
DSTYK Enhances Chemoresistance in Triple-Negative Breast Cancer Cells

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Xenografts Experiments in Mice
2.2. Cell Culture and Reagents
2.3. Annexin V-FITC/7-AAD Double-Staining Assay
2.4. MTT Assay
2.5. Western Blotting
2.6. Real Time-Quantitative PCR (RT-qPCR)
2.7. Terminal Deoxynucleotidyl Transferase-Mediated dUTP Nick End-Labeling (TUNEL) Assay
2.8. DSTYK Knockout (DSTYKKO) via Crispr/Cas9
2.9. Immunohistochemistry
2.10. Statistical Analysis
3. Results
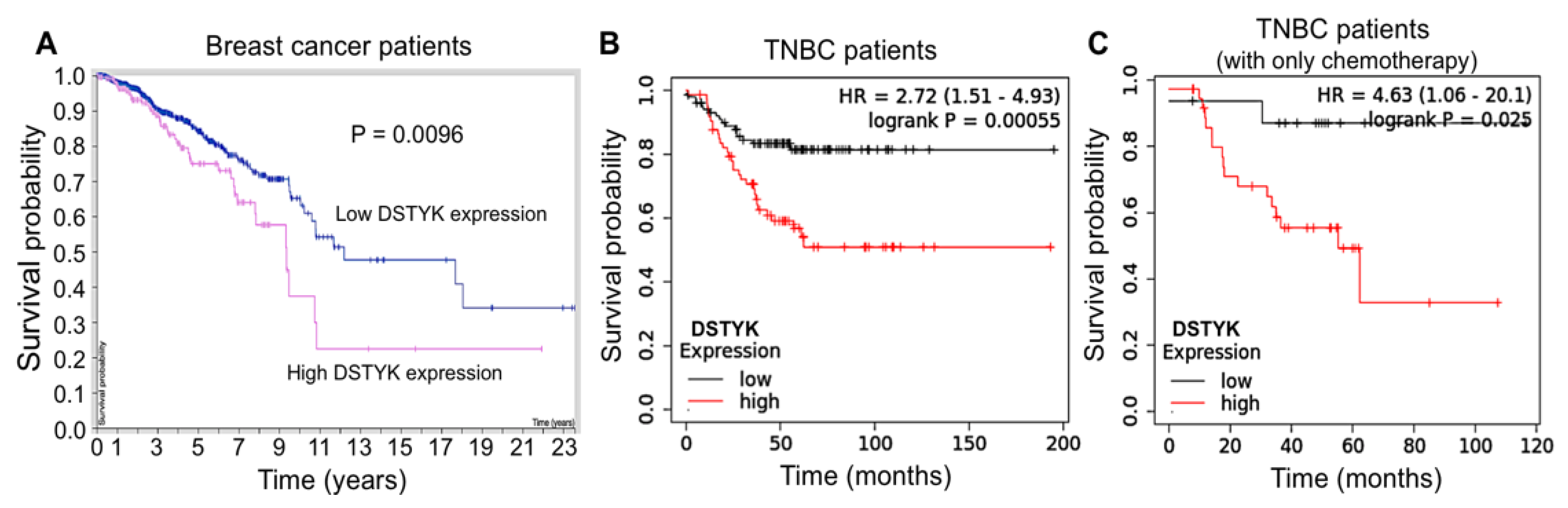
3.1. The Expression of DSTYK Is Related to the Survival Probability of TNBC Patients
3.2. Establishment of Chemoresistant Cell Lines
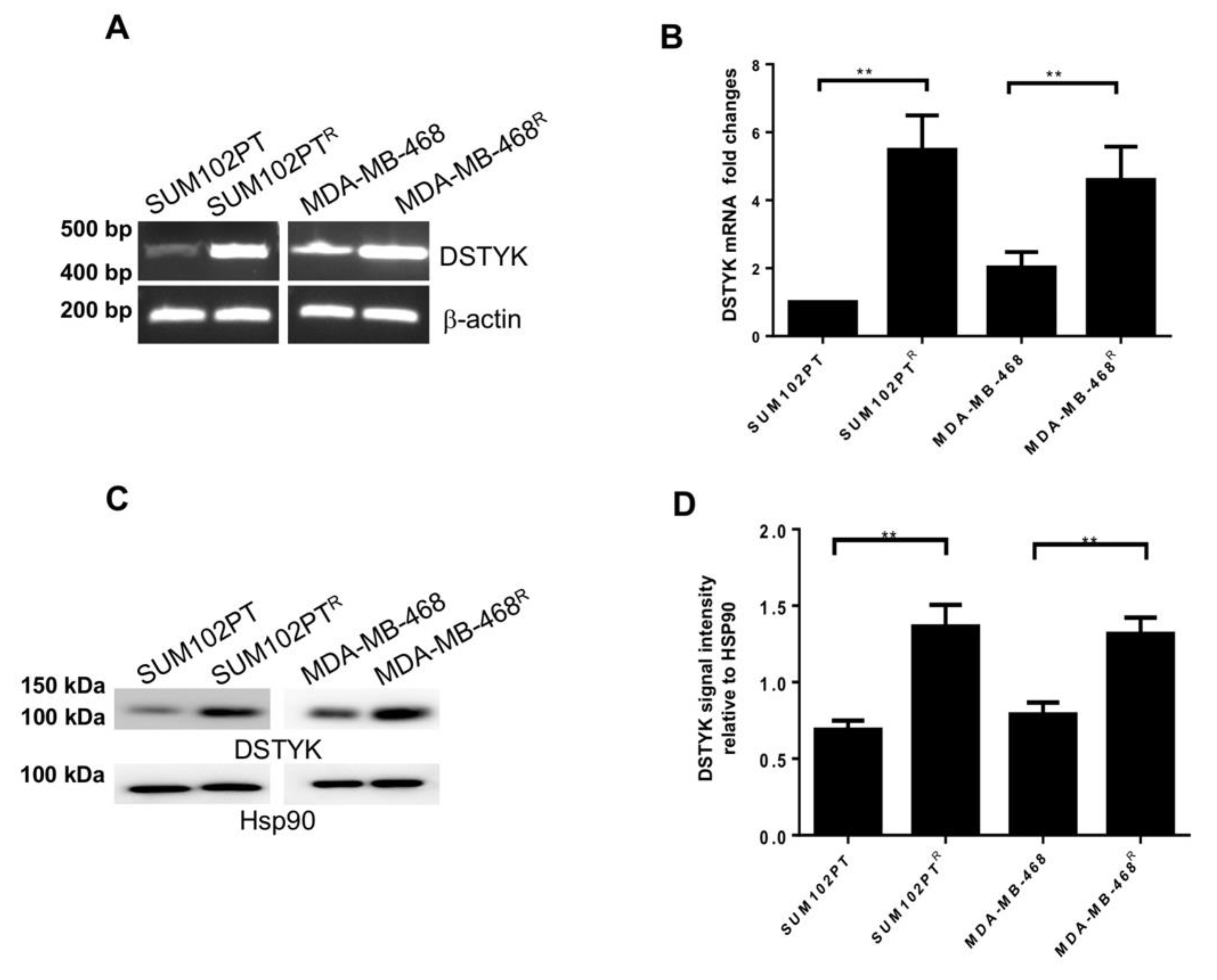
3.3. DSTYK Expression Is Upregulated in Chemoresistant Cells
3.4. DSTYKKO Attenuates Chemoresistance in Chemoresistant Cells
3.5. DSTYK Plays a Key Role in Chemoresistance through Inhibiting Apoptosis
3.6. Knockout of DSTYK Facilitates Tumor Regression during Drug Treatment in an In Vivo Orthotopic Mouse Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ji, X.; Lu, Y.; Tian, H.; Meng, X.; Wei, M.; Cho, W.C. Chemoresistance mechanisms of breast cancer and their countermeasures. Biomed. Pharmacother. 2019, 114, 108800. [Google Scholar] [CrossRef]
- Bordoloi, D.; Roy, N.K.; Monisha, J.; Padmavathi, G.; Kunnumakkara, A.B. Multi-Targeted Agents in Cancer Cell Chemosensitization: What We Learnt from Curcumin Thus Far. Recent Pat. Anticancer Drug Discov. 2016, 11, 67–97. [Google Scholar] [CrossRef]
- da Silva, J.L.; Cardoso Nunes, N.C.; Izetti, P.; de Mesquita, G.G.; de Melo, A.C. Triple negative breast cancer: A thorough review of biomarkers. Crit. Rev. Oncol. Hematol. 2020, 145, 102855. [Google Scholar] [CrossRef]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef]
- Omarini, C.; Guaitoli, G.; Pipitone, S.; Moscetti, L.; Cortesi, L.; Cascinu, S.; Piacentini, F. Neoadjuvant treatments in triple-negative breast cancer patients: Where we are now and where we are going. Cancer Manag. Res. 2018, 10, 91–103. [Google Scholar] [CrossRef] [Green Version]
- Narayan, V.; Vaughn, D. Pharmacokinetic and toxicity considerations in the use of neoadjuvant chemotherapy for bladder cancer. Expert Opin. Drug Metab. Toxicol. 2015, 11, 731–742. [Google Scholar] [CrossRef]
