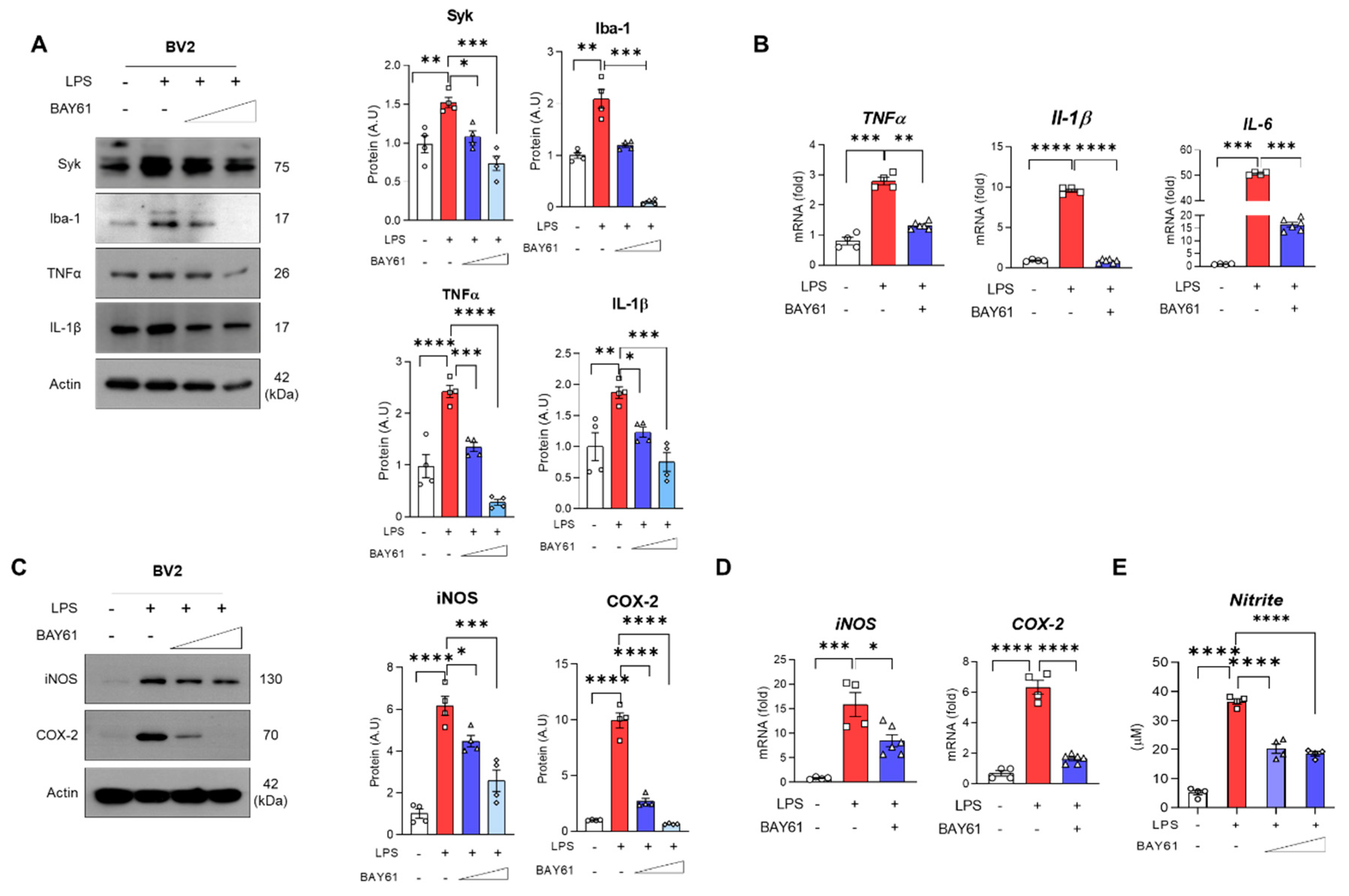
3.3. Pharmacological Inhibition of Syk Mitigated LPS-Induced Inflammation in BV2 Cells
3.4. Pharmacological Inhibition of Syk Mitigated the Apoptotic Cell Death in LPS-Induced Mice
3.5. Pharmacological Inhibition of Syk Protected against Neurotoxin-Mediated Neuronal Cell Death
3.6. Pharmacological Inhibition of Syk Restored Synaptic Dysfunctions, Cognitive, and Spatial Working Memory Impairment in LPS-Injected Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Streit, W.J.; Mrak, R.E.; Griffin, W.S.T. Microglia and neuroinflammation: A pathological perspective. J. Neuroinflamm. 2004, 1, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Dyken, P.; Lacoste, B. Impact of metabolic syndrome on neuroinflammation and the blood-brain barrier. Front. Neurosci. 2018, 12, 930. [Google Scholar] [CrossRef] [PubMed]
- Ullah, R.; Park, T.J.; Huang, X.; Kim, M.O. Abnormal amyloid beta metabolism in systemic abnormalities and alzheimer’s pathology: Insights and therapeutic approaches from periphery. Ageing Res. Rev. 2021, 71, 101451. [Google Scholar] [CrossRef]
- Butovsky, O.; Weiner, H.L. Microglial signatures and their role in health and disease. Nat. Rev. Neurosci. 2018, 19, 622–635. [Google Scholar] [CrossRef]
- Block, M.L.; Zecca, L.; Hong, J.-S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Fricker, M.; Tolkovsky, A.M.; Borutaite, V.; Coleman, M.; Brown, G.C. Neuronal cell death. Physiol. Rev. 2018, 98, 813–880. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.-C.; Yeh, W.-C.; Ohashi, P.S. Lps/tlr4 signal transduction pathway. Cytokine 2008, 42, 145–151. [Google Scholar] [CrossRef]
- Fiebich, B.L.; Batista, C.R.A.; Saliba, S.W.; Yousif, N.M.; de Oliveira, A.C.P. Role of microglia tlrs in neurodegeneration. Front. Cell. Neurosci. 2018, 12, 329. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Bi, W.; Xiao, S.; Lan, X.; Cheng, X.; Zhang, J.; Lu, D.; Wei, W.; Wang, Y.; Li, H.; et al. Neuroinflammation induced by lipopolysaccharide causes cognitive impairment in mice. Sci. Rep. 2019, 9, 5790. [Google Scholar] [CrossRef] [Green Version]
- Combs, C.K.; Johnson, D.E.; Cannady, S.B.; Lehman, T.M.; Landreth, G.E. Identification of microglial signal transduction pathways mediating a neurotoxic response to amyloidogenic fragments of beta-amyloid and prion proteins. J. Neurosci. Off. J. Soc. Neurosci. 1999, 19, 928–939. [Google Scholar] [CrossRef] [Green Version]
- Schweig, J.E.; Yao, H.; Coppola, K.; Jin, C.; Crawford, F.; Mullan, M.; Paris, D. Spleen tyrosine kinase (syk) blocks autophagic tau degradation in vitro and in vivo. J. Biol. Chem. 2019, 294, 13378–13395. [Google Scholar] [CrossRef] [PubMed]
