
Fluorescence Microscopy—An Outline of Hardware, Biological Handling, and Fluorophore Considerations

 ,
,  ,
,  ,
,  ,
,  , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Fluorescence Microscope Hardware Systems
2.1. Multiphoton and Other Advanced Microscopy Techniques
2.2. Super Resolution Microscopy (SRM); Going beyond the Limits of Light
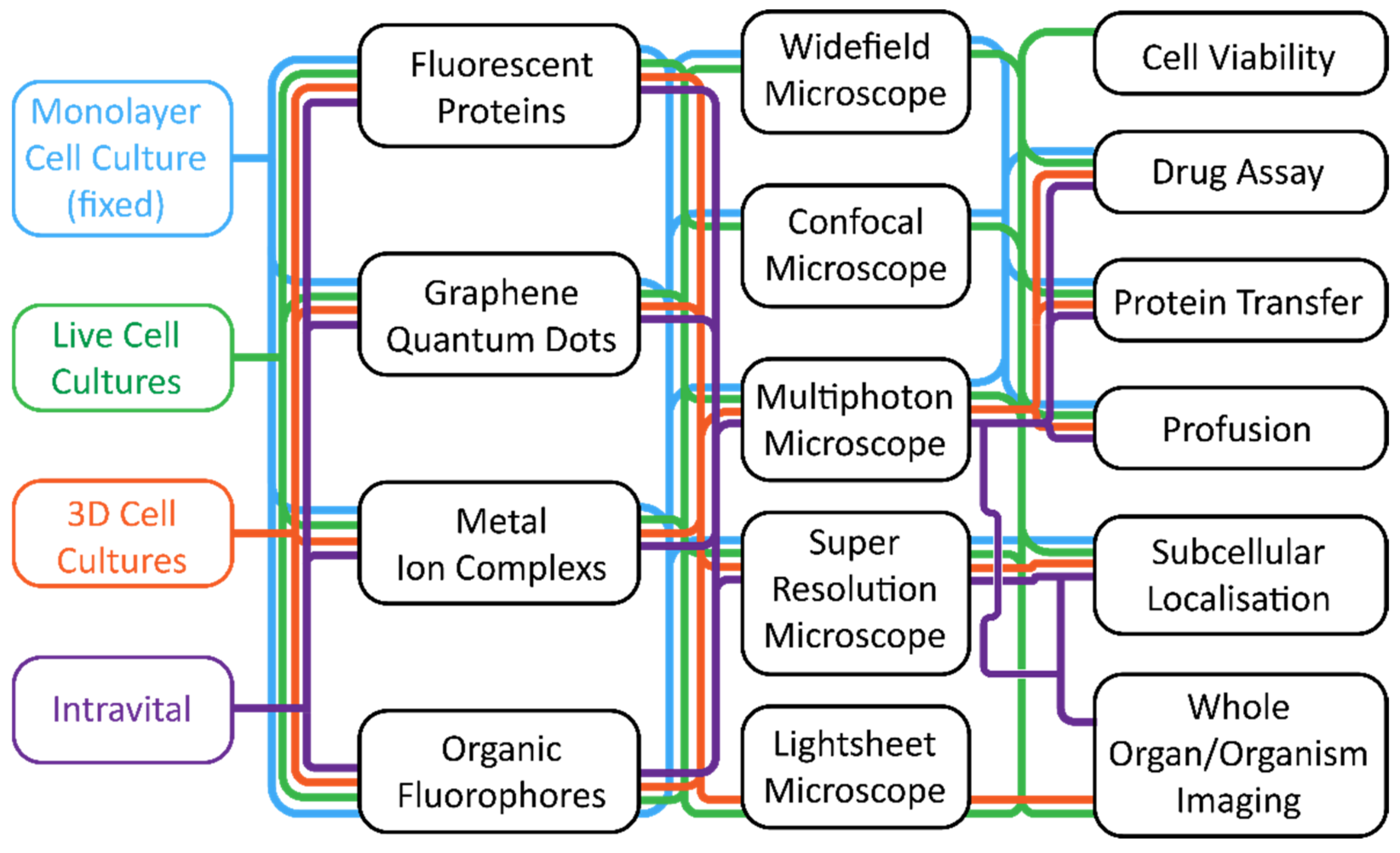
3. Biological Models for Fluorescence Imaging; from the Monolayer Culture to the Whole Organ
3.1. Monolayer Cell Culture
3.2. 3D Cell Cultures
3.3. Tissue Sections
3.4. Intravital Imaging
4. Imaging Agents Used for Fluorescence Microscopy
4.1. Fluorescent Proteins
4.2. Graphene Quantum Dots
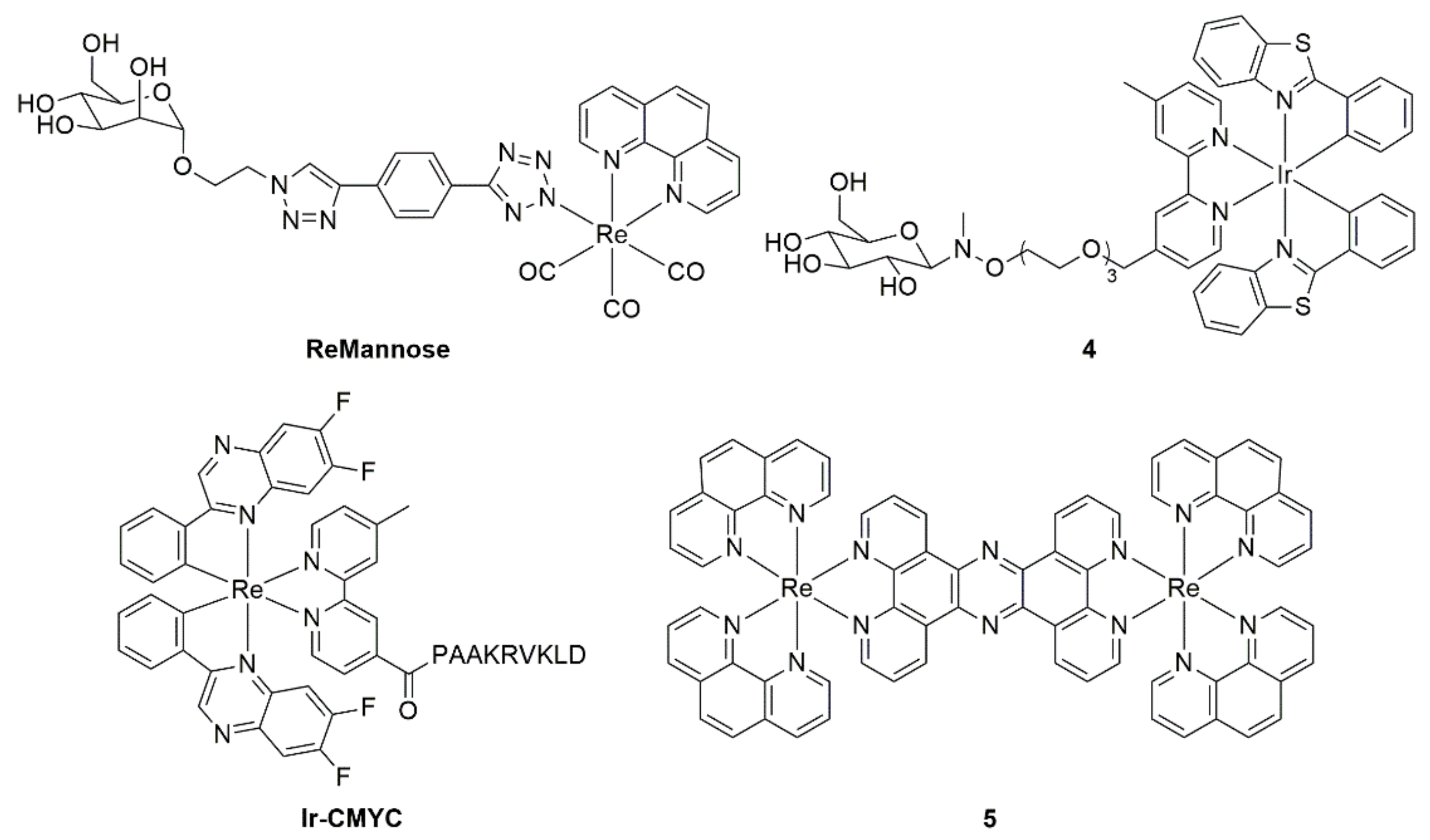
4.3. Metal Ion Complexes
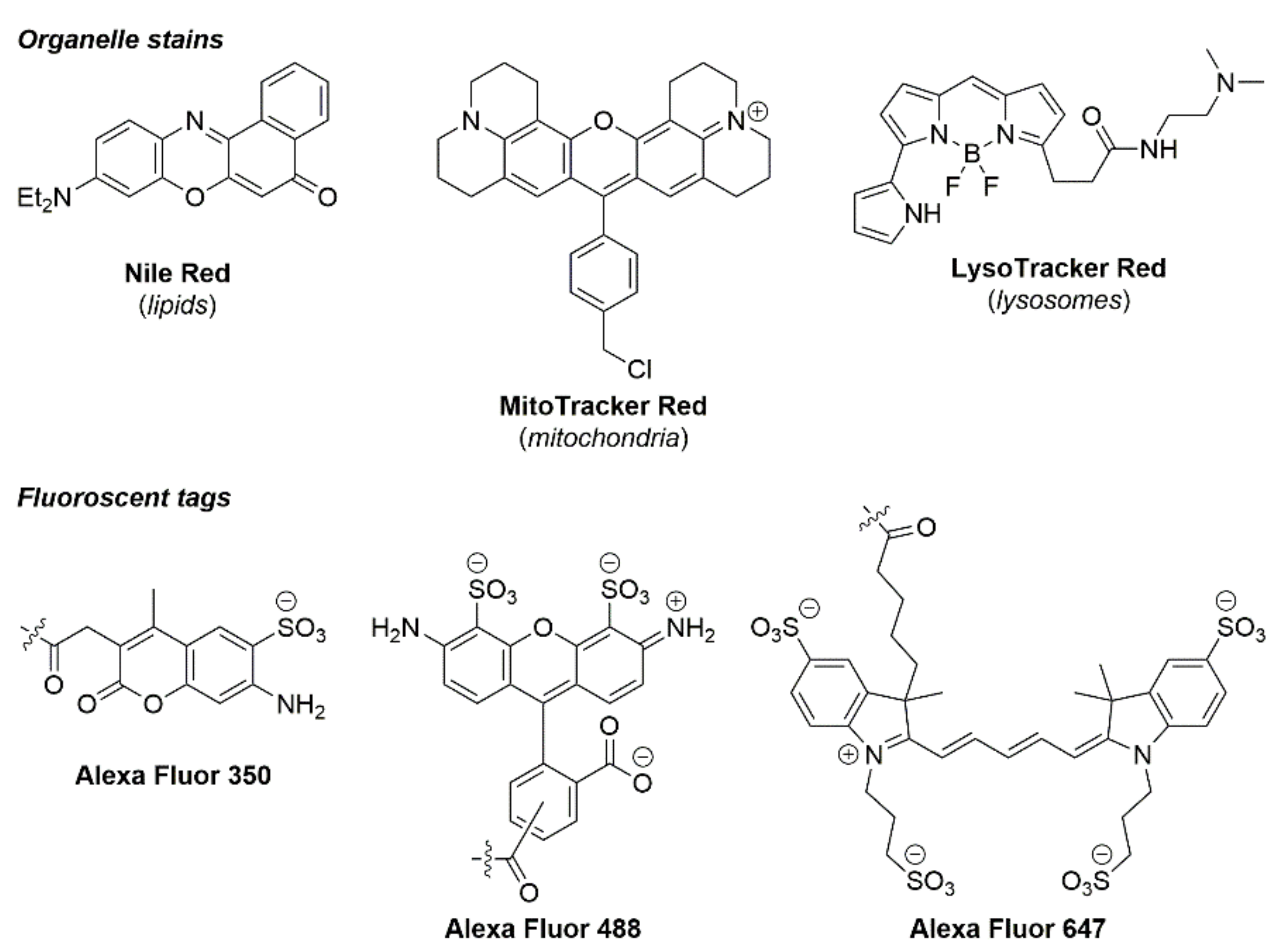
4.4. Organic Fluorophores
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Renz, M. Fluorescence microscopy—A historical and technical perspective. Cytom. Part A 2013, 83, 767–779. [Google Scholar] [CrossRef] [PubMed]
- Dunst, S.; Tomancak, P. Imaging flies by fluorescence microscopy: Principles, technologies, and applications. Genetics 2018, 211, 15–34. [Google Scholar] [CrossRef] [PubMed]
- Chalfie, M.; Tu, Y.; Euskirchen, G.; Ward, W.W.; Prasher, D.C. Green fluorescent protein as a marker for gene expression. Science 1994, 263, 802–805. [Google Scholar] [CrossRef] [PubMed]
- Prasher, D.C.; Eckenrode, V.K.; Ward, W.W.; Prendergast, F.G.; Cormier, M.J. Primary structure of the aequorea victoria green-fluorescent protein. Gene 1992, 111, 229–233. [Google Scholar] [CrossRef]
- Marvin, M. Microscopy Apparatus. U.S. Patent 3,013,467, 19 December 1961. [Google Scholar]
- Minsky, M. Memoir on inventing the confocal scanning microscope. Scanning 1988, 10, 128–138. [Google Scholar] [CrossRef]
- Pawley, J. Handbook of Biological Confocal Microscopy; Springer Science & Business Media: Heidelberg, Germany, 2006; Volume 236. [Google Scholar]
- Murphy, D.B.; Michael, D. Two-photon excitation fluorescence microscopy. In Fundamentals of Light Microscopy and Electronic Imaging; John Wiley & Sons: Hoboken, NJ, USA, 2012; pp. 307–329. [Google Scholar]
- Ishikawa-Ankerhold, H.C.; Ankerhold, R.; Drummen, G.P.C. Advanced fluorescence microscopy techniques—Frap, flip, flap, fret and flim. Molecules 2012, 17, 4047–4132. [Google Scholar] [CrossRef]
- De Los Santos, C.; Chang, C.-W.; Mycek, M.-A.; Cardullo, R.A. Frap, flim, and fret: Detection and analysis of cellular dynamics on a molecular scale using fluorescence microscopy. Mol. Reprod. Dev. 2015, 82, 587–604. [Google Scholar] [CrossRef]
- Blacker, T.S.; Mann, Z.F.; Gale, J.E.; Ziegler, M.; Bain, A.J.; Szabadkai, G.; Duchen, M.R. Separating nadh and nadph fluorescence in live cells and tissues using flim. Nat. Commun. 2014, 5, 3936. [Google Scholar] [CrossRef]
- Sekar, R.B.; Periasamy, A. Fluorescence resonance energy transfer (fret) microscopy imaging of live cell protein localizations. J. Cell Biol. 2003, 160, 629–633. [Google Scholar] [CrossRef]
- Margineanu, A.; Chan, J.J.; Kelly, D.J.; Warren, S.C.; Flatters, D.; Kumar, S.; Katan, M.; Dunsby, C.W.; French, P.M.W. Screening for protein-protein interactions using förster resonance energy transfer (fret) and fluorescence lifetime imaging microscopy (flim). Sci. Rep. 2016, 6, 28186. [Google Scholar] [CrossRef]
- Blumenthal, D.; Goldstien, L.; Edidin, M.; Gheber, L.A. Universal approach to frap analysis of arbitrary bleaching patterns. Sci. Rep. 2015, 5, 11655. [Google Scholar] [CrossRef]
- Ji, N.; Magee, J.C.; Betzig, E. High-speed, low-photodamage nonlinear imaging using passive pulse splitters. Nat. Methods 2008, 5, 197–202. [Google Scholar] [CrossRef]
- Sakaguchi, H.; Ozaki, Y.; Ashida, T.; Matsubara, T.; Oishi, N.; Kihara, S.; Takahashi, J. Self-organized synchronous calcium transients in a cultured human neural network derived from cerebral organoids. Stem Cell Rep. 2019, 13, 458–473. [Google Scholar] [CrossRef]
- Au-Strobl, F.; Au-Klees, S.; Au-Stelzer, E.H.K. Light sheet-based fluorescence microscopy of living or fixed and stained tribolium castaneum embryos. JoVE 2017, 122, e55629. [Google Scholar]
- Ovečka, M.; von Wangenheim, D.; Tomančák, P.; Šamajová, O.; Komis, G.; Šamaj, J. Multiscale imaging of plant development by light-sheet fluorescence microscopy. Nat. Plants 2018, 4, 639–650. [Google Scholar] [CrossRef]
- Chakraborty, T.; Driscoll, M.K.; Jeffery, E.; Murphy, M.M.; Roudot, P.; Chang, B.-J.; Vora, S.; Wong, W.M.; Nielson, C.D.; Zhang, H.; et al. Light-sheet microscopy of cleared tissues with isotropic, subcellular resolution. Nat. Methods 2019, 16, 1109–1113. [Google Scholar] [CrossRef]
- Ueda, H.R.; Dodt, H.-U.; Osten, P.; Economo, M.N.; Chandrashekar, J.; Keller, P.J. Whole-brain profiling of cells and circuits in mammals by tissue clearing and light-sheet microscopy. Neuron 2020, 106, 369–387. [Google Scholar] [CrossRef]
- Chen, B.-C.; Legant, W.R.; Wang, K.; Shao, L.; Milkie, D.E.; Davidson, M.W.; Janetopoulos, C.; Wu, X.S.; Hammer, J.A.; Liu, Z.; et al. Lattice light-sheet microscopy: Imaging molecules to embryos at high spatiotemporal resolution. Science 2014, 346, 1257998. [Google Scholar] [CrossRef]
- Abbe, E. Contributions to the theory of the microscope and that microscopic perception. Arch. Microsc. Anat. 1873, 9, 413–468. [Google Scholar] [CrossRef]
- Perez, V.; Chang, B.-J.; Stelzer, E.H.K. Optimal 2d-sim reconstruction by two filtering steps with richardson-lucy deconvolution. Sci. Rep. 2016, 6, 37149. [Google Scholar] [CrossRef]
- Vicidomini, G.; Bianchini, P.; Diaspro, A. Sted super-resolved microscopy. Nat. Methods 2018, 15, 173–182. [Google Scholar] [CrossRef]
- Huff, J. The airyscan detector from zeiss: Confocal imaging with improved signal-to-noise ratio and super-resolution. Nat. Methods 2015, 12. [Google Scholar] [CrossRef]
- Wu, X.; Hammer, J.A. Zeiss airyscan: Optimizing usage for fast, gentle, super-resolution imaging. In Confocal Microscopy: Methods and Protocols; Brzostowski, J., Sohn, H., Eds.; Springer US: New York, NY, USA, 2021; pp. 111–130. [Google Scholar]
- Gustafsson, M.G.L. Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. J. Microsc. 2000, 198, 82–87. [Google Scholar] [CrossRef]
- Minner-Meinen, R.; Weber, J.-N.; Albrecht, A.; Matis, R.; Behnecke, M.; Tietge, C.; Frank, S.; Schulze, J.; Buschmann, H.; Walla, P.J.; et al. Split-halotag imaging assay for sophisticated microscopy of protein–protein interactions in planta. Plant Commun. 2021, 2, 100212. [Google Scholar] [CrossRef]
- Hinsdale, T.A.; Stallinga, S.; Rieger, B. High-speed multicolor structured illumination microscopy using a hexagonal single mode fiber array. Biomed. Opt. Express 2021, 12, 1181–1194. [Google Scholar] [CrossRef]
- Wu, Y.; Christensen, R.; Colón-Ramos, D.; Shroff, H. Advanced optical imaging techniques for neurodevelopment. Curr. Opin. Neurobiol. 2013, 23, 1090–1097. [Google Scholar] [CrossRef]
- Lin, R.; Kipreos, E.T.; Zhu, J.; Khang, C.H.; Kner, P. Subcellular three-dimensional imaging deep through multicellular thick samples by structured illumination microscopy and adaptive optics. Nat. Commun. 2021, 12, 3148. [Google Scholar] [CrossRef]
- Betzig, G.; Patterson, R.S.; OW, L.S. Olenych, js bonifacino, mw davidson, j. Lippincott-Schwartz HF Hess Sci. 2006, 313, 1642. [Google Scholar]
- Verma, A.; Verma, M.; Singh, A. Animal tissue culture principles and applications. Anim. Biotechnol. 2020, 269–293. [Google Scholar] [CrossRef]
- Mirabelli, P.; Coppola, L.; Salvatore, M. Cancer cell lines are useful model systems for medical research. Cancers 2019, 11, 1098. [Google Scholar] [CrossRef]
- Monici, M. Cell and tissue autofluorescence research and diagnostic applications. In Biotechnology Annual Review; Elsevier: Amsterdam, The Netherlands, 2005; Volume 11, pp. 227–256. [Google Scholar]
- Wang, C.; Han, B.; Zhou, R.; Zhuang, X. Real-time imaging of translation on single mrna transcripts in live cells. Cell 2016, 165, 990–1001. [Google Scholar] [CrossRef] [PubMed]
- Bandaria, J.N.; Qin, P.; Berk, V.; Chu, S.; Yildiz, A. Shelterin protects chromosome ends by compacting telomeric chromatin. Cell 2016, 164, 735–746. [Google Scholar] [CrossRef] [PubMed]
- Carragher, N.O. Profiling distinct mechanisms of tumour invasion for drug discovery: Imaging adhesion, signalling and matrix turnover. Clin. Exp. Metastasis 2009, 26, 381–397. [Google Scholar] [CrossRef] [PubMed]
- Johnson, I.R.D.; Parkinson-Lawrence, E.J.; Shandala, T.; Weigert, R.; Butler, L.M.; Brooks, D.A. Altered endosome biogenesis in prostate cancer has biomarker potential. Mol. Cancer Res. 2014, 12, 1851. [Google Scholar] [CrossRef] [PubMed]