- Collignon, J.; Lousberg, L.; Schroeder, H.; Jerusalem, G. Triple-negative breast cancer: Treatment challenges and solutions. Breast Cancer (Dove Med. Press) 2016, 8, 93–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nedeljkovic, M.; Damjanovic, A. Mechanisms of Chemotherapy Resistance in Triple-Negative Breast Cancer-How We Can Rise to the Challenge. Cells 2019, 8, 957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrido-Castro, A.C.; Lin, N.U.; Polyak, K. Insights into Molecular Classifications of Triple-Negative Breast Cancer: Improving Patient Selection for Treatment. Cancer Discov. 2019, 9, 176–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Strietz, J.; Bleilevens, A.; Stickeler, E.; Maurer, J. Chemotherapeutic Stress Influences Epithelial-Mesenchymal Transition and Stemness in Cancer Stem Cells of Triple-Negative Breast Cancer. Int. J. Mol. Sci. 2020, 21, 404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.; Settleman, J. EMT, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene 2010, 29, 4741–4751. [Google Scholar] [CrossRef] [Green Version]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Maugeri-Sacca, M.; Vigneri, P.; De Maria, R. Cancer stem cells and chemosensitivity. Clin. Cancer Res. 2011, 17, 4942–4947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayob, A.Z.; Ramasamy, T.S. Cancer stem cells as key drivers of tumour progression. J. Biomed. Sci. 2018, 25, 20. [Google Scholar] [CrossRef] [PubMed]
- Shimono, Y.; Zabala, M.; Cho, R.W.; Lobo, N.; Dalerba, P.; Qian, D.; Diehn, M.; Liu, H.; Panula, S.P.; Chiao, E.; et al. Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell 2009, 138, 592–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desai, A.; Yan, Y.; Gerson, S.L. Concise Reviews: Cancer Stem Cell Targeted Therapies: Toward Clinical Success. Stem Cells Transl. Med. 2019, 8, 75–81. [Google Scholar] [CrossRef] [Green Version]
