
The Molecular Subtype of Adult Acute Lymphoblastic Leukemia Samples Determines the Engraftment Site and Proliferation Kinetics in Patient-Derived Xenograft Models

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Human Tissue Collection
2.2. Tumor Cell Purification and Storage
2.3. Sample Preparation for Xenotransplantation
2.4. Orthotopic Xenografting of Tumor Cells
2.5. Monitoring of Tumor Cell Proliferation
2.6. Tissue Collection and Sterile Preparation
2.7. Preparation and Storage of Backup Sampls
2.8. Determination of Tumor Cell Frequency by Flow Cytometry
2.9. Cytospin Preparation and Pappenheim Staining
2.10. Short Tandem Repeat (STR) Fingerprint Analysis
2.11. Analysis of Genomic Hotspot Single Nucleotide Variants
2.12. Statistical Analysis
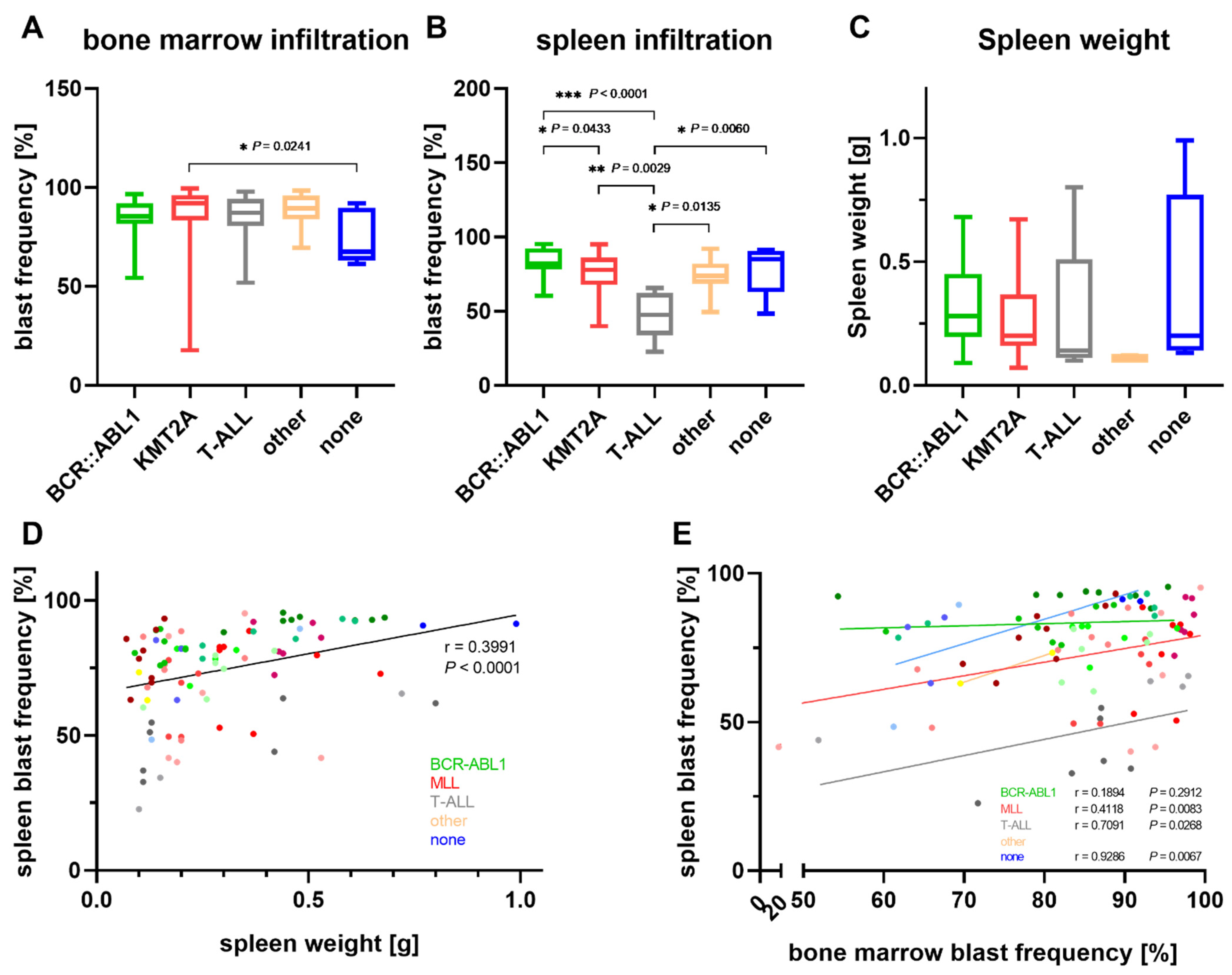
3. Results
3.1. Characterization of the Patient Cohort
3.2. Establishment of Patient Derived Xenograft Models
3.3. Characterization of PDX Models throughout Serial Transplantation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kola, I.; Landis, J. Can the pharmaceutical industry reduce attrition rates? Nat. Rev. Drug Discov. 2004, 3, 711–715. [Google Scholar] [CrossRef]
- Ledford, B.Y.H. Clinical trials are crumbling under modern economic and scientific pressures. Nature looks at ways they might be saved. Nature 2011, 477, 7–9. [Google Scholar]
- Mak, I.W.Y.; Evaniew, N.; Ghert, M. Lost in translation: Animal models and clinical trials in cancer treatment. Am. J. Transl. Res. 2014, 6, 114–118. [Google Scholar]
- Gillet, J.P.; Calcagno, A.M.; Varma, S.; Marino, M.; Green, L.J.; Vora, M.I.; Patel, C.; Orina, J.N.; Eliseeva, T.A.; Singal, V.; et al. Redefining the relevance of established cancer cell lines to the study of mechanisms of clinical anti-cancer drug resistance. Proc. Natl. Acad. Sci. USA 2011, 108, 18708–18713. [Google Scholar] [CrossRef] [PubMed]
- Hausser, H.J.; Brenner, R.E. Phenotypic instability of Saos-2 cells in long-term culture. Biochem. Biophys. Res. Commun. 2005, 333, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Cucchi, D.G.J.; Groen, R.W.J.; Janssen, J.J.W.M.; Cloos, J. Ex vivo cultures and drug testing of primary acute myeloid leukemia samples: Current techniques and implications for experimental design and outcome. Drug Resist. Updat. 2020, 53, 100730. [Google Scholar] [CrossRef]
- Nijmeijer, B.A.; Szuhai, K.; Goselink, H.M.; van Schie, M.L.J.; van der Burg, M.; de Jong, D.; Marijt, E.W.; Ottmann, O.G.; Willemze, R.; Falkenburg, J.H.F. Long-term culture of primary human lymphoblastic leukemia cells in the absence of serum or hematopoietic growth factors. Exp. Hematol. 2009, 37, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Bruserud, Ø.; Glenjen, N.; Ryningen, A.; Ulvestad, E. In vitro culture of human acute lymphoblastic leukemia (ALL) cells in serum-free media; a comparison of native ALL blasts, ALL cell lines and virus-transformed B cell lines. Leuk. Res. 2003, 27, 455–464. [Google Scholar] [CrossRef]
- Williams, M.T.S.; Yousafzai, Y.; Cox, C.; Blair, A.; Carmody, R.; Sai, S.; Chapman, K.E.; McAndrew, R.; Thomas, A.; Spence, A.; et al. Interleukin-15 enhances cellular proliferation and upregulates CNS homing molecules in pre-B acute lymphoblastic leukemia. Blood 2014, 123, 3116–3127. [Google Scholar] [CrossRef]
