
Elucidating T Cell and B Cell Responses to SARS-CoV-2 in Humans: Gaining Insights into Protective Immunity and Immunopathology


Abstract
:1. Introduction
2. T Cell Responses in the Setting of SARS-CoV-2 Infection
2.1. Correlating T Cell Responses with COVID-19 Disease Phenotypes
2.2. Cross-Reactive T Cells
2.3. T Cell Responses in Multi-Inflammatory Syndrome in Children (MIS-C)
2.4. T Cell Responses in COVID-19 Vaccine Recipients
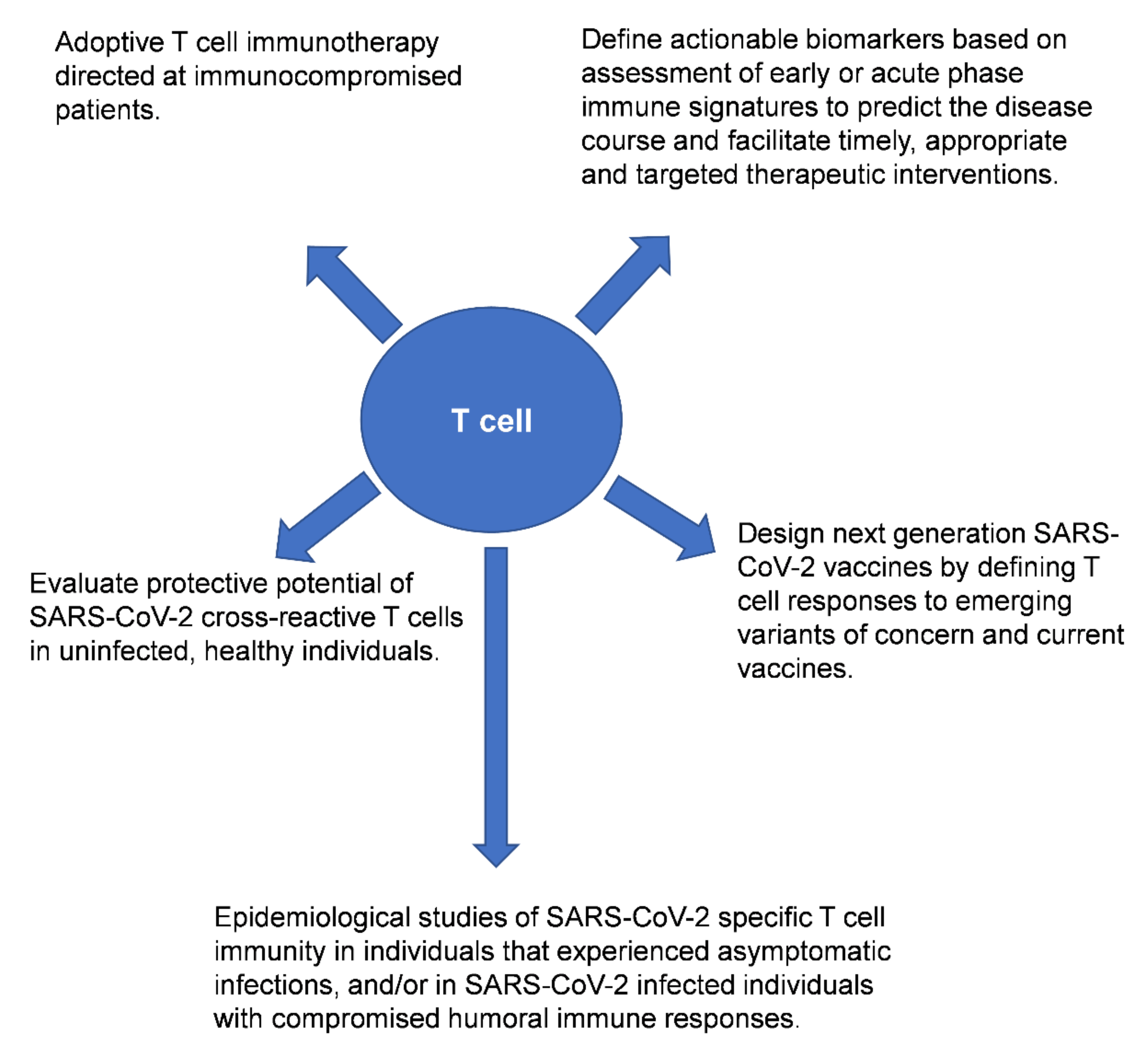
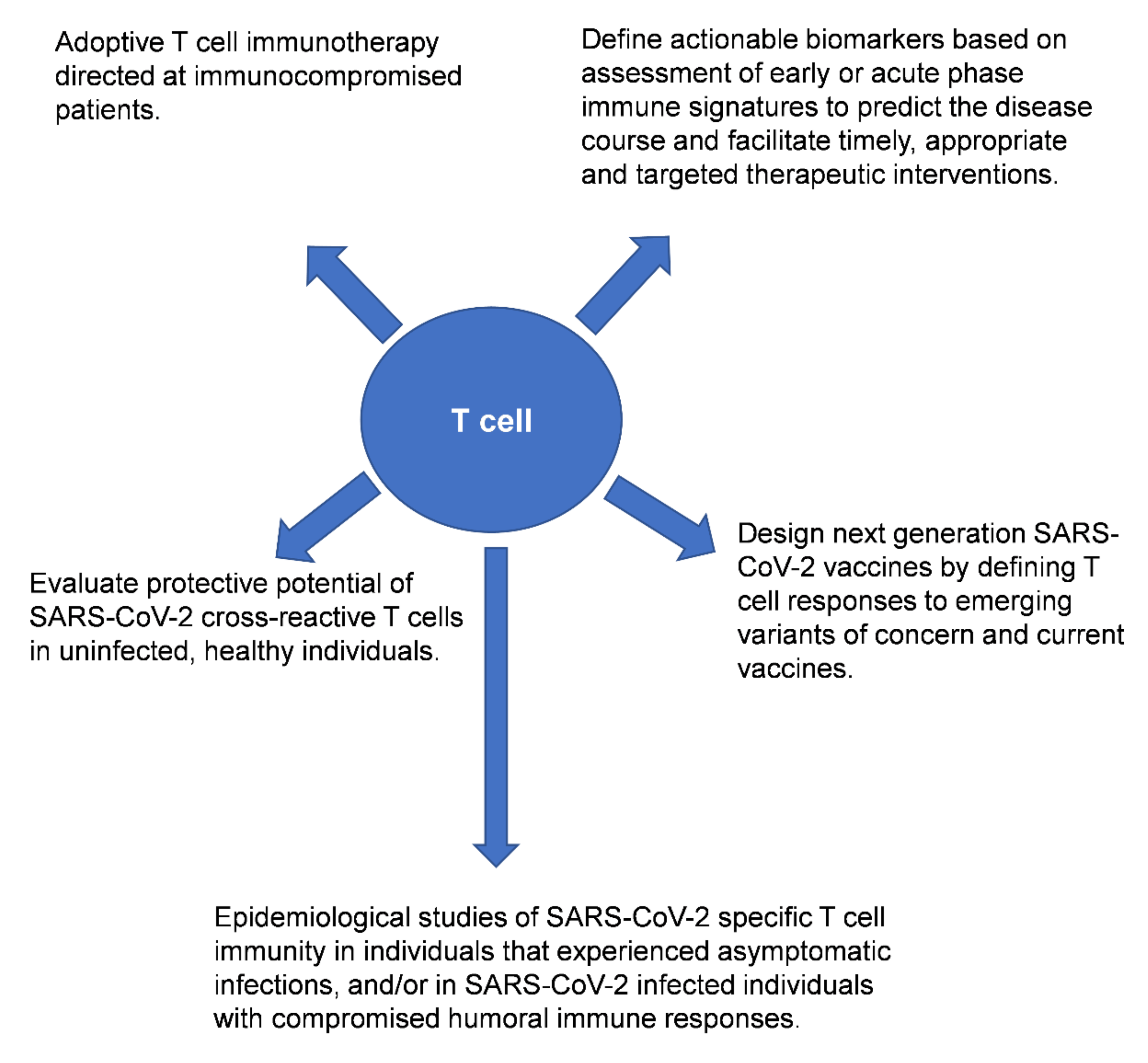
2.5. Adoptive Transfer Therapy Using SARS-CoV-2 Specific T Cells
3. B Cell Responses to SARS-CoV-2
3.1. Detecting Ag-Specific B Cells in SARS-CoV-2 Infected Individuals
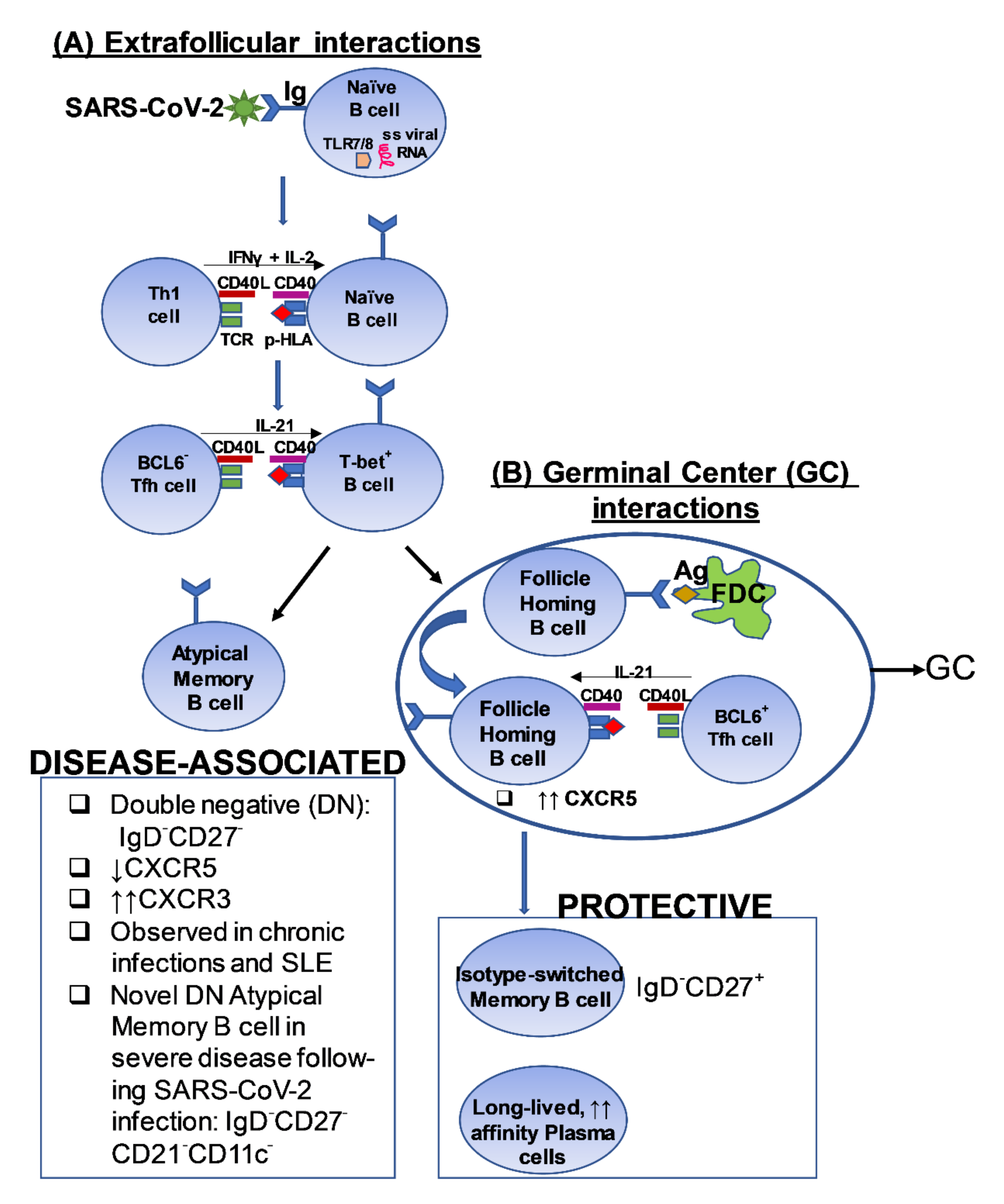
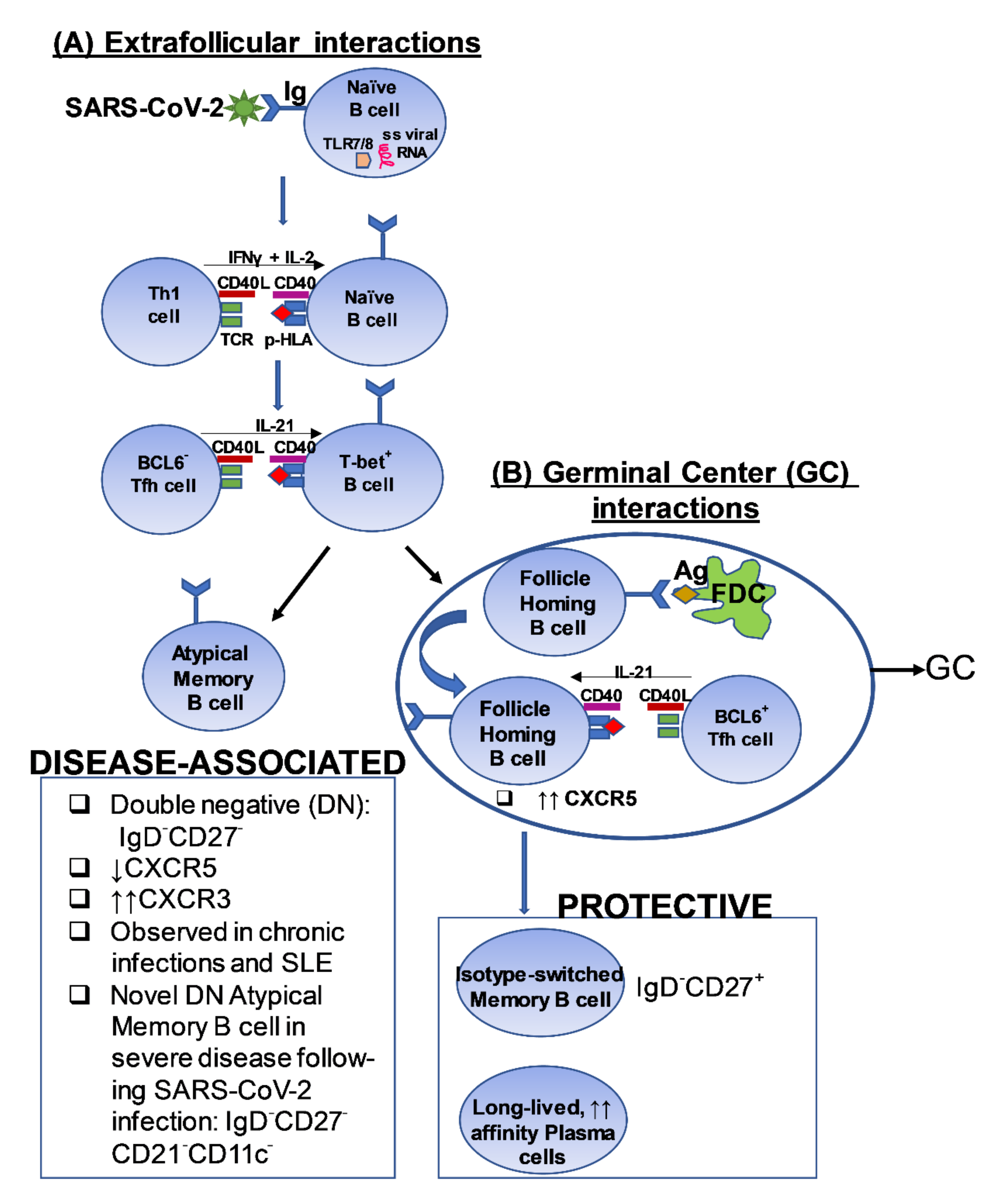
3.2. B Cell Responses in the Setting of Mild COVID-19 Disease
3.3. B Cell Responses in the Setting of Severe COVID-19 Disease
3.4. B Cell Responses in MIS-C
3.5. B Cell Response in COVID-19 Vaccine Recipients
4. Evaluation of the SARS-CoV-2-Related Immune Response in the Immune-Suppressed State
5. Conclusions
Funding
Conflicts of Interest
References
- Ksiazek, T.G.; Erdman, D.; Goldsmith, C.S.; Zaki, S.R.; Peret, T.; Emery, S.; Tong, S.; Urbani, C.; Comer, J.A.; Lim, W.; et al. A Novel Coronavirus Associated with Severe Acute Respiratory Syndrome. N. Engl. J. Med. 2003, 348, 1953–1966. [Google Scholar] [CrossRef] [PubMed]
- Zhong, N.S.; Zheng, B.J.; Li, Y.M.; Poon, L.L.M.; Xie, Z.H.; Chan, K.H.; Li, P.H.; Tan, S.Y.; Chang, Q.; Xie, J.P.; et al. Epidemiology and cause of severe acute respiratory syndrome (SARS) in Guangdong, People’s Republic of China, in February, 2003. Lancet 2003, 362, 1353–1358. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.F.-W.; Lau, S.K.P.; To, K.K.W.; Cheng, V.C.C.; Woo, P.C.Y.; Yuen, K.-Y. Middle East Respiratory Syndrome Coronavirus: Another Zoonotic Betacoronavirus Causing SARS-Like Disease. Clin. Microbiol. Rev. 2015, 28, 465–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Breedon, A.M.E.; Saldanha, R.J.; Salisbury, R.L.; Metzger, D.E.; Werry, M.P.; McPherson, C.J.; Irvin, A.P.; Davis, C.M.; Bogner, C.A.; Braddock, A.M.; et al. COVID-19 Seroprevalence and Active Infection in an Asymptomatic Population. Front. Med. 2021, 8, 1580. [Google Scholar] [CrossRef]
- Abbas, A.K.; Lichtman, A.; Pillai, S. Cellular and Molecular Immunology, 9th ed.; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Khanolkar, A.; Fuller, M.J.; Zajac, A.J. T Cell Responses to Viral Infections: Lessons from Lymphocytic Choriomeningitis Virus. Immunol. Res. 2002, 26, 309–322. [Google Scholar] [CrossRef]
- Lucas, C.; Wong, P.; Klein, J.; Castro, T.B.R.; Silva, J.; Sundaram, M.; Ellingson, M.K.; Mao, T.; Oh, J.E.; Israelow, B.; et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature 2020, 584, 463–469. [Google Scholar] [CrossRef]
- Mathew, D.; Giles, J.R.; Baxter, A.E.; Oldridge, D.A.; Greenplate, A.R.; Wu, J.E.; Alanio, C.; Kuri-Cervantes, L.; Pampena, M.B.; D’Andrea, K.; et al. Deep immune profiling of COVID-19 patients reveals distinct immunotypes with therapeutic implications. Science 2020, 369, eabc8511. [Google Scholar] [CrossRef]
- Carter, M.J.; Fish, M.; Jennings, A.; Doores, K.J.; Wellman, P.; Seow, J.; Acors, S.; Graham, C.; Timms, E.; Kenny, J.; et al. Peripheral immunophenotypes in children with multisystem inflammatory syndrome associated with SARS-CoV-2 infection. Nat. Med. 2020, 26, 1701–1707. [Google Scholar] [CrossRef]
- Rodriguez, L.; Pekkarinen, P.T.; Lakshmikanth, T.; Tan, Z.; Consiglio, C.R.; Pou, C.; Chen, Y.; Mugabo, C.H.; Nguyen, N.A.; Nowlan, K.; et al. Systems-Level Immunomonitoring from Acute to Recovery Phase of Severe COVID-19. Cell Rep. Med. 2020, 1, 100078. [Google Scholar] [CrossRef]
- Laing, A.G.; Lorenc, A.; del Barrio, I.D.M.; Das, A.; Fish, M.; Monin, L.; Muñoz-Ruiz, M.; McKenzie, D.R.; Hayday, T.S.; Francos-Quijorna, I.; et al. A dynamic COVID-19 immune signature includes associations with poor prognosis. Nat. Med. 2020, 26, 1623–1635. [Google Scholar] [CrossRef]
- Carissimo, G.; Xu, W.; Kwok, I.; Abdad, M.Y.; Chan, Y.-H.; Fong, S.-W.; Puan, K.J.; Lee, C.Y.-P.; Yeo, N.K.-W.; Amrun, S.N.; et al. Whole blood immunophenotyping uncovers immature neutrophil-to-VD2 T-cell ratio as an early marker for severe COVID-19. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef]
- Takahashi, T.; Ellingson, M.K.; Wong, P.; Israelow, B.; Lucas, C.; Klein, J.; Silva, J.; Mao, T.; Oh, J.E.; Tokuyama, M.; et al. Sex differences in immune responses that underlie COVID-19 disease outcomes. Nature 2020, 588, 315–320. [Google Scholar] [CrossRef]
