
PPARγ Regulates Triclosan Induced Placental Dysfunction

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture and Transfection
2.3. Animal Treatment
2.4. Cell Viability Assay
2.5. Cell Migration Assay
2.6. Tube Formation Assay
2.7. Real Time PCR (RT-PCR)
2.8. Western Blot Analysis
2.9. Statistical Analysis
3. Results
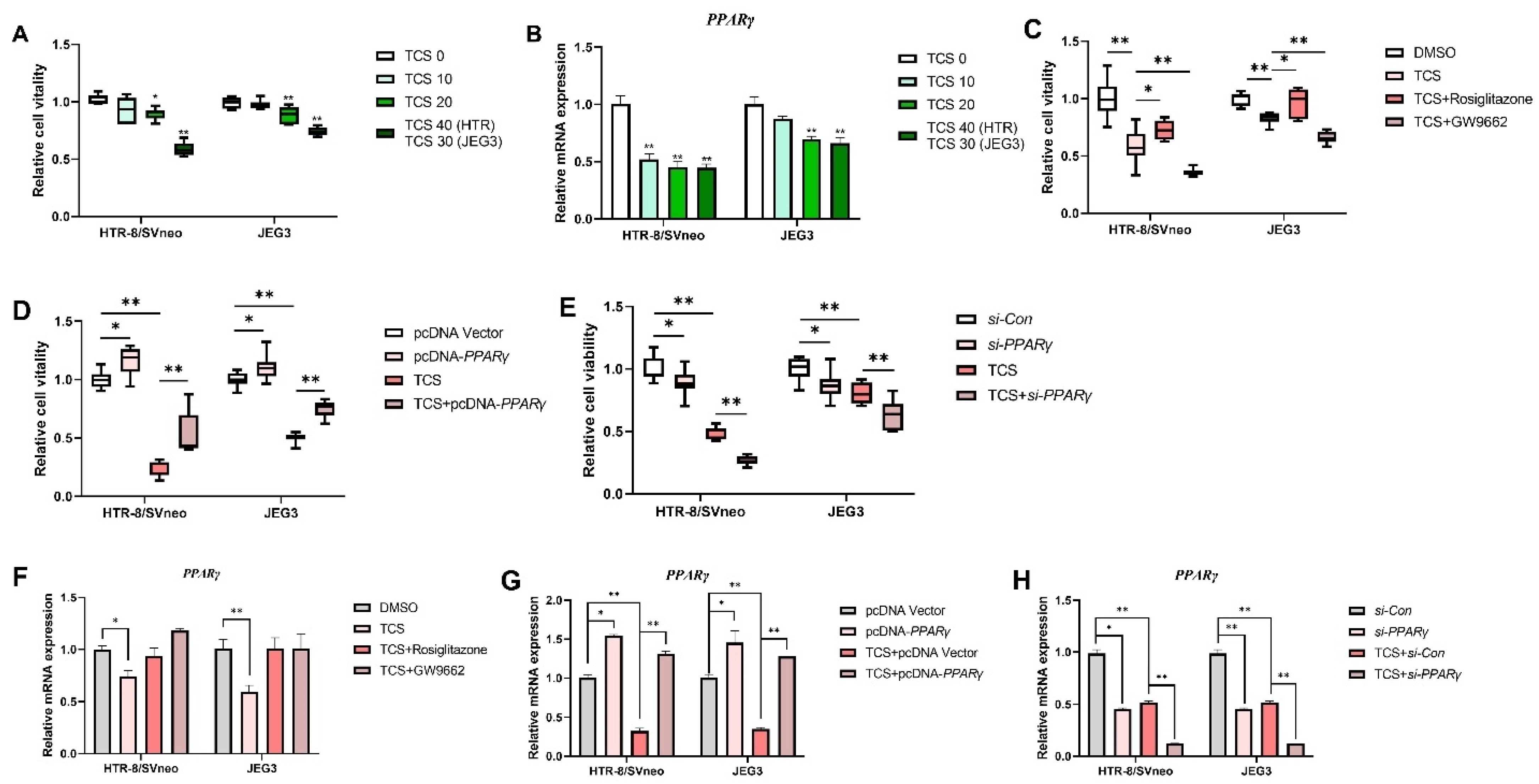
3.1. PPARγ Is Crucial in Cell Viability Inhibition Induced by TCS
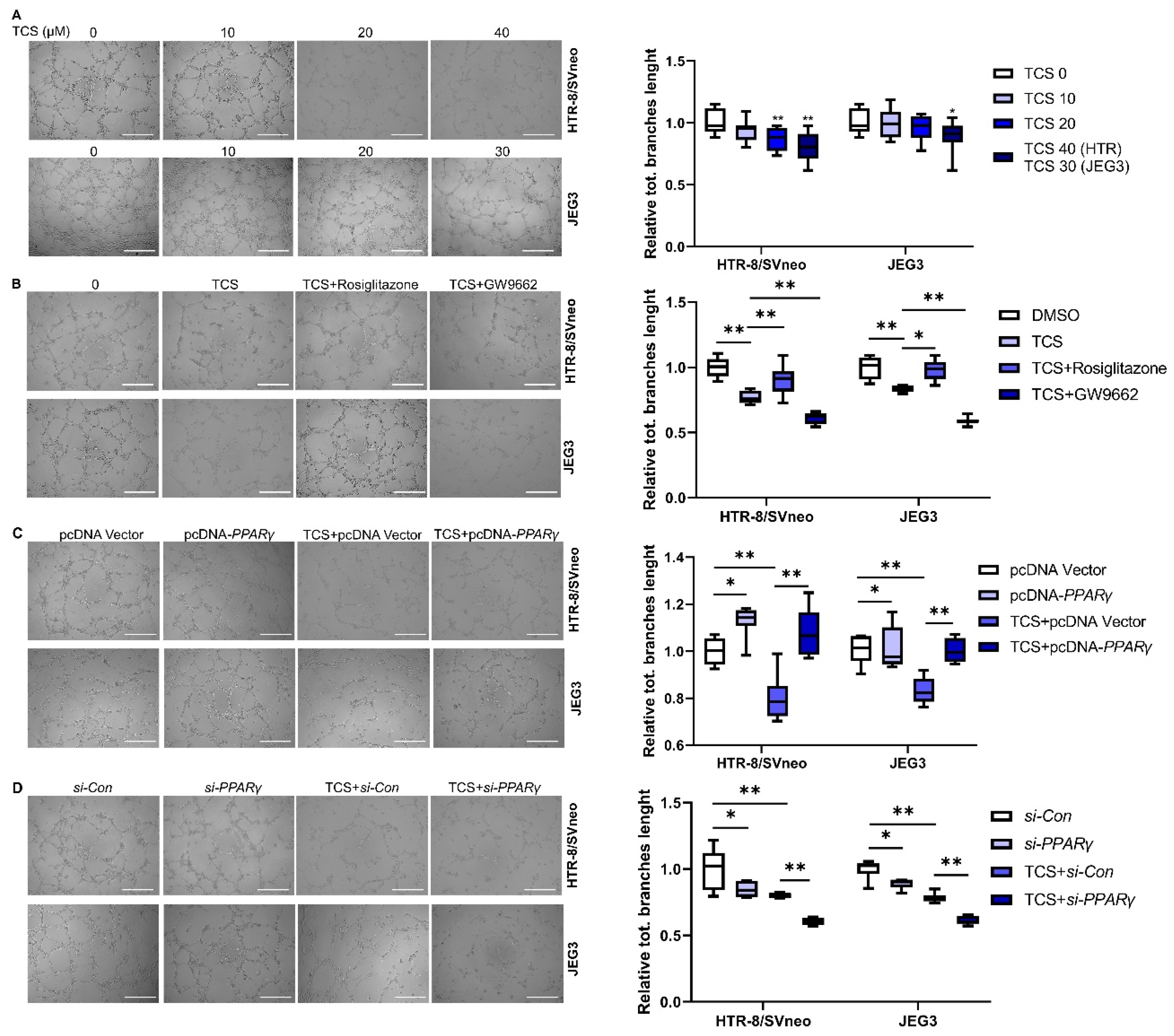
3.2. PPARγ Is Involved in TCS-Elicited Impaired Migration
3.3. PPARγ Alleviates TCS-Elicited Angiogenesis Inhibition
3.4. TCS Alters the Expression of PPARγ Target Genes Associated with Viability, Angiogenesis, and Migration
3.5. TCS Changes Expression Level of PPARγ-Regulated Inflammation Genes
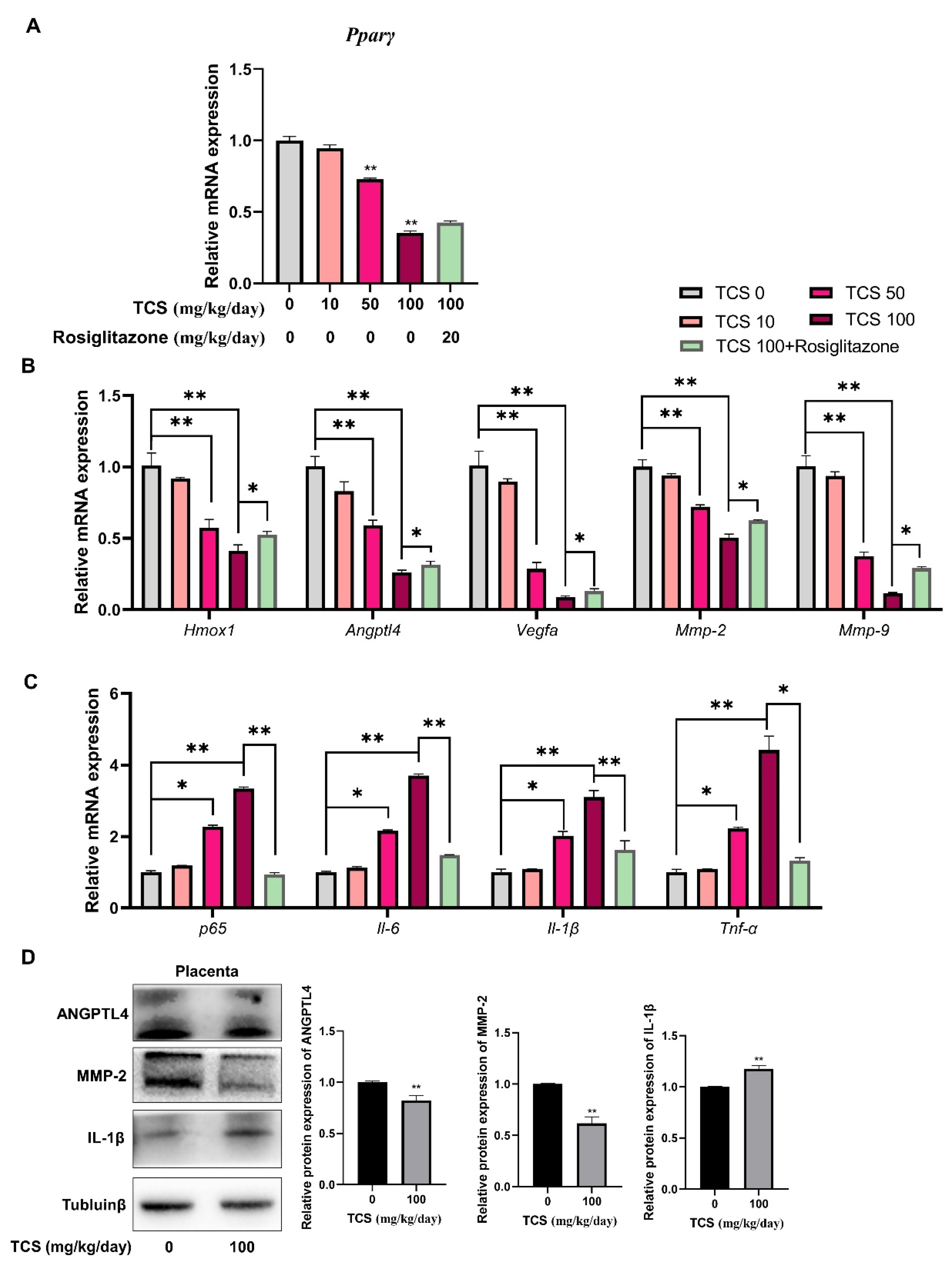
3.6. TCS Induces Placenta Dysfunction through PPARγ Pathway in Mice
3.7. TCS Alters Expression of PPARγ-Regulated Genes in Mice Placenta
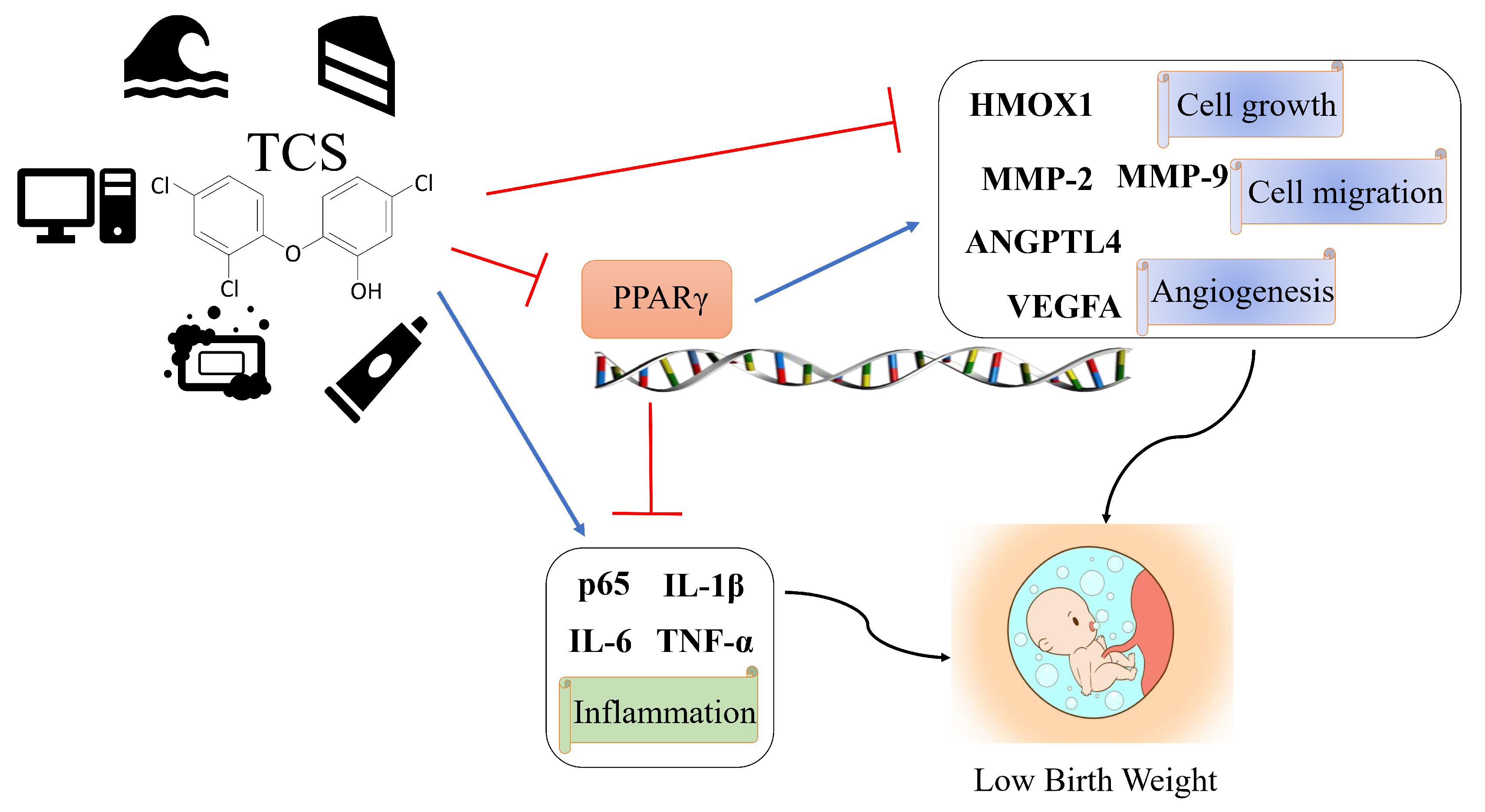
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Weatherly, L.M.; Gosse, J.A. Triclosan exposure, transformation, and human health effects. J. Toxicol. Environ. Health B. Crit. Rev. 2017, 20, 447–469. [Google Scholar] [CrossRef]
- Alfhili, M.A.; Lee, M.-H. Triclosan: An Update on Biochemical and Molecular Mechanisms. Oxid. Med. Cell Longev. 2019, 2019, 1607304. [Google Scholar] [CrossRef]
- Rodricks, J.V.; Swenberg, J.A.; Borzelleca, J.F.; Maronpot, R.R.; Shipp, A.M. Triclosan: A critical review of the experimental data and development of margins of safety for consumer products. Crit. Rev. Toxicol. 2010, 40, 422–484. [Google Scholar] [CrossRef]
- Ribeiro, E.; Ladeira, C.; Viegas, S. EDCs Mixtures: A Stealthy Hazard for Human Health? Toxics 2017, 5, 5. [Google Scholar] [CrossRef]