- Cooperman, J.; Neely, R.; Teachey, D.T.; Grupp, S.; Choi, J.K. Cell division rates of primary human precursor B cells in culture reflect in vivo rates. Stem Cells 2004, 22, 1111–1120. [Google Scholar] [CrossRef]
- Jiang, Z.; Wu, D.; Ye, W.; Weng, J.; Lai, P.; Shi, P.; Guo, X.; Huang, G.; Deng, Q.; Tang, Y.; et al. Defined, serum/feeder-free conditions for expansion and drug screening of primary B-acute lymphoblastic leukemia. Oncotarget 2017, 8, 106382–106392. [Google Scholar] [CrossRef][Green Version]
- Richter, A.; Fischer, E.; Holz, C.; Schulze, J.; Lange, S.; Sekora, A.; Knuebel, G.; Henze, L.; Roolf, C.; Murua Escobar, H.; et al. Combined Application of Pan-AKT Inhibitor MK-2206 and BCL-2 Antagonist Venetoclax in B-Cell Precursor Acute Lymphoblastic Leukemia. Int. J. Mol. Sci. 2021, 22, 2771. [Google Scholar] [CrossRef]
- Kohnken, R.; Porcu, P.; Mishra, A. Overview of the use of murine models in leukemia and lymphoma research. Front. Oncol. 2017, 7, 22. [Google Scholar] [CrossRef]
- Milan, T.; Canaj, H.; Villeneuve, C.; Ghosh, A.; Barabé, F.; Cellot, S.; Wilhelm, B.T. Pediatric leukemia: Moving toward more accurate models. Exp. Hematol. 2019, 74, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Malaney, P.; Nicosia, S.V.; Davé, V. One mouse, one patient paradigm: New avatars of personalized cancer therapy. Cancer Lett. 2014, 344, 1–12. [Google Scholar] [CrossRef]
- Aparicio, S.; Hidalgo, M.; Kung, A.L. Examining the utility of patient-derived xenograft mouse models. Nat. Rev. Cancer 2015, 15, 311–316. [Google Scholar] [CrossRef]
- Alruwetei, A.M.; Bendak, K.; Yadav, B.D.; Carol, H.; Evans, K.; Mayoh, C.; Sutton, R.; Marshall, G.M.; Lock, R.B. Examining treatment responses of diagnostic marrow in murine xenografts to predict relapse in children with acute lymphoblastic leukaemia. Br. J. Cancer 2020, 123, 742–751. [Google Scholar] [CrossRef] [PubMed]
- Richter-Pechańska, P.; Kunz, J.B.; Bornhauser, B.; Knebel Doeberitz, C.; Rausch, T.; Erarslan-Uysal, B.; Assenov, Y.; Frismantas, V.; Marovca, B.; Waszak, S.M.; et al. PDX models recapitulate the genetic and epigenetic landscape of pediatric T-cell leukemia. EMBO Mol. Med. 2018, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Uzozie, A.C.; Ergin, E.K.; Rolf, N.; Tsui, J.; Lorentzian, A.; Weng, S.S.H.; Nierves, L.; Smith, T.G.; Lim, C.J.; Maxwell, C.A.; et al. PDX models reflect the proteome landscape of pediatric acute lymphoblastic leukemia but divert in select pathways. J. Exp. Clin. Cancer Res. 2021, 40, 1–24. [Google Scholar] [CrossRef]
- Woiterski, J.; Ebinger, M.; Witte, K.E.; Goecke, B.; Heininger, V.; Philippek, M.; Bonin, M.; Schrauder, A.; Röttgers, S.; Herr, W.; et al. Engraftment of low numbers of pediatric acute lymphoid and myeloid leukemias into NOD/SCID/IL2Rcγnull mice reflects individual leukemogenecity and highly correlates with clinical outcome. Int. J. Cancer 2013, 133, 1547–1556. [Google Scholar] [CrossRef] [PubMed]
