
Potassium Effects on NCC Are Attenuated during Inhibition of Cullin E3–Ubiquitin Ligases

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Procedures
2.1. Animal Experiments and Tissue Isolation
2.2. Studies in Cul3-Het/Δ9 Mice
2.3. Kidney Tubule Suspensions
2.4. Antibodies and Immunoblotting
2.5. Statistics
3. Results
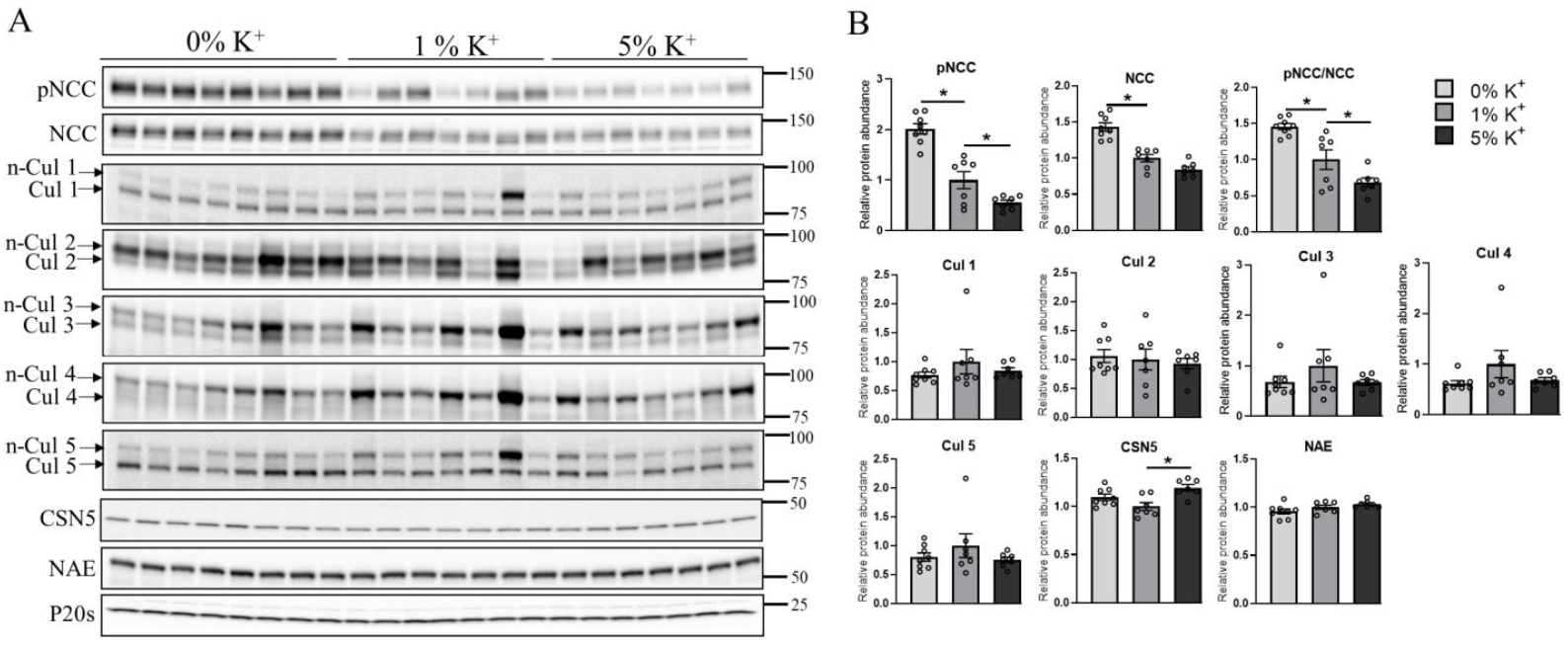
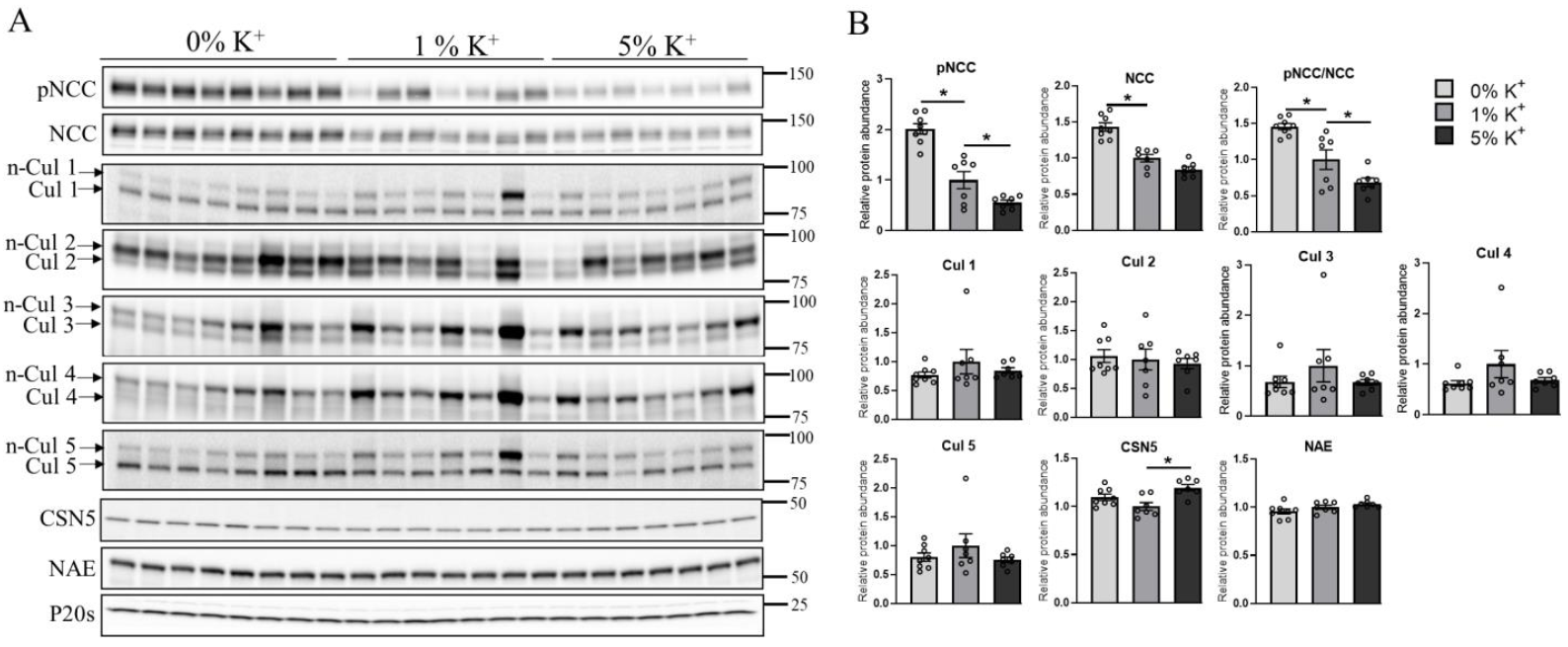
3.1. High Dietary K+ Intake Decreases NCC without Major Alterations in Cullin Expression
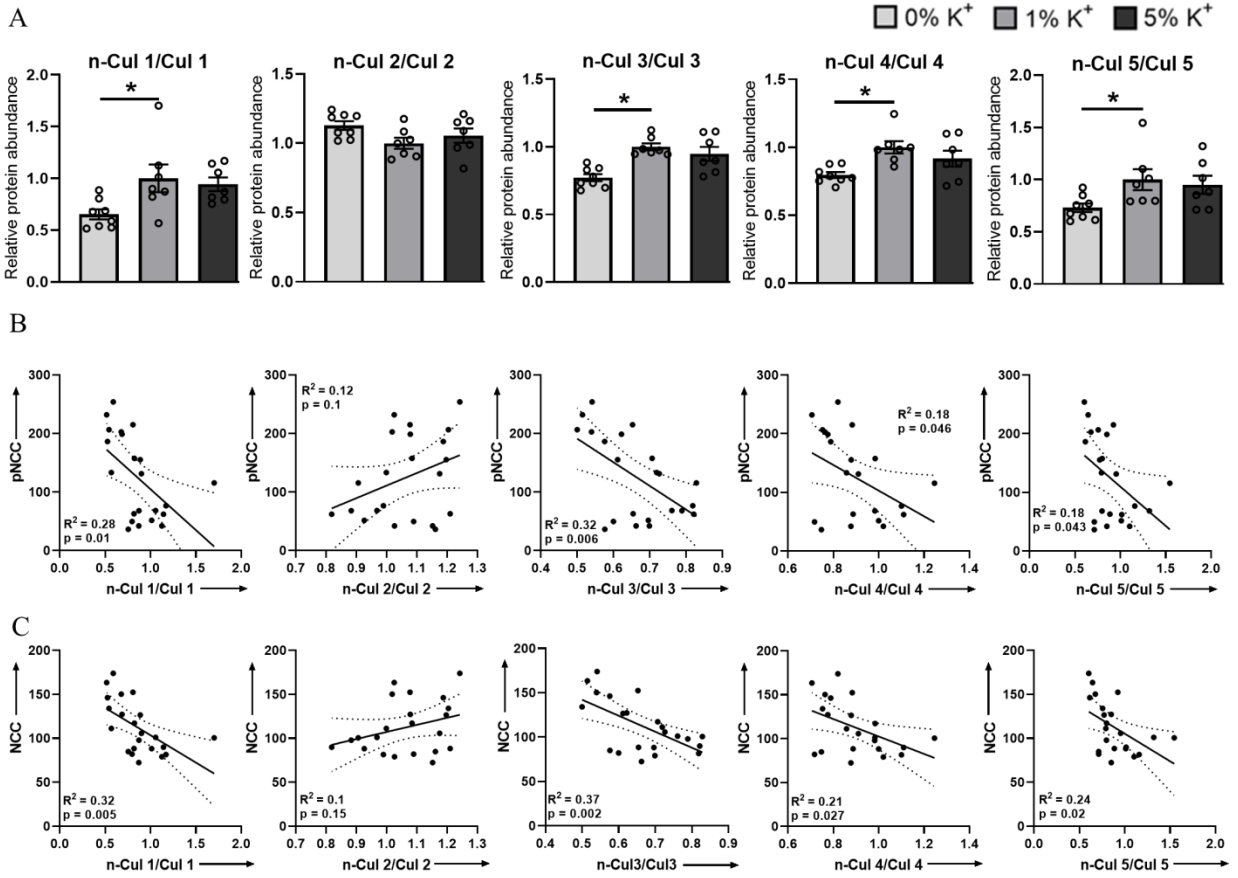
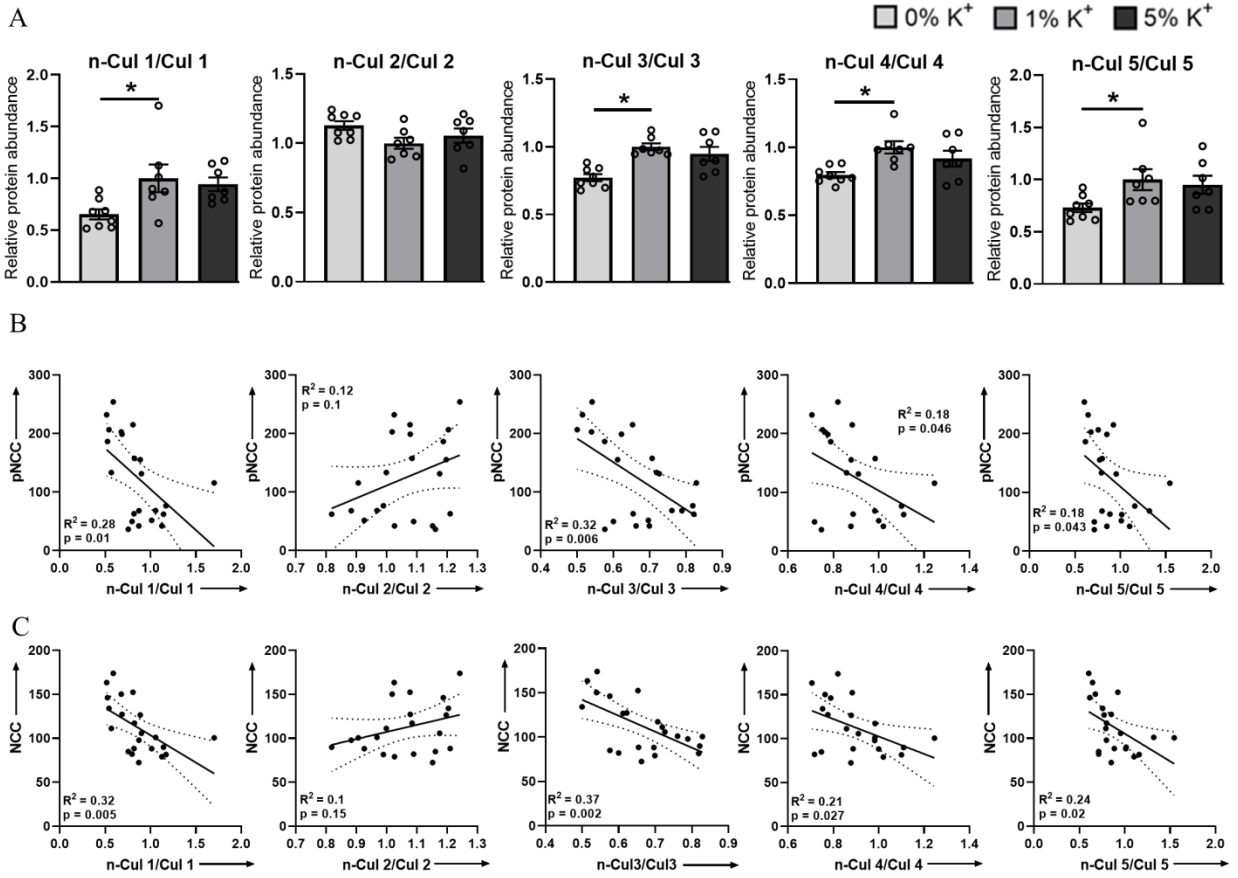
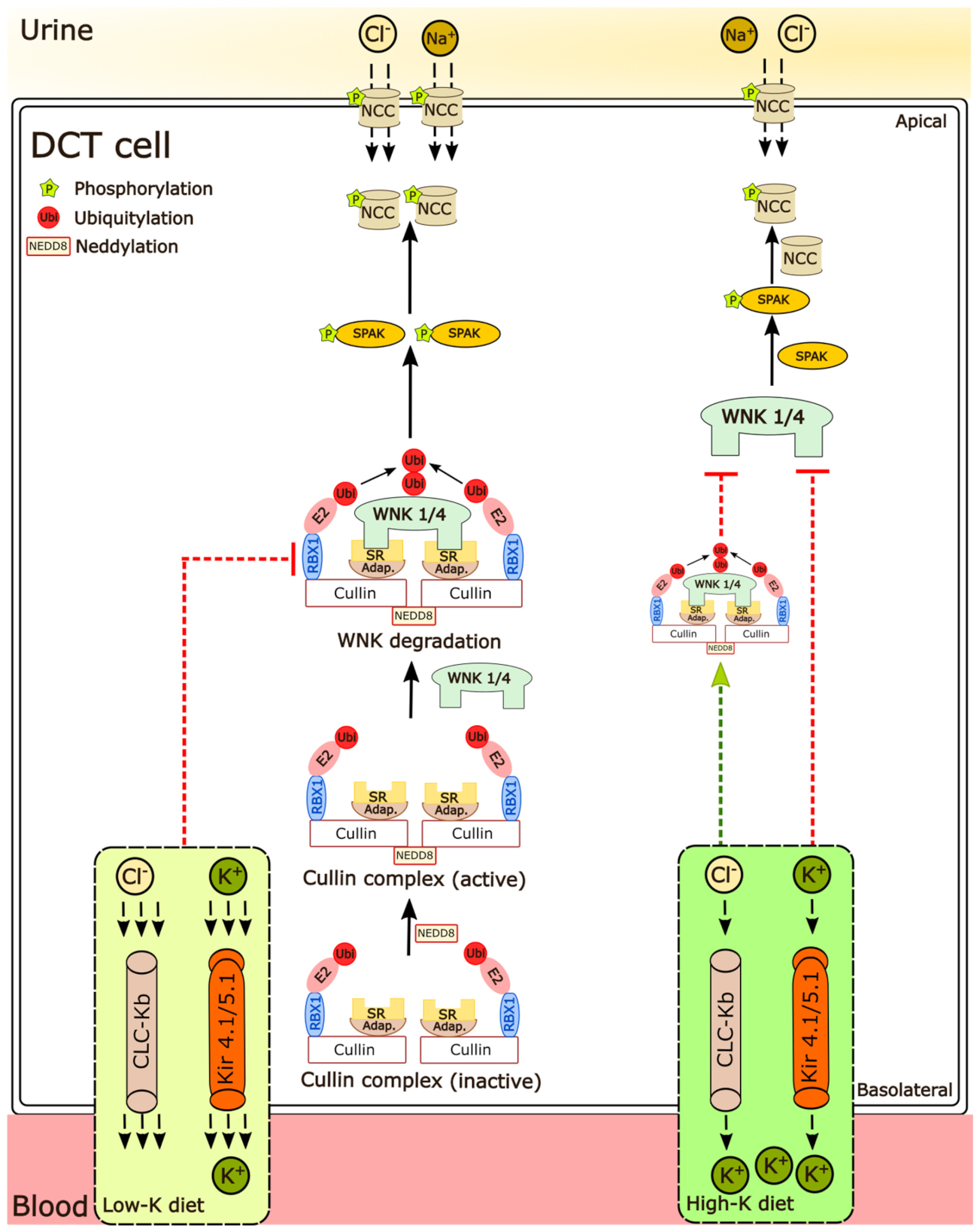
3.2. Low Dietary K+ Intake Is Associated with Decreased Neddylation of Cullins
3.3. The Effects of a High Dietary K+ Intake on NCC Phosphorylation Are Attenuated in Cul3-Het/Δ9 Mice
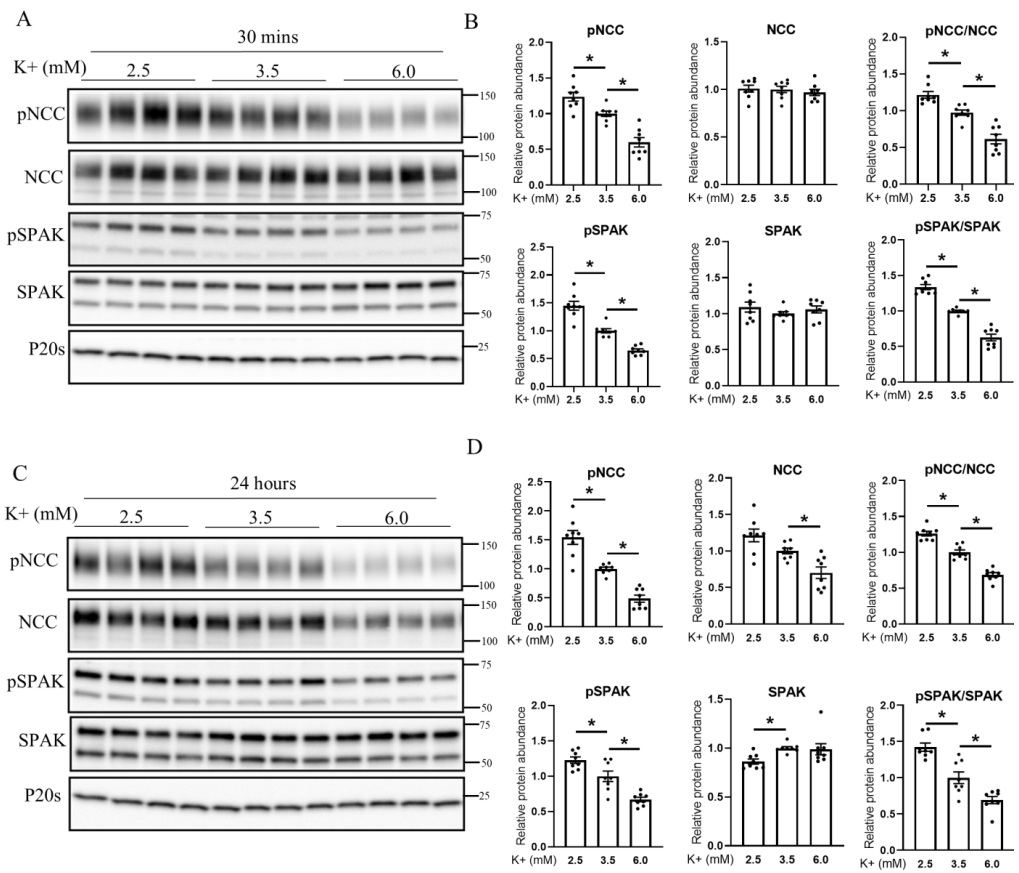
3.4. Extracellular K+ Changes NCC Phosphorylation and Abundance in Isolated Ex Vivo Renal Tubules
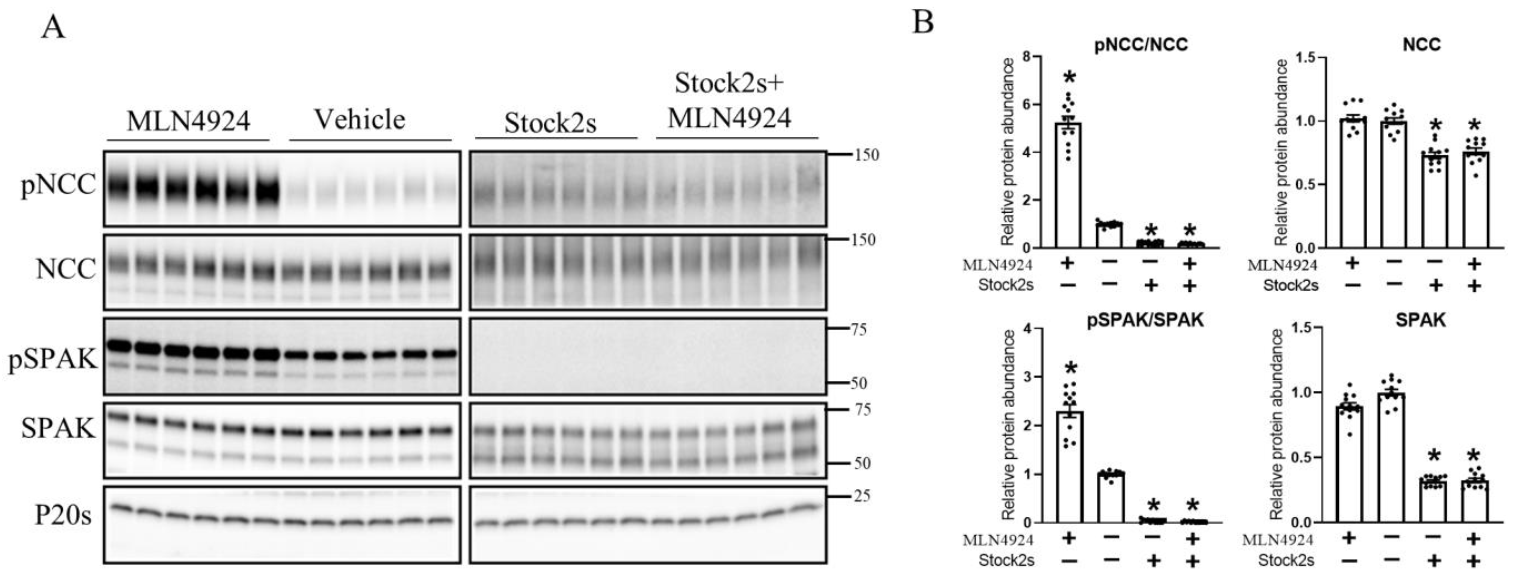
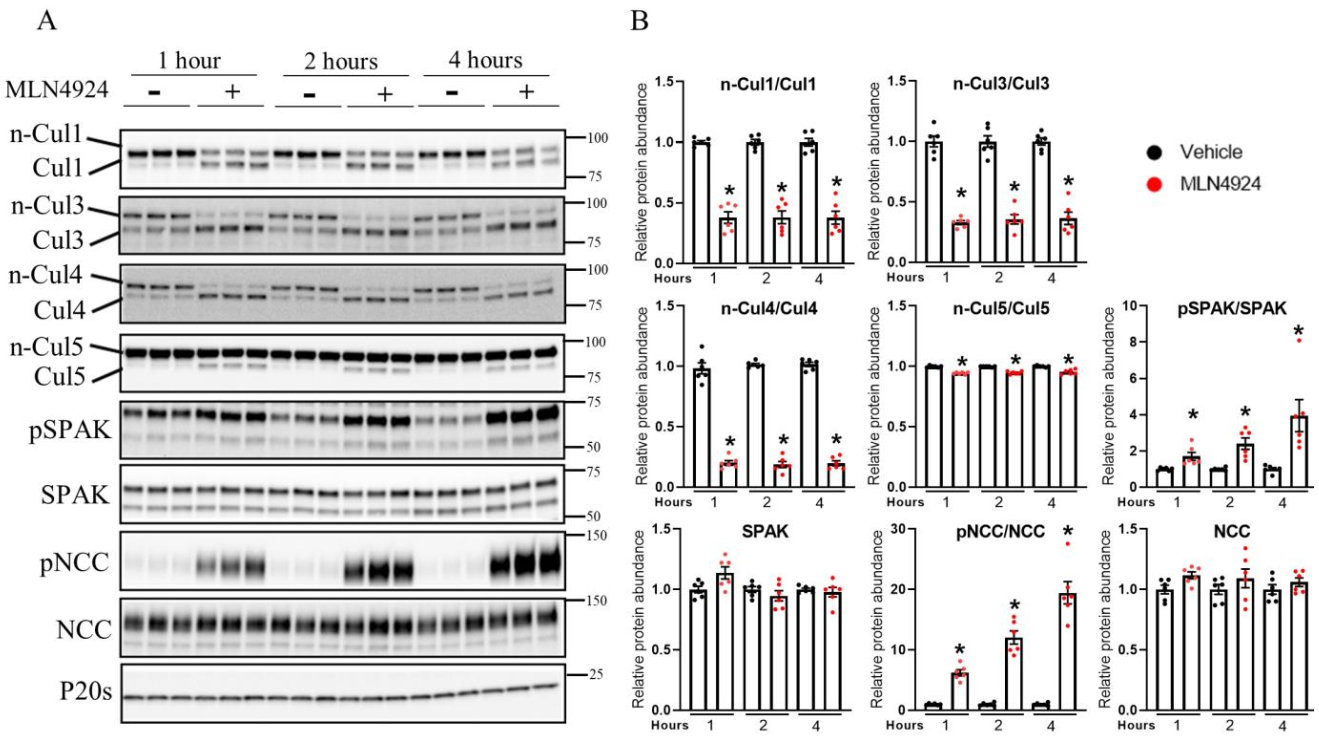
3.5. Pharmacological Inhibition of Cullin Activity Ex Vivo Increases NCC Phosphorylation
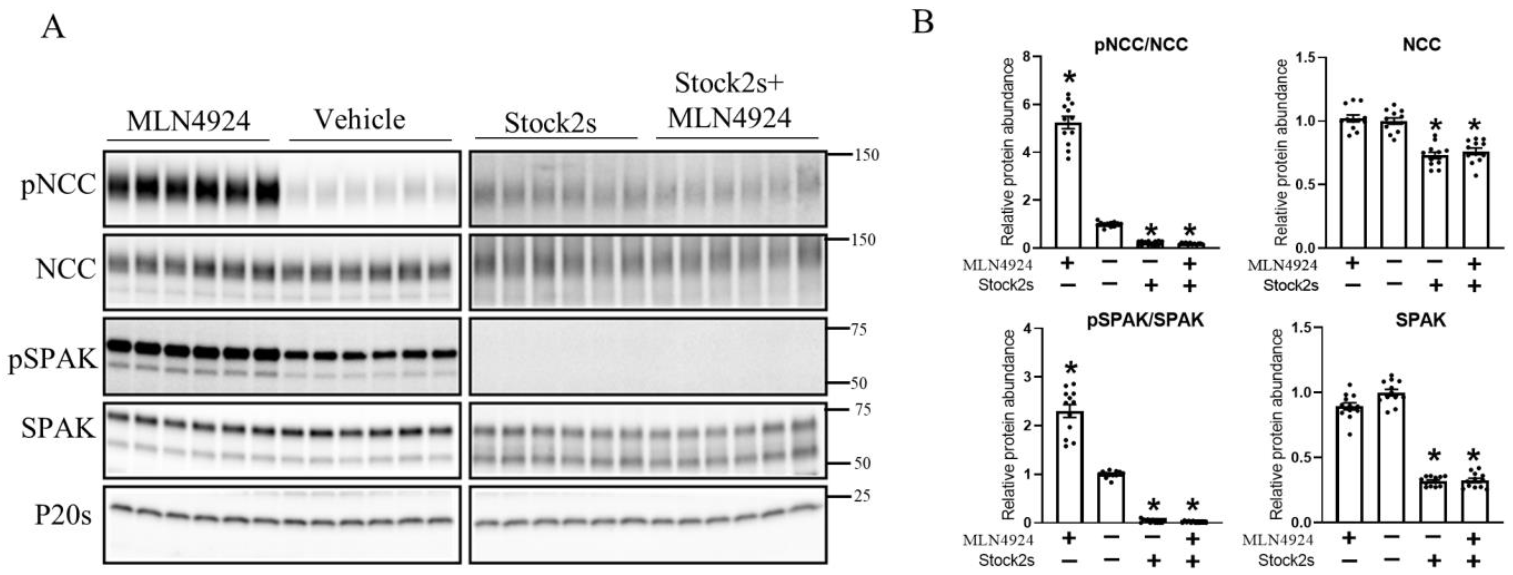
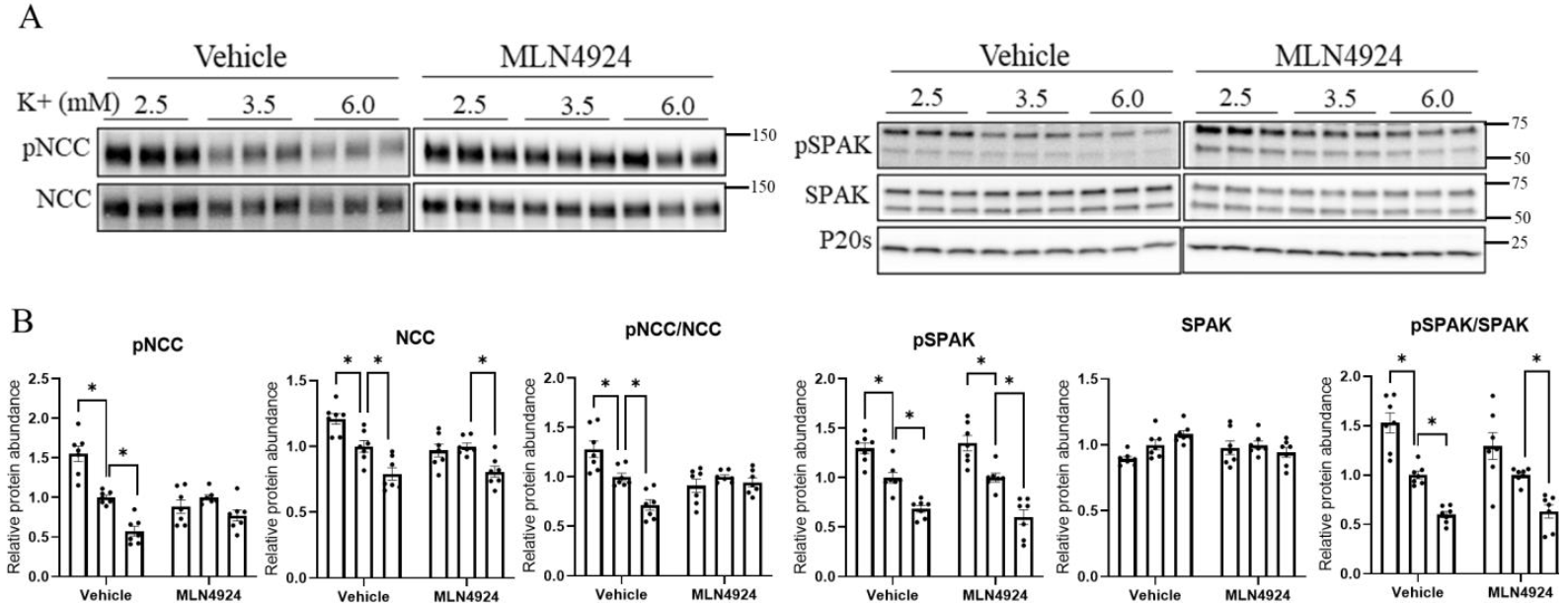
3.6. The Effects of K+ on NCC Are Attenuated during Long-Term Cullin Inhibition
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Egan, B.M.; Kjeldsen, S.E.; Grassi, G.; Esler, M.; Mancia, G. The global burden of hypertension exceeds 1.4 billion people: Should a systolic blood pressure target below 130 become the universal standard? J. Hypertens. 2019, 37, 1148–1153. [Google Scholar] [CrossRef]
- Mente, A.; O’Donnell, M.J.; Rangarajan, S.; McQueen, M.J.; Poirier, P.; Wielgosz, A.; Morrison, H.; Li, W.; Wang, X.; Di, C.; et al. Association of Urinary Sodium and Potassium Excretion with Blood Pressure. N. Engl. J. Med. 2014, 371, 601–611. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, M.; Mente, A.; Rangarajan, S.; McQueen, M.J.; Wang, X.; Liu, L.; Yan, H.; Lee, S.F.; Mony, P.; Devanath, A.; et al. Urinary sodium and potassium excretion, mortality, and cardiovascular events. N. Engl. J. Med. 2014, 371, 612–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, F.; Capasso, G.; Schwab, M.; Waldegger, S. Renal tubular transport and the genetic basis of hypertensive disease. Clin. Exp. Nephrol. 2005, 9, 91–99. [Google Scholar] [CrossRef]