- Losavio, B.E.; Liang, Y.; Santamaría-Pang, A.; Kakadiaris, I.A.; Colbert, C.M.; Saggau, P. Live neuron morphology automatically reconstructed from multiphoton and confocal imaging data. J. Neurophysiol. 2008, 100, 2422–2429. [Google Scholar] [CrossRef] [PubMed]
- Bodeen, W.J.; Marada, S.; Truong, A.; Ogden, S.K. A fixation method to preserve cultured cell cytonemes facilitates mechanistic interrogation of morphogen transport. Development (Camb. Engl.) 2017, 144, 3612–3624. [Google Scholar] [CrossRef] [PubMed]
- Bhat, S.; Ljubojevic, N.; Zhu, S.; Fukuda, M.; Echard, A.; Zurzolo, C. Rab35 and its effectors promote formation of tunneling nanotubes in neuronal cells. Sci. Rep. 2020, 10, 16803. [Google Scholar] [CrossRef]
- Zani, B.G.; Indolfi, L.; Edelman, E.R. Tubular bridges for bronchial epithelial cell migration and communication. PLoS ONE 2010, 5, e8930. [Google Scholar] [CrossRef]
- Shabbir, A.; Cox, A.; Rodriguez-Menocal, L.; Salgado, M.; Van Badiavas, E. Mesenchymal stem cell exosomes induce proliferation and migration of normal and chronic wound fibroblasts, and enhance angiogenesis in vitro. Stem Cells Dev. 2015, 24, 1635–1647. [Google Scholar] [CrossRef]
- Sigal, Y.M.; Zhou, R.; Zhuang, X. Visualizing and discovering cellular structures with super-resolution microscopy. Science 2018, 361, 880–887. [Google Scholar] [CrossRef]
- Valm, A.M.; Cohen, S.; Legant, W.R.; Melunis, J.; Hershberg, U.; Wait, E.; Cohen, A.R.; Davidson, M.W.; Betzig, E.; Lippincott-Schwartz, J. Applying systems-level spectral imaging and analysis to reveal the organelle interactome. Nature 2017, 546, 162–167. [Google Scholar] [CrossRef]
- Jensen, E.C. Overview of live-cell imaging: Requirements and methods used. Anat. Rec. 2013, 296, 1–8. [Google Scholar] [CrossRef]
- Ettinger, A.; Wittmann, T. Chapter 5-fluorescence live cell imaging. In Methods in Cell Biology; Waters, J.C., Wittman, T., Eds.; Academic Press: Cambridge, MA, USA, 2014; Volume 123, pp. 77–94. [Google Scholar]
- Bogdanov, A.M.; Kudryavtseva, E.I.; Lukyanov, K.A. Anti-fading media for live cell gfp imaging. PLoS ONE 2012, 7, e53004. [Google Scholar] [CrossRef]
- Sasmal, M.; Bhowmick, R.; Musha Islam, A.S.; Bhuiya, S.; Das, S.; Ali, M. Domain-specific association of a phenanthrene–pyrene-based synthetic fluorescent probe with bovine serum albumin: Spectroscopic and molecular docking analysis. ACS Omega 2018, 3, 6293–6304. [Google Scholar] [CrossRef]
- Lazniewska, J.; Agostino, M.; Hickey, S.M.; Parkinson-Lawrence, E.; Stagni, S.; Massi, M.; Brooks, D.A.; Plush, S.E. Spectroscopic and molecular docking study of the interaction between neutral re(i) tetrazolate complexes and bovine serum albumin. Chem. –A Eur. J. 2021, 27, 11406–11417. [Google Scholar] [CrossRef]
- Gopalakrishnan, J. The emergence of stem cell-based brain organoids: Trends and challenges. BioEssays 2019, 41, 1900011. [Google Scholar] [CrossRef]
- Rosenbluth, J.M.; Schackmann, R.C.J.; Gray, G.K.; Selfors, L.M.; Li, C.M.-C.; Boedicker, M.; Kuiken, H.J.; Richardson, A.; Brock, J.; Garber, J.; et al. Organoid cultures from normal and cancer-prone human breast tissues preserve complex epithelial lineages. Nat. Commun. 2020, 11, 1711. [Google Scholar] [CrossRef]
- Drost, J.; Karthaus, W.R.; Gao, D.; Driehuis, E.; Sawyers, C.L.; Chen, Y.; Clevers, H. Organoid culture systems for prostate epithelial and cancer tissue. Nat. Protoc. 2016, 11, 347–358. [Google Scholar] [CrossRef]
- Jensen, C.; Teng, Y. Is it time to start transitioning from 2d to 3d cell culture? Front. Mol. Biosci. 2020, 7, 33. [Google Scholar] [CrossRef]
- Kim, M.; Mun, H.; Sung, C.O.; Cho, E.J.; Jeon, H.-J.; Chun, S.-M.; Jung, D.J.; Shin, T.H.; Jeong, G.S.; Kim, D.K.; et al. Patient-derived lung cancer organoids as in vitro cancer models for therapeutic screening. Nat. Commun. 2019, 10, 3991. [Google Scholar] [CrossRef]
- Loong, H.H.F.; Wong, A.M.; Chan, D.T.M.; Cheung, M.S.H.; Chow, C.; Ding, X.; Chan, A.K.Y.; Johnston, P.A.; Lau, J.Y.W.; Poon, W.S.; et al. Patient-derived tumor organoid predicts drugs response in glioblastoma: A step forward in personalized cancer therapy? J. Clin. Neurosci. 2020, 78, 400–402. [Google Scholar] [CrossRef]
- Lee, S.Y.; Ju, M.K.; Jeon, H.M.; Jeong, E.K.; Lee, Y.J.; Kim, C.H.; Park, H.G.; Han, S.I.; Kang, H.S. Regulation of tumor progression by programmed necrosis. Oxidative Med. Cell. Longev. 2018, 2018, 3537471. [Google Scholar] [CrossRef]
- Pontes Soares, C.; Midlej, V.; de Oliveira, M.E.; Benchimol, M.; Costa, M.L.; Mermelstein, C. 2d and 3d-organized cardiac cells shows differences in cellular morphology, adhesion junctions, presence of myofibrils and protein expression. PLoS ONE 2012, 7, e38147. [Google Scholar]
- Badder, L.M.; Hollins, A.J.; Herpers, B.; Yan, K.; Ewan, K.B.; Thomas, M.; Shone, J.R.; Badder, D.A.; Naven, M.; Ashelford, K.E.; et al. 3d imaging of colorectal cancer organoids identifies responses to tankyrase inhibitors. PLoS ONE 2020, 15, e0235319. [Google Scholar] [CrossRef] [PubMed]
- Bowers, H.J.; Fannin, E.E.; Thomas, A.; Weis, J.A. Characterization of multicellular breast tumor spheroids using image data-driven biophysical mathematical modeling. Sci. Rep. 2020, 10, 11583. [Google Scholar] [CrossRef] [PubMed]
- Hof, L.; Moreth, T.; Koch, M.; Liebisch, T.; Kurtz, M.; Tarnick, J.; Lissek, S.M.; Verstegen, M.M.A.; van der Laan, L.J.W.; Huch, M.; et al. Long-term live imaging and multiscale analysis identify heterogeneity and core principles of epithelial organoid morphogenesis. BMC Biol. 2021, 19, 37. [Google Scholar] [CrossRef]
- Ritter, A.; Roth, S.; Kreis, N.N.; Friemel, A.; Hoock, S.C.; Steglich Souto, A.; Eichbaum, C.; Neuhoff, A.; Chen, Q.; Solbach, C.; et al. Primary cilia in trophoblastic cells: Potential involvement in preeclampsia. Hypertension (Dallas Tex.: 1979) 2020, 76, 1491–1505. [Google Scholar] [CrossRef]
- Al-Ghadban, S.; Pursell, I.A.; Diaz, Z.T.; Herbst, K.L.; Bunnell, B.A. 3d spheroids derived from human lipedema ascs demonstrated similar adipogenic differentiation potential and ecm remodeling to non-lipedema ascs in vitro. Int. J. Mol. Sci. 2020, 21, 8350. [Google Scholar] [CrossRef]
- Boyle, M.A.; Sequeira, D.J.; McNeill, E.P.; Criss, Z.K., 2nd; Shroyer, N.F.; Speer, A.L. In vivo transplantation of human intestinal organoids enhances select tight junction gene expression. J. Surg. Res. 2021, 259, 500–508. [Google Scholar] [CrossRef]
- Park, M.; Kwon, J.; Shin, H.J.; Moon, S.M.; Kim, S.B.; Shin, U.S.; Han, Y.H.; Kim, Y. Butyrate enhances the efficacy of radiotherapy via foxo3a in colorectal cancer patient-derived organoids. Int. J. Oncol. 2020, 57, 1307–1318. [Google Scholar] [CrossRef]
- Higa, K.; Higuchi, J.; Kimoto, R.; Miyashita, H.; Shimazaki, J.; Tsubota, K.; Shimmura, S. Human corneal limbal organoids maintaining limbal stem cell niche function. Stem Cell Res. 2020, 49, 102012. [Google Scholar] [CrossRef]
- Johnsen, S.; Widder, E.A. The physical basis of transparency in biological tissue: Ultrastructure and the minimization of light scattering. J. Theor. Biol. 1999, 199, 181–198. [Google Scholar] [CrossRef]
- Steelman, Z.A.; Eldridge, W.J.; Weintraub, J.B.; Wax, A. Is the nuclear refractive index lower than cytoplasm? Validation of phase measurements and implications for light scattering technologies. J. Biophotonics 2017, 10, 1714–1722. [Google Scholar] [CrossRef]
- Costa, E.C.; Silva, D.N.; Moreira, A.F.; Correia, I.J. Optical clearing methods: An overview of the techniques used for the imaging of 3d spheroids. Biotechnol. Bioeng. 2019, 116, 2742–2763. [Google Scholar] [CrossRef]
- Susaki, E.A.; Tainaka, K.; Perrin, D.; Kishino, F.; Tawara, T.; Watanabe, T.M.; Yokoyama, C.; Onoe, H.; Eguchi, M.; Yamaguchi, S.; et al. Whole-brain imaging with single-cell resolution using chemical cocktails and computational analysis. Cell 2014, 157, 726–739. [Google Scholar] [CrossRef]
- Ertürk, A.; Becker, K.; Jährling, N.; Mauch, C.P.; Hojer, C.D.; Egen, J.G.; Hellal, F.; Bradke, F.; Sheng, M.; Dodt, H.-U. Three-dimensional imaging of solvent-cleared organs using 3disco. Nat. Protoc. 2012, 7, 1983–1995. [Google Scholar] [CrossRef]
- Nürnberg, E.; Vitacolonna, M.; Klicks, J.; von Molitor, E.; Cesetti, T.; Keller, F.; Bruch, R.; Ertongur-Fauth, T.; Riedel, K.; Scholz, P.; et al. Routine optical clearing of 3d-cell cultures: Simplicity forward. Front. Mol. Biosci. 2020, 7, 20. [Google Scholar] [CrossRef]
- Van Ineveld, R.L.; Ariese, H.C.R.; Wehrens, E.J.; Dekkers, J.F.; Rios, A.C. Single-cell resolution three-dimensional imaging of intact organoids. J. Vis. Exp. 2020, 160, e60709. [Google Scholar] [CrossRef]
- Held, M.; Santeramo, I.; Wilm, B.; Murray, P.; Lévy, R. Ex vivo live cell tracking in kidney organoids using light sheet fluorescence microscopy. PLoS ONE 2018, 13, e0199918. [Google Scholar] [CrossRef]