- Moderbacher, C.R.; Ramirez, S.I.; Dan, J.M.; Grifoni, A.; Hastie, K.M.; Weiskopf, D.; Belanger, S.; Abbott, R.K.; Kim, C.; Choi, J.; et al. Antigen-Specific Adaptive Immunity to SARS-CoV-2 in Acute COVID-19 and Associations with Age and Disease Severity. Cell 2020, 183, 996–1012.e19. [Google Scholar] [CrossRef]
- Blanchard-Rohner, G.; Didierlaurent, A.; Tilmanne, A.; Smeesters, P.; Marchant, A. Pediatric COVID-19: Immunopathogenesis, Transmission and Prevention. Vaccines 2021, 9, 1002. [Google Scholar] [CrossRef]
- Cummings, M.J.; Baldwin, M.R.; Abrams, D.; Jacobson, S.D.; Meyer, B.J.; Balough, E.M.; Aaron, J.G.; Claassen, J.; Rabbani, L.E.; Hastie, J.; et al. Epidemiology, clinical course, and outcomes of critically ill adults with COVID-19 in New York City: A prospective cohort study. Lancet 2020, 395, 1763–1770. [Google Scholar] [CrossRef]
- Garibaldi, B.T.; Fiksel, J.; Muschelli, J.; Robinson, M.L.; Rouhizadeh, M.; Perin, J.; Schumock, B.G.; Nagy, P.; Gray, B.J.H.; Malapati, B.H.; et al. Patient Trajectories Among Persons Hospitalized for COVID-19: A Cohort Study. Ann. Intern. Med. 2021, 174, 33–41. [Google Scholar] [CrossRef]
- Bastard, P.; Rosen, L.B.; Zhang, Q.; Michailidis, E.; Hoffmann, H.-H.; Zhang, Y.; Dorgham, K.; Philippot, Q.; Rosain, J.; Béziat, V.; et al. Auto-antibodies against type I IFNs in patients with life-threatening COVID-19. Science 2020, 370, eabd4585. [Google Scholar] [CrossRef]
- Grifoni, A.; Weiskopf, D.; Ramirez, S.I.; Mateus, J.; Dan, J.M.; Moderbacher, C.R.; Rawlings, S.A.; Sutherland, A.; Premkumar, L.; Jadi, R.S.; et al. Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans with COVID-19 Disease and Unexposed Individuals. Cell 2020, 181, 1489–1501.e1415. [Google Scholar] [CrossRef]
- Mateus, J.; Grifoni, A.; Tarke, A.; Sidney, J.; Ramirez, S.I.; Dan, J.M.; Burger, Z.C.; Rawlings, S.A.; Smith, D.M.; Phillips, E.; et al. Selective and cross-reactive SARS-CoV-2 T cell epitopes in unexposed humans. Science 2020, 370, 89–94. [Google Scholar] [CrossRef]
- Weiskopf, D.; Schmitz, K.S.; Raadsen, M.P.; Grifoni, A.; Okba, N.M.A.; Endeman, H.; Van den Akker, J.P.C.; Molenkamp, R.; Koopmans, M.P.G.; Van Gorp, E.C.M.; et al. Phenotype and kinetics of SARS-CoV-2-specific T cells in COVID-19 patients with acute respiratory distress syndrome. Sci. Immunol. 2020, 5, eabd2071. [Google Scholar] [CrossRef]
- Braun, J.; Loyal, L.; Frentsch, M.; Wendisch, D.; Georg, P.; Kurth, F.; Hippenstiel, S.; Dingeldey, M.; Kruse, B.; Fauchere, F.; et al. SARS-CoV-2-reactive T cells in healthy donors and patients with COVID-19. Nature 2020, 587, 270–274. [Google Scholar] [CrossRef]
- Sekine, T.; Perez-Potti, A.; Rivera-Ballesteros, O.; Strålin, K.; Gorin, J.-B.; Olsson, A.; Llewellyn-Lacey, S.; Kamal, H.; Bogdanovic, G.; Muschiol, S.; et al. Robust T Cell Immunity in Convalescent Individuals with Asymptomatic or Mild COVID-19. Cell 2020, 183, 158–168.e14. [Google Scholar] [CrossRef]
- Le Bert, N.; Tan, A.T.; Kunasegaran, K.; Tham, C.Y.L.; Hafezi, M.; Chia, A.; Chng, M.H.Y.; Lin, M.; Tan, N.; Linster, M.; et al. SARS-CoV-2-specific T cell immunity in cases of COVID-19 and SARS, and uninfected controls. Nature 2020, 584, 457–462. [Google Scholar] [CrossRef]
- Kusnadi, A.; Ramírez-Suástegui, C.; Fajardo, V.; Chee, S.J.; Meckiff, B.J.; Simon, H.; Pelosi, E.; Seumois, G.; Ay, F.; Vijayanand, P.; et al. Severely ill patients with COVID-19 display impaired exhaustion features in SARS-CoV-2–reactive CD8 + T cells. Sci. Immunol. 2021, 6. [Google Scholar] [CrossRef]
- Peng, Y.; Mentzer, A.J.; Liu, G.; Yao, X.; Yin, Z.; Dong, D.; Dejnirattisai, W.; Rostron, T.; Supasa, P.; Liu, C.; et al. Broad and strong memory CD4+ and CD8+ T cells induced by SARS-CoV-2 in UK convalescent individuals following COVID-19. Nat. Immunol. 2020, 21, 1336–1345. [Google Scholar] [CrossRef]
- Szabo, P.A.; Dogra, P.; Gray, J.I.; Wells, S.B.; Connors, T.J.; Weisberg, S.P.; Krupska, I.; Matsumoto, R.; Poon, M.M.L.; Idzikowski, E.; et al. Longitudinal profiling of respiratory and systemic immune responses reveals myeloid cell-driven lung inflammation in severe COVID-19. Immunity 2021, 54, 797–814.e796. [Google Scholar] [CrossRef]
- Zhao, Y.; Kilian, C.; Turner, J.-E.; Bosurgi, L.; Roedl, K.; Bartsch, P.; Gnirck, A.-C.; Cortesi, F.; Schultheiß, C.; Hellmig, M.; et al. Clonal expansion and activation of tissue-resident memory-like T H 17 cells expressing GM-CSF in the lungs of patients with severe COVID-19. Sci. Immunol. 2021, 6. [Google Scholar] [CrossRef]
- De Luca, G.; Cavalli, G.; Campochiaro, C.; Della-Torre, E.; Angelillo, P.; Tomelleri, A.; Boffini, N.; Tentori, S.; Mette, F.; Farina, N.; et al. GM-CSF blockade with mavrilimumab in severe COVID-19 pneumonia and systemic hyperinflammation: A single-centre, prospective cohort study. Lancet Rheumatol. 2020, 2, e465–e473. [Google Scholar] [CrossRef]