- Bever, C.S.; Rand, A.A.; Nording, M.; Taft, D.; Kalanetra, K.M.; Mills, D.A.; Breck, M.A.; Smilowitz, J.T.; German, J.B.; Hammock, B.D. Effects of triclosan in breast milk on the infant fecal microbiome. Chemosphere 2018, 203, 467–473. [Google Scholar] [CrossRef]
- Etzel, T.; Muckle, G.; Arbuckle, T.E.; Fraser, W.D.; Ouellet, E.; Séguin, J.R.; Lanphear, B.; Braun, J.M. Prenatal urinary triclosan concentrations and child neurobehavior. Environ. Int. 2018, 114, 152–159. [Google Scholar] [CrossRef]
- Ashley-Martin, J.; Dodds, L.; Arbuckle, T.E.; Marshall, J. Prenatal triclosan exposure and cord blood immune system biomarkers. Int. J. Hyg. Environ. Health 2016, 219, 454–457. [Google Scholar] [CrossRef][Green Version]
- Wang, X.; Chen, X.; Feng, X.; Chang, F.; Chen, M.; Xia, Y.; Chen, L. Triclosan causes spontaneous abortion accompanied by decline of estrogen sulfotransferase activity in humans and mice. Sci. Rep. 2015, 5, 18252. [Google Scholar] [CrossRef]
- Lassen, T.H.; Frederiksen, H.; Kyhl, H.B.; Swan, S.H.; Main, K.M.; Andersson, A.-M.; Lind, D.V.; Husby, S.; Wohlfahrt-Veje, C.; Skakkebæk, N.E.; et al. Prenatal Triclosan Exposure and Anthropometric Measures Including Anogenital Distance in Danish Infants. Environ. Health Perspect. 2016, 124, 1261–1268. [Google Scholar] [CrossRef]
- Zhao, J.-L.; Zhang, Q.-Q.; Chen, F.; Wang, L.; Ying, G.-G.; Liu, Y.-S.; Yang, B.; Zhou, L.-J.; Liu, S.; Su, H.-C.; et al. Evaluation of triclosan and triclocarban at river basin scale using monitoring and modeling tools: Implications for controlling of urban domestic sewage discharge. Water Res. 2013, 47, 395–405. [Google Scholar] [CrossRef]
- Hong, F.; Xu, P.; Zhai, Y. The Opportunities and Challenges of Peroxisome Proliferator-Activated Receptors Ligands in Clinical Drug Discovery and Development. Int. J. Mol. Sci. 2018, 19, 2189. [Google Scholar] [CrossRef]