- O’Donnell, N.; Dmitriev, R.I. Three-dimensional tissue models and available probes for multi-parametric live cell microscopy: A brief overview. Multi-Parametr. Live Cell Microsc. 3D Tissue Models 2017, 1035, 49–67. [Google Scholar]
- Theocharis, A.D.; Skandalis, S.S.; Gialeli, C.; Karamanos, N.K. Extracellular matrix structure. Adv. Drug Deliv. Rev. 2016, 97, 4–27. [Google Scholar] [CrossRef]
- Mahajan, U.M.; Langhoff, E.; Goni, E.; Costello, E.; Greenhalf, W.; Halloran, C.; Ormanns, S.; Kruger, S.; Boeck, S.; Ribback, S.; et al. Immune cell and stromal signature associated with progression-free survival of patients with resected pancreatic ductal adenocarcinoma. Gastroenterology 2018, 155, 1625–1639.e1622. [Google Scholar] [CrossRef]
- Wang, X.; Dong, B.; Zhang, K.; Ji, Z.; Cheng, C.; Zhao, H.; Sheng, Y.; Li, X.; Fan, L.; Xue, W.; et al. E-cadherin bridges cell polarity and spindle orientation to ensure prostate epithelial integrity and prevent carcinogenesis in vivo. PLOS Genet. 2018, 14, e1007609. [Google Scholar] [CrossRef]
- Bass, B.P.; Engel, K.B.; Greytak, S.R.; Moore, H.M. A review of preanalytical factors affecting molecular, protein, and morphological analysis of formalin-fixed, paraffin-embedded (ffpe) tissue: How well do you know your ffpe specimen? Arch. Pathol. Lab. Med. 2014, 138, 1520–1530. [Google Scholar] [CrossRef]
- Fujimoto, D.; Akiba, K.-Y.; Nakamura, N. Isolation and characterization of a fluorescent material in bovine achilles tendon collagen. Biochem. Biophys. Res. Commun. 1977, 76, 1124–1129. [Google Scholar] [CrossRef]
- Blomfield, J.; Farrar, J.F. The fluorescent properties of maturing arterial elastin. Cardiovasc. Res. 1969, 3, 161–170. [Google Scholar] [CrossRef]
- Davis, A.S.; Richter, A.; Becker, S.; Moyer, J.E.; Sandouk, A.; Skinner, J.; Taubenberger, J.K. Characterizing and diminishing autofluorescence in formalin-fixed paraffin-embedded human respiratory tissue. J. Histochem. Cytochem. 2014, 62, 405–423. [Google Scholar] [CrossRef]
- Carriel, V.; Campos, F.; Aneiros-Fernández, J.; Kiernan, J.A. Tissue fixation and processing for the histological identification of lipids. In Histochemistry of Single Molecules; Springer: Berlin/Heidelberg, Germany, 2017; pp. 197–206. [Google Scholar]
- Scalia, C.R.; Boi, G.; Bolognesi, M.M.; Riva, L.; Manzoni, M.; DeSmedt, L.; Bosisio, F.M.; Ronchi, S.; Leone, B.E.; Cattoretti, G. Antigen masking during fixation and embedding, dissected. J. Histochem. Cytochem. 2016, 65, 5–20. [Google Scholar] [CrossRef]
- Gao, X.H.; Li, J.; Gong, H.F.; Yu, G.Y.; Liu, P.; Hao, L.Q.; Liu, L.J.; Bai, C.G.; Zhang, W. Comparison of fresh frozen tissue with formalin-fixed paraffin-embedded tissue for mutation analysis using a multi-gene panel in patients with colorectal cancer. Front. Oncol. 2020, 10, 310. [Google Scholar] [CrossRef] [PubMed]
- Jensen, E. Technical review: In situ hybridization. Anat. Rec. 2014, 297, 1349–1353. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S. Artefacts in histopathology. J. Oral Maxillofac. Pathol. 2014, 18, S111–S116. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.-R.; Liu, C.; Pootrakul, L.; Tang, L.; Young, A.; Chen, R.; Cote, R.J.; Taylor, C.R. Evaluation of the value of frozen tissue section used as “gold standard” for immunohistochemistry. Am. J. Clin. Pathol. 2008, 129, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Gaffney, E.F.; Riegman, P.H.; Grizzle, W.E.; Watson, P.H. Factors that drive the increasing use of ffpe tissue in basic and translational cancer research. Biotech. Histochem. 2018, 93, 373–386. [Google Scholar] [CrossRef] [PubMed]
- Mulligan, S.J.; MacVicar, B.A. Calcium transients in astrocyte endfeet cause cerebrovascular constrictions. Nature 2004, 431, 195–199. [Google Scholar] [CrossRef]
- Takano, T.; Tian, G.-F.; Peng, W.; Lou, N.; Libionka, W.; Han, X.; Nedergaard, M. Astrocyte-mediated control of cerebral blood flow. Nat. Neurosci. 2006, 9, 260–267. [Google Scholar] [CrossRef]
- Chen, Z.; Ross, J.L.; Hambardzumyan, D. Intravital 2-photon imaging reveals distinct morphology and infiltrative properties of glioblastoma-associated macrophages. Proc. Natl. Acad. Sci. USA 2019, 116, 14254–14259. [Google Scholar] [CrossRef]
- Lau, D.; Garçon, F.; Chandra, A.; Lechermann, L.M.; Aloj, L.; Chilvers, E.R.; Corrie, P.G.; Okkenhaug, K.; Gallagher, F.A. Intravital imaging of adoptive t-cell morphology, mobility and trafficking following immune checkpoint inhibition in a mouse melanoma model. Front. Immunol. 2020, 11, 11514. [Google Scholar] [CrossRef]
- Sugano, Y.; Siegfried, H.; Merkel, E.; Drummond, I.A. Novel transgenic lines to analyze renal glutathione redox potential in vivo. Zebrafish 2020, 17, 153–155. [Google Scholar] [CrossRef]
- Brighton, H.E.; Angus, S.P.; Bo, T.; Roques, J.; Tagliatela, A.C.; Darr, D.; Karagoz, K.; Sciaky, N.; Gatza, M.; Sharpless, N.E.; et al. New mechanisms of resistance to mek inhibitors in melanoma revealed by intravital imaging. Cancer Res. 2018, 78, 542–557. [Google Scholar] [CrossRef]
- Huang, Q.; Cohen, M.A.; Alsina, F.C.; Devlin, G.; Garrett, A.; McKey, J.; Havlik, P.; Rakhilin, N.; Wang, E.; Xiang, K.; et al. Intravital imaging of mouse embryos. Science 2020, 368, 181–186. [Google Scholar] [CrossRef]
- Dondossola, E.; Alexander, S.; Holzapfel, B.M.; Filippini, S.; Starbuck, M.W.; Hoffman, R.M.; Navone, N.; De-Juan-Pardo, E.M.; Logothetis, C.J.; Hutmacher, D.W.; et al. Intravital microscopy of osteolytic progression and therapy response of cancer lesions in the bone. Sci. Transl. Med. 2018, 10, 452. [Google Scholar] [CrossRef]
- Alieva, M.; Leidgens, V.; Riemenschneider, M.J.; Klein, C.A.; Hau, P.; van Rheenen, J. Intravital imaging of glioma border morphology reveals distinctive cellular dynamics and contribution to tumor cell invasion. Sci. Rep. 2019, 9, 2054. [Google Scholar] [CrossRef]
- You, S.; Tu, H.; Chaney, E.J.; Sun, Y.; Zhao, Y.; Bower, A.J.; Liu, Y.-Z.; Marjanovic, M.; Sinha, S.; Pu, Y.; et al. Intravital imaging by simultaneous label-free autofluorescence-multiharmonic microscopy. Nat. Commun. 2018, 9, 2125. [Google Scholar] [CrossRef]
- Seynhaeve, A.L.B.; Oostinga, D.; van Haperen, R.; Eilken, H.M.; Adams, S.; Adams, R.H.; ten Hagen, T.L.M. Spatiotemporal endothelial cell–pericyte association in tumors as shown by high resolution 4d intravital imaging. Sci. Rep. 2018, 8, 9596. [Google Scholar] [CrossRef]
- Boulch, M.; Grandjean, C.L.; Cazaux, M.; Bousso, P. Tumor immunosurveillance and immunotherapies: A fresh look from intravital imaging. Trends Immunol. 2019, 40, 1022–1034. [Google Scholar] [CrossRef]
- Nezu, A.; Morita, T.; Nagai, T.; Tanimura, A. Simultaneous monitoring of ca(2+) responses and salivary secretion in live animals reveals a threshold intracellular ca(2+) concentration for salivation. Exp. Physiol. 2019, 104, 61–69. [Google Scholar] [CrossRef]
- Tvrdik, P.; Kearns, K.N.; Sharifi, K.A.; Sluzewski, M.F.; Acton, S.T.; Kalani, M.Y.S. Calcium imaging of microglial network activity in stroke. In Microglia: Methods and Protocols; Garaschuk, O., Verkhratsky, A., Eds.; Springer New York: New York, NY, USA, 2019; pp. 267–279. [Google Scholar]
- Archambault, L.S.; Trzilova, D.; Gonia, S.; Gale, C.; Wheeler, R.T.; Gacser, A.; Hube, B. Intravital imaging reveals divergent cytokine and cellular immune responses to candida albicans and candida parapsilosis. mBio 2019, 10, e00266-19. [Google Scholar] [CrossRef]
- Shandala, T.; Lim, C.; Sorvina, A.; Brooks, D.A. A drosophila model to image phagosome maturation. Cells 2013, 2, 188–201. [Google Scholar] [CrossRef]
- Koyama, L.A.J.; Aranda-Díaz, A.; Su, Y.H.; Balachandra, S.; Martin, J.L.; Ludington, W.B.; Huang, K.C.; O’Brien, L.E. Bellymount enables longitudinal, intravital imaging of abdominal organs and the gut microbiota in adult drosophila. PLoS Biol. 2020, 18, e3000567. [Google Scholar] [CrossRef]
- Sorvina, A.; Brooks, D.A.; Ng, Y.S.; Bader, C.A.; Weigert, R.; Shandala, T. Bacterial challenge initiates endosome-lysosome response in drosophila immune tissues. IntraVital 2013, 2, e23889. [Google Scholar] [CrossRef][Green Version]
- Asokan, N.; Daetwyler, S.; Bernas, S.N.; Schmied, C.; Vogler, S.; Lambert, K.; Wobus, M.; Wermke, M.; Kempermann, G.; Huisken, J.; et al. Long-term in vivo imaging reveals tumor-specific dissemination and captures host tumor interaction in zebrafish xenografts. Sci. Rep. 2020, 10, 13254. [Google Scholar] [CrossRef]
- Revskoy, S.; Blair, M.E.; Powell, S.M.; Hausman, E.S.; Blackburn, J.S. In vivo imaging defines vascular interplay in the development of lymphocytic leukemia in zebrafish models. bioRxiv 2019, 806562. [Google Scholar] [CrossRef]
- Sedin, J.; Giraud, A.; Steiner, S.E.; Ahl, D.; Persson, A.E.G.; Melican, K.; Richter-Dahlfors, A.; Phillipson, M. High resolution intravital imaging of the renal immune response to injury and infection in mice. Front. Immunol. 2019, 10, 2744. [Google Scholar] [CrossRef]