- Nunes, T.; Hamdan, D.; Leboeuf, C.; El Bouchtaoui, M.; Gapihan, G.; Nguyen, T.T.; Meles, S.; Angeli, E.; Ratajczak, P.; Lu, H.; et al. Targeting Cancer Stem Cells to Overcome Chemoresistance. Int. J. Mol. Sci. 2018, 19, 4036. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.L.; Kuo, Y.C.; Ho, Y.S.; Huang, Y.H. Triple-Negative Breast Cancer: Current Understanding and Future Therapeutic Breakthrough Targeting Cancer Stemness. Cancers 2019, 11, 1334. [Google Scholar] [CrossRef] [Green Version]
- Tang, G.; Yang, Y.; Shang, L.; Jun, F.; Liu, Q. A DSTYK mutation activates ERK1/2 signaling to promote intraspinal dissemination in a case of solitary fibrous tumor/hemangiopericytoma. Lab. Investig. 2019, 99, 1501–1514. [Google Scholar] [CrossRef]
- Zhang, J.; Miller, Z.; Musich, P.R.; Thomas, A.E.; Yao, Z.Q.; Xie, Q.; Howe, P.H.; Jiang, Y. DSTYK Promotes Metastasis and Chemoresistance via EMT in Colorectal Cancer. Front. Pharmacol. 2020, 11, 1250. [Google Scholar] [CrossRef]
- Baltali, E.; Ozisik, Y.; Guler, N.; Firat, D.; Altundag, K. Combination of docetaxel and doxorubicin as first-line chemotherapy in metastatic breast cancer. Tumori 2001, 87, 18–19. [Google Scholar] [CrossRef]
- Evans, T.R.; Yellowlees, A.; Foster, E.; Earl, H.; Cameron, D.A.; Hutcheon, A.W.; Coleman, R.E.; Perren, T.; Gallagher, C.J.; Quigley, M.; et al. Phase III randomized trial of doxorubicin and docetaxel versus doxorubicin and cyclophosphamide as primary medical therapy in women with breast cancer: An anglo-celtic cooperative oncology group study. J. Clin. Oncol. 2005, 23, 2988–2995. [Google Scholar] [CrossRef]
- von Minckwitz, G.; Raab, G.; Caputo, A.; Schutte, M.; Hilfrich, J.; Blohmer, J.U.; Gerber, B.; Costa, S.D.; Merkle, E.; Eidtmann, H.; et al. Doxorubicin with cyclophosphamide followed by docetaxel every 21 days compared with doxorubicin and docetaxel every 14 days as preoperative treatment in operable breast cancer: The GEPARDUO study of the German Breast Group. J. Clin. Oncol. 2005, 23, 2676–2685. [Google Scholar] [CrossRef]
- Wali, V.B.; Langdon, C.G.; Held, M.A.; Platt, J.T.; Patwardhan, G.A.; Safonov, A.; Aktas, B.; Pusztai, L.; Stern, D.F.; Hatzis, C. Systematic Drug Screening Identifies Tractable Targeted Combination Therapies in Triple-Negative Breast Cancer. Cancer Res. 2017, 77, 566–578. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.W.; Hsu, C.K.; Michael, M.; Nanda, A.; Liu, L.; McMillan, J.R.; Pourreyron, C.; Takeichi, T.; Tolar, J.; Reid, E.; et al. Large Intragenic Deletion in DSTYK Underlies Autosomal-Recessive Complicated Spastic Paraparesis, SPG23. Am. J. Hum. Genet. 2017, 100, 364–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rieger, A.M.; Nelson, K.L.; Konowalchuk, J.D.; Barreda, D.R. Modified annexin V/propidium iodide apoptosis assay for accurate assessment of cell death. J. Vis. Exp. 2011, 50, e2597. [Google Scholar] [CrossRef]
- Wang, Q.; Liang, D.; Shen, P.; Yu, Y.; Yan, Y.; You, W. Hsa_circ_0092276 promotes doxorubicin resistance in breast cancer cells by regulating autophagy via miR-348/ATG7 axis. Transl. Oncol. 2021, 14, 101045. [Google Scholar] [CrossRef]
- Jiang, Y.; Woosley, A.N.; Sivalingam, N.; Natarajan, S.; Howe, P.H. Cathepsin-B-mediated cleavage of Disabled-2 regulates TGF-beta-induced autophagy. Nat. Cell Biol. 2016, 18, 851–863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quereda, V.; Bayle, S.; Vena, F.; Frydman, S.M.; Monastyrskyi, A.; Roush, W.R.; Duckett, D.R. Therapeutic Targeting of CDK12/CDK13 in Triple-Negative Breast Cancer. Cancer Cell 2019, 36, 545–558.e547. [Google Scholar] [CrossRef] [PubMed]
- Barbie, T.U.; Alexe, G.; Aref, A.R.; Li, S.; Zhu, Z.; Zhang, X.; Imamura, Y.; Thai, T.C.; Huang, Y.; Bowden, M.; et al. Targeting an IKBKE cytokine network impairs triple-negative breast cancer growth. J. Clin. Investig. 2014, 124, 5411–5423. [Google Scholar] [CrossRef]
- Walsh, L.A.; Alvarez, M.J.; Sabio, E.Y.; Reyngold, M.; Makarov, V.; Mukherjee, S.; Lee, K.W.; Desrichard, A.; Turcan, S.; Dalin, M.G.; et al. An Integrated Systems Biology Approach Identifies TRIM25 as a Key Determinant of Breast Cancer Metastasis. Cell Rep. 2017, 20, 1623–1640. [Google Scholar] [CrossRef] [Green Version]
- Sartor, C.I.; Dziubinski, M.L.; Yu, C.L.; Jove, R.; Ethier, S.P. Role of epidermal growth factor receptor and STAT-3 activation in autonomous proliferation of SUM-102PT human breast cancer cells. Cancer Res. 1997, 57, 978–987. [Google Scholar] [PubMed]