- Sandén, C.; Lilljebjörn, H.; Orsmark Pietras, C.; Henningsson, R.; Saba, K.H.; Landberg, N.; Thorsson, H.; von Palffy, S.; Peña-Martinez, P.; Högberg, C.; et al. Clonal competition within complex evolutionary hierarchies shapes AML over time. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Klco, J.M.; Spencer, D.H.; Miller, C.A.; Griffith, M.; Lamprecht, T.L.; O’Laughlin, M.; Fronick, C.; Magrini, V.; Demeter, R.T.; Fulton, R.S.; et al. Functional heterogeneity of genetically defined subclones in acute myeloid leukemia. Cancer Cell 2014, 25, 379–392. [Google Scholar] [CrossRef]
- Rokita, J.L.; Rathi, K.S.; Cardenas, M.F.; Upton, K.A.; Jayaseelan, J.; Cross, K.L.; Pfeil, J.; Egolf, L.E.; Way, G.P.; Farrel, A.; et al. Genomic Profiling of Childhood Tumor Patient-Derived Xenograft Models to Enable Rational Clinical Trial Design. Cell Rep. 2019, 29, 1675–1689. [Google Scholar] [CrossRef] [PubMed]
- Townsend, E.C.; Murakami, M.A.; Christodoulou, A.; Christie, A.L.; Köster, J.; DeSouza, T.A.; Morgan, E.A.; Kallgren, S.P.; Liu, H.; Wu, S.-C.; et al. The Public Repository of Xenografts Enables Discovery and Randomized Phase II-like Trials in Mice. Cancer Cell 2016, 29, 574–586. [Google Scholar] [CrossRef]
- Vick, B.; Rothenberg, M.; Sandhöfer, N.; Carlet, M.; Finkenzeller, C.; Krupka, C.; Grunert, M.; Trumpp, A.; Corbacioglu, S.; Ebinger, M.; et al. An advanced preclinical mouse model for acute myeloid leukemia using patients’ cells of various genetic subgroups and in vivo bioluminescence imaging. PLoS ONE 2015, 10, 1–20. [Google Scholar] [CrossRef]
- Lumkul, R.; Gorin, N.C.; Malehorn, M.T.; Hoehn, G.T.; Zheng, R.; Baldwin, B.; Small, D.; Gore, S.; Smith, D.; Meltzer, P.S.; et al. Human AML cells in NOD/SCID mice: Engraftment potential and gene expression. Leukemia 2002, 16, 1818–1826. [Google Scholar] [CrossRef][Green Version]
- Belderbos, M.E.; Koster, T.; Ausema, B.; Jacobs, S.; Sowdagar, S.; Zwart, E.; De Bont, E.; De Haan, G.; Bystrykh, L.V. Clonal selection and asymmetric distribution of human leukemia in murine xenografts revealed by cellular barcoding. Blood 2017, 129, 3210–3220. [Google Scholar] [CrossRef]
- Nowak, D.; Liem, N.L.M.; Mossner, M.; Klaumünzer, M.; Papa, R.A.; Nowak, V.; Jann, J.C.; Akagi, T.; Kawamata, N.; Okamoto, R.; et al. Variegated clonality and rapid emergence of new molecular lesions in xenografts of acute lymphoblastic leukemia are associated with drug resistance. Exp. Hematol. 2015, 43, 32–43. [Google Scholar] [CrossRef]
- El-Hoss, J.; Jing, D.; Evans, K.; Toscan, C.; Xie, J.; Lee, H.; Taylor, R.A.; Lawrence, M.G.; Risbridger, G.P.; MacKenzie, K.L.; et al. A single nucleotide polymorphism genotyping platform for the authentication of patient derived xenografts. Oncotarget 2016, 7, 60475–60490. [Google Scholar] [CrossRef]
- Malard, F.; Mohty, M. Acute lymphoblastic leukaemia. Lancet (London, England) 2020, 395, 1146–1162. [Google Scholar] [CrossRef]
- Wang, K.; Sanchez-Martin, M.; Wang, X.; Knapp, K.M.; Koche, R.; Vu, L.; Nahas, M.K.; He, J.; Hadler, M.; Stein, E.M.; et al. Patient-derived xenotransplants can recapitulate the genetic driver landscape of acute leukemias. Leukemia 2017, 31, 151–158. [Google Scholar] [CrossRef]
- Terziyska, N.; Castro Alves, C.; Groiss, V.; Schneider, K.; Farkasova, K.; Ogris, M.; Wagner, E.; Ehrhardt, H.; Brentjens, R.J.; zur Stadt, U.; et al. In vivo imaging enables high resolution preclinical trials on patients’ leukemia cells growing in mice. PLoS ONE 2012, 7, e52798. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, 1062–1067. [Google Scholar] [CrossRef]