- Upadhaya, S.; Krichevsky, O.; Akhmetzyanova, I.; Sawai, C.M.; Fooksman, D.R.; Reizis, B. Intravital imaging reveals motility of adult hematopoietic stem cells in the bone marrow niche. Cell Stem Cell 2020, 27, 336–345.e334. [Google Scholar] [CrossRef]
- Lai, C.W.; Chen, H.L.; Yen, C.C.; Wang, J.L.; Yang, S.H.; Chen, C.M. Using dual fluorescence reporting genes to establish an in vivo imaging model of orthotopic lung adenocarcinoma in mice. Mol. Imaging Biol. 2016, 18, 849–859. [Google Scholar] [CrossRef]
- Ikeda, W.; Sasai, K.; Akagi, T. Imaging window device for subcutaneous implantation tumor. Methods Mol. Biol. (Clifton N. J.) 2018, 1763, 153–163. [Google Scholar]
- Ryu, C.M.; Yu, H.Y.; Lee, H.Y.; Shin, J.H.; Lee, S.; Ju, H.; Paulson, B.; Lee, S.; Kim, S.; Lim, J.; et al. Longitudinal intravital imaging of transplanted mesenchymal stem cells elucidates their functional integration and therapeutic potency in an animal model of interstitial cystitis/bladder pain syndrome. Theranostics 2018, 8, 5610–5624. [Google Scholar] [CrossRef]
- Fumagalli, A.; Bruens, L.; Scheele, C.; van Rheenen, J. Capturing stem cell behavior using intravital and live cell microscopy. Cold Spring Harb. Perspect. Biol. 2020, 12, a035949. [Google Scholar] [CrossRef] [PubMed]
- Balan, M.; Trusohamn, M.; Ning, F.C.; Jacob, S.; Pietras, K.; Eriksson, U.; Berggren, P.-O.; Nyqvist, D. Noninvasive intravital high-resolution imaging of pancreatic neuroendocrine tumours. Sci. Rep. 2019, 9, 14636. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.; Kong, E.; Lee, J.; Jung, J.; Kim, E.; Park, S.B.; Kim, P. Intravital longitudinal imaging of hepatic lipid droplet accumulation in a murine model for nonalcoholic fatty liver disease. Biomed. Opt. Express 2020, 11, 5132–5146. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, T.; Tamaki, S.; Fujita, S.; Suematsu, M.; Takubo, K. Identification and local manipulation of bone marrow vasculature during intravital imaging. Sci. Rep. 2020, 10, 6422. [Google Scholar] [CrossRef]
- Wu, J.; Lu, Z.; Jiang, D.; Guo, Y.; Qiao, H.; Zhang, Y.; Zhu, T.; Cai, Y.; Zhang, X.; Zhanghao, K.; et al. Iterative tomography with digital adaptive optics permits hour-long intravital observation of 3d subcellular dynamics at millisecond scale. Cell 2021, 184, 3318–3332.e3317. [Google Scholar] [CrossRef]
- Hato, T.; Winfree, S.; Day, R.; Sandoval, R.M.; Molitoris, B.A.; Yoder, M.C.; Wiggins, R.C.; Zheng, Y.; Dunn, K.W.; Dagher, P.C. Two-photon intravital fluorescence lifetime imaging of the kidney reveals cell-type specific metabolic signatures. J. Am. Soc. Nephrol. JASN 2017, 28, 2420–2430. [Google Scholar] [CrossRef]
- Rhodes, G.J. Surgical preparation of rats and mice for intravital microscopic imaging of abdominal organs. Methods (San Diego Calif.) 2017, 128, 129–138. [Google Scholar] [CrossRef]
- Rompolas, P.; Deschene, E.R.; Zito, G.; Gonzalez, D.G.; Saotome, I.; Haberman, A.M.; Greco, V. Live imaging of stem cell and progeny behaviour in physiological hair-follicle regeneration. Nature 2012, 487, 496–499. [Google Scholar] [CrossRef]
- Rompolas, P.; Mesa, K.R.; Greco, V. Spatial organization within a niche as a determinant of stem-cell fate. Nature 2013, 502, 513–518. [Google Scholar] [CrossRef]
- Masedunskas, A.; Sramkova, M.; Parente, L.; Weigert, R. Intravital microscopy to image membrane trafficking in live rats. In Cell Imaging Techniques: Methods and Protocols; Taatjes, D.J., Roth, J., Eds.; Humana Press: Totowa, NJ, USA, 2013; pp. 153–167. [Google Scholar]
- Shitara, A.; Bleck, C.K.E.; Weigert, R. Cdc42 controls secretory granules morphology in rodent salivary glands in vivo. Commun. Integr. Biol. 2020, 13, 22–26. [Google Scholar] [CrossRef]
- Deane, J.A.; Abeynaike, L.D.; Norman, M.U.; Wee, J.L.; Kitching, A.R.; Kubes, P.; Hickey, M.J. Endogenous regulatory t cells adhere in inflamed dermal vessels via icam-1: Association with regulation of effector leukocyte adhesion. J. Immunol. (Baltimore Md. 1950) 2012, 188, 2179–2188. [Google Scholar] [CrossRef]
- Liou, H.L.; Myers, J.T.; Barkauskas, D.S.; Huang, A.Y. Intravital imaging of the mouse popliteal lymph node. J. Vis. Exp. 2012, 60, e3720. [Google Scholar] [CrossRef]
- Jang, W.H.; Park, A.; Wang, T.; Kim, C.J.; Chang, H.; Yang, B.-G.; Kim, M.J.; Myung, S.-J.; Im, S.-H.; Jang, M.H.; et al. Two-photon microscopy of paneth cells in the small intestine of live mice. Sci. Rep. 2018, 8, 14174. [Google Scholar] [CrossRef]
- Jenne, C.N.; Wong, C.H.; Petri, B.; Kubes, P. The use of spinning-disk confocal microscopy for the intravital analysis of platelet dynamics in response to systemic and local inflammation. PLoS ONE 2011, 6, e25109. [Google Scholar] [CrossRef]
- Alieva, M.; Ritsma, L.; Giedt, R.J.; Weissleder, R.; van Rheenen, J. Imaging windows for long-term intravital imaging: General overview and technical insights. Intravital 2014, 3, e29917. [Google Scholar] [CrossRef]
- Prunier, C.; Chen, N.; Ritsma, L.; Vrisekoop, N. Procedures and applications of long-term intravital microscopy. Methods (San Diego Calif.) 2017, 128, 52–64. [Google Scholar] [CrossRef]
- Rouffiac, V.; Ser-Le Roux, K.; Salomé-Desnoulez, S.; Leguerney, I.; Ginefri, J.C.; Sébrié, C.; Jourdain, L.; Lécluse, Y.; Laplace-Builhé, C. Multimodal imaging for tumour characterization from micro- to macroscopic level using a newly developed dorsal chamber designed for long-term follow-up. J. Biophotonics 2020, 13, e201900217. [Google Scholar]
- Ritsma, L.; Steller, E.J.; Ellenbroek, S.I.; Kranenburg, O.; Borel Rinkes, I.H.; van Rheenen, J. Surgical implantation of an abdominal imaging window for intravital microscopy. Nat. Protoc. 2013, 8, 583–594. [Google Scholar] [CrossRef]
- Lefrançais, E.; Ortiz-Muñoz, G.; Caudrillier, A.; Mallavia, B.; Liu, F.; Sayah, D.M.; Thornton, E.E.; Headley, M.B.; David, T.; Coughlin, S.R.; et al. The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature 2017, 544, 105–109. [Google Scholar] [CrossRef]
- Thanabalasuriar, A.; Neupane, A.S.; Wang, J.; Krummel, M.F.; Kubes, P. Inkt cell emigration out of the lung vasculature requires neutrophils and monocyte-derived dendritic cells in inflammation. Cell Rep. 2016, 16, 3260–3272. [Google Scholar] [CrossRef]
- Seynhaeve, A.L.B.; Ten Hagen, T.L.M. Intravital microscopy of tumor-associated vasculature using advanced dorsal skinfold window chambers on transgenic fluorescent mice. J. Vis. Exp. 2018, 131, 55115. [Google Scholar] [CrossRef]
- Kilarski, W.W.; Güç, E.; Swartz, M.A. Dorsal ear skin window for intravital imaging and functional analysis of lymphangiogenesis. Methods Mol. Biol. (Clifton N. J.) 2018, 1846, 261–277. [Google Scholar]
- Haghayegh Jahromi, N.; Tardent, H.; Enzmann, G.; Deutsch, U.; Kawakami, N.; Bittner, S.; Vestweber, D.; Zipp, F.; Stein, J.V.; Engelhardt, B. A novel cervical spinal cord window preparation allows for two-photon imaging of t-cell interactions with the cervical spinal cord microvasculature during experimental autoimmune encephalomyelitis. Front. Immunol. 2017, 8, 406. [Google Scholar] [CrossRef] [PubMed]
- Askoxylakis, V.; Badeaux, M.; Roberge, S.; Batista, A.; Kirkpatrick, N.; Snuderl, M.; Amoozgar, Z.; Seano, G.; Ferraro, G.B.; Chatterjee, S.; et al. A cerebellar window for intravital imaging of normal and disease states in mice. Nat. Protoc. 2017, 12, 2251–2262. [Google Scholar] [CrossRef] [PubMed]
- Barretto, R.P.J.; Ko, T.H.; Jung, J.C.; Wang, T.J.; Capps, G.; Waters, A.C.; Ziv, Y.; Attardo, A.; Recht, L.; Schnitzer, M.J. Time-lapse imaging of disease progression in deep brain areas using fluorescence microendoscopy. Nat. Med. 2011, 17, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Vyas, K.; Yang, G.Z. Line scanning, fiber bundle fluorescence hilo endomicroscopy with confocal slit detection. J. Biomed. Opt. 2019, 24, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Paulson, B.; Kim, I.H.; Namgoong, J.M.; Kim, Y.G.; Lee, S.; Moon, Y.; Shin, D.M.; Choo, M.S.; Kim, J.K. Longitudinal micro-endoscopic monitoring of high-success intramucosal xenografts for mouse models of colorectal cancer. Int. J. Med. Sci. 2019, 16, 1453–1460. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.M.; Bae, D.-J.; Do, E.-J.; Oh, G.; Yoo, S.W.; Lee, G.-J.; Chae, J.S.; Yun, Y.; Kim, S.; Kim, K.H.; et al. Multi-spectral fluorescence imaging of colon dysplasia in vivo using a multi-spectral endoscopy system. Transl. Oncol. 2019, 12, 226–235. [Google Scholar] [CrossRef]
- Boerwinkel, D.F.; Holz, J.A.; Hawkins, D.M.; Curvers, W.L.; Aalders, M.C.; Weusten, B.L.; Visser, M.; Meijer, S.L.; Bergman, J.J. Fluorescence spectroscopy incorporated in an optical biopsy system for the detection of early neoplasia in barrett’s esophagus. Dis. Esophagus Off. J. Int. Soc. Dis. Esophagus 2015, 28, 345–351. [Google Scholar] [CrossRef]