- Xu, P.; Zhai, Y.; Wang, J. The Role of PPAR and Its Cross-Talk with CAR and LXR in Obesity and Atherosclerosis. Int. J. Mol. Sci. 2018, 19, 1260. [Google Scholar] [CrossRef]
- Hong, F.; Pan, S.; Guo, Y.; Xu, P.; Zhai, Y. PPARs as Nuclear Receptors for Nutrient and Energy Metabolism. Molecules 2019, 24, 2545. [Google Scholar] [CrossRef]
- Xu, P.; Hong, F.; Wang, J.; Wang, J.; Zhao, X.; Wang, S.; Xue, T.; Xu, J.; Zheng, X.; Zhai, Y. DBZ is a putative PPARγ agonist that prevents high fat diet-induced obesity, insulin resistance and gut dysbiosis. Biochim. Biophys. Acta Gen. Subj. 2017, 11 Pt A, 2690–2701. [Google Scholar] [CrossRef]
- Xi, Y.; Zhang, Y.; Zhu, S.; Luo, Y.; Xu, P.; Huang, Z. PPAR-Mediated Toxicology and Applied Pharmacology. Cells 2020, 9, 200. [Google Scholar] [CrossRef]
- Nadra, K.; Quignodon, L.; Sardella, C.; Joye, E.; Mucciolo, A.; Chrast, R.; Desvergne, B. PPARgamma in placental angiogenesis. Endocrinology 2010, 151, 4969–4981. [Google Scholar] [CrossRef]
- Fournier, T.; Tsatsaris, V.; Handschuh, K.; Evain-Brion, D. PPARs and the placenta. Placenta 2007, 28, 65–76. [Google Scholar] [CrossRef]
- Debril, M.B.; Renaud, J.P.; Fajas, L.; Auwerx, J. The pleiotropic functions of peroxisome proliferator-activated receptor gamma. J. Mol. Med. 2001, 79, 30–47. [Google Scholar] [CrossRef]
- Wagner, N.; Wagner, K.-D. PPARs and Angiogenesis-Implications in Pathology. Int. J. Mol. Sci. 2020, 21, 5723. [Google Scholar] [CrossRef]
- Barak, Y.; Nelson, M.C.; Ong, E.S.; Jones, Y.Z.; Ruiz-Lozano, P.; Chien, K.R.; Koder, A.; Evans, R.M. PPAR gamma is required for placental, cardiac, and adipose tissue development. Mol. Cell 1999, 4, 585–595. [Google Scholar] [CrossRef]