- Simon, D.B.; Nelson-Williams, C.; Bia, M.J.; Ellison, D.; Karet, F.E.; Molina, A.M.; Vaara, I.; Iwata, F.; Cushner, H.M.; Koolen, M.; et al. Gitelman’s variant of Bartter’s syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nat. Genet. 1996, 12, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Vitzthum, H.; Seniuk, A.; Schulte, L.H.; Müller, M.L.; Hetz, H.; Ehmke, H. Functional coupling of renal K + and Na + handling causes high blood pressure in Na + replete mice. J. Physiol. 2014, 592, 1139–1157. [Google Scholar] [CrossRef]
- Terker, A.S.; Zhang, C.; McCormick, J.; Lazelle, R.A.; Zhang, C.; Meermeier, N.P.; Siler, D.A.; Park, H.J.; Fu, Y.; Cohen, D.M.; et al. Potassium Modulates Electrolyte Balance and Blood Pressure through Effects on Distal Cell Voltage and Chloride. Cell Metab. 2015, 21, 39–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, F.H.; Disse-Nicodème, S.; Choate, K.A.; Ishikawa, K.; Nelson-Williams, C.; Desitter, I.; Gunel, M.; Milford, D.V.; Lipkin, G.W.; Achard, J.-M.; et al. Human Hypertension Caused by Mutations in WNK Kinases. Science 2001, 293, 1107–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCormick, J.A.; Ellison, D.H. Distal convoluted tubule. Compr. Physiol. 2015, 5, 45–98. [Google Scholar]
- Boyden, L.M.; Choi, M.; Choate, K.A.; Nelson-Williams, C.J.; Farhi, A.; Toka, H.R.; Tikhonova, I.R.; Bjornson, R.; Mane, S.M.; Colussi, G.; et al. Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities. Nature 2012, 482, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Piala, A.T.; Moon, T.M.; Akella, R.; He, H.; Cobb, M.H.; Goldsmith, E.J. Chloride Sensing by WNK1 Involves Inhibition of Autophosphorylation. Sci. Signal. 2014, 7, ra41. [Google Scholar] [CrossRef] [Green Version]
- Bazúa-Valenti, S.; Chávez-Canales, M.; Rojas, L.; González-Rodríguez, X.; Vázquez, N.; Rodríguez-Gama, A.; Argaiz, E.; Melo, Z.; Plata, C.; Ellison, D.; et al. The Effect of WNK4 on the Na+–Cl−Cotransporter Is Modulated by Intracellular Chloride. J. Am. Soc. Nephrol. 2014, 26, 1781–1786. [Google Scholar] [CrossRef] [Green Version]
- Cuevas, C.; Su, X.-T.; Wang, M.-X.; Terker, A.S.; Lin, D.-H.; McCormick, J.; Yang, C.-L.; Ellison, D.H.; Wang, W.-H. Potassium Sensing by Renal Distal Tubules Requires Kir4.1. J. Am. Soc. Nephrol. 2017, 28, 1814–1825. [Google Scholar] [CrossRef]