- Patel, B.; Dey, A.; Castleton, A.Z.; Schwab, C.; Samuel, E.; Sivakumaran, J.; Beaton, B.; Zareian, N.; Zhang, C.Y.; Rai, L.; et al. Mouse xenograft modeling of human adult acute lymphoblastic leukemia provides mechanistic insights into adult LIC biology. Blood 2014, 124, 96–105. [Google Scholar] [CrossRef][Green Version]
- Jones, L.; Richmond, J.; Evans, K.; Carol, H.; Jing, D.; Kurmasheva, R.T.; Billups, C.A.; Houghton, P.J.; Smith, M.A.; Lock, R.B. Bioluminescence imaging enhances analysis of drug responses in a patient-derived xenograft model of pediatric ALL. Clin. Cancer Res. 2017, 23, 3744–3755. [Google Scholar] [CrossRef]
- Richter, A.; Sender, S.; Lenz, A.; Schwarz, R.; Hinz, B.; Knuebel, G.; Sekora, A.; Murua Escobar, H.; Junghanss, C.; Roolf, C. Influence of Casein kinase II inhibitor CX-4945 on BCL6-mediated apoptotic signaling in B-ALL in vitro and in vivo. BMC Cancer 2020, 20, 184. [Google Scholar] [CrossRef]
- Aoki, Y.; Watanabe, T.; Saito, Y.; Kuroki, Y.; Hijikata, A.; Takagi, M.; Tomizawa, D.; Eguchi, M.; Eguchi-Ishimae, M.; Kaneko, A.; et al. Identification of CD34+ and CD34- Leukemia-initiating cells in MLL-rearranged human acute lymphoblastic Leukemia. Blood 2015, 125, 967–980. [Google Scholar] [CrossRef] [PubMed]
- Dialynas, D.P.; Shao, L.; Billman, G.F.; Yu, J. Engraftment of Human T-Cell Acute Lymphoblastic Leukemia in Immunodeficient NOD/SCID Mice Which Have Been Preconditioned by Injection of Human Cord Blood. Stem Cells 2001, 19, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Dialynas, D.P.; Lee, M.J.; Gold, D.P.; Shao, L.E.; Yu, A.L.; Borowitz, M.J.; Yu, J. Preconditioning with fetal cord blood facilitates engraftment of primary childhood T-cell acute lymphoblastic leukemia in immunodeficient mice. Blood 2001, 97, 3218–3225. [Google Scholar] [CrossRef]
- Mitchell, P.; Lee, F.T.; Hall, C.; Rigopoulos, A.; Smyth, F.E.; Hekman, A.M.; Van Schijndel, G.M.; Powles, R.; Brechbiel, M.W.; Scott, A.M. Targeting primary human Ph+ B-cell precursor leukemia-engrafted SCID mice using radiolabeled anti-CD19 monoclonal antibodies. J. Nucl. Med. 2003, 44, 1105–1112. [Google Scholar] [PubMed]
- Gopalakrishnapillai, A.; Kolb, E.A.; Dhanan, P.; Bojja, A.S.; Mason, R.W.; Corao, D.; Barwe, S.P. Generation of pediatric leukemia xenograft models in NSG-B2m mice: Comparison with NOD/SCID mice. Front. Oncol. 2016, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Woo, X.Y.; Giordano, J.; Srivastava, A.; Zhao, Z.M.; Lloyd, M.W.; de Bruijn, R.; Suh, Y.S.; Patidar, R.; Chen, L.; Scherer, S.; et al. Conservation of copy number profiles during engraftment and passaging of patient-derived cancer xenografts. Nat. Genet. 2021, 53, 86–99. [Google Scholar] [CrossRef] [PubMed]
- Morita, K.; Wang, F.; Jahn, K.; Hu, T.; Tanaka, T.; Sasaki, Y.; Kuipers, J.; Loghavi, S.; Wang, S.A.; Yan, Y.; et al. Clonal evolution of acute myeloid leukemia revealed by high-throughput single-cell genomics. Nat. Commun. 2020, 11, 1–17. [Google Scholar] [CrossRef]