- Jun, E.J.; Song, H.Y.; Park, J.H.; Bae, Y.S.; Paulson, B.; Lee, S.; Cho, Y.C.; Tsauo, J.; Kim, M.T.; Kim, K.Y.; et al. In vivo fluorescence microendoscopic monitoring of stent-induced fibroblast cell proliferation in an esophageal mouse model. J. Vasc. Interv. Radiol. JVIR 2018, 29, 1756–1763. [Google Scholar] [CrossRef]
- Trindade, J.R.M.; Figueiredo, V.R.; Pêgo-Fernandes, P.M.; Terra, R.M. Probe-based confocal laser endomicroscopy of the airways: Physiological and pathological characteristics of preneoplastic and neoplastic lesions. J. Bras. Pneumol. 2019, 45, e20180103. [Google Scholar] [CrossRef]
- Lee, S.; Vinegoni, C.; Sebas, M.; Weissleder, R. Automated motion artifact removal for intravital microscopy, without a priori information. Sci. Rep. 2014, 4, 4507. [Google Scholar] [CrossRef]
- Soulet, D.; Lamontagne-Proulx, J.; Aube, B.; Davalos, D. Multiphoton intravital microscopy in small animals: Motion artefact challenges and technical solutions. J. Microsc. 2020, 278, 3–17. [Google Scholar] [CrossRef]
- Chatalic, K.L.S.; Kwekkeboom, D.J.; de Jong, M. Radiopeptides for imaging and therapy: A radiant future. J. Nucl. Med. 2015, 56, 1809. [Google Scholar] [CrossRef]
- Wadas, T.J.; Wong, E.H.; Weisman, G.R.; Anderson, C.J. Coordinating radiometals of copper, gallium, indium, yttrium, and zirconium for pet and spect imaging of disease. Chem. Rev. 2010, 110, 2858–2902. [Google Scholar] [CrossRef]
- Thakor, A.S.; Jokerst, J.V.; Ghanouni, P.; Campbell, J.L.; Mittra, E.; Gambhir, S.S. Clinically approved nanoparticle imaging agents. J. Nucl. Med. 2016, 57, 1833. [Google Scholar] [CrossRef]
- Torchilin, V.P. Recent advances with liposomes as pharmaceutical carriers. Nat. Rev. Drug Discov. 2005, 4, 145–160. [Google Scholar] [CrossRef]
- Gindy, M.E.; Prud’homme, R.K. Multifunctional nanoparticles for imaging, delivery and targeting in cancer therapy. Expert Opin. Drug Deliv. 2009, 6, 865–878. [Google Scholar] [CrossRef]
- Forte, E.; Fiorenza, D.; Torino, E.; Costagliola di Polidoro, A.; Cavaliere, C.; Netti, P.A.; Salvatore, M.; Aiello, M. Radiolabeled pet/mri nanoparticles for tumor imaging. J. Clin. Med. 2020, 9, 89. [Google Scholar] [CrossRef]
- Bouziotis, P.; Stellas, D.; Thomas, E.; Truillet, C.; Tsoukalas, C.; Lux, F.; Tsotakos, T.; Xanthopoulos, S.; Paravatou-Petsotas, M.; Gaitanis, A. 68ga-radiolabeled aguix nanoparticles as dual-modality imaging agents for pet/mri-guided radiation therapy. Nanomedicine 2017, 12, 1561–1574. [Google Scholar] [CrossRef]
- Motiei, M.; Dreifuss, T.; Sadan, T.; Omer, N.; Blumenfeld-Katzir, T.; Fragogeorgi, E.; Loudos, G.; Popovtzer, R.; Ben-Eliezer, N. Trimodal nanoparticle contrast agent for ct, mri and spect imaging: Synthesis and characterization of radiolabeled core/shell iron oxide@ gold nanoparticles. Chem. Lett. 2019, 48, 291–294. [Google Scholar] [CrossRef]
- Imlimthan, S.; Otaru, S.; Keinaänen, O.; Correia, A.; Lintinen, K.; Santos, H.A.; Airaksinen, A.J.; Kostiainen, M.A.; Sarparanta, M. Radiolabeled molecular imaging probes for the in vivo evaluation of cellulose nanocrystals for biomedical applications. Biomacromolecules 2018, 20, 674–683. [Google Scholar] [CrossRef]
- Köker, T.; Tang, N.; Tian, C.; Zhang, W.; Wang, X.; Martel, R.; Pinaud, F. Cellular imaging by targeted assembly of hot-spot sers and photoacoustic nanoprobes using split-fluorescent protein scaffolds. Nat. Commun. 2018, 9, 607. [Google Scholar] [CrossRef]
- Qian, X.M.; Nie, S.M. Single-molecule and single-nanoparticle sers: From fundamental mechanisms to biomedical applications. Chem. Soc. Rev. 2008, 37, 912–920. [Google Scholar] [CrossRef]
- Chow, K.; Tyrrell, D.; Yang, M.; Abraham, L.; Anderson, G.; Mansfield, C. Scintigraphic assessment of deposition of radiolabeled fluticasone delivered from a nebulizer and metered dose inhaler in 10 healthy dogs. J. Vet. Intern. Med. 2017, 31, 1849–1857. [Google Scholar] [CrossRef]
- Shimomura, O.; Johnson, F.H.; Saiga, Y. Extraction, purification and properties of aequorin, a bioluminescent protein from the luminous hydromedusan, aequorea. J. Cell. Comp. Physiol. 1962, 59, 223–239. [Google Scholar] [CrossRef]
- Kremers, G.J.; Gilbert, S.G.; Cranfill, P.J.; Davidson, M.W.; Piston, D.W. Fluorescent proteins at a glance. J. Cell Sci. 2011, 124, 157–160. [Google Scholar] [CrossRef]
- Chudakov, D.M.; Matz, M.V.; Lukyanov, S.; Lukyanov, K.A. Fluorescent proteins and their applications in imaging living cells and tissues. Physiol. Rev. 2010, 90, 1103–1163. [Google Scholar] [CrossRef]
- Miyawaki, A.; Niino, Y. Molecular spies for bioimaging—Fluorescent protein-based probes. Mol. Cell 2015, 58, 632–643. [Google Scholar] [CrossRef]
- Patterson, G.H.; Lippincott-Schwartz, J. A photoactivatable gfp for selective photolabeling of proteins and cells. Science 2002, 297, 1873. [Google Scholar] [CrossRef]
- Ganini, D.; Leinisch, F.; Kumar, A.; Jiang, J.; Tokar, E.J.; Malone, C.C.; Petrovich, R.M.; Mason, R.P. Fluorescent proteins such as egfp lead to catalytic oxidative stress in cells. Redox Biol. 2017, 12, 462–468. [Google Scholar] [CrossRef]
- Stadler, C.; Rexhepaj, E.; Singan, V.R.; Murphy, R.F.; Pepperkok, R.; Uhlén, M.; Simpson, J.C.; Lundberg, E. Immunofluorescence and fluorescent-protein tagging show high correlation for protein localization in mammalian cells. Nat. Methods 2013, 10, 315–323. [Google Scholar] [CrossRef]
- Cranfill, P.J.; Sell, B.R.; Baird, M.A.; Allen, J.R.; Lavagnino, Z.; de Gruiter, H.M.; Kremers, G.J.; Davidson, M.W.; Ustione, A.; Piston, D.W. Quantitative assessment of fluorescent proteins. Nat. Methods 2016, 13, 557–562. [Google Scholar] [CrossRef]
- Zacharias, D.A.; Violin, J.D.; Newton, A.C.; Tsien, R.Y. Partitioning of lipid-modified monomeric gfps into membrane microdomains of live cells. Science 2002, 296, 913–916. [Google Scholar] [CrossRef] [PubMed]
- Snapp, E.L. Fluorescent proteins: A cell biologist’s user guide. Trends Cell Biol. 2009, 19, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Jaffrey, S.R. Imaging mrna trafficking in living cells using fluorogenic proteins. Curr. Opin. Chem. Biol. 2020, 57, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Shaner, N.C.; Steinbach, P.A.; Tsien, R.Y. A guide to choosing fluorescent proteins. Nat. Methods 2005, 2, 905–909. [Google Scholar] [CrossRef]
- Megason, S.G.; Fraser, S.E. Imaging in systems biology. Cell 2007, 130, 784–795. [Google Scholar] [CrossRef]
- Oliva Trejo, J.A.; Tanida, I.; Suzuki, C.; Kakuta, S.; Tada, N.; Uchiyama, Y. Characterization of starvation-induced autophagy in cerebellar purkinje cells of phluorin-mkate2-human lc3b transgenic mice. Sci. Rep. 2020, 10, 9643. [Google Scholar] [CrossRef]
- Bozhanova, N.G.; Baranov, M.S.; Klementieva, N.V.; Sarkisyan, K.S.; Gavrikov, A.S.; Yampolsky, I.V.; Zagaynova, E.V.; Lukyanov, S.A.; Lukyanov, K.A.; Mishin, A.S. Protein labeling for live cell fluorescence microscopy with a highly photostable renewable signal. Chem. Sci. 2017, 8, 7138–7142. [Google Scholar] [CrossRef]
- Wu, J.; Zaccara, S.; Khuperkar, D.; Kim, H.; Tanenbaum, M.E.; Jaffrey, S.R. Live imaging of mrna using rna-stabilized fluorogenic proteins. Nat. Methods 2019, 16, 862–865. [Google Scholar] [CrossRef]
- Bertrand, E.; Chartrand, P.; Schaefer, M.; Shenoy, S.M.; Singer, R.H.; Long, R.M. Localization of ash1 mrna particles in living yeast. Mol. Cell 1998, 2, 437–445. [Google Scholar] [CrossRef]
- Vera, M.; Tutucci, E.; Singer, R.H. Imaging single mrna molecules in mammalian cells using an optimized ms2-mcp system. In Imaging Gene Expression; Springer: Berlin/Heidelberg, Germany, 2019; pp. 3–20. [Google Scholar]
- Tutucci, E.; Vera, M.; Biswas, J.; Garcia, J.; Parker, R.; Singer, R.H. An improved ms2 system for accurate reporting of the mrna life cycle. Nat. Methods 2018, 15, 81–89. [Google Scholar] [CrossRef]
- Filonov, G.S.; Moon, J.D.; Svensen, N.; Jaffrey, S.R. Broccoli: Rapid selection of an rna mimic of green fluorescent protein by fluorescence-based selection and directed evolution. J. Am. Chem. Soc. 2014, 136, 16299–16308. [Google Scholar] [CrossRef]
- Paige, J.S.; Wu, K.Y.; Jaffrey, S.R. Rna mimics of green fluorescent protein. Science 2011, 333, 642. [Google Scholar] [CrossRef]
- Weissman, T.A.; Pan, Y.A. Brainbow: New resources and emerging biological applications for multicolor genetic labeling and analysis. Genetics 2015, 199, 293–306. [Google Scholar] [CrossRef]
- Roo, J.J.d.; Vloemans, S.A.; Vrolijk, H.; Haas, E.F.d.; Staal, F.J. Development of an in vivo model to study clonal lineage relationships in hematopoietic cells using brainbow2.1/confetti mice. Future Sci. OA 2019, 5, FSO427. [Google Scholar] [CrossRef]