- Wu, P.; Gao, Z.-X.; Zhang, D.-D.; Su, X.-T.; Wang, W.-H.; Lin, D.-H. Deletion of Kir5.1 Impairs Renal Ability to Excrete Potassium during Increased Dietary Potassium Intake. J. Am. Soc. Nephrol. 2019, 30, 1425–1438. [Google Scholar] [CrossRef]
- Hoorn, E.J.; Gritter, M.; Cuevas, C.A.; Fenton, R.A. Regulation of the Renal NaCl Cotransporter and Its Role in Potassium Homeostasis. Physiol. Rev. 2020, 100, 321–356. [Google Scholar] [CrossRef]
- Ishizawa, K.; Xu, N.; Loffing, J.; Lifton, R.P.; Fujita, T.; Uchida, S.; Shibata, S. Potassium depletion stimulates Na-Cl cotransporter via phosphorylation and inactivation of the ubiquitin ligase Kelch-like 3. Biochem. Biophys. Res. Commun. 2016, 480, 745–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Zhou, P. Cullins and cancer. Genes Cancer 2010, 1, 690–699. [Google Scholar] [CrossRef] [PubMed]
- Enchev, R.I.; Schulman, B.; Peter, M. Protein neddylation: Beyond cullin–RING ligases. Nat. Rev. Mol. Cell Biol. 2015, 16, 30–44. [Google Scholar] [CrossRef] [PubMed]
- Merlet, J.; Burger, J.; Gomes, J.-E.; Pintard, L. Regulation of cullin-RING E3 ubiquitin-ligases by neddylation and dimerization. Cell. Mol. Life Sci. 2009, 66, 1924–1938. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Yeh, E.T.H. Identification of the Activating and Conjugating Enzymes of the NEDD8 Conjugation Pathway. J. Biol. Chem. 1999, 274, 12036–12042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deshaies, R.J.; Emberley, E.D.; Saha, A. Control of Cullin-Ring Ubiquitin Ligase Activity by Nedd8. Subcell Biochem. 2010, 54, 41–56. [Google Scholar] [CrossRef]
- Wei, N.; Serino, G.; Deng, X.-W. The COP9 signalosome: More than a protease. Trends Biochem. Sci. 2008, 33, 592–600. [Google Scholar] [CrossRef]
- Schmaler, T.; Dubiel, W. Control of Deneddylation by the COP9 Signalosome. Subcell Biochem. 2010, 54, 57–68. [Google Scholar] [CrossRef]
- Kortenoeven, M.L.A.; Cheng, L.; Wu, Q.; Fenton, R.A. An in vivo protein landscape of the mouse DCT during high dietary K+ or low dietary Na+ intake. Am. J. Physiol. Physiol. 2021, 320, F908–F921. [Google Scholar] [CrossRef]
- Ferdaus, M.; Miller, L.N.; Agbor, L.N.; Saritas, T.; Singer, J.D.; Sigmund, C.; McCormick, J.A. Mutant Cullin 3 causes familial hyperkalemic hypertension via dominant effects. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Murali, S.K.; Aroankins, T.S.; Moeller, H.B.; Fenton, R.A. The Deubiquitylase USP4 Interacts with the Water Channel AQP2 to Modulate Its Apical Membrane Accumulation and Cellular Abundance. Cells 2019, 8, 265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kortenoeven, M.L.; Esteva-Font, C.; Dimke, H.; Poulsen, S.B.; Murali, S.K.; Fenton, R.A. High dietary potassium causes ubiquitin-dependent degradation of the kidney sodium-chloride cotransporter. J. Biol. Chem. 2021, 297, 100915. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, N.B.; Hofmeister, M.V.; Rosenbaek, L.L.; Nielsen, J.; Fenton, R.A. Vasopressin induces phosphorylation of the thiazide-sensitive sodium chloride cotransporter in the distal convoluted tubule. Kidney Int. 2010, 78, 160–169. [Google Scholar] [CrossRef] [Green Version]
- Moriguchi, T.; Urushiyama, S.; Hisamoto, N.; Iemura, S.-I.; Uchida, S.; Natsume, T.; Matsumoto, K.; Shibuya, H. WNK1 Regulates Phosphorylation of Cation-Chloride-coupled Cotransporters via the STE20-related Kinases, SPAK and OSR1. J. Biol. Chem. 2005, 280, 42685–42693. [Google Scholar] [CrossRef] [Green Version]
- McCormick, J.A.; Nelson, J.H.; Yang, C.L.; Curry, J.N.; Ellison, D.H. Overexpression of the sodium chloride cotransporter is not sufficient to cause familial hyperkalemic hypertension. Hypertension 2011, 58, 888–894. [Google Scholar] [CrossRef]
- Wu, K.; Chen, A.; Pan, Z.Q. Conjugation of Nedd8 to CUL1 enhances the ability of the ROC1-CUL1 complex to promote ubiquitin polymerization. J. Biol. Chem. 2000, 275, 32317–32324. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Xu, J.; Li, H.; Xu, M.; Chen, Z.; Weihua, Z.; Pan, Z.; Sun, Y. Neddylation E2 UBE2F Promotes the Survival of Lung Cancer Cells by Activating CRL5 to Degrade NOXA via the K11 Linkage. Clin. Cancer Res. 2016, 23, 1104–1116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duda, D.M.; Borg, L.A.; Scott, D.C.; Hunt, H.W.; Hammel, M.; Schulman, B.A. Structural Insights into NEDD8 Activation of Cullin-RING Ligases: Conformational Control of Conjugation. Cell 2008, 134, 995–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, A.; Deshaies, R.J. Multimodal Activation of the Ubiquitin Ligase SCF by Nedd8 Conjugation. Mol. Cell 2008, 32, 21–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Reichermeier, K.M.; Liu, X. Quantitative analyses for effects of neddylation on CRL2 VHL substrate ubiquitination and degradation. Protein Sci. 2021, 30, 2338–2345. [Google Scholar] [CrossRef]