- Melchor, L.; Brioli, A.; Wardell, C.P.; Murison, A.; Potter, N.E.; Kaiser, M.F.; Fryer, R.A.; Johnson, D.C.; Begum, D.B.; Wilson, S.H.; et al. Single-cell genetic analysis reveals the composition of initiating clones and phylogenetic patterns of branching and parallel evolution in myeloma. Leukemia 2014, 28, 1705–1715. [Google Scholar] [CrossRef] [PubMed]
- Clappier, E.; Gerby, B.; Sigaux, F.; Delord, M.; Touzri, F.; Hernandez, L.; Ballerini, P.; Baruchel, A.; Pflumio, F.; Soulier, J. Clonal selection in xenografted human T cell acute lymphoblastic leukemia recapitulates gain of malignancy at relapse. J. Exp. Med. 2011, 208, 653–661. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Laboratory ID | Subtype | Sex | Age at Sample Collection | Diagnosis | Age at Diagnosis | Sample Type | Cytogenetic Aberrations | Survival | Xeno | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ph+ | MLL | p16 | Other | Condition | Days Since dx | ||||||||
| 0054 | pro B-ALL | f | 70 | initial | 70 | bone marrow | dead | 358 | ✓ | ||||
| 0054 | pro B-ALL | f | 71 | relapse | 70 | blood | dead | 358 | |||||
| 0072 | pro B-ALL | m | 63 | initial | 63 | blood | near tetraploid | dead | 77 | ||||
| 0122 | pro B-ALL | m | 47 | initial | 47 | blood | +X, −, +21 | unknown | 2018 | ✓ | |||
| 0130 | pro B-ALL | f | 18 | initial | 18 | bone marrow | del(12)(p13) | dead | 160 | ||||
| 0134 | pro B-ALL | m | 43 | initial | 43 | bone marrow | +22 | dead | 205 | ✓ | |||
| 0152 | pro B-ALL, biphenotypic | f | 52 | initial | 52 | blood | dead | 66 | ✓ | ||||
| 0210 | pro B-ALL, biphenotypic | m | 67 | initial | 67 | bone marrow | del(6)(p21p25), ins(2;6)(p22;?p22?p23?), r(6)(p12q13), dic(16;17)(q11;p11) | alive | 237 | ||||
| 0012 | common B-ALL | f | 77 | relapse | 76 | blood | dead | 285 | |||||
| 0031 | common B-ALL | f | 37 | initial | 37 | unknown | IGH rearrangement | dead | 1186 | X | |||
| 0043 | common B-ALL | f | 40 | initial | 40 | blood | dead | 81 | ✓ | ||||
| 0062 | common B-ALL | m | 21 | initial | 21 | blood | del(6)(q?16q23), +19, add(20)(p) | alive | 4155 | ||||
| 0065 | common B-ALL | f | 20 | initial | 20 | blood | unknown | 4 | |||||
| 0071 | common B-ALL | m | 73 | initial | 73 | blood | dead | 12 | |||||
| 0082 | common B-ALL | m | 63 | initial | 63 | bone marrow | inv(3)(p21;q26), der(7;8)(q10;q10), del(7)(q22), del(13)(q14q31), −14, −15, −Y | alive | 3713 | ||||
| 0100 | common B-ALL | m | 42 | initial | 42 | bone marrow | +der(5), −22, +der(22)x2 | alive | 3457 | ||||
| 0125 | common B-ALL | f | 75 | initial | 75 | blood | unknown | - | |||||
| 0147 | common B-ALL | f | 55 | initial | 55 | blood | +6, +8, +11, +14, +21, +22 | unknown | 1784 | ||||
| 0154 | common B-ALL | m | 78 | initial | 78 | blood | +1, del(6)(q21q23), del(11)(q21q23), +19, +21, +21 | dead | 79 | ||||
| 0161 | common B-ALL | m | 23 | initial | 23 | bone marrow | +6, +11, +14, +21, +21 | unknown | 1853 | ||||
| 0168 | common B-ALL | f | 78 | initial | 78 | blood | −4, −7, −9, t(12;17)(q24;q21), −17, +mar | alive | 1997 | ||||
| 0183 | common B-ALL | f | 59 | initial | 59 | bone marrow | dead | 1396 | |||||
| 0188 | common B-ALL | f | 80 | relapse | 78 | blood | t(4;9)(p15;q21), −4, −7, −9, t(12;17)(q24;q12), −17, +mar | alive | 1994 | ||||
| 0202 | common B-ALL | m | 36 | initial | 36 | bone marrow | unknown | 343 | ✓ | ||||