- Temesgen, Z.; Assi, M.; Shweta, F.N.U.; Vergidis, P.; Rizza, S.A.; Bauer, P.R.; Pickering, B.W.; Razonable, R.R.; Libertin, C.R.; Burger, C.D.; et al. GM-CSF Neutralization with Lenzilumab in Severe COVID-19 Pneumonia: A Case-Cohort Study. Mayo Clin. Proc. 2020, 95, 2382–2394. [Google Scholar] [CrossRef]
- Lipsitch, M.; Grad, Y.H.; Sette, A.; Crotty, S. Cross-reactive memory T cells and herd immunity to SARS-CoV-2. Nat. Rev. Immunol. 2020, 20, 709–713. [Google Scholar] [CrossRef] [PubMed]
- De Vries, R.D. SARS-CoV-2-specific T-cells in unexposed humans: Presence of cross-reactive memory cells does not equal protective immunity. Signal Transduct. Target. Ther. 2020, 5, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Sette, A.; Crotty, S. Pre-existing immunity to SARS-CoV-2: The knowns and unknowns. Nat. Rev. Immunol. 2020, 20, 457–458. [Google Scholar] [CrossRef] [PubMed]
- Sagar, M.; Reifler, K.; Rossi, M.; Miller, N.S.; Sinha, P.; White, L.F.; Mizgerd, J.P. Recent endemic coronavirus infection is associated with less-severe COVID-19. J. Clin. Investig. 2021, 131, e143380. [Google Scholar] [CrossRef] [PubMed]
- Loyal, L.; Braun, J.; Henze, L.; Kruse, B.; Dingeldey, M.; Reimer, U.; Kern, F.; Schwarz, T.; Mangold, M.; Unger, C.; et al. Cross-reactive CD4 + T cells enhance SARS-CoV-2 immune responses upon infection and vaccination. Science 2021, 374, eabh1823. [Google Scholar] [CrossRef] [PubMed]
- Papayanni, P.-G.; Chasiotis, D.; Koukoulias, K.; Georgakopoulou, A.; Iatrou, A.; Gavriilaki, E.; Giannaki, C.; Bitzani, M.; Geka, E.; Tasioudis, P.; et al. Vaccinated and Convalescent Donor–Derived Severe Acute Respiratory Syndrome Coronavirus 2–Specific T Cells as Adoptive Immunotherapy for High-Risk Coronavirus Disease 2019 Patients. Clin. Infect. Dis. 2021, 73, 2073–2082. [Google Scholar] [CrossRef] [PubMed]
- Woldemeskel, B.A.; Garliss, C.C.; Blankson, J.N. SARS-CoV-2 mRNA vaccines induce broad CD4+ T cell responses that recognize SARS-CoV-2 variants and HCoV-NL63. J. Clin. Investig. 2021, 131, e149335. [Google Scholar] [CrossRef]
- Sahin, U.; Muik, A.; Vogler, I.; Derhovanessian, E.; Kranz, L.M.; Vormehr, M.; Quandt, J.; Bidmon, N.; Ulges, A.; Baum, A.; et al. BNT162b2 vaccine induces neutralizing antibodies and poly-specific T cells in humans. Nature 2021, 595, 572–577. [Google Scholar] [CrossRef]
- Keller, M.D.; Harris, K.M.; Jensen-Wachspress, M.A.; Kankate, V.V.; Lang, H.; Lazarski, C.A.; Durkee-Shock, J.R.; Lee, P.-H.; Chaudhry, K.; Webber, K.; et al. SARS-CoV-2–specific T cells are rapidly expanded for therapeutic use and target conserved regions of the membrane protein. Blood 2020, 136, 2905–2917. [Google Scholar] [CrossRef]
- Pérez-Martínez, A.; Mora-Rillo, M.; Ferreras, C.; Guerra-García, P.; Pascual-Miguel, B.; Mestre-Durán, C.; Borobia, A.M.; Carcas, A.; Queiruga-Parada, J.; García, I.; et al. Phase I dose-escalation single centre clinical trial to evaluate the safety of infusion of memory T cells as adoptive therapy in COVID-19 (RELEASE). EClinicalMedicine 2021, 39, 101086. [Google Scholar] [CrossRef]
- Basar, R.; Uprety, N.; Ensley, E.; Daher, M.; Klein, K.; Martinez, F.; Aung, F.; Shanley, M.; Hu, B.; Gokdemir, E.; et al. Generation of glucocorticoid-resistant SARS-CoV-2 T cells for adoptive cell therapy. Cell Rep. 2021, 36, 109432. [Google Scholar] [CrossRef]
- Wang, H.; Wang, Z.; Cao, W.; Wu, Q.; Yuan, Y.; Zhang, X. Regulatory T cells in COVID-19. Aging Dis. 2021, 12, 1545–1553. [Google Scholar] [CrossRef]
- Elkoshi, Z. The Binary Model of Chronic Diseases Applied to COVID-19. Front. Immunol. 2021, 12, 716084. [Google Scholar] [CrossRef]
- Baeten, P.; Van Zeebroeck, L.; Kleinewietfeld, M.; Hellings, N.; Broux, B. Improving the Efficacy of Regulatory T Cell Therapy. Clin. Rev. Allergy Immunol. 2021, 1–19. [Google Scholar] [CrossRef]
- Gladstone, D.E.; Kim, B.S.; Mooney, R.K.; Karaba, A.H.; D’Alessio, F.R. Regulatory T Cells for Treating Patients With COVID-19 and Acute Respiratory Distress Syndrome: Two Case Reports. Ann. Intern. Med. 2020, 173, 852–853. [Google Scholar] [CrossRef]
- Hadjadj, J.; Yatim, N.; Barnabei, L.; Corneau, A.; Boussier, J.; Smith, N.; Péré, H.; Charbit, B.; Bondet, V.; Chenevier-Gobeaux, C.; et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 2020, 369, 718–724. [Google Scholar] [CrossRef]
- Silvin, A.; Chapuis, N.; Dunsmore, G.; Goubet, A.-G.; Dubuisson, A.; Derosa, L.; Almire, C.; Hénon, C.; Kosmider, O.; Droin, N.; et al. Elevated Calprotectin and Abnormal Myeloid Cell Subsets Discriminate Severe from Mild COVID-19. Cell 2020, 182, 1401–1418.e18. [Google Scholar] [CrossRef]