- Hall-Roberts, H.; Agarwal, D.; Obst, J.; Smith, T.B.; Monzon-Sandoval, J.; Di Daniel, E.; Webber, C.; James, W.S.; Mead, E.; Davis, J.B.; et al. Trem2 alzheimer’s variant r47h causes similar transcriptional dysregulation to knockout, yet only subtle functional phenotypes in human ipsc-derived macrophages. Alzheimers Res. Ther. 2020, 12, 151. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Geahlen, R.L. Stress granules modulate syk to cause microglial cell dysfunction in alzheimer’s disease. EBioMedicine 2015, 2, 1785–1798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikram, M.; Park, T.J.; Ali, T.; Kim, M.O. Antioxidant and neuroprotective effects of caffeine against alzheimer’s and parkinson’s disease: Insight into the role of nrf-2 and a2ar signaling. Antioxidants 2020, 9, 902. [Google Scholar] [CrossRef] [PubMed]
- Combs, C.K.; Karlo, J.C.; Kao, S.C.; Landreth, G.E. Beta-amyloid stimulation of microglia and monocytes results in tnfalpha-dependent expression of inducible nitric oxide synthase and neuronal apoptosis. J. Neurosci. Off. J. Soc. Neurosci. 2001, 21, 1179–1188. [Google Scholar] [CrossRef] [Green Version]
- Xu, P.; Zhang, X.; Liu, Q.; Xie, Y.; Shi, X.; Chen, J.; Li, Y.; Guo, H.; Sun, R.; Hong, Y.; et al. Microglial trem-1 receptor mediates neuroinflammatory injury via interaction with syk in experimental ischemic stroke. Cell Death Dis. 2019, 10, 555. [Google Scholar] [CrossRef]
- Paris, D.; Ait-Ghezala, G.; Bachmeier, C.; Laco, G.; Beaulieu-Abdelahad, D.; Lin, Y.; Jin, C.; Crawford, F.; Mullan, M. The spleen tyrosine kinase (syk) regulates alzheimer amyloid-beta production and tau hyperphosphorylation. J. Biol. Chem. 2014, 289, 33927–33944. [Google Scholar] [CrossRef] [Green Version]
- Unsal, D.; Kacan, M.; Temiz-Resitoglu, M.; Guden, D.S.; Korkmaz, B.; Sari, A.N.; Buharalioglu, C.K.; Yildirim-Yaroglu, H.; Tamer-Gumus, L.; Tunctan, B.; et al. The role of syk/ikb-alpha/nf-kb pathway activation in the reversal effect of bay 61-3606, a selective syk inhibitor, on hypotension and inflammation in a rat model of zymosan-induced non-septic shock. Clin. Exp. Pharmacol. Physiol. 2018, 45, 155–165. [Google Scholar] [CrossRef]
- Tan, R.Z.; Li, J.C.; Liu, J.; Lei, X.Y.; Zhong, X.; Wang, C.; Yan, Y.; Linda Ye, L.; Darrel Duan, D.; Lan, H.Y.; et al. Bay61-3606 protects kidney from acute ischemia/reperfusion injury through inhibiting spleen tyrosine kinase and suppressing inflammatory macrophage response. FASEB J. 2020, 34, 15029–15046. [Google Scholar] [CrossRef]