- Kubota, N.; Terauchi, Y.; Miki, H.; Tamemoto, H.; Yamauchi, T.; Komeda, K.; Satoh, S.; Nakano, R.; Ishii, C.; Sugiyama, T.; et al. PPAR gamma mediates high-fat diet-induced adipocyte hypertrophy and insulin resistance. Mol. Cell 1999, 4, 597–609. [Google Scholar] [CrossRef]
- Kadam, L.; Kohan-Ghadr, H.R.; Drewlo, S. The balancing act—PPAR-γ’s roles at the maternal-fetal interface. Syst. Biol. Reprod. Med. 2015, 61, 65–71. [Google Scholar] [CrossRef]
- Hua, X.; Cao, X.-Y.; Wang, X.-L.; Sun, P.; Chen, L. Exposure of Pregnant Mice to Triclosan Causes Insulin Resistance via Thyroxine Reduction. Toxicol. Sci. 2017, 160, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Szychowski, K.A.; Skóra, B.; Wójtowicz, A.K. Triclosan affects the expression of nitric oxide synthases (NOSs), peroxisome proliferator-activated receptor gamma (PPARγ), and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) in mouse neocortical neurons in vitro. Toxicol. Vitro 2021, 73, 105143. [Google Scholar] [CrossRef]
- La Paglia, L.; Listì, A.; Caruso, S.; Amodeo, V.; Passiglia, F.; Bazan, V.; Fanale, D. Potential Role of ANGPTL4 in the Cross Talk between Metabolism and Cancer through PPAR Signaling Pathway. PPAR Res. 2017, 2017, 8187235. [Google Scholar] [CrossRef]
- Shyu, L.-Y.; Chen, K.-M.; Lu, C.-Y.; Lai, S.-C. Regulation of Proinflammatory Enzymes by Peroxisome Proliferator-Activated Receptor Gamma in Astroglia Infected with Toxoplasma gondii. J. Parasitol. 2020, 106, 564–571. [Google Scholar] [CrossRef]
- Shen, B.; Zhao, C.; Wang, Y.; Peng, Y.; Cheng, J.; Li, Z.; Wu, L.; Jin, M.; Feng, H. Aucubin inhibited lipid accumulation and oxidative stress via Nrf2/HO-1 and AMPK signalling pathways. J. Cell Mol. Med. 2019, 23, 4063–4075. [Google Scholar] [CrossRef]
- Kelleher, A.M.; DeMayo, F.J.; Spencer, T.E. Uterine Glands: Developmental Biology and Functional Roles in Pregnancy. Endocr. Rev. 2019, 40, 1424–1445. [Google Scholar] [CrossRef]