| 0207 | common B-ALL | m | 46 | initial | 46 | bone marrow | unknown | 959 | |||||
| 0212 | common B-ALL | f | 62 | relapse | 62 | bone marrow | alive | 172 | ✓ | ||||
| 0213 | common B-ALL | m | 53 | initial | 53 | bone marrow | alive | 44 | |||||
| P67 | common B-ALL | m | 18 | initial | 18 | bone marrow | alive | 5180 | ✓ | ||||
| P74 | common B-ALL | f | 76 | initial | 76 | unknown | unknown | 287 | |||||
| 0141 | common B-ALL, biphenotypic | m | 43 | initial | 43 | bone marrow | +21 | alive | 2880 | ✓ | |||
| P33 | pre-B-ALL | m | 28 | initial | 28 | unknown | unknown | 274 | ✓ | ||||
| 0094 | mature B-ALL | f | 85 | relapse | 84 | blood | dead | 226 | ✓ | ||||
| 0074 | B-ALL, not further specified | m | 31 | initial | 31 | blood | del(1)(q23), del(12)(p13), +21 | dead | 35 | ✓ | |||
| 0138 | B-ALL, not further specified | f | 49 | initial | 49 | bone marrow | der(15), MYC rearrangement, TCF3 deletion | dead | 46 | ||||
| 0151 | B-ALL, not further specified | m | 84 | initial | 84 | blood | dead | 97 | ✓ | ||||
| 0159 | B-ALL, not further specified | f | 74 | initial | 74 | bone marrow | unknown | - | ✓ | ||||
| 0200 | B-ALL, not further specified | f | 37 | initial | 37 | blood | unknown | 5 | ✓ | ||||
| 0204 | B-ALL, not further specified | m | 20 | initial | 20 | blood | CRLF2 and IGH rearrangement | alive | 724 | ||||
| P21 | B-ALL, not further specified | f | 76 | relapse | 75 | blood | unknown | 2 | |||||
| P11 | B-ALL, not further specified | f | 22 | relapse | 19 | bone marrow | dead | 1384 | |||||
| P25 | pre T-ALL | m | 22 | initial | 22 | bone marrow | add(3)(q), del(7)(q31), +21 | unknown | 3561 | ||||
| 0019 | T-ALL, not further specified | m | 26 | initial | 26 | bone marrow | unknown | 2933 | ✓ | ||||
| 0070 | T-ALL, not further specified | m | 43 | initial | 43 | blood | unknown | 2094 | |||||
| 0170 | T-ALL, not further specified | f | 57 | initial | 57 | bone marrow | alive | 1890 | ✓ | ||||
| #0054 | #0094 | #0122 | #0134 | #0152 | #0159 | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Base Change | AA Change | Prim | P1 | Prim | P1 | P2 | P3 | P4 | Prim | P1 | P2 | P3 | Prim | P1 | P2 | P3 | Prim | P1 | P2 | Prim | P1 | P2 | P3 |
| APC | c.4326T > A | p.(=) | 0 | 0 | 51 | 45 | 45 | 45 | 46 | 0 | 0 | 0 | 48 | 48 | 45 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||
| 0 | 0 | 45 | 45 | |||||||||||||||||||||
| APC | c.4479G > a | p.(=) | 0 | 0 | 100 | 100 | 100 | 100 | 100 | 49 | 52 | 50 | 100 | 99 | 100 | 100 | 100 | 100 | 0 | 0 | 0 | 0 | ||
| 50 | 51 | 100 | 100 | |||||||||||||||||||||
| ATM | c.*29C > G | p.? | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 49 | 50 | 50 | 53 | ||||
| 0 | 0 | 0 | 0 | 0 | 0 | |||||||||||||||||||
| ATM | c.5793T > C | p.(=) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 52 | 50 | 50 | 51 | ||||
| 0 | 0 | 0 | 0 | 0 | 0 | |||||||||||||||||||
| BRAF | c.1789C > T | p.(=) | 47 | 52 | 51 | 49 | 47 | 0 | 0 | 0 | 51 | 48 | 49 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||||
| 0 | 0 | 0 | 0 | 46 | 50 | |||||||||||||||||||
| CSF1R | c.*36CA > TC | p.? | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 34 | 29 | 31 | 100 | 100 | 100 | 36 | 31 | 28 | 100 | 100 | 100 | 100 | ||