- Boh, B.K.; Smith, P.G.; Hagen, T. Neddylation-Induced Conformational Control Regulates Cullin RING Ligase Activity In Vivo. J. Mol. Biol. 2011, 409, 136–145. [Google Scholar] [CrossRef]
- Ferdaus, M.Z.; McCormick, J.A. The CUL3/KLHL3-WNK-SPAK/OSR1 pathway as a target for antihypertensive therapy. Am. J. Physiol. Ren. Physiol. 2016, 310, F1389–F1396. [Google Scholar] [CrossRef] [Green Version]
- McCormick, J.; Yang, C.-L.; Zhang, C.; Davidge, B.; Blankenstein, K.I.; Terker, A.; Yarbrough, B.; Meermeier, N.P.; Park, H.J.; McCully, B.; et al. Hyperkalemic hypertension–associated cullin 3 promotes WNK signaling by degrading KLHL3. J. Clin. Investig. 2014, 124, 4723–4736. [Google Scholar] [CrossRef] [Green Version]
- Ferdaus, M.Z.; McCormick, J. Mechanisms and controversies in mutant Cul3-mediated familial hyperkalemic hypertension. Am. J. Physiol. Physiol. 2018, 314, F915–F920. [Google Scholar] [CrossRef] [Green Version]
- Vallon, V.; Schroth, J.; Lang, F.; Kuhl, D.; Uchida, S. Expression and phosphorylation of the Na+-Cl- cotransporter NCC in vivo is regulated by dietary salt, potassium, and SGK1. Am. J. Physiol. Ren. Physiol. 2009, 297, F704–F712. [Google Scholar] [CrossRef] [Green Version]
- Van Der Lubbe, N.; Lim, C.H.; Fenton, R.A.; Meima, M.E.; Danser, A.H.J.; Zietse, R.; Hoorn, E.J. Angiotensin II induces phosphorylation of the thiazide-sensitive sodium chloride cotransporter independent of aldosterone. Kidney Int. 2011, 79, 66–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Lubbe, N.; Lim, C.H.; Meima, M.E.; van Veghel, R.; Rosenbaek, L.L.; Mutig, K. Aldosterone does not require angiotensin II to activate NCC through a WNK4-SPAK-dependent pathway. Pflug. Arch. 2012, 463, 853–863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poulsen, S.B.; Cheng, L.; Penton, D.; Kortenoeven, M.L.; Matchkov, V.V.; Loffing, J.; Little, R.; Murali, S.K.; Fenton, R.A. Activation of the kidney sodium chloride cotransporter by the beta2-adrenergic receptor agonist salbutamol increases blood pressure. Kidney Int. 2021, 100, 321–335. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Poulsen, S.B.; Wu, Q.; Esteva-Font, C.; Olesen, E.T.B.; Peng, L.; Olde, B.; Leeb-Lundberg, L.M.F.; Pisitkun, T.; Rieg, T.; et al. Rapid Aldosterone-Mediated Signaling in the DCT Increases Activity of the Thiazide-Sensitive NaCl Cotransporter. J. Am. Soc. Nephrol. 2019, 30, 1454–1470. [Google Scholar] [CrossRef]