- Bost, P.; Giladi, A.; Liu, Y.; Bendjelal, Y.; Xu, G.; David, E.; Blecher-Gonen, R.; Cohen, M.; Medaglia, C.; Li, H.; et al. Host-Viral Infection Maps Reveal Signatures of Severe COVID-19 Patients. Cell 2020, 181, 1475–1488.e12. [Google Scholar] [CrossRef]
- Khanolkar, A.; Hartwig, S.M.; Haag, B.A.; Meyerholz, D.K.; Epping, L.L.; Haring, J.S.; Varga, S.M.; Harty, J.T. Protective and Pathologic Roles of the Immune Response to Mouse Hepatitis Virus Type 1: Implications for Severe Acute Respiratory Syndrome. J. Virol. 2009, 83, 9258–9272. [Google Scholar] [CrossRef] [Green Version]
- Khanolkar, A.; Fulton, R.B.; Epping, L.L.; Pham, N.-L.; Tifrea, D.; Varga, S.M.; Harty, J.T. T Cell Epitope Specificity and Pathogenesis of Mouse Hepatitis Virus-1–Induced Disease in Susceptible and Resistant Hosts. J. Immunol. 2010, 185, 1132–1141. [Google Scholar] [CrossRef]
- Hewagama, A.; Patel, D.; Yarlagadda, S.P.; Strickland, F.M.; Richardson, B.C. Stronger inflammatory/cytotoxic T-cell response in women identified by microarray analysis. Genes Immun. 2009, 10, 509–516. [Google Scholar] [CrossRef]
- Abdullah, M.; Chai, P.-S.; Chong, P.P.; Tohit, E.R.M.; Ramasamy, R.; Pei, C.P.; Vidyadaran, S. Gender effect on in vitro lymphocyte subset levels of healthy individuals. Cell. Immunol. 2012, 272, 214–219. [Google Scholar] [CrossRef]
- Furman, D.; Hejblum, B.P.; Simon, N.; Jojic, V.; Dekker, C.L.; Thiébaut, R.; Tibshirani, R.J.; Davis, M.M. Systems analysis of sex differences reveals an immunosuppressive role for testosterone in the response to influenza vaccination. Proc. Natl. Acad. Sci. USA 2014, 111, 869–874. [Google Scholar] [CrossRef] [Green Version]
- Sohn, E. Why autoimmunity is most common in women. Nature 2021, 595, S51–S53. [Google Scholar] [CrossRef]
- Burbelo, P.D.; Seam, N.; Groot, S.; Ching, K.H.; Han, B.L.; Meduri, G.U.; Iadarola, M.J.; Suffredini, A.F. Rapid induction of autoantibodies during ARDS and septic shock. J. Transl. Med. 2010, 8, 97. [Google Scholar] [CrossRef] [Green Version]
- Trahtemberg, U.; Fritzler, M.J.; Rottapel, R.; dos Santos, C.C.; Di Battista, A.P.; Slutsky, A.S.; Baker, A.J. On behalf of the COVID-19 chapter of the “Longitudinal Biomarkers in Lung Injury” study group COVID-19-associated autoimmunity as a feature of acute respiratory failure. Intensive Care Med. 2021, 47, 801–804. [Google Scholar] [CrossRef]
- Dan, J.M.; Mateus, J.; Kato, Y.; Hastie, K.M.; Yu, E.D.; Faliti, C.E.; Grifoni, A.; Ramirez, S.I.; Haupt, S.; Frazier, A.; et al. Immunological memory to SARS-CoV-2 assessed for up to 8 months after infection. Science 2021, 371, eabf4063. [Google Scholar] [CrossRef]
- Nguyen, T.H.O.; Rowntree, L.C.; Petersen, J.; Chua, B.Y.; Hensen, L.; Kedzierski, L.; van de Sandt, C.E.; Chaurasia, P.; Tan, H.-X.; Habel, J.R.; et al. CD8+ T cells specific for an immunodominant SARS-CoV-2 nucleocapsid epitope display high naive precursor frequency and TCR promiscuity. Immunity 2021, 54, 1066–1082.e5. [Google Scholar] [CrossRef]
- Habel, J.R.; Nguyen, T.H.O.; van de Sandt, C.E.; Juno, J.A.; Chaurasia, P.; Wragg, K.; Koutsakos, M.; Hensen, L.; Jia, X.; Chua, B.; et al. Suboptimal SARS-CoV-2−specific CD8+ T cell response associated with the prominent HLA-A*02:01 phenotype. Proc. Natl. Acad. Sci. USA 2020, 117, 24384–24391. [Google Scholar] [CrossRef]
- Sidney, J.; Peters, B.; Frahm, N.; Brander, C.; Sette, A. HLA class I supertypes: A revised and updated classification. BMC Immunol. 2008, 9, 1. [Google Scholar] [CrossRef] [Green Version]
- Campbell, K.M.; Steiner, G.; Wells, D.K.; Ribas, A.; Kalbasi, A. Prioritization of SARS-CoV-2 epitopes using a pan-HLA and global population inference approach. bioRxiv 2020. preprint. [Google Scholar] [CrossRef] [Green Version]
- Weingarten-Gabbay, S.; Klaeger, S.; Sarkizova, S.; Pearlman, L.R.; Chen, D.-Y.; Gallagher, K.M.E.; Bauer, M.R.; Taylor, H.B.; Dunn, W.A.; Tarr, C.; et al. Profiling SARS-CoV-2 HLA-I peptidome reveals T cell epitopes from out-of-frame ORFs. Cell 2021, 184, 3962–3980.e17. [Google Scholar] [CrossRef] [PubMed]
- Poon, M.M.L.; Rybkina, K.; Kato, Y.; Kubota, M.; Matsumoto, R.; Bloom, N.I.; Zhang, Z.; Hastie, K.M.; Grifoni, A.; Weiskopf, D.; et al. SARS-CoV-2 infection generates tissue-localized immunological memory in humans. Sci. Immunol. 2021, 6, eabl9105. [Google Scholar] [CrossRef] [PubMed]
- Niessl, J.; Sekine, T.; Lange, J.; Konya, V.; Forkel, M.; Maric, J.; Rao, A.; Mazzurana, L.; Kokkinou, E.; Weigel, W.; et al. Identification of resident memory CD8 + T cells with functional specificity for SARS-CoV-2 in unexposed oropharyngeal lymphoid tissue. Sci. Immunol. 2021, 6, eabk0894. [Google Scholar] [CrossRef]