- Zhang, N.; Wang, W.; Li, W.; Liu, C.; Chen, Y.; Yang, Q.; Wang, Y.; Sun, K. Inhibition of 11β-HSD2 expression by triclosan via induction of apoptosis in human placental syncytiotrophoblasts. J. Clin. Endocrinol. Metab. 2015, 100, E542–E549. [Google Scholar] [CrossRef][Green Version]
- Honkisz, E.; Zieba-Przybylska, D.; Wojtowicz, A.K. The effect of triclosan on hormone secretion and viability of human choriocarcinoma JEG-3 cells. Reprod. Toxicol. 2012, 34, 385–392. [Google Scholar] [CrossRef]
- Burton, G.J.; Jauniaux, E. Pathophysiology of placental-derived fetal growth restriction. Am. J. Obstet. Gynecol. 2018, 218, S745–S761. [Google Scholar] [CrossRef]
- Zhang, M.; Zhu, R.; Zhang, L. Triclosan stimulates human vascular endothelial cell injury via repression of the PI3K/Akt/mTOR axis. Chemosphere 2020, 241, 125077. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, M.; Liu, J.; Wang, X.; Wang, C.; Ai, W.; Chen, S.; Wang, H. Combined toxicity of triclosan, 2,4-dichlorophenol and 2,4,6-trichlorophenol to zebrafish (Danio rerio). Environ. Toxicol. Pharmacol. 2018, 57, 9–18. [Google Scholar] [CrossRef]
- Feng, Y.; Zhang, P.; Zhang, Z.; Shi, J.; Jiao, Z.; Shao, B. Endocrine Disrupting Effects of Triclosan on the Placenta in Pregnant Rats. PLoS ONE 2016, 11, e0154758. [Google Scholar] [CrossRef] [PubMed]
- Garnier, V.; Traboulsi, W.; Salomon, A.; Brouillet, S.; Fournier, T.; Winkler, C.; Desvergne, B.; Hoffmann, P.; Zhou, Q.-Y.; Congiu, C.; et al. PPARγ controls pregnancy outcome through activation of EG-VEGF: New insights into the mechanism of placental development. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E357–E369. [Google Scholar] [CrossRef]
- Shibuya, M. Vascular endothelial growth factor and its receptor system: Physiological functions in angiogenesis and pathological roles in various diseases. J. Biochem. 2013, 153, 13–19. [Google Scholar] [CrossRef]