- Jiao, X.; Wang, M.; Zhang, Z.; Li, Z.; Ni, D.; Ashton, A.W.; Tang, H.Y.; Speicher, D.W.; Pestell, R.G. Leronlimab, a humanized monoclonal antibody to CCR5, blocks breast cancer cellular metastasis and enhances cell death induced by DNA damaging chemotherapy. Breast Cancer Res. 2021, 23, 11. [Google Scholar] [CrossRef]
- Hu, M.H.; Wu, T.Y.; Huang, Q.; Jin, G. New substituted quinoxalines inhibit triple-negative breast cancer by specifically downregulating the c-MYC transcription. Nucleic Acids Res. 2019, 47, 10529–10542. [Google Scholar] [CrossRef]
- Montinaro, A.; Areso Zubiaur, I.; Saggau, J.; Kretz, A.L.; Ferreira, R.M.M.; Hassan, O.; Kitzig, E.; Muller, I.; El-Bahrawy, M.A.; von Karstedt, S.; et al. Potent pro-apoptotic combination therapy is highly effective in a broad range of cancers. Cell Death Differ. 2021. [Google Scholar] [CrossRef]
- Dewangan, J.; Srivastava, S.; Mishra, S.; Pandey, P.K.; Divakar, A.; Rath, S.K. Chetomin induces apoptosis in human triple-negative breast cancer cells by promoting calcium overload and mitochondrial dysfunction. Biochem. Biophys. Res. Commun. 2018, 495, 1915–1921. [Google Scholar] [CrossRef]
- Shen, L.W.; Jiang, X.X.; Li, Z.Q.; Li, J.; Wang, M.; Jia, G.F.; Ding, X.; Lei, L.; Gong, Q.H.; Gao, N. Cepharanthine sensitizes human triple negative breast cancer cells to chemotherapeutic agent epirubicin via inducing cofilin oxidation-mediated mitochondrial fission and apoptosis. Acta Pharmacol. Sin. 2021. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, W.; Miyazaki, K.; Asano, Y.; Kubota, S.; Tanaka, N. Kruppel-Like Factor 4 and Its Activator APTO-253 Induce NOXA-Mediated, p53-Independent Apoptosis in Triple-Negative Breast Cancer Cells. Genes 2021, 12, 539. [Google Scholar] [CrossRef]
- Wu, Q.; Siddharth, S.; Sharma, D. Triple Negative Breast Cancer: A Mountain Yet to Be Scaled Despite the Triumphs. Cancers 2021, 13, 3697. [Google Scholar] [CrossRef]
- Neophytou, C.M.; Trougakos, I.P.; Erin, N.; Papageorgis, P. Apoptosis Deregulation and the Development of Cancer Multi-Drug Resistance. Cancers 2021, 13, 4363. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Zhou, Y.; Zhang, R.; Wang, Z.; Xu, M.; Zhang, D.; Huang, J.; Luo, F.; Li, F.; Ni, Z.; et al. Dstyk mutation leads to congenital scoliosis-like vertebral malformations in zebrafish via dysregulated mTORC1/TFEB pathway. Nat. Commun. 