- Consiglio, C.R.; Cotugno, N.; Sardh, F.; Pou, C.; Amodio, D.; Rodriguez, L.; Tan, Z.; Zicari, S.; Ruggiero, A.; Pascucci, G.R.; et al. The Immunology of Multisystem Inflammatory Syndrome in Children with COVID-19. Cell 2020, 183, 968–981.e967. [Google Scholar] [CrossRef]
- Vella, L.A.; Giles, J.R.; Baxter, A.E.; Oldridge, D.A.; Diorio, C.; Kuri-Cervantes, L.; Alanio, C.; Pampena, M.B.; Wu, J.E.; Chen, Z.; et al. Deep immune profiling of MIS-C demonstrates marked but transient immune activation compared to adult and pediatric COVID-19. Sci. Immunol. 2021, 6, eabf7570. [Google Scholar] [CrossRef]
- Ramaswamy, A.; Brodsky, N.N.; Sumida, T.S.; Comi, M.; Asashima, H.; Hoehn, K.B.; Li, N.; Liu, Y.; Shah, A.; Ravindra, N.G.; et al. Immune dysregulation and autoreactivity correlate with disease severity in SARS-CoV-2-associated multisystem inflammatory syndrome in children. Immunity 2021, 54, 1083–1095.e1087. [Google Scholar] [CrossRef]
- Pierce, C.A.; Preston-Hurlburt, P.; Dai, Y.; Aschner, C.B.; Cheshenko, N.; Galen, B.; Garforth, S.J.; Herrera, N.G.; Jangra, R.K.; Morano, N.C.; et al. Immune responses to SARS-CoV-2 infection in hospitalized pediatric and adult patients. Sci. Transl. Med. 2020, 12, 564. [Google Scholar] [CrossRef]
- Bhatt, S.; Holmes, E.C.; Pybus, O.G. The Genomic Rate of Molecular Adaptation of the Human Influenza A Virus. Mol. Biol. Evol. 2011, 28, 2443–2451. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.-R.T.; Zarnitsyna, V.I.; Lowen, A.C.; Weissman, D.; Koelle, K.; Kohlmeier, J.E.; Antia, R. Why Are CD8 T Cell Epitopes of Human Influenza A Virus Conserved? J. Virol. 2019, 93, e01534-18. [Google Scholar] [CrossRef] [Green Version]
- Caccamo, N.; Sullivan, L.C.; Brooks, A.G.; Dieli, F. Harnessing HLA-E-restricted CD8 T lymphocytes for adoptive cell therapy of patients with severe COVID-19. Br. J. Haematol. 2020, 190, e185–e187. [Google Scholar] [CrossRef]
- Cooper, R.S.; Fraser, A.R.; Smith, L.; Burgoyne, P.; Imlach, S.N.; Jarvis, L.M.; Turner, D.M.; Zahra, S.; Turner, M.L.; Campbell, J.D.M. Rapid GMP-Compliant Expansion of SARS-CoV-2–Specific T Cells from Convalescent Donors for Use as an Allogeneic Cell Therapy for COVID-19. Front. Immunol. 2021, 11, 598402. [Google Scholar] [CrossRef]
- Rodda, L.B.; Netland, J.; Shehata, L.; Pruner, K.B.; Morawski, P.A.; Thouvenel, C.D.; Takehara, K.K.; Eggenberger, J.; Hemann, E.A.; Waterman, H.R.; et al. Functional SARS-CoV-2-Specific Immune Memory Persists after Mild COVID-19. Cell 2021, 184, 169–183.e117. [Google Scholar] [CrossRef]
- Turner, J.S.; Kim, W.; Kalaidina, E.; Goss, C.W.; Rauseo, A.M.; Schmitz, A.J.; Hansen, L.; Haile, A.; Klebert, M.K.; Pusic, I.; et al. SARS-CoV-2 infection induces long-lived bone marrow plasma cells in humans. Nature 2021, 595, 421–425. [Google Scholar] [CrossRef]
- Turner, J.S.; O’Halloran, J.A.; Kalaidina, E.; Kim, W.; Schmitz, A.J.; Zhou, J.Q.; Lei, T.; Thapa, M.; Chen, R.E.; Case, J.B.; et al. SARS-CoV-2 mRNA vaccines induce persistent human germinal centre responses. Nature 2021, 596, 109–113. [Google Scholar] [CrossRef]
- Hartley, G.E.; Edwards, E.S.J.; Aui, P.M.; Varese, N.; Stojanovic, S.; McMahon, J.; Peleg, A.Y.; Boo, I.; Drummer, H.E.; Hogarth, P.M.; et al. Rapid Generation of Durable B Cell Memory to SARS-CoV-2 Spike and Nucleocapsid Proteins in COVID-19 and Convalescence. Sci. Immunol. 2020, 5, eabf8891. [Google Scholar] [CrossRef]
- Berkowska, M.A.; Driessen, G.J.A.; Bikos, V.; Grosserichter-Wagener, C.; Stamatopoulos, K.; Cerutti, A.; He, B.; Biermann, K.; Lange, J.F.; van der Burg, M.; et al. Human memory B cells originate from three distinct germinal center-dependent and -independent maturation pathways. Blood 2011, 118, 2150–2158. [Google Scholar] [CrossRef] [Green Version]
- Berkowska, M.A.; Schickel, J.-N.; Grosserichter-Wagener, C.; De Ridder, D.; Ng, Y.S.; Van Dongen, J.; Meffre, E.; van Zelm, M. Circulating Human CD27−IgA+Memory B Cells Recognize Bacteria with Polyreactive Igs. J. Immunol. 2015, 195, 1417–1426. [Google Scholar] [CrossRef] [Green Version]
- Amanna, I.J.; Carlson, N.E.; Slifka, M.K. Duration of Humoral Immunity to Common Viral and Vaccine Antigens. N. Engl. J. Med. 2007, 357, 1903–1915. [Google Scholar] [CrossRef] [Green Version]
- Cirelli, K.M.; Crotty, S. Germinal center enhancement by extended antigen availability. Curr. Opin. Immunol. 2017, 47, 64–69. [Google Scholar] [CrossRef]