- El-Nachef, D.; Shi, K.; Beussman, K.M.; Martinez, R.; Regier, M.C.; Everett, G.W.; Murry, C.E.; Stevens, K.R.; Young, J.E.; Sniadecki, N.J.; et al. A rainbow reporter tracks single cells and reveals heterogeneous cellular dynamics among pluripotent stem cells and their differentiated derivatives. Stem Cell Rep. 2020, 15, 226–241. [Google Scholar] [CrossRef]
- Cook, Z.T.; Brockway, N.L.; Tobias, Z.J.C.; Pajarla, J.; Boardman, I.S.; Ippolito, H.; Nkoula, S.N.; Weissman, T.A. Combining near-infrared fluorescence with brainbow to visualize expression of specific genes within a multicolor context. Mol. Biol. Cell 2019, 30, 491–505. [Google Scholar] [CrossRef]
- Specht, E.A.; Braselmann, E.; Palmer, A.E. A critical and comparative review of fluorescent tools for live-cell imaging. Annu. Rev. Physiol. 2017, 79, 93–117. [Google Scholar] [CrossRef]
- Wei, L.; Chen, Z.; Shi, L.; Long, R.; Anzalone, A.V.; Zhang, L.; Hu, F.; Yuste, R.; Cornish, V.W.; Min, W. Super-multiplex vibrational imaging. Nature 2017, 544, 465–470. [Google Scholar] [CrossRef]
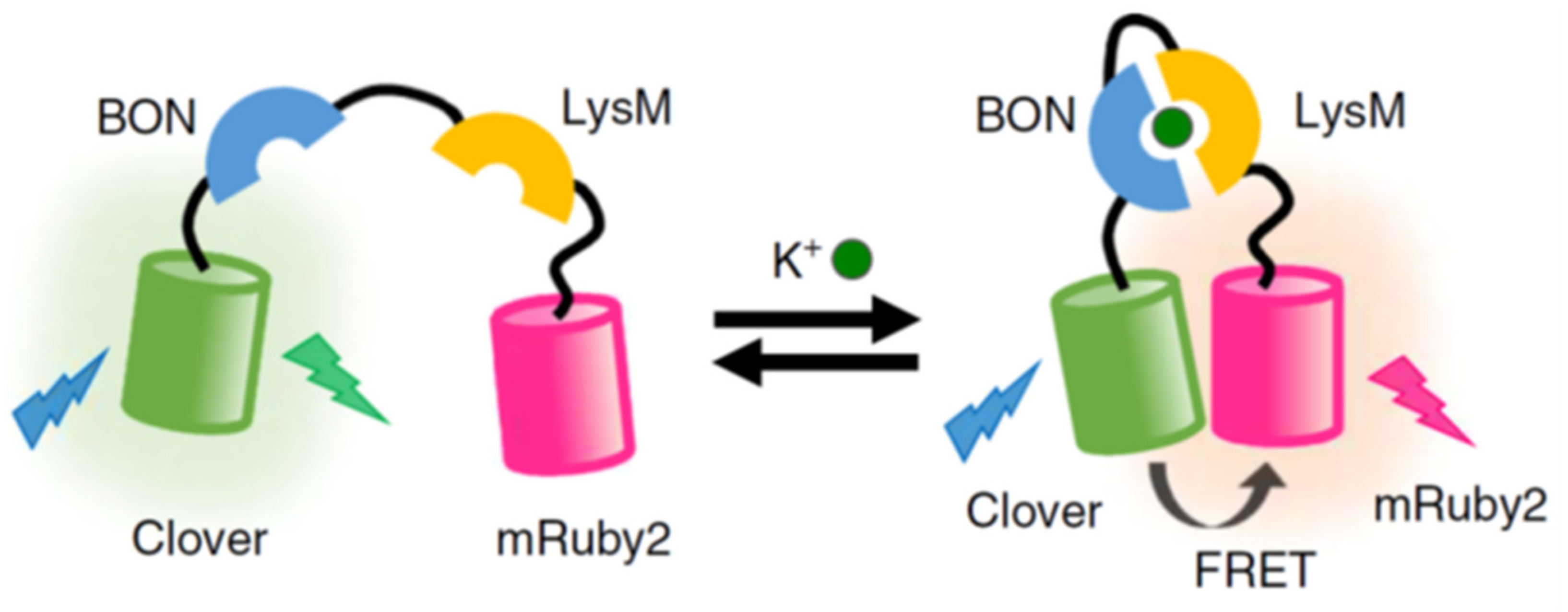
- Shen, Y.; Wu, S.-Y.; Rancic, V.; Aggarwal, A.; Qian, Y.; Miyashita, S.-I.; Ballanyi, K.; Campbell, R.E.; Dong, M. Genetically encoded fluorescent indicators for imaging intracellular potassium ion concentration. Commun. Biol. 2019, 2, 18. [Google Scholar] [CrossRef]
- Udensi, U.K.; Tchounwou, P.B. Potassium homeostasis, oxidative stress, and human disease. Int. J. Clin. Exp. Physiol. 2017, 4, 111–122. [Google Scholar]
- Angres, B.; Steuer, H.; Weber, P.; Wagner, M.; Schneckenburger, H. A membrane-bound fret-based caspase sensor for detection of apoptosis using fluorescence lifetime and total internal reflection microscopy. Cytom. Part A 2009, 75A, 420–427. [Google Scholar] [CrossRef]
- Savitsky, A.P.; Rusanov, A.L.; Zherdeva, V.V.; Gorodnicheva, T.V.; Khrenova, M.G.; Nemukhin, A.V. Flim-fret imaging of caspase-3 activity in live cells using pair of red fluorescent proteins. Theranostics 2012, 2, 215–226. [Google Scholar] [CrossRef]
- Heim, R.; Tsien, R.Y. Engineering green fluorescent protein for improved brightness, longer wavelengths and fluorescence resonance energy transfer. Curr. Biol. 1996, 6, 178–182. [Google Scholar] [CrossRef]
- Erismann-Ebner, K.; Marowsky, A.; Arand, M. In-vitro characterization of mcerulean3_mruby3 as a novel fret pair with favorable bleed-through characteristics. Biosensors 2019, 9, 33. [Google Scholar] [CrossRef]
- George Abraham, B.; Sarkisyan, K.S.; Mishin, A.S.; Santala, V.; Tkachenko, N.V.; Karp, M. Fluorescent protein based fret pairs with improved dynamic range for fluorescence lifetime measurements. PLoS ONE 2015, 10, e0134436. [Google Scholar] [CrossRef]
- Umrao, S.; Jain, V.; Anusha; Chakraborty, B.; Roy, R. Protein-induced fluorescence enhancement as aptamer sensing mechanism for thrombin detection. Sens. Actuators B Chem. 2018, 267, 294–301. [Google Scholar] [CrossRef]
- Vanderklish, P.W.; Krushel, L.A.; Holst, B.H.; Gally, J.A.; Crossin, K.L.; Edelman, G.M. Marking synaptic activity in dendritic spines with a calpain substrate exhibiting fluorescence resonance energy transfer. Proc. Natl. Acad. Sci. USA 2000, 97, 2253. [Google Scholar] [CrossRef]
- Stockholm, D.; Bartoli, M.; Sillon, G.; Bourg, N.; Davoust, J.; Richard, I. Imaging calpain protease activity by multiphoton fret in living mice. J. Mol. Biol. 2005, 346, 215–222. [Google Scholar] [CrossRef]
- Mo, G.C.; Posner, C.; Rodriguez, E.A.; Sun, T.; Zhang, J. A rationally enhanced red fluorescent protein expands the utility of fret biosensors. Nat. Commun. 2020, 11, 1–9. [Google Scholar] [CrossRef]
- Duwé, S.; Dedecker, P. Optimizing the fluorescent protein toolbox and its use. Curr. Opin. Biotechnol. 2019, 58, 183–191. [Google Scholar] [CrossRef]
- Kleeman, B.; Olsson, A.; Newkold, T.; Kofron, M.; DeLay, M.; Hildeman, D.; Grimes, H.L. A guide to choosing fluorescent protein combinations for flow cytometric analysis based on spectral overlap. Cytom. Part A 2018, 93, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Richards, R.; Mason, D.; Kelly, R.; Lévy, R.; Bearon, R.; Sée, V. 4d imaging and analysis of multicellular tumour spheroid cell migration and invasion. bioRxiv 2019, 443648. [Google Scholar] [CrossRef]
- Shashkova, S.; Leake, M.C. Single-molecule fluorescence microscopy review: Shedding new light on old problems. Biosci. Rep. 2017, 37, BSR20170031. [Google Scholar] [CrossRef] [PubMed]
- Johnson, N.; Glick, B.S. 4D microscopy of yeast. J. Vis. Exp. 2019, 146. [Google Scholar] [CrossRef]
- Descloux, A.; Grußmayer, K.S.; Bostan, E.; Lukes, T.; Bouwens, A.; Sharipov, A.; Geissbuehler, S.; Mahul-Mellier, A.L.; Lashuel, H.A.; Leutenegger, M.; et al. Combined multi-plane phase retrieval and super-resolution optical fluctuation imaging for 4d cell microscopy. Nat. Photonics 2018, 12, 165–172. [Google Scholar] [CrossRef]
- Politi, A.Z.; Cai, Y.; Walther, N.; Hossain, M.J.; Koch, B.; Wachsmuth, M.; Ellenberg, J. Quantitative mapping of fluorescently tagged cellular proteins using fcs-calibrated four-dimensional imaging. Nat. Protoc. 2018, 13, 1445–1464. [Google Scholar] [CrossRef]
- Yong, K.-T.; Law, W.-C.; Hu, R.; Ye, L.; Liu, L.; Swihart, M.T.; Prasad, P.N. Nanotoxicity assessment of quantum dots: From cellular to primate studies. Chem. Soc. Rev. 2013, 42, 1236–1250. [Google Scholar] [CrossRef]
- Han, X.; Xu, K.; Taratula, O.; Farsad, K. Applications of nanoparticles in biomedical imaging. Nanoscale 2019, 11, 799–819. [Google Scholar] [CrossRef]
- Goreham, R.V.; Schroeder, K.L.; Holmes, A.; Bradley, S.J.; Nann, T. Demonstration of the lack of cytotoxicity of unmodified and folic acid modified graphene oxide quantum dots, and their application to fluorescence lifetime imaging of hacat cells. Microchim. Acta 2018, 185, 128. [Google Scholar] [CrossRef]
- Mei, K.-C.; Rubio, N.; Costa, P.M.; Kafa, H.; Abbate, V.; Festy, F.; Bansal, S.S.; Hider, R.C.; Al-Jamal, K.T. Synthesis of double-clickable functionalised graphene oxide for biological applications. Chem. Commun. 2015, 51, 14981–14984. [Google Scholar] [CrossRef]
- Jiang, D.; Chen, Y.; Li, N.; Li, W.; Wang, Z.; Zhu, J.; Zhang, H.; Liu, B.; Xu, S. Synthesis of luminescent graphene quantum dots with high quantum yield and their toxicity study. PLoS ONE 2016, 10, e0144906. [Google Scholar] [CrossRef]
- Mitchell, B.; Bradley, S.J.; Thomas, N. Graphene quantum dots. Part. Part. Syst. Charact. 2014, 31, 415–428. [Google Scholar]
- Shen, J.; Zhu, Y.; Yang, X.; Li, C. Graphene quantum dots: Emergent nanolights for bioimaging, sensors, catalysis and photovoltaic devices. Chem. Commun. 2012, 48, 3686–3699. [Google Scholar] [CrossRef]
- Chung, S.; Revia, R.A.; Zhang, M. Graphene quantum dots and their applications in bioimaging, biosensing, and therapy. Adv. Mater. 2021, 33, 1904362. [Google Scholar] [CrossRef]
- Sweetman, M.J.; Hickey, S.M.; Brooks, D.A.; Hayball, J.D.; Plush, S.E. A practical guide to prepare and synthetically modify graphene quantum dots. Adv. Funct. Mater. 2019, 29, 1808740–1808758. [Google Scholar] [CrossRef]
- Georgakilas, V.; Otyepka, M.; Bourlinos, A.B.; Chandra, V.; Kim, N.; Kemp, K.C.; Hobza, P.; Zboril, R.; Kim, K.S. Functionalization of graphene: Covalent and non-covalent approaches, derivatives and applications. Chem. Rev. 2012, 112, 6156–6214. [Google Scholar] [CrossRef]
- Narayanan, T.N.; Gupta, B.K.; Vithayathil, S.A.; Aburto, R.R.; Mani, S.A.; Taha-Tijerina, J.; Xie, B.; Kaipparettu, B.A.; Torti, S.V.; Ajayan, P.M. Hybrid 2d nanomaterials as dual-mode contrast agents in cellular imaging. Adv. Mater. 2012, 24, 2992–2998. [Google Scholar] [CrossRef]
- Peng, J.; Gao, W.; Gupta, B.K.; Liu, Z.; Romero-Aburto, R.; Ge, L.; Song, L.; Alemany, L.B.; Zhan, X.; Gao, G.; et al. Graphene quantum dots derived from carbon fibers. Nano Lett. 2012, 12, 844–849. [Google Scholar] [CrossRef]
- Chen, T.; Yu, H.; Yang, N.; Wang, M.; Ding, C.; Fu, J. Graphene quantum dot-capped mesoporous silica nanoparticles through an acid-cleavable acetal bond for intracellular drug delivery and imaging. J. Mater. Chem. B 2014, 2, 4979–4982. [Google Scholar] [CrossRef]
- Hai, X.; Wang, Y.; Hao, X.; Chen, X.; Wang, J. Folic acid encapsulated graphene quantum dots for ratiometric ph sensing and specific multicolor imaging in living cells. Sens. Actuators B Chem. 2018, 268, 61–69. [Google Scholar] [CrossRef]
- Lee, B.H.; McKinney, R.L.; Hasan, M.T.; Naumov, A.V. Graphene quantum dots as intracellular imaging-based temperature sensors. Materials 2021, 14, 616. [Google Scholar] [CrossRef]
- Malek, A.; Aouad, K.; El Khoury, R.; Halabi-Tawil, M.; Choucair, J. Chronic mercury intoxication masquerading as systemic disease: A case report and review of the literature. Eur. J. Case Rep. Intern. Med. 2017, 4, 000632. [Google Scholar] [CrossRef]