- Hato, T.; Tabata, M.; Oike, Y. The role of angiopoietin-like proteins in angiogenesis and metabolism. Trends Cardiovasc. Med. 2008, 18, 6–14. [Google Scholar] [CrossRef]
- Goh, Y.Y.; Pal, M.; Chong, H.C.; Zhu, P.; Tan, M.J.; Punugu, L.; Tan, C.K.; Huang, R.-L.; Sze, S.K.; Tang, M.B.Y.; et al. Angiopoietin-like 4 interacts with matrix proteins to modulate wound healing. J. Biol. Chem. 2010, 285, 32999–33009. [Google Scholar] [CrossRef]
- Liu, L.; Zhuang, X.; Jiang, M.; Guan, F.; Fu, Q.; Lin, J. ANGPTL4 mediates the protective role of PPARγ activators in the pathogenesis of preeclampsia. Cell Death Dis. 2017, 8, e3054. [Google Scholar] [CrossRef]
- Woeckel, V.J.; Bruedigam, C.; Koedam, M.; Chiba, H.; van der Eerden, B.C.J.; van Leeuwen, J.P.T.M. 1α,25-dihydroxyvitamin D3 and rosiglitazone synergistically enhance osteoblast-mediated mineralization. Gene 2013, 512, 438–443. [Google Scholar] [CrossRef]
- Tian, F.-J.; Qin, C.-M.; Li, X.-C.; Wu, F.; Liu, X.-R.; Xu, W.-M.; Lin, Y. Decreased stathmin-1 expression inhibits trophoblast proliferation and invasion and is associated with recurrent miscarriage. Am. J. Pathol. 2015, 185, 2709–2721. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, J.; Gong, Y.; Liu, X.; Bai, Y.; Xu, W.; Zhou, R. Increased expression of prostasin contributes to early-onset severe preeclampsia through inhibiting trophoblast invasion. J. Perinatol. 2015, 35, 16–22. [Google Scholar] [CrossRef]
- Vannuccini, S.; Clifton, V.L.; Fraser, I.S.; Taylor, H.S.; Critchley, H.; Giudice, L.C.; Petraglia, F. Infertility and reproductive disorders: Impact of hormonal and inflammatory mechanisms on pregnancy outcome. Hum. Reprod. Update 2016, 22, 104–115. [Google Scholar] [CrossRef]