- Folegatti, P.M.; Ewer, K.J.; Aley, P.K.; Angus, B.; Becker, S.; Belij-Rammerstorfer, S.; Bellamy, D.; Bibi, S.; Bittaye, M.; Clutterbuck, E.A.; et al. Safety and immunogenicity of the ChAdOx1 nCoV-19 vaccine against SARS-CoV-2: A preliminary report of a phase 1/2, single-blind, randomised controlled trial. Lancet 2020, 396, 467–478, Addendum in 2020, 396, E89. [Google Scholar] [CrossRef]
- Pegu, A.; O’Connell, S.E.; Schmidt, S.D.; O’Dell, S.; Talana, C.A.; Lai, L.; Albert, J.; Anderson, E.; Bennett, H.; Corbett, K.S.; et al. Durability of mRNA-1273 vaccine–induced antibodies against SARS-CoV-2 variants. Science 2021, 373, 1372–1377. [Google Scholar] [CrossRef] [PubMed]
- Lopez Bernal, J.; Andrews, N.; Gower, C.; Gallagher, E.; Simmons, R.; Thelwall, S.; Stowe, J.; Tessier, E.; Groves, N.; Dabrera, G.; et al. Effectiveness of COVID-19 Vaccines against the B.1.617.2 (Delta) Variant. N. Engl. J. Med. 2021, 385, 585–594. [Google Scholar] [CrossRef]
- Planas, D.; Veyer, D.; Baidaliuk, A.; Staropoli, I.; Guivel-Benhassine, F.; Rajah, M.M.; Planchais, C.; Porrot, F.; Robillard, N.; Puech, J.; et al. Reduced sensitivity of SARS-CoV-2 variant Delta to antibody neutralization. Nature 2021, 596, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Amanat, F.; Thapa, M.; Lei, T.; Ahmed, S.M.S.; Adelsberg, D.C.; Carreño, J.M.; Strohmeier, S.; Schmitz, A.J.; Zafar, S.; Zhou, J.Q.; et al. SARS-CoV-2 mRNA vaccination induces functionally diverse antibodies to NTD, RBD, and S2. Cell 2021, 184, 3936–3948.e3910. [Google Scholar] [CrossRef]
- Liu, J.; Liu, Y.; Xia, H.; Zou, J.; Weaver, S.C.; Swanson, K.A.; Cai, H.; Cutler, M.; Cooper, D.; Muik, A.; et al. BNT162b2-elicited neutralization of B.1.617 and other SARS-CoV-2 variants. Nature 2021, 596, 273–275. [Google Scholar] [CrossRef]
- Wu, K.; Werner, A.P.; Koch, M.; Choi, A.; Narayanan, E.; Stewart-Jones, G.B.E.; Colpitts, T.; Bennett, H.; Boyoglu-Barnum, S.; Shi, W.; et al. Serum Neutralizing Activity Elicited by mRNA-1273 Vaccine. N. Engl. J. Med. 2021, 384, 1468–1470. [Google Scholar] [CrossRef]
- Krause, P.R.; Fleming, T.R.; Longini, I.M.; Peto, R.; Briand, S.; Heymann, D.L.; Beral, V.; Snape, M.D.; Rees, H.; Ropero, A.-M.; et al. SARS-CoV-2 Variants and Vaccines. N. Engl. J. Med. 2021, 385, 179–186. [Google Scholar] [CrossRef]
- Wall, E.C.; Wu, M.; Harvey, R.; Kelly, G.; Warchal, S.; Sawyer, C.; Daniels, R.; Adams, L.; Hobson, P.; Hatipoglu, E.; et al. AZD1222-induced neutralising antibody activity against SARS-CoV-2 Delta VOC. Lancet 2021, 398, 207–209. [Google Scholar] [CrossRef]
- Wall, E.C.; Wu, M.; Harvey, R.; Kelly, G.; Warchal, S.; Sawyer, C.; Daniels, R.; Hobson, P.; Hatipoglu, E.; Ngai, Y.; et al. Neutralising antibody activity against SARS-CoV-2 VOCs B.1.617.2 and B.1.351 by BNT162b2 vaccination. Lancet 2021, 397, 2331–2333. [Google Scholar] [CrossRef]
- Seow, J.; Graham, C.; Merrick, B.; Acors, S.; Pickering, S.; Steel, K.J.A.; Hemmings, O.; O’Byrne, A.; Kouphou, N.; Galao, R.P.; et al. Longitudinal observation and decline of neutralizing antibody responses in the three months following SARS-CoV-2 infection in humans. Nat. Microbiol. 2020, 5, 1598–1607. [Google Scholar] [CrossRef]
- Sterlin, D.; Mathian, A.; Miyara, M.; Mohr, A.; Anna, F.; Claër, L.; Quentric, P.; Fadlallah, J.; Devilliers, H.; Ghillani, P.; et al. IgA dominates the early neutralizing antibody response to SARS-CoV-2. Sci. Transl. Med. 2021, 13, 577. [Google Scholar] [CrossRef]
- Starr, T.N.; Czudnochowski, N.; Liu, Z.; Zatta, F.; Park, Y.-J.; Addetia, A.; Pinto, D.; Beltramello, M.; Hernandez, P.; Greaney, A.J.; et al. SARS-CoV-2 RBD antibodies that maximize breadth and resistance to escape. Nature 2021, 597, 97–102. [Google Scholar] [CrossRef]
- Marot, S.; Malet, I.; Leducq, V.; Zafilaza, K.; Sterlin, D.; Planas, D.; Gothland, A.; Jary, A.; Dorgham, K.; Bruel, T.; et al. Rapid decline of neutralizing antibodies against SARS-CoV-2 among infected healthcare workers. Nat. Commun. 2021, 12, 844. [Google Scholar] [CrossRef]
- Wajnberg, A.; Amanat, F.; Firpo, A.; Altman, D.R.; Bailey, M.J.; Mansour, M.; McMahon, M.; Meade, P.; Mendu, D.R.; Muellers, K.; et al. Robust neutralizing antibodies to SARS-CoV-2 infection persist for months. Science 2020, 370, 1227–1230. [Google Scholar] [CrossRef]
- Asarnow, D.; Wang, B.; Lee, W.-H.; Hu, Y.; Huang, C.-W.; Faust, B.; Ng, P.M.L.; Ngoh, E.Z.X.; Bohn, M.; Bulkley, D.; et al. Structural insight into SARS-CoV-2 neutralizing antibodies and modulation of syncytia. Cell 2021, 184, 3192–3204.e16. [Google Scholar] [CrossRef]
- Hoepel, W.; Chen, H.-J.; Geyer, C.E.; Allahverdiyeva, S.; Manz, X.D.; de Taeye, S.W.; Aman, J.; Mes, L.; Steenhuis, M.; Griffith, G.R.; et al. High titers and low fucosylation of early human anti–SARS-CoV-2 IgG promote inflammation by alveolar macrophages. Sci. Transl. Med. 2021, 13. [Google Scholar] [CrossRef]