2020, 11, 479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zha, J.; Zhou, Q.; Xu, L.G.; Chen, D.; Li, L.; Zhai, Z.; Shu, H.B. RIP5 is a RIP-homologous inducer of cell death. Biochem. Biophys. Res. Commun. 2004, 319, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Torres-Adorno, A.M.; Lee, J.; Kogawa, T.; Ordentlich, P.; Tripathy, D.; Lim, B.; Ueno, N.T. Histone Deacetylase Inhibitor Enhances the Efficacy of MEK Inhibitor through NOXA-Mediated MCL1 Degradation in Triple-Negative and Inflammatory Breast Cancer. Clin. Cancer Res. 2017, 23, 4780–4792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.X.; Feng, J.M.; Wang, Y.; Li, X.H.; Chen, X.X.; Su, Y.; Shen, Y.Y.; Chen, Y.; Xiong, B.; Yang, C.H.; et al. The B-Raf(V600E) inhibitor dabrafenib selectively inhibits RIP3 and alleviates acetaminophen-induced liver injury. Cell Death Dis. 2014, 5, e1278. [Google Scholar] [CrossRef] [Green Version]
- Pawlowski, J.; Kraft, A.S. Bax-induced apoptotic cell death. Proc. Natl. Acad. Sci. USA 2000, 97, 529–531. [Google Scholar] [CrossRef] [Green Version]
- Westphal, D.; Dewson, G.; Czabotar, P.E.; Kluck, R.M. Molecular biology of Bax and Bak activation and action. Biochim. Biophys. Acta 2011, 1813, 521–531. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ogbu, S.C.; Rojas, S.; Weaver, J.; Musich, P.R.; Zhang, J.; Yao, Z.Q.; Jiang, Y. DSTYK Enhances Chemoresistance in Triple-Negative Breast Cancer Cells. Cells 2022, 11, 97. https://doi.org/10.3390/cells11010097
Ogbu SC, Rojas S, Weaver J, Musich PR, Zhang J, Yao ZQ, Jiang Y. DSTYK Enhances Chemoresistance in Triple-Negative Breast Cancer Cells. Cells. 2022; 11(1):97. https://doi.org/10.3390/cells11010097
Chicago/Turabian StyleOgbu, Stella C., Samuel Rojas, John Weaver, Phillip R. Musich, Jinyu Zhang, Zhi Q. Yao, and Yong Jiang. 2022. "DSTYK Enhances Chemoresistance in Triple-Negative Breast Cancer Cells" Cells 11, no. 1: 97. https://doi.org/10.3390/cells11010097
APA StyleOgbu, S. C., Rojas, S., Weaver, J., Musich, P. R., Zhang, J., Yao, Z. Q., & Jiang, Y. (2022). DSTYK Enhances Chemoresistance in Triple-Negative Breast Cancer Cells. Cells, 11(1), 97. https://doi.org/10.3390/cells11010097