- Remels, A.H.V.; Langen, R.C.J.; Gosker, H.R.; Russell, A.P.; Spaapen, F.; Voncken, J.W.; Schrauwen, P.; Schols, A.M.W.J. PPARgamma inhibits NF-kappaB-dependent transcriptional activation in skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E174–E183. [Google Scholar] [CrossRef] [PubMed]
- Challis, J.R.; Lockwood, C.J.; Myatt, L.; Norman, J.E.; Strauss, J.F.; Petraglia, F. Inflammation and pregnancy. Reprod. Sci. 2009, 16, 206–215. [Google Scholar] [CrossRef]
- Jiang, C.; Ting, A.T.; Seed, B. PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature 1998, 391, 82–86. [Google Scholar] [CrossRef]
- Lappas, M.; Permezel, M.; Georgiou, H.M.; Rice, G.E. Regulation of proinflammatory cytokines in human gestational tissues by peroxisome proliferator-activated receptor-gamma: Effect of 15-deoxy-Delta(12,14)-PGJ(2) and troglitazone. J. Clin. Endocrinol. Metab. 2002, 87, 4667–4672. [Google Scholar] [CrossRef] [PubMed]
- Kadam, L.; Kilburn, B.; Baczyk, D.; Kohan-Ghadr, H.R.; Kingdom, J.; Drewlo, S. Rosiglitazone blocks first trimester in-vitro placental injury caused by NF-κB-mediated inflammation. Sci. Rep. 2019, 9, 2018. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Quan, X.; Lei, S.; Huang, Z.; Wang, Q.; Xu, P. PFOS Inhibited Normal Functional Development of Placenta Cells via Signaling. Biomedicines 2021, 9, 276. [Google Scholar] [CrossRef] [PubMed]
- Yeligar, S.M.; Kang, B.-Y.; Bijli, K.M.; Kleinhenz, J.M.; Murphy, T.C.; Torres, G.; San Martin, A.; Sutliff, R.L.; Hart, C.M. PPARγ Regulates Mitochondrial Structure and Function and Human Pulmonary Artery Smooth Muscle Cell Proliferation. Am. J. Respir. Cell Mol. Biol. 2018, 58, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Teplova, V.V.; Belosludtsev, K.N.; Kruglov, A.G. Mechanism of triclosan toxicity: Mitochondrial dysfunction including complex II inhibition, superoxide release and uncoupling of oxidative phosphorylation. Toxicol. Lett. 2017, 275, 108–117. [Google Scholar] [CrossRef]