- Zhao, X.; Gao, J.; He, X.; Cong, L.; Zhao, H.; Li, X.; Tan, F. DNA-modified graphene quantum dots as a sensing platform for detection of hg2+ in living cells. RSC Adv. 2015, 5, 39587–39591. [Google Scholar] [CrossRef]
- Liu, M.; Liu, T.; Li, Y.; Xu, H.; Zheng, B.; Wang, D.; Du, J.; Xiao, D. A fret chemsensor based on graphene quantum dots for detecting and intracellular imaging of hg2+. Talanta 2015, 143, 442–449. [Google Scholar] [CrossRef]
- Zhu, A.; Ding, C.; Tian, Y. A two-photon ratiometric fluorescence probe for cupric ions in live cells and tissues. Sci. Rep. 2013, 3, 2933. [Google Scholar] [CrossRef]
- Fang, B.-Y.; Li, C.; Song, Y.-Y.; Tan, F.; Cao, Y.-C.; Zhao, Y.-D. Nitrogen-doped graphene quantum dot for direct fluorescence detection of al3+ in aqueous media and living cells. Biosens. Bioelectron. 2018, 100, 41–48. [Google Scholar] [CrossRef]
- Liu, R.; Zhao, J.; Huang, Z.; Zhang, L.; Zou, M.; Shi, B.; Zhao, S. Nitrogen and phosphorus co-doped graphene quantum dots as a nano-sensor for highly sensitive and selective imaging detection of nitrite in live cell. Sens. Actuators B Chem. 2017, 240, 604–612. [Google Scholar] [CrossRef]
- Ma, L.; Sun, S.; Wang, Y.; Jiang, K.; Zhu, J.; Li, J.; Lin, H. A graphene quantum dot-based fluorescent nanoprobe for hypochlorite detection in water and in living cells. Microchim. Acta 2017, 184, 3833–3840. [Google Scholar] [CrossRef]
- Zhao, W.; Li, Y.; Yang, S.; Chen, Y.; Zheng, J.; Liu, C.; Qing, Z.; Li, J.; Yang, R. Target-activated modulation of dual-color and two-photon fluorescence of graphene quantum dots for in vivo imaging of hydrogen peroxide. Anal. Chem. 2016, 88, 4833–4840. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Y.; He, H.; Chen, X.; Sun, X.; Sun, Y.; Zhou, G.; Xu, H.; Huang, F. Steering graphene quantum dots in living cells: Lighting up the nucleolus. J. Mater. Chem. B 2016, 4, 779–784. [Google Scholar] [CrossRef]
- Yang, K.; Hou, L.; Li, Z.; Lin, T.; Tian, J.; Zhao, S. A mitochondria-targeted ratiometric fluorescent nanoprobe for imaging of peroxynitrite in living cells. Talanta 2021, 231, 122421. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.T.; Than, A.; Ananthanaraya, A.; Kim, D.-H.; Chen, P. Graphene quantum dots as universal fluorophores and their use in revealing regulated trafficking of insulin receptors in adipocytes. Acs Nano 2013, 7, 6278–6286. [Google Scholar] [CrossRef] [PubMed]
- Ananthanarayanan, A.; Wang, Y.; Routh, P.; Sk, M.A.; Than, A.; Lin, M.; Zhang, J.; Chen, J.; Sun, H.; Chen, P. Nitrogen and phosphorus co-doped graphene quantum dots: Synthesis from adenosine triphosphate, optical properties, and cellular imaging. Nanoscale 2015, 7, 8159–8165. [Google Scholar] [CrossRef] [PubMed]
- Nurunnabi, M.; Khatun, Z.; Reeck, G.R.; Lee, D.Y.; Lee, Y.-k. Near infra-red photoluminescent graphene nanoparticles greatly expand their use in noninvasive biomedical imaging. Chem. Commun. 2013, 49, 5079–5081. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.T.; Gonzalez-Rodriguez, R.; Lin, C.-W.; Campbell, E.; Vasireddy, S.; Tsedev, U.; Belcher, A.M.; Naumov, A.V. Rare-earth metal ions doped graphene quantum dots for near-ir in vitro/in vivo/ex vivo imaging applications. Adv. Opt. Mater. 2020, 8, 2000897. [Google Scholar] [CrossRef]
- Zhang, M.; Bai, L.; Shang, W.; Xie, W.; Ma, H.; Fu, Y.; Fang, D.; Sun, H.; Fan, L.; Han, M.; et al. Facile synthesis of water-soluble, highly fluorescent graphene quantum dots as a robust biological label for stem cells. J. Mater. Chem. 2012, 22, 7461–7467. [Google Scholar] [CrossRef]
- Ko, N.; Nafiujjaman, M.; Lee, J.; Lim, H.-N.; Lee, Y.-k.; Kwon, I. Graphene quantum dot-based theranostic agents for active targeting of breast cancer. RSC Adv. 2017, 7, 11420–11427. [Google Scholar] [CrossRef]
- Nigam, P.; Waghmode, S.; Louis, M.; Wangnoo, S.; Chavan, P.; Sarkar, D. Graphene quantum dots conjugated albumin nanoparticles for targeted drug delivery and imaging of pancreatic cancer. J. Mater. Chem. B 2014, 2, 3190–3195. [Google Scholar] [CrossRef]
- Wang, X.; Sun, X.; Lao, J.; He, H.; Cheng, T.; Wang, M.; Wang, S.; Huang, F. Multifunctional graphene quantum dots for simultaneous targeted cellular imaging and drug delivery. Colloids Surf. B: Biointerfaces 2014, 122, 638–644. [Google Scholar] [CrossRef]
- Vahedi, N.; Tabandeh, F.; Mahmoudifard, M. Hyaluronic acid–graphene quantum dot nanocomposite: Potential target drug delivery and cancer cell imaging. Biotechnol. Appl. Biochem. 2021. [Google Scholar] [CrossRef]
- Zhao, Q.; Huang, C.; Li, F. Phosphorescent heavy-metal complexes for bioimaging. Chem. Soc. Rev. 2011, 40, 2508–2524. [Google Scholar] [CrossRef]
- Suhling, K.; French, P.M.W.; Phillips, D. Time-resolved fluorescence microscopy. Photochem. Photobiol. Sci. 2005, 4, 13–22. [Google Scholar] [CrossRef]
- Gillam, T.A.; Sweetman, M.J.; Bader, C.A.; Morrison, J.L.; Hayball, J.D.; Brooks, D.A.; Plush, S.E. Bright lights down under: Metal ion complexes turning the spotlight on metabolic processes at the cellular level. Coord. Chem. Rev. 2018, 375, 234–255. [Google Scholar] [CrossRef]
- Lo, K.K.-W.; Zhang, K.Y. Iridium(iii) complexes as therapeutic and bioimaging reagents for cellular applications. RSC Adv. 2012, 2, 12069–12083. [Google Scholar] [CrossRef]
- Coogan, M.P.; Fernández-Moreira, V. Progress with, and prospects for, metal complexes in cell imaging. Chem. Commun. 2014, 50, 384–399. [Google Scholar] [CrossRef]
- Hostachy, S.; Policar, C.; Delsuc, N. Re(i) carbonyl complexes: Multimodal platforms for inorganic chemical biology. Coord. Chem. Rev. 2017, 351, 172–188. [Google Scholar] [CrossRef]
- Amoroso, A.J.; Coogan, M.P.; Dunne, J.E.; Fernández-Moreira, V.; Hess, J.B.; Hayes, A.J.; Lloyd, D.; Millet, C.; Pope, S.J.A.; Williams, C. Rhenium fac tricarbonyl bisimine complexes: Biologically useful fluorochromes for cell imaging applications. Chem. Commun. 2007, 29, 3066–3068. [Google Scholar] [CrossRef]
- Amoroso, A.J.; Arthur, R.J.; Coogan, M.P.; Court, J.B.; Fernández-Moreira, V.; Hayes, A.J.; Lloyd, D.; Millet, C.; Pope, S.J.A. 3-chloromethylpyridyl bipyridine fac-tricarbonyl rhenium: A thiol-reactive luminophore for fluorescence microscopy accumulates in mitochondria. New J. Chem. 2008, 32, 1097–1102. [Google Scholar] [CrossRef]
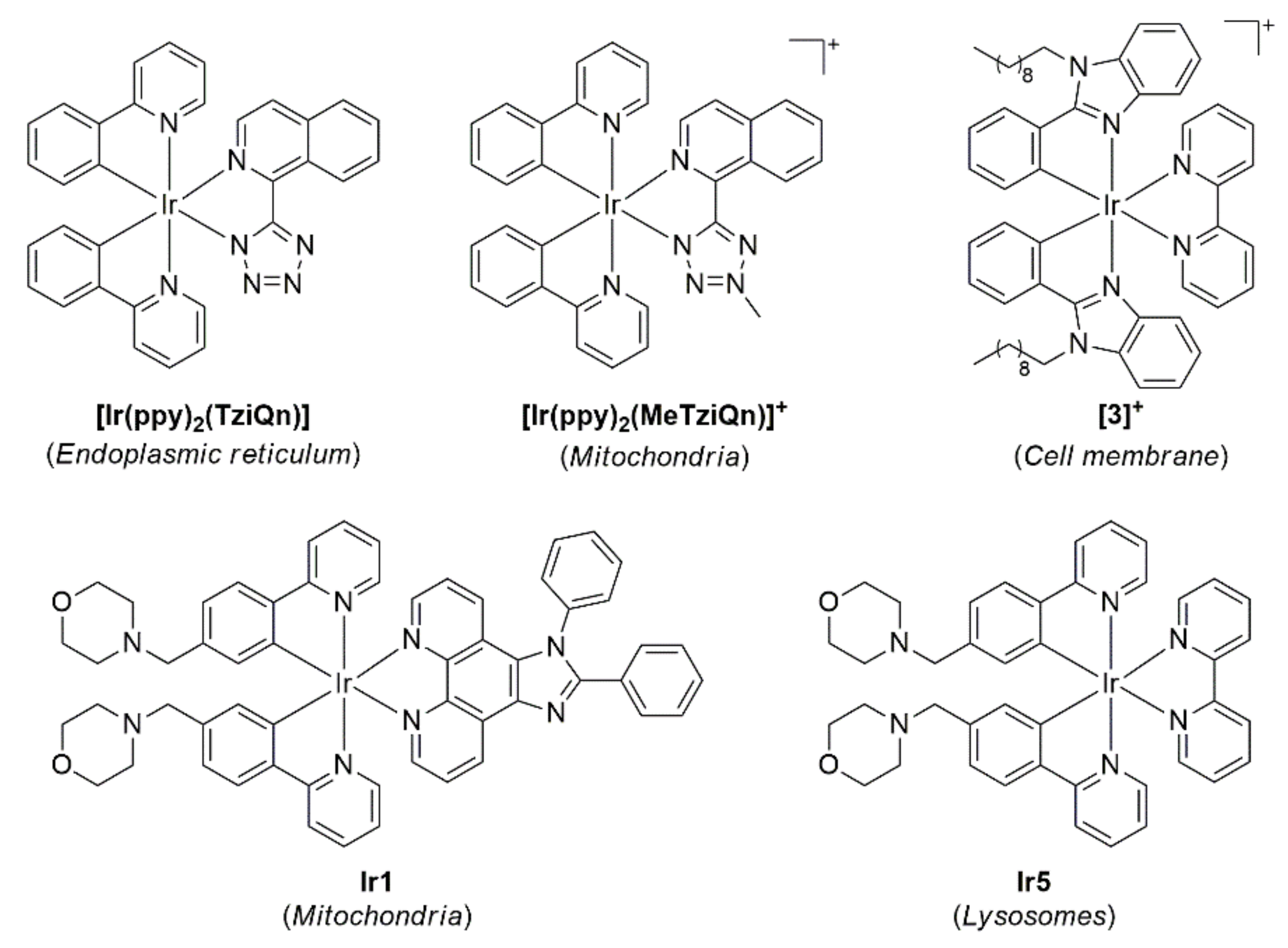
- Bader, C.A.; Brooks, R.D.; Ng, Y.S.; Sorvina, A.; Werrett, M.V.; Wright, P.J.; Anwer, A.G.; Brooks, D.A.; Stagni, S.; Muzzioli, S.; et al. Modulation of the organelle specificity in re(i) tetrazolato complexes leads to labeling of lipid droplets. RSC Adv. 2014, 4, 16345–16351. [Google Scholar] [CrossRef]
- Bader, C.A.; Shandala, T.; Carter, E.A.; Ivask, A.; Guinan, T.; Hickey, S.M.; Werrett, M.V.; Wright, P.J.; Simpson, P.V.; Stagni, S. A molecular probe for the detection of polar lipids in live cells. PLoS ONE 2016, 11, e0161557. [Google Scholar] [CrossRef]
- Bader, C.A.; Sorvina, A.; Simpson, P.V.; Wright, P.J.; Stagni, S.; Plush, S.E.; Massi, M.; Brooks, D.A. Imaging nuclear, endoplasmic reticulum and plasma membrane events in real time. FEBS Lett. 2016, 590, 3051–3060. [Google Scholar] [CrossRef]
- Caporale, C.; Bader, C.A.; Sorvina, A.; MaGee, K.D.M.; Skelton, B.W.; Gillam, T.A.; Wright, P.J.; Raiteri, P.; Stagni, S.; Morrison, J.L.; et al. Investigating intracellular localisation and cytotoxicity trends for neutral and cationic iridium tetrazolato complexes in live cells. Chem.—A Eur. J. 2017, 23, 15666–15679. [Google Scholar] [CrossRef]
- Laha, P.; De, U.; Chandra, F.; Dehury, N.; Khullar, S.; Kim, H.S.; Patra, S. Alkyl chain-modified cyclometalated iridium complexes as tunable anticancer and imaging agents. Dalton Trans. 2018, 47, 15873–15881. [Google Scholar] [CrossRef]
- Liu, J.-B.; Vellaisamy, K.; Li, G.; Yang, C.; Wong, S.-Y.; Leung, C.-H.; Pu, S.-Z.; Ma, D.-L. A long-lifetime iridium(iii) complex for lysosome tracking with high specificity and a large stokes shift. J. Mater. Chem. B 2018, 6, 3855–3858. [Google Scholar] [CrossRef]
- Qiu, K.; Liu, Y.; Huang, H.; Liu, C.; Zhu, H.; Chen, Y.; Ji, L.; Chao, H. Biscylometalated iridium(iii) complexes target mitochondria or lysosomes by regulating the lipophilicity of the main ligands. Dalton Trans. 2016, 45, 16144–16147. [Google Scholar] [CrossRef]
- Qiu, K.; Huang, H.; Liu, B.; Liu, Y.; Huang, Z.; Chen, Y.; Ji, L.; Chao, H. Long-term lysosomes tracking with a water-soluble two-photon phosphorescent iridium(iii) complex. ACS Appl. Mater. Interfaces 2016, 8, 12702–12710. [Google Scholar] [CrossRef]
- Li, J.; Chen, H.; Zeng, L.; Rees, T.W.; Xiong, K.; Chen, Y.; Ji, L.; Chao, H. Mitochondria-targeting cyclometalated iridium(iii) complexes for tumor hypoxic imaging and therapy. Inorg. Chem. Front. 2019, 6, 1003–1010. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, M.; Liu, S.; Yang, H.; Zhao, Q.; Liu, Z.; He, W. Phosphorescence lifetime imaging of labile zn2+ in mitochondria via a phosphorescent iridium(iii) complex. Inorg. Chem. 2018, 57, 10625–10632. [Google Scholar] [CrossRef]