- Brownell, J.E.; Sintchak, M.D.; Gavin, J.M.; Liao, H.; Bruzzese, F.J.; Bump, N.J.; Soucy, T.A.; Milhollen, M.A.; Yang, X.; Burkhardt, A.L.; et al. Substrate-Assisted Inhibition of Ubiquitin-like Protein-Activating Enzymes: The NEDD8 E1 Inhibitor MLN4924 Forms a NEDD8-AMP Mimetic In Situ. Mol. Cell 2010, 37, 102–111. [Google Scholar] [CrossRef]
- Sacks, F.M.; Svetkey, L.P.; Vollmer, W.M.; Appel, L.J.; Bray, G.A.; Harsha, D.; Obarzanek, E.; Conlin, P.R.; Miller, E.R.; Simons-Morton, D.G.; et al. Effects on Blood Pressure of Reduced Dietary Sodium and the Dietary Approaches to Stop Hypertension (DASH) Diet. N. Engl. J. Med. 2001, 344, 3–10. [Google Scholar] [CrossRef]
- Sorensen, M.V.; Grossmann, S.; Roesinger, M.; Gresko, N.; Todkar, A.P.; Barmettler, G.; Ziegler, U.; Odermatt, A.; Loffing-Cueni, D.; Loffing, J. Rapid dephosphorylation of the renal sodium chloride cotransporter in response to oral potassium intake in mice. Kidney Int. 2013, 83, 811–824. [Google Scholar] [CrossRef] [Green Version]
- Rengarajan, S.; Lee, D.H.; Oh, Y.T.; Delpire, E.; Youn, J.H.; McDonough, A.A. Increasing plasma [K+] by intravenous potassium infusion reduces NCC phosphorylation and drives kaliuresis and natriuresis. Am. J. Physiol. Physiol. 2014, 306, F1059–F1068. [Google Scholar] [CrossRef] [Green Version]
- Frindt, G.; Palmer, L.G. Effects of dietary K on cell-surface expression of renal ion channels and transporters. Am. J. Physiol. Physiol. 2010, 299, F890–F897. [Google Scholar] [CrossRef] [Green Version]
- Akita, S.; Sacks, F.M.; Svetkey, L.P.; Conlin, P.R.; Kimura, G. Effects of the Dietary Approaches to Stop Hypertension (DASH) Diet on the Pressure-Natriuresis Relationship. Hypertension 2003, 42, 8–13. [Google Scholar] [CrossRef] [Green Version]
- Terker, A.S.; Zhang, C.; Erspamer, K.J.; Gamba, G.; Yang, C.-L.; Ellison, D.H. Unique chloride-sensing properties of WNK4 permit the distal nephron to modulate potassium homeostasis. Kidney Int. 2016, 89, 127–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, D.; Mori, T.; Nomura, N.; Khan, M.Z.H.; Araki, Y.; Zeniya, M.; Sohara, E.; Rai, T.; Sasaki, S.; Uchida, S. WNK4 is the major WNK positively regulating NCC in the mouse kidney. Biosci. Rep. 2014, 34, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.-X.; Cuevas, C.; Su, X.-T.; Wu, P.; Gao, Z.-X.; Lin, D.-H.; McCormick, J.; Yang, C.-L.; Wang, W.-H.; Ellison, D.H. Potassium intake modulates the thiazide-sensitive sodium-chloride cotransporter (NCC) activity via the Kir4.1 potassium channel. Kidney Int. 2018, 93, 893–902. [Google Scholar] [CrossRef] [PubMed]