- He, X.; Huang, Y.; Liu, Y.; Zhang, X.; Yue, P.; Ma, X.; Miao, Z.; Long, X.; Yang, Y.; Wan, X.; et al. Bay613606 attenuates neuroinflammation and neurofunctional damage by inhibiting microglial mincle/syk signaling response after traumatic brain injury. Int. J. Mol. Med. 2022, 49, 5. [Google Scholar] [CrossRef]
- Schweig, J.E.; Yao, H.; Beaulieu-Abdelahad, D.; Ait-Ghezala, G.; Mouzon, B.; Crawford, F.; Mullan, M.; Paris, D. Alzheimer’s disease pathological lesions activate the spleen tyrosine kinase. Acta Neuropathol. Commun. 2017, 5, 69. [Google Scholar] [CrossRef] [PubMed]
- Mandelkow, E.M.; Mandelkow, E. Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb. Perspect. Med. 2012, 2, a006247. [Google Scholar] [CrossRef] [PubMed]
- Ali, T.; Rehman, S.U.; Khan, A.; Badshah, H.; Abid, N.B.; Kim, M.W.; Jo, M.H.; Chung, S.S.; Lee, H.G.; Rutten, B.P.F.; et al. Adiponectin-mimetic novel nonapeptide rescues aberrant neuronal metabolic-associated memory deficits in alzheimer’s disease. Mol. Neurodegener. 2021, 16, 23. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.A.; Yoon, G.H.; Chung, S.S.; Abid, M.N.; Kim, T.H.; Lee, H.Y.; Kim, M.O. Novel osmotin inhibits srebp2 via the adipor1/ampk/sirt1 pathway to improve alzheimer’s disease neuropathological deficits. Mol. Psychiatry 2017, 22, 407–416. [Google Scholar] [CrossRef]
- Ali, T.; Badshah, H.; Kim, T.H.; Kim, M.O. Melatonin attenuates d-galactose-induced memory impairment, neuroinflammation and neurodegeneration via rage/nf-k b/jnk signaling pathway in aging mouse model. J. Pineal Res. 2015, 58, 71–85. [Google Scholar] [CrossRef]
- Kim, M.W.; Abid, N.B.; Jo, M.H.; Jo, M.G.; Yoon, G.H.; Kim, M.O. Suppression of adiponectin receptor 1 promotes memory dysfunction and alzheimer’s disease-like pathologies. Sci. Rep. 2017, 7, 12435. [Google Scholar] [CrossRef]
- Ali, T.; Kim, M.O. Melatonin ameliorates amyloid beta-induced memory deficits, tau hyperphosphorylation and neurodegeneration via pi3/akt/gsk3beta pathway in the mouse hippocampus. J. Pineal Res. 2015, 59, 47–59. [Google Scholar] [CrossRef]
- Jeong, G.W.; Lee, H.H.; Lee-Kwon, W.; Kwon, H.M. Microglial tonebp mediates lps-induced inflammation and memory loss as transcriptional cofactor for nf-kappab and ap-1. J. Neuroinflamm. 2020, 17, 372. [Google Scholar] [CrossRef]
- Ye, X.C.; Hao, Q.; Ma, W.J.; Zhao, Q.C.; Wang, W.W.; Yin, H.H.; Zhang, T.; Wang, M.; Zan, K.; Yang, X.X.; et al. Dectin-1/syk signaling triggers neuroinflammation after ischemic stroke in mice. J. Neuroinflamm. 2020, 17, 17. [Google Scholar] [CrossRef] [Green Version]