- Pasha, S.S.; Das, P.; Rath, N.P.; Bandyopadhyay, D.; Jana, N.R.; Laskar, I.R. Water soluble luminescent cyclometalated platinum(ii) complex—A suitable probe for bio-imaging applications. Inorg. Chem. Commun. 2016, 67, 107–111. [Google Scholar] [CrossRef]
- Botchway, S.W.; Charnley, M.; Haycock, J.W.; Parker, A.W.; Rochester, D.L.; Weinstein, J.A.; Williams, J.A.G. Time-resolved and two-photon emission imaging microscopy of live cells with inert platinum complexes. Proc. Natl. Acad. Sci. USA 2008, 105, 16071. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.-j.; Zhao, W.; Xie, M.; Li, X.; Sun, M.; He, J.; Wang, L.; Yu, L. Real-time monitoring of self-aggregation of β-amyloid by a fluorescent probe based on ruthenium complex. Anal. Chem. 2020, 92, 2953–2960. [Google Scholar] [CrossRef] [PubMed]
- Estalayo-Adrián, S.; Blasco, S.; Bright, S.A.; McManus, G.J.; Orellana, G.; Williams, D.C.; Kelly, J.M.; Gunnlaugsson, T. Water-soluble amphiphilic ruthenium(ii) polypyridyl complexes as potential light-activated therapeutic agents. Chem. Commun. 2020, 56, 9332–9335. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Ye, Z.; Wang, G.; Zhang, W.; Yuan, J. Development of a ruthenium(ii) complex based luminescent probe for imaging nitric oxide production in living cells. Chem.—A Eur. J. 2010, 16, 6884–6891. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.; Guo, H.; Yuan, X.; Li, X.; Ding, H.; Gao, P.; Zhao, C.; Wu, W.; Wu, W.; Zhao, J. A highly selective off-on red-emitting phosphorescent thiol probe with large stokes shift and long luminescent lifetime. Org. Lett. 2010, 12, 2876–2879. [Google Scholar] [CrossRef]
- Eliseeva, S.V.; Bünzli, J.-C.G. Lanthanide luminescence for functional materials and bio-sciences. Chem. Soc. Rev. 2010, 39, 189–227. [Google Scholar] [CrossRef]
- Bunzli, J.-C.G.; Eliseeva, S.V. Intriguing aspects of lanthanide luminescence. Chem. Sci. 2013, 4, 1939–1949. [Google Scholar] [CrossRef]
- Mathieu, E.; Sipos, A.; Demeyere, E.; Phipps, D.; Sakaveli, D.; Borbas, K.E. Lanthanide-based tools for the investigation of cellular environments. Chem. Commun. 2018, 54, 10021–10035. [Google Scholar] [CrossRef]
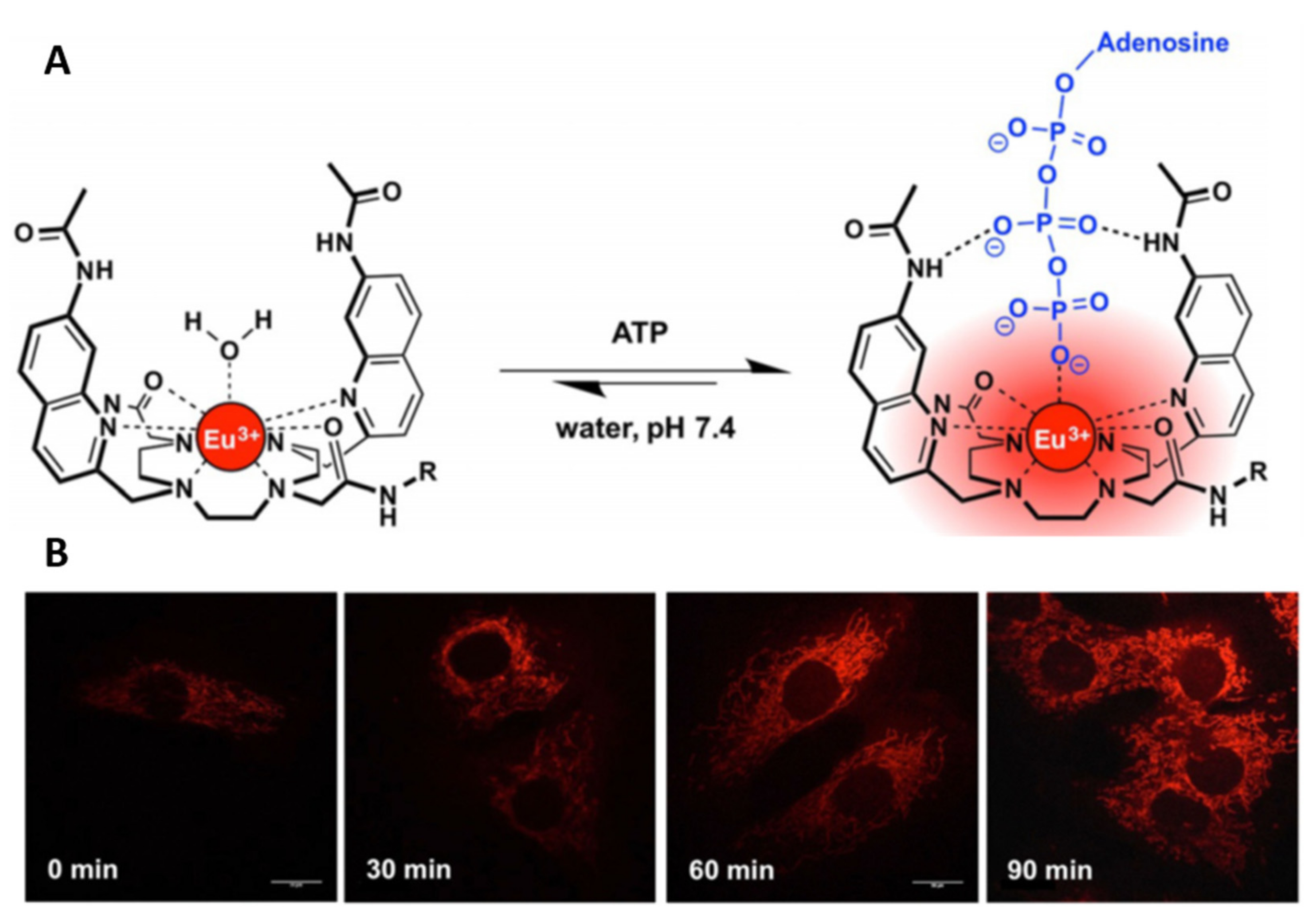
- Mailhot, R.; Traviss-Pollard, T.; Pal, R.; Butler, S.J. Cationic europium complexes for visualizing fluctuations in mitochondrial atp levels in living cells. Chemistry 2018, 24, 10745–10755. [Google Scholar] [CrossRef]
- Wu, J.; Che, W.; Liu, H.; Min, P.; Zheng, D. Development of a lysosome-targetable visible-light-excited europium(iii) complex-based luminescent probe to image hypochlorous acid in living cells. Opt. Mater. 2020, 109, 110273. [Google Scholar] [CrossRef]
- Gillam, T.A.; Caporale, C.; Brooks, R.D.; Bader, C.A.; Sorvina, A.; Werrett, M.V.; Wright, P.J.; Morrison, J.L.; Massi, M.; Brooks, D.A.; et al. Neutral re(i) complex platform for live intracellular imaging. Inorg. Chem. 2021, 60, 10173–10185. [Google Scholar] [CrossRef]
- Law, W.H.-T.; Lee, L.C.-C.; Louie, M.-W.; Liu, H.-W.; Ang, T.W.-H.; Lo, K.K.-W. Phosphorescent cellular probes and uptake indicators derived from cyclometalated iridium(iii) bipyridine complexes appended with a glucose or galactose entity. Inorg. Chem. 2013, 52, 13029–13041. [Google Scholar] [CrossRef]
- Day, A.H.; Übler, M.H.; Best, H.L.; Lloyd-Evans, E.; Mart, R.J.; Fallis, I.A.; Allemann, R.K.; Al-Wattar, E.A.H.; Keymer, N.I.; Buurma, N.J.; et al. Targeted cell imaging properties of a deep red luminescent iridium(iii) complex conjugated with a c-myc signal peptide. Chem. Sci. 2020, 11, 1599–1606. [Google Scholar] [CrossRef]
- Gill, M.R.; Garcia-Lara, J.; Foster, S.J.; Smythe, C.; Battaglia, G.; Thomas, J.A. A ruthenium(ii) polypyridyl complex for direct imaging of DNA structure in living cells. Nat. Chem. 2009, 1, 662–667. [Google Scholar] [CrossRef]
- Lavis, L.D.; Raines, R.T. Bright ideas for chemical biology. ACS Chem. Biol. 2008, 3, 142–155. [Google Scholar] [CrossRef]
- Wu, D.; Sedgwick, A.C.; Gunnlaugsson, T.; Akkaya, E.U.; Yoon, J.; James, T.D. Fluorescent chemosensors: The past, present and future. Chem. Soc. Rev. 2017, 46, 7105–7123. [Google Scholar] [CrossRef]
- Miller, D.R.; Jarrett, J.W.; Hassan, A.M.; Dunn, A.K. Deep tissue imaging with multiphoton fluorescence microscopy. Curr. Opin. Biomed. Eng. 2017, 4, 32–39. [Google Scholar] [CrossRef]
- Zheng, Q.; Juette, M.F.; Jockusch, S.; Wasserman, M.R.; Zhou, Z.; Altman, R.B.; Blanchard, S.C. Ultra-stable organic fluorophores for single-molecule research. Chem. Soc. Rev. 2014, 43, 1044–1056. [Google Scholar] [CrossRef]
- Lei, Z.; Li, X.; Luo, X.; He, H.; Zheng, J.; Qian, X.; Yang, Y. Bright, stable, and biocompatible organic fluorophores absorbing/emitting in the deep near-infrared spectral region. Angew. Chem. Int. Ed. 2017, 56, 2979–2983. [Google Scholar] [CrossRef]
- Hickey, S.M.; Ashton, T.D.; Boer, G.; Bader, C.A.; Thomas, M.; Elliott, A.G.; Schmuck, C.; Yu, H.Y.; Li, J.; Nation, R.L.; et al. Norbornane-based cationic antimicrobial peptidomimetics targeting the bacterial membrane. Eur. J. Med. Chem. 2018, 160, 9–22. [Google Scholar] [CrossRef]
- Knorr, G.; Kozma, E.; Schaart, J.M.; Németh, K.; Török, G.; Kele, P. Bioorthogonally applicable fluorogenic cyanine-tetrazines for no-wash super-resolution imaging. Bioconjugate Chem. 2018, 29, 1312–1318. [Google Scholar] [CrossRef]
- Jana, P.; Siva, A.; Soppina, V.; Kanvah, S. Live-cell imaging of lipid droplets using solvatochromic coumarin derivatives. Org. Biomol. Chem. 2020, 18, 5608–5616. [Google Scholar] [CrossRef]
- Collot, M.; Kreder, R.; Tatarets, A.L.; Patsenker, L.D.; Mely, Y.; Klymchenko, A.S. Bright fluorogenic squaraines with tuned cell entry for selective imaging of plasma membrane vs. Endoplasmic reticulum. Chem. Commun. 2015, 51, 17136–17139. [Google Scholar] [CrossRef]
- Chao, S.; Krejci, E.; Bernard, V.; Leroy, J.; Jean, L.; Renard, P.-Y. A selective and sensitive near-infrared fluorescent probe for acetylcholinesterase imaging. Chem. Commun. 2016, 52, 11599–11602. [Google Scholar] [CrossRef]
- Holmila, R.J.; Vance, S.A.; Chen, X.; Wu, H.; Shukla, K.; Bharadwaj, M.S.; Mims, J.; Wary, Z.; Marrs, G.; Singh, R.; et al. Mitochondria-targeted probes for imaging protein sulfenylation. Sci. Rep. 2018, 8, 6635. [Google Scholar] [CrossRef]
- Calatrava-Pérez, E.; Bright, S.A.; Achermann, S.; Moylan, C.; Senge, M.O.; Veale, E.B.; Williams, D.C.; Gunnlaugsson, T.; Scanlan, E.M. Glycosidase activated release of fluorescent 1,8-naphthalimide probes for tumor cell imaging from glycosylated ‘pro-probes’. Chem. Commun. 2016, 52, 13086–13089. [Google Scholar] [CrossRef]
- Ramu, V.; Kundu, P.; Kondaiah, P.; Chakravarty, A.R. Maloplatin-b, a cisplatin-based bodipy-tagged mito-specific “chemo-pdt” agent active in red light. Inorg. Chem. 2021, 60, 6410–6420. [Google Scholar] [CrossRef]
- Shi, C.; Wu, J.B.; Pan, D. Review on near-infrared heptamethine cyanine dyes as theranostic agents for tumor imaging, targeting, and photodynamic therapy. J. Biomed. Opt. 2016, 21, 050901. [Google Scholar] [CrossRef]
- Pham, T.C.; Nguyen, V.-N.; Choi, Y.; Lee, S.; Yoon, J. Recent strategies to develop innovative photosensitizers for enhanced photodynamic therapy. Chem. Rev. 2021, 121, 13454–13619. [Google Scholar] [CrossRef]













Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hickey, S.M.; Ung, B.; Bader, C.; Brooks, R.; Lazniewska, J.; Johnson, I.R.D.; Sorvina, A.; Logan, J.; Martini, C.; Moore, C.R.; et al. Fluorescence Microscopy—An Outline of Hardware, Biological Handling, and Fluorophore Considerations. Cells 2022, 11, 35. https://doi.org/10.3390/cells11010035
Hickey SM, Ung B, Bader C, Brooks R, Lazniewska J, Johnson IRD, Sorvina A, Logan J, Martini C, Moore CR, et al. Fluorescence Microscopy—An Outline of Hardware, Biological Handling, and Fluorophore Considerations. Cells. 2022; 11(1):35. https://doi.org/10.3390/cells11010035
Chicago/Turabian StyleHickey, Shane M., Ben Ung, Christie Bader, Robert Brooks, Joanna Lazniewska, Ian R. D. Johnson, Alexandra Sorvina, Jessica Logan, Carmela Martini, Courtney R. Moore, and et al. 2022. "Fluorescence Microscopy—An Outline of Hardware, Biological Handling, and Fluorophore Considerations" Cells 11, no. 1: 35. https://doi.org/10.3390/cells11010035
APA StyleHickey, S. M., Ung, B., Bader, C., Brooks, R., Lazniewska, J., Johnson, I. R. D., Sorvina, A., Logan, J., Martini, C., Moore, C. R., Karageorgos, L., Sweetman, M. J., & Brooks, D. A. (2022). Fluorescence Microscopy—An Outline of Hardware, Biological Handling, and Fluorophore Considerations. Cells, 11(1), 35. https://doi.org/10.3390/cells11010035