- Su, X.-T.; Klett, N.J.; Sharma, A.; Allen, C.N.; Wang, W.-H.; Yang, C.-L.; Ellison, D.H. Distal convoluted tubule Cl− concentration is modulated via K+ channels and transporters. Am. J. Physiol. Physiol. 2020, 319, F534–F540. [Google Scholar] [CrossRef]
- Chen, J.-C.; Lo, Y.-F.; Lin, Y.-W.; Lin, S.-H.; Huang, C.-L.; Cheng, C.-J. WNK4 kinase is a physiological intracellular chloride sensor. Proc. Natl. Acad. Sci. USA 2019, 116, 4502–4507. [Google Scholar] [CrossRef] [Green Version]
- Ishizawa, K.; Wang, Q.; Li, J.; Yamazaki, O.; Tamura, Y.; Fujigaki, Y.; Uchida, S.; Lifton, R.P.; Shibata, S. Calcineurin dephosphorylates Kelch-like 3, reversing phosphorylation by angiotensin II and regulating renal electrolyte handling. Proc. Natl. Acad. Sci. USA 2019, 116, 3155–3160. [Google Scholar] [CrossRef] [Green Version]
- Shibata, S.; Arroyo, J.P.; Castañeda-Bueno, M.; Puthumana, J.; Zhang, J.; Uchida, S.; Stone, K.L.; Lam, T.T.; Lifton, R.P. Angiotensin II signaling via protein kinase C phosphorylates Kelch-like 3, preventing WNK4 degradation. Proc. Natl. Acad. Sci. USA 2014, 111, 15556–15561. [Google Scholar] [CrossRef] [Green Version]
- Nkashama, L.J.; Roy, A.; Carrisoza-Gaytan, R.; Ray, E.C.; Marciszyn, A.; Kleyman, T.R. The Nedd8 Co-E3 DCNL4 is regulated by dietary potassium in renal intercalated cells and promotes WNK kinase degradation via the KLHL3/CUL3 complex. FASEB J. 2018, 32, 747–748. [Google Scholar] [CrossRef]
- Yu, C.; Ji, S.-Y.; Sha, Q.-Q.; Sun, Q.-Y.; Fan, H.-Y. CRL4–DCAF1 ubiquitin E3 ligase directs protein phosphatase 2A degradation to control oocyte meiotic maturation. Nat. Commun. 2015, 6, 8017. [Google Scholar] [CrossRef]
- Xu, J.; Zhou, J.-Y.; Xu, Z.; Kho, D.-H.; Zhuang, Z.; Raz, A.; Wu, G.S. The role of Cullin3-mediated ubiquitination of the catalytic subunit of PP2A in TRAIL signaling. Cell Cycle 2014, 13, 3750–3758. [Google Scholar] [CrossRef] [Green Version]
- Flores-Delgado, G.; Liu, C.W.Y.; Sposto, A.R.; Berndt, N. A Limited Screen for Protein Interactions Reveals New Roles for Protein Phosphatase 1 in Cell Cycle Control and Apoptosis. J. Proteome Res. 2007, 6, 1165–1175. [Google Scholar] [CrossRef]
- Li, X.; Liu, J.; Gao, T. β-TrCP-mediated ubiquitination and degradation of PHLPP1 are negatively regulated by Akt. Mol. Cell. Biol. 2009, 29, 6192–6205. [Google Scholar] [CrossRef] [Green Version]