- Buffolo, F.; Petrosino, V.; Albini, M.; Moschetta, M.; Carlini, F.; Floss, T.; Kerlero de Rosbo, N.; Cesca, F.; Rocchi, A.; Uccelli, A.; et al. Neuroinflammation induces synaptic scaling through il-1beta-mediated activation of the transcriptional repressor rest/nrsf. Cell Death Dis. 2021, 12, 180. [Google Scholar] [CrossRef]
- Badshah, H.; Ikram, M.; Ali, W.; Ahmad, S.; Hahm, J.R.; Kim, M.O. Caffeine may abrogate lps-induced oxidative stress and neuroinflammation by regulating nrf2/tlr4 in adult mouse brains. Biomolecules 2019, 9, 719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muhammad, T.; Ikram, M.; Ullah, R.; Rehman, S.U.; Kim, M.O. Hesperetin, a citrus flavonoid, attenuates lps-induced neuroinflammation, apoptosis and memory impairments by modulating tlr4/nf-kappab signaling. Nutrients 2019, 11, 648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, J.; Zhao, Y.; Su, J.; Liu, Z.; Fang, S.; Li, L.; Deng, J.; Fan, G. Toddalolactone protects lipopolysaccharide-induced sepsis and attenuates lipopolysaccharide-induced inflammatory response by modulating hmgb1-nf-kappab translocation. Front. Pharmacol. 2020, 11, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, X.; Jiang, X.; Jiang, X.; Wang, Y.; Miao, Z.; He, W.; Yang, G.; Lv, Z.; Yu, Y.; Zheng, Y. Micheliolide inhibits lps-induced inflammatory response and protects mice from lps challenge. Sci. Rep. 2016, 6, 23240. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.; Ali, T.; Rehman, S.U.; Khan, M.S.; Alam, S.I.; Ikram, M.; Muhammad, T.; Saeed, K.; Badshah, H.; Kim, M.O. Neuroprotective effect of quercetin against the detrimental effects of lps in the adult mouse brain. Front. Pharmacol. 2018, 9, 1383. [Google Scholar] [CrossRef]
- Khan, M.S.; Muhammad, T.; Ikram, M.; Kim, M.O. Dietary supplementation of the antioxidant curcumin halts systemic lps-induced neuroinflammation-associated neurodegeneration and memory/synaptic impairment via the jnk/nf-kappab/akt signaling pathway in adult rats. Oxidative Med. Cell. Longev. 2019, 2019, 7860650. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, A.; Ali, T.; Rehman, S.U.; Kim, M.O. Phytomedicine-based potent antioxidant, fisetin protects cns-insult lps-induced oxidative stress-mediated neurodegeneration and memory impairment. J. Clin. Med. 2019, 8, 850. [Google Scholar] [CrossRef] [Green Version]
- Ikram, M.; Ullah, R.; Khan, A.; Kim, M.O. Ongoing research on the role of gintonin in the management of neurodegenerative disorders. Cells 2020, 9, 1464. [Google Scholar] [CrossRef]
- Ullah, R.; Ikram, M.; Park, T.J.; Ahmad, R.; Saeed, K.; Alam, S.I.; Rehman, I.U.; Khan, A.; Khan, I.; Jo, M.G.; et al. Vanillic acid, a bioactive phenolic compound, counteracts lps-induced neurotoxicity by regulating c-jun n-terminal kinase in mouse brain. Int. J. Mol. Sci. 2020, 22, 361. [Google Scholar] [CrossRef]