- Belosludtsev, K.N.; Belosludtseva, N.V.; Tenkov, K.S.; Penkov, N.V.; Agafonov, A.V.; Pavlik, L.L.; Yashin, V.A.; Samartsev, V.N.; Dubinin, M.V. Study of the mechanism of permeabilization of lecithin liposomes and rat liver mitochondria by the antimicrobial drug triclosan. Biochim. Biophys. Acta Biomembr. 2018, 1860, 264–271. [Google Scholar] [CrossRef]
- Tossetta, G.; Fantone, S.; Giannubilo, S.R.; Marzioni, D. The Multifaced Actions of Curcumin in Pregnancy Outcome. Antioxidants 2021, 10, 126. [Google Scholar] [CrossRef]
- Blanquicett, C.; Kang, B.-Y.; Ritzenthaler, J.D.; Jones, D.P.; Hart, C.M. Oxidative stress modulates PPAR gamma in vascular endothelial cells. Free Radic. Biol. Med. 2010, 48, 1618–1625. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Quan, X.; Zhang, Y.; Yu, T.; Lei, S.; Huang, Z.; Wang, Q.; Song, W.; Yang, X.; Xu, P. PPARγ Regulates Triclosan Induced Placental Dysfunction. Cells 2022, 11, 86. https://doi.org/10.3390/cells11010086
Li J, Quan X, Zhang Y, Yu T, Lei S, Huang Z, Wang Q, Song W, Yang X, Xu P. PPARγ Regulates Triclosan Induced Placental Dysfunction. Cells. 2022; 11(1):86. https://doi.org/10.3390/cells11010086
Chicago/Turabian StyleLi, Jing, Xiaojie Quan, Yue Zhang, Ting Yu, Saifei Lei, Zhenyao Huang, Qi Wang, Weiyi Song, Xinxin Yang, and Pengfei Xu. 2022. "PPARγ Regulates Triclosan Induced Placental Dysfunction" Cells 11, no. 1: 86. https://doi.org/10.3390/cells11010086
APA StyleLi, J., Quan, X., Zhang, Y., Yu, T., Lei, S., Huang, Z., Wang, Q., Song, W., Yang, X., & Xu, P. (2022). PPARγ Regulates Triclosan Induced Placental Dysfunction. Cells, 11(1), 86. https://doi.org/10.3390/cells11010086