| 13 | 30 | 100 | 100 | |||||||||||||||||||||
| EGFR | c.2361G > A | p.(=) | 51 | 51 | 100 | 100 | 100 | 100 | 100 | 53 | 44 | 52 | 100 | 100 | 100 | 100 | 100 | 100 | 51 | 49 | 49 | 48 | ||
| 48 | 47 | 100 | 100 | |||||||||||||||||||||
| ERBB4 | c.421+58A > G | p.? | 59 | 59 | 0 | 0 | 0 | 0 | 0 | 100 | 100 | 100 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||
| 100 | 100 | 0 | 0 | |||||||||||||||||||||
| ERBB4 | c.884-20T > C | p.? | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||
| 0 | 0 | 0 | 0 | |||||||||||||||||||||
| FBXW7 | c.1524A > G | p.(=) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||
| 0 | 0 | 0 | 0 | |||||||||||||||||||||
| FGFR3 | c.1953G > A | p.(=) | 100 | 100 | 0 | 0 | 0 | 0 | 0 | 100 | 100 | 100 | 100 | 100 | 100 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||
| 100 | 100 | 100 | 100 | |||||||||||||||||||||
| FGFR3 | c.1959+22G > A | p.? | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||
| 49 | 50 | 0 | 0 | |||||||||||||||||||||
| FLT3 | c.1310-3T > C | p.? | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 0 | 0 | 0 | 100 | 100 | 100 | 100 | 100 | 100 | 51 | 52 | 52 | 52 | ||
| 0 | 0 | 100 | 100 | |||||||||||||||||||||
| FLT3 | c.1775T > C | p.Val592Ala | 0 | 0 | 44 | 50 | 51 | 51 | 49 | 0 | 0 | 0 | 51 | 52 | 49 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||
| 0 | 0 | 56 | 52 | |||||||||||||||||||||
| FLT3 | c.2504A > T | p.As p835Val | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||
| 0 | 0 | 0 | 0 | |||||||||||||||||||||
| HRAS | c.81T > C | p.(=) | 0 | 0 | 60 | 49 | 54 | 48 | 48 | 52 | 49 | 52 | 48 | 53 | 51 | 55 | 55 | 46 | 0 | 0 | 0 | 0 | ||
| 50 | 51 | 50 | 50 | |||||||||||||||||||||
| IDH1 | c.315C > T | p.(=) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 51 | 53 | 53 | 52 | ||
| 0 | 0 | 0 | 0 | |||||||||||||||||||||
| JAK3 | c.2164G > A | p.Val722le | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 51 | 49 | 49 | 52 | ||
| 0 | 0 | 0 | 0 | |||||||||||||||||||||
| KDR | c.798+54G > A | p.? | 0 | 0 | 52 | 44 | 48 | 45 | 47 | 49 | 50 | 45 | 46 | 48 | 52 | 100 | 100 | 100 | 100 | 98 | 98 | 100 | ||
| 48 | 46 | 50 | 51 | |||||||||||||||||||||
| KDR | c.4008C > T | p.(=) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 49 | 53 | 48 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||
| 49 | 48 | 0 | 0 | |||||||||||||||||||||
| KIT | c.1638A > G | p.(=) | 51 | 50 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||
| 0 | 0 | 0 | 0 | |||||||||||||||||||||
| KRAS | c.34G > A | p.Gly 12Ser | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 31 | 24 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||
| 6 | 7 | 0 | 0 | |||||||||||||||||||||
| MET | c.3029C > T | p.Thr 1010lle | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 51 | 50 | 50 | 50 | ||
| 0 | 0 | 0 | 0 | |||||||||||||||||||||
| NRAS | c.183A > C | p.Gln61His | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 6 | 0 | 0 | 0 | ||
| 0 | 0 | 0 | 0 | |||||||||||||||||||||
| PDGFRA | c.1701A > G | p.(=) | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | ||
| 100 | 100 | 100 | 100 | |||||||||||||||||||||
| PDGFRA | c.2472C > T | p.(=) | 49 | 47 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||
| 0 | 0 | 0 | 0 | |||||||||||||||||||||