- Ahmad, S.; Khan, A.; Ali, W.; Jo, M.H.; Park, J.; Ikram, M.; Kim, M.O. Fisetin rescues the mice brains against d-galactose-induced oxidative stress, neuroinflammation and memory impairment. Front. Pharmacol. 2021, 12, 612078. [Google Scholar] [CrossRef]
- Neher, J.J.; Neniskyte, U.; Zhao, J.W.; Bal-Price, A.; Tolkovsky, A.M.; Brown, G.C. Inhibition of microglial phagocytosis is sufficient to prevent inflammatory neuronal death. J. Immunol. 2011, 186, 4973–4983. [Google Scholar] [CrossRef] [PubMed]
- Cherry, J.D.; Olschowka, J.A.; O’Banion, M.K. Neuroinflammation and m2 microglia: The good, the bad, and the inflamed. J. Neuroinflamm. 2014, 11, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akhmetzyanova, E.; Kletenkov, K.; Mukhamedshina, Y.; Rizvanov, A. Different approaches to modulation of microglia phenotypes after spinal cord injury. Front. Syst. Neurosci. 2019, 13, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.; Masuda, E.S.; Payan, D.G. Discovery and development of spleen tyrosine kinase (syk) inhibitors. J. Med. Chem. 2012, 55, 3614–3643. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Mamorska-Dyga, A. Syk inhibitors in clinical development for hematological malignancies. J. Hematol. Oncol. 2017, 10, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, M.W.; Choe, K.; Park, J.S.; Lee, H.J.; Kang, M.H.; Ahmad, R.; Kim, M.O. Pharmacological Inhibition of Spleen Tyrosine Kinase Suppressed Neuroinflammation and Cognitive Dysfunction in LPS-Induced Neurodegeneration Model. Cells 2022, 11, 1777. https://doi.org/10.3390/cells11111777
Kim MW, Choe K, Park JS, Lee HJ, Kang MH, Ahmad R, Kim MO. Pharmacological Inhibition of Spleen Tyrosine Kinase Suppressed Neuroinflammation and Cognitive Dysfunction in LPS-Induced Neurodegeneration Model. Cells. 2022; 11(11):1777. https://doi.org/10.3390/cells11111777
Chicago/Turabian StyleKim, Min Woo, Kyonghwan Choe, Jun Sung Park, Hyeon Jin Lee, Min Hwa Kang, Riaz Ahmad, and Myeong Ok Kim. 2022. "Pharmacological Inhibition of Spleen Tyrosine Kinase Suppressed Neuroinflammation and Cognitive Dysfunction in LPS-Induced Neurodegeneration Model" Cells 11, no. 11: 1777. https://doi.org/10.3390/cells11111777
APA StyleKim, M. W., Choe, K., Park, J. S., Lee, H. J., Kang, M. H., Ahmad, R., & Kim, M. O. (2022). Pharmacological Inhibition of Spleen Tyrosine Kinase Suppressed Neuroinflammation and Cognitive Dysfunction in LPS-Induced Neurodegeneration Model. Cells, 11(11), 1777. https://doi.org/10.3390/cells11111777