- Dugan, H.L.; Stamper, C.T.; Li, L.; Changrob, S.; Asby, N.W.; Halfmann, P.J.; Zheng, N.-Y.; Huang, M.; Shaw, D.G.; Cobb, M.S.; et al. Profiling B cell immunodominance after SARS-CoV-2 infection reveals antibody evolution to non-neutralizing viral targets. Immunity 2021, 54, 1290–1303.e1297. [Google Scholar] [CrossRef]
- Krishnamurty, A.T.; Thouvenel, C.D.; Portugal, S.; Keitany, G.J.; Kim, K.S.; Holder, A.; Crompton, P.D.; Rawlings, D.J.; Pepper, M. Somatically Hypermutated Plasmodium-Specific IgM+ Memory B Cells Are Rapid, Plastic, Early Responders upon Malaria Rechallenge. Immunity 2016, 45, 402–414. [Google Scholar] [CrossRef] [Green Version]
- Goel, R.R.; Apostolidis, S.A.; Painter, M.M.; Mathew, D.; Pattekar, A.; Kuthuru, O.; Gouma, S.; Hicks, P.; Meng, W.; Rosenfeld, A.M.; et al. Distinct antibody and memory B cell responses in SARS-CoV-2 naïve and recovered individuals after mRNA vaccination. Sci. Immunol. 2021, 6, eabi6950. [Google Scholar] [CrossRef]
- Kaneko, N.; Kuo, H.-H.; Boucau, J.; Farmer, J.R.; Allard-Chamard, H.; Mahajan, V.S.; Piechocka-Trocha, A.; Lefteri, K.; Osborn, M.; Bals, J.; et al. Loss of Bcl-6-Expressing T Follicular Helper Cells and Germinal Centers in COVID-19. Cell 2020, 183, 143–157.e13. [Google Scholar] [CrossRef]
- Woodruff, M.C.; Ramonell, R.P.; Nguyen, D.C.; Cashman, K.S.; Saini, A.S.; Haddad, N.S.; Ley, A.M.; Kyu, S.; Howell, J.C.; Ozturk, T.; et al. Extrafollicular B cell responses correlate with neutralizing antibodies and morbidity in COVID-19. Nat. Immunol. 2020, 21, 1506–1516. [Google Scholar] [CrossRef]
- Portugal, S.; Obeng-Adjei, N.; Moir, S.; Crompton, P.D.; Pierce, S.K. Atypical memory B cells in human chronic infectious diseases: An interim report. Cell. Immunol. 2017, 321, 18–25. [Google Scholar] [CrossRef]
- Jenks, S.A.; Cashman, K.S.; Woodruff, M.C.; Lee, F.E.; Sanz, I. Extrafollicular responses in humans and SLE. Immunol. Rev. 2019, 288, 136–148. [Google Scholar] [CrossRef]
- Zumaquero, E.; Stone, S.L.; Scharer, C.D.; Jenks, S.A.; Nellore, A.; Mousseau, B.; Rosal-Vela, A.; Botta, D.; Bradley, J.E.; Wojciechowski, W.; et al. IFNγ induces epigenetic programming of human T-bethi B cells and promotes TLR7/8 and IL-21 induced differentiation. eLife 2019, 8, e41641. [Google Scholar] [CrossRef]
- Martines, R.B.; Ng, D.L.; Greer, P.W.; Rollin, P.E.; Zaki, S.R. Tissue and cellular tropism, pathology and pathogenesis of Ebola and Marburg viruses. J. Pathol. 2014, 235, 153–174. [Google Scholar] [CrossRef]
- Rippey, J.J.; Schepers, N.J.; Gear, J.H. The pathology of Marburg virus disease. S. Afr. Med. J. 1984, 66, 50–54. [Google Scholar]
- Gao, R.; Dong, L.; Dong, J.; Wen, L.; Zhang, Y.; Yu, H.; Feng, Z.; Chen, M.; Tan, Y.; Mo, Z.; et al. A Systematic Molecular Pathology Study of a Laboratory Confirmed H5N1 Human Case. PLoS ONE 2010, 5, e13315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, M.; Xie, Z.-G.; Gao, Z.-C.; Wang, C.; Li, N.; Li, M.; Shao, H.-Q.; Wang, Y.-P.; Gao, Z.-F. Histopathologic study of avian influenza H5N1 infection in humans. Zhonghua Bing Li Xue Za Zhi Chin. J. Pathol. 2008, 37, 145–149. [Google Scholar]
- Buja, L.M.; Wolf, D.A.; Zhao, B.; Akkanti, B.; McDonald, M.; Lelenwa, L.; Reilly, N.; Ottaviani, G.; Elghetany, M.T.; Trujillo, D.O.; et al. The emerging spectrum of cardiopulmonary pathology of the coronavirus disease 2019 (COVID-19): Report of 3 autopsies from Houston, Texas, and review of autopsy findings from other United States cities. Cardiovasc. Pathol. 2020, 48, 107233. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Chang, X.N.; Pan, H.X.; Su, H.; Huang, B.; Yang, M.; Luo, D.J.; Weng, M.X.; Ma, L.; Nie, X. Pathological changes of the spleen in ten patients with coronavirus disease 2019(COVID-19) by postmortem needle autopsy. Zhonghua Bing Li Xue Za Zhi 2020, 49, 576–582. [Google Scholar] [CrossRef]
- Gu, J.; Gong, E.; Zhang, B.; Zheng, J.; Gao, Z.; Zhong, Y.; Zou, W.; Zhan, J.; Wang, S.; Xie, Z.; et al. Multiple organ infection and the pathogenesis of SARS. J. Exp. Med. 2005, 202, 415–424. [Google Scholar] [CrossRef]
- Ryg-Cornejo, V.; Ioannidis, L.J.; Ly, A.; Chiu, Y.H.C.; Tellier, J.; Hill, D.; Preston, S.; Pellegrini, M.; Yu, D.; Nutt, S.; et al. Severe Malaria Infections Impair Germinal Center Responses by Inhibiting T Follicular Helper Cell Differentiation. Cell Rep. 2015, 14, 68–81. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://www.fda.gov/news-events/press-announcements/coronavirus-COVID-19-update-fda-expands-eligibility-pfizer-biontech-COVID-19-booster-dose-16-and-17 (accessed on 9 December 2021).