| PIK3CA | c.352+40A > G | p.? | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 51 | 53 | 53 | 51 | ||
| 0 | 0 | 0 | 0 | |||||||||||||||||||||
| PIK3CA | c.1636C > A | p.Gln546Lys | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 4 | 0 | 0 | 0 | ||
| 0 | 0 | 0 | 0 | |||||||||||||||||||||
| PIK3CA | c.3075C > T | p.(=) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 51 | 50 | 50 | 52 | ||
| 0 | 0 | 0 | 0 | |||||||||||||||||||||
| RET | c.2307G > T | p.(=) | 48 | 51 | 54 | 49 | 52 | 54 | 50 | 0 | 0 | 0 | 51 | 50 | 54 | 54 | 50 | 49 | 100 | 100 | 100 | 100 | ||
| 0 | 0 | 51 | 52 | |||||||||||||||||||||
| RET | c.2712C > G | p.(=) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 51 | 50 | 50 | 51 | 47 | 47 | 49 | ||
| 0 | 0 | 0 | 0 | |||||||||||||||||||||
| SMAD4 | c.955+58C > T | p.? | 54 | 49 | 48 | 51 | 54 | 54 | 56 | 0 | 0 | 0 | 53 | 53 | 48 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||
| 0 | 0 | 51 | 48 | |||||||||||||||||||||
| STK11 | c.126G > C | p.(=) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 5 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||
| 0 | 0 | 0 | 0 | |||||||||||||||||||||
| STK11 | c.465-51T > C | p.? | 100 | 100 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 100 | 100 | 100 | 100 | ||
| 0 | 0 | 0 | 0 | |||||||||||||||||||||
| TP53 | c.215C > G | p.Pro72Arg | 99 | 100 | 94 | 49 | 49 | 50 | 50 | 100 | 100 | 100 | 52 | 53 | 53 | 100 | 100 | 99 | 56 | 52 | 52 | 49 | ||
| 98 | 99 | 51 | 61 | |||||||||||||||||||||
| TP53 | c.847C > A | p.Arg283Ser | 0 | 0 | 86 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||
| 0 | 0 | 0 | 0 | |||||||||||||||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Richter, A.; Roolf, C.; Sekora, A.; Knuebel, G.; Krohn, S.; Lange, S.; Krebs, V.; Schneider, B.; Lakner, J.; Wittke, C.; et al. The Molecular Subtype of Adult Acute Lymphoblastic Leukemia Samples Determines the Engraftment Site and Proliferation Kinetics in Patient-Derived Xenograft Models. Cells 2022, 11, 150. https://doi.org/10.3390/cells11010150
Richter A, Roolf C, Sekora A, Knuebel G, Krohn S, Lange S, Krebs V, Schneider B, Lakner J, Wittke C, et al. The Molecular Subtype of Adult Acute Lymphoblastic Leukemia Samples Determines the Engraftment Site and Proliferation Kinetics in Patient-Derived Xenograft Models. Cells. 2022; 11(1):150. https://doi.org/10.3390/cells11010150
Chicago/Turabian StyleRichter, Anna, Catrin Roolf, Anett Sekora, Gudrun Knuebel, Saskia Krohn, Sandra Lange, Vivien Krebs, Bjoern Schneider, Johannes Lakner, Christoph Wittke, and et al. 2022. "The Molecular Subtype of Adult Acute Lymphoblastic Leukemia Samples Determines the Engraftment Site and Proliferation Kinetics in Patient-Derived Xenograft Models" Cells 11, no. 1: 150. https://doi.org/10.3390/cells11010150
APA StyleRichter, A., Roolf, C., Sekora, A., Knuebel, G., Krohn, S., Lange, S., Krebs, V., Schneider, B., Lakner, J., Wittke, C., Kiefel, C., Jeremias, I., Murua Escobar, H., Vollmar, B., & Junghanss, C. (2022). The Molecular Subtype of Adult Acute Lymphoblastic Leukemia Samples Determines the Engraftment Site and Proliferation Kinetics in Patient-Derived Xenograft Models. Cells, 11(1), 150. https://doi.org/10.3390/cells11010150