- Dushukyan, N.; Dunn, D.; Sager, R.A.; Woodford, M.; Loiselle, D.R.; Daneshvar, M.; Baker-Williams, A.J.; Chisholm, J.; Truman, A.; Vaughan, C.K.; et al. Phosphorylation and Ubiquitination Regulate Protein Phosphatase 5 Activity and Its Prosurvival Role in Kidney Cancer. Cell Rep. 2017, 21, 1883–1895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, A.; Yang, C.-L.; McCormick, J.A.; Martz, K.; Sharma, A.; Ellison, D.H. Roles of WNK4 and SPAK in K+-mediated dephosphorylation of the NaCl cotransporter. Am. J. Physiol. Physiol. 2021, 320, F719–F733. [Google Scholar] [CrossRef] [PubMed]
- Shoda, W.; Nomura, N.; Ando, F.; Mori, Y.; Mori, T.; Sohara, E.; Rai, T.; Uchida, S. Calcineurin inhibitors block sodium-chloride cotransporter dephosphorylation in response to high potassium intake. Kidney Int. 2016, 91, 402–411. [Google Scholar] [CrossRef]
- Penton, D.; Czogalla, J.; Wengi, A.; Himmerkus, N.; Loffing-Cueni, D.; Carrel, M.; Rajaram, R.D.; Staub, O.; Bleich, M.; Schweda, F.; et al. Extracellular K+ rapidly controls NaCl cotransporter phosphorylation in the native distal convoluted tubule by Cl−-dependent and independent mechanisms. J. Physiol. 2016, 594, 6319–6331. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murali, S.K.; Little, R.; Poulsen, S.B.; Ferdaus, M.Z.; Ellison, D.H.; McCormick, J.A.; Fenton, R.A. Potassium Effects on NCC Are Attenuated during Inhibition of Cullin E3–Ubiquitin Ligases. Cells 2022, 11, 95. https://doi.org/10.3390/cells11010095
Murali SK, Little R, Poulsen SB, Ferdaus MZ, Ellison DH, McCormick JA, Fenton RA. Potassium Effects on NCC Are Attenuated during Inhibition of Cullin E3–Ubiquitin Ligases. Cells. 2022; 11(1):95. https://doi.org/10.3390/cells11010095
Chicago/Turabian StyleMurali, Sathish K., Robert Little, Søren B. Poulsen, Mohammed Z. Ferdaus, David H. Ellison, James A. McCormick, and Robert A. Fenton. 2022. "Potassium Effects on NCC Are Attenuated during Inhibition of Cullin E3–Ubiquitin Ligases" Cells 11, no. 1: 95. https://doi.org/10.3390/cells11010095
APA StyleMurali, S. K., Little, R., Poulsen, S. B., Ferdaus, M. Z., Ellison, D. H., McCormick, J. A., & Fenton, R. A. (2022). Potassium Effects on NCC Are Attenuated during Inhibition of Cullin E3–Ubiquitin Ligases. Cells, 11(1), 95. https://doi.org/10.3390/cells11010095