- Shields, A.M.; Burns, S.O.; Savic, S.; Richter, A.G.; Anantharachagan, A.; Arumugakani, G.; Baker, K.; Bahal, S.; Bermingham, W.; Bhole, M.; et al. COVID-19 in patients with primary and secondary immunodeficiency: The United Kingdom experience. J. Allergy Clin. Immunol. 2021, 147, 870–875.e1. [Google Scholar] [CrossRef]
- Delavari, S.; Abolhassani, H.; Abolnezhadian, F.; Babaha, F.; Iranparast, S.; Ahanchian, H.; Moazzen, N.; Nabavi, M.; Arshi, S.; Fallahpour, M.; et al. Impact of SARS-CoV-2 Pandemic on Patients with Primary Immunodeficiency. J. Clin. Immunol. 2020, 41, 345–355. [Google Scholar] [CrossRef]
- Ameratunga, R.; Longhurst, H.; Steele, R.; Lehnert, K.; Leung, E.; Brooks, A.E.S.; Woon, S.-T. Common Variable Immunodeficiency Disorders, T-Cell Responses to SARS-CoV-2 Vaccines, and the Risk of Chronic COVID-19. J. Allergy Clin. Immunol. Pract. 2021, 9, 3575–3583. [Google Scholar] [CrossRef]
- Hagin, D.; Freund, T.; Navon, M.; Halperin, T.; Adir, D.; Marom, R.; Levi, I.; Benor, S.; Alcalay, Y.; Freund, N.T. Immunogenicity of Pfizer-BioNTech COVID-19 vaccine in patients with inborn errors of immunity. J. Allergy Clin. Immunol. 2021, 148, 739–749. [Google Scholar] [CrossRef]
- Danziger-Isakov, L.; Blumberg, E.A.; Manuel, O.; Sester, M. Impact of COVID-19 in solid organ transplant recipients. Am J Transpl. 2021, 21, 925–937. [Google Scholar] [CrossRef]
- Azzi, Y.; Bartash, R.; Scalea, J.; Loarte-Campos, P.; Akalin, E. COVID-19 and Solid Organ Transplantation: A Review Article. Transplantation 2020, 105, 37–55. [Google Scholar] [CrossRef]
- Nair, V.; Jandovitz, N.; Hirsch, J.S.; Nair, G.; Abate, M.; Bhaskaran, M.; Grodstein, E.; Berlinrut, I.; Hirschwerk, D.; Cohen, S.L.; et al. COVID-19 in kidney transplant recipients. Am. J. Transplant. 2020, 20, 1819–1825. [Google Scholar] [CrossRef]
- Caillard, S.; Chavarot, N.; Francois, H.; Matignon, M.; Greze, C.; Kamar, N.; Gatault, P.; Thaunat, O.; Legris, T.; Frimat, L.; et al. Is COVID-19 infection more severe in kidney transplant recipients? Am. J. Transplant. 2021, 21, 1295–1303. [Google Scholar] [CrossRef] [PubMed]
- Rincon-Arevalo, H.; Choi, M.; Stefanski, A.-L.; Halleck, F.; Weber, U.; Szelinski, F.; Jahrsdörfer, B.; Schrezenmeier, H.; Ludwig, C.; Sattler, A.; et al. Impaired humoral immunity to SARS-CoV-2 BNT162b2 vaccine in kidney transplant recipients and dialysis patients. Sci. Immunol. 2021, 6, eabj1031. [Google Scholar] [CrossRef] [PubMed]
- Fung, M.; Chiu, C.Y.; DeVoe, C.; Doernberg, S.B.; Schwartz, B.S.; Langelier, C.; Henrich, T.J.; Yokoe, D.; Davis, J.; Hays, S.R.; et al. Clinical outcomes and serologic response in solid organ transplant recipients with COVID-19: A case series from the United States. Am. J. Transplant. 2020, 20, 3225–3233. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.X.; Cardona, O.Q.; Ho, D.Y.; Busque, S.; Lenihan, C.R. Influence of immunosuppression on seroconversion against SARS-CoV-2 in two kidney transplant recipients. Transpl. Infect. Dis. 2020, 23, e13423. [Google Scholar] [CrossRef]
- Babel, N.; Anft, M.; Blazquez-Navarro, A.; Doevelaar, A.A.N.; Seibert, F.S.; Bauer, F.; Rohn, B.J.; Hoelzer, B.; Thieme, C.J.; Roch, T.; et al. Immune monitoring facilitates the clinical decision in multifocal COVID-19 of a pancreas-kidney transplant patient. Am. J. Transplant. 2020, 20, 3210–3215. [Google Scholar] [CrossRef]
- Candon, S.; Guerrot, D.; Drouot, L.; Lemoine, M.; Lebourg, L.; Hanoy, M.; Boyer, O.; Bertrand, D. T cell and antibody responses to SARS-CoV-2: Experience from a French transplantation and hemodialysis center during the COVID-19 pandemic. Am. J. Transplant. 2021, 21, 854–863. [Google Scholar] [CrossRef]
- Kamar, N.; Abravanel, F.; Marion, O.; Romieu-Mourez, R.; Couat, C.; Del Bello, A.; Izopet, J. Assessment of 4 Doses of SARS-CoV-2 Messenger RNA–Based Vaccine in Recipients of a Solid Organ Transplant. JAMA Netw. Open 2021, 4, e2136030. [Google Scholar] [CrossRef]
- Alejo, J.L.; Mitchell, J.; Chiang, T.P.-Y.; Abedon, A.T.; Boyarsky, B.J.; Avery, R.K.; Tobian, A.A.R.; Levan, M.L.; Massie, A.B.; Garonzik-Wang, J.M.; et al. Antibody Response to a Fourth Dose of a SARS-CoV-2 Vaccine in Solid Organ Transplant Recipients: A Case Series. Transplantation 2021, 105, e280–e281. [Google Scholar] [CrossRef]
- Taylor, P.C.; Adams, A.C.; Hufford, M.M.; de la Torre, I.; Winthrop, K.; Gottlieb, R.L. Neutralizing monoclonal antibodies for treatment of COVID-19. Nat. Rev. Immunol. 2021, 21, 382–393. [Google Scholar] [CrossRef]
- Korley, F.K.; Durkalski-Mauldin, V.; Yeatts, S.D.; Schulman, K.; Davenport, R.D.; Dumont, L.J.; El Kassar, N.; Foster, L.D.; Hah, J.M.; Jaiswal, S.; et al. Early Convalescent Plasma for High-Risk Outpatients with COVID-19. N. Engl. J. Med. 2021, 385, 1951–1960. [Google Scholar] [CrossRef]
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khanolkar, A. Elucidating T Cell and B Cell Responses to SARS-CoV-2 in Humans: Gaining Insights into Protective Immunity and Immunopathology. Cells 2022, 11, 67. https://doi.org/10.3390/cells11010067
Khanolkar A. Elucidating T Cell and B Cell Responses to SARS-CoV-2 in Humans: Gaining Insights into Protective Immunity and Immunopathology. Cells. 2022; 11(1):67. https://doi.org/10.3390/cells11010067
Chicago/Turabian StyleKhanolkar, Aaruni. 2022. "Elucidating T Cell and B Cell Responses to SARS-CoV-2 in Humans: Gaining Insights into Protective Immunity and Immunopathology" Cells 11, no. 1: 67. https://doi.org/10.3390/cells11010067
APA StyleKhanolkar, A. (2022). Elucidating T Cell and B Cell Responses to SARS-CoV-2 in Humans: Gaining Insights into Protective Immunity and Immunopathology. Cells, 11(1), 67. https://doi.org/10.3390/cells11010067