
Insights into Modern Therapeutic Approaches in Pediatric Acute Leukemias


Abstract
1. Introduction
2. FDA-Approved Drugs to Treat ALL or AML
2.1. CAR-T Immunotherapy—Genetically Modified T Lymphocytes
2.1.1. Chimeric Antigen Receptor
2.1.2. Vectors
2.1.3. Treatment by CAR-T
2.2. Bispecific Antibodies
2.3. Antibody–Drug Conjugates (ADCs)
2.3.1. Inotuzumab Ozogamicin
2.3.2. Gemtuzumab Ozogamicin (Approved in 2017 for Pediatric >2 y.o. and Adult Cases of AML)
3. Future Perspectives in the Treatment of ALL or AML
3.1. Monoclonal Antibodies
3.1.1. Daratumumab
3.1.2. Alemtuzumab
3.1.3. Rituximab
3.1.4. Ofatumumab
3.1.5. Moxetumomab Pasudotox
3.1.6. Combotox
3.1.7. Denintuzumab Mafodotin (SGN-CD19A)
3.1.8. Loncastuximab Tesirine (ADCT-402)
3.1.9. Camidanlumab Tesirine (Cami-T or ADCT-301)
3.1.10. Coltuximab Ravtansine (SAR3419)
3.1.11. Epratuzumab
3.2. Immune Checkpoint Inhibitors
3.2.1. CTLA-4 Antibodies
3.2.2. Programmed Cell Death Protein (PD-1, CD279)
3.2.3. PD-1 Inhibitors
3.2.4. PD-L1 Inhibitors
3.3. Other Targets for Immune Checkpoint Inhibitors
3.3.1. Anti-CD47 Antibodies
3.3.2. Anti T-Cell Immunoglobulin and a Mucin-Domain Containing-3 (Tim-3) Antibody
3.4. Pattern Recognition Receptors (PRRs)
3.4.1. Toll-like Receptors (TLRs)
3.4.2. NOD-Like Receptors (NLRs)
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cancer Facts & Figures. 2020. Available online: https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/cancer-facts-figures-2020.html (accessed on 12 October 2021).
- Malard, F.; Mohty, M. Acute lymphoblastic leukaemia. Lancet 2020, 395, 1146–1162. [Google Scholar] [CrossRef]
- Inaba, H.; Pui, C.-H. Advances in the Diagnosis and Treatment of Pediatric Acute Lymphoblastic Leukemia. J. Clin. Med. 2021, 10, 1926. [Google Scholar] [CrossRef] [PubMed]
- Paulsson, K.; Lilljebjörn, H.; Biloglav, A.; Olsson, L.; Rissler, M.; Castor, A.; Barbany, G.; Fogelstrand, L.; Nordgren, A.; Sjögren, H.; et al. The genomic landscape of high hyperdiploid childhood acute lymphoblastic leukemia. Nat. Genet. 2015, 47, 672–676. [Google Scholar] [CrossRef]
- Paulsson, K.; Johansson, B. High hyperdiploid childhood acute lymphoblastic leukemia. Genes Chromosomes Cancer 2009, 48, 637–660. [Google Scholar] [CrossRef]
- Pui, C.-H. Precision medicine in acute lymphoblastic leukemia. Front. Med. 2020, 14, 689–700. [Google Scholar] [CrossRef]
- Safavi, S.; Paulsson, K. Near-haploid and low-hypodiploid acute lymphoblastic leukemia: Two distinct subtypes with consistently poor prognosis. Blood 2017, 129, 420–423. [Google Scholar] [CrossRef] [PubMed]
- Pui, C.-H.; Rebora, P.; Schrappe, M.; Attarbaschi, A.; Baruchel, A.; Basso, G.; Cavé, H.; Elitzur, S.; Koh, K.; Liu, H.-C.; et al. Outcome of Children With Hypodiploid Acute Lymphoblastic Leukemia: A Retrospective Multinational Study. J. Clin. Oncol. 2019, 37, 770–779. [Google Scholar] [CrossRef] [PubMed]
- Jeha, S.; Pei, D.; Raimondi, S.C.; Onciu, M.; Campana, D.; Cheng, C.; Sandlund, J.T.; Ribeiro, R.C.; Rubnitz, J.E.; Howard, S.C.; et al. Increased risk for CNS relapse in pre-B cell leukemia with the t(1;19)/TCF3-PBX1. Leukemia 2009, 23, 1406–1409. [Google Scholar] [CrossRef]
- Meyer, C.; Burmeister, T.; Gröger, D.; Tsaur, G.; Fechina, L.; Renneville, A.; Sutton, R.; Venn, N.C.; Emerenciano, M.; Pombo-De-Oliveira, M.S.; et al. The MLL recombinome of acute leukemias in 2017. Leukemia 2018, 32, 273–284. [Google Scholar] [CrossRef]
- Chaer, F.E.; Keng, M.; Ballen, K.K. MLL-Rearranged Acute Lymphoblastic Leukemia. Curr. Hematol. Malig. Rep. 2020, 15, 83–89. [Google Scholar] [CrossRef]
- Wang, H.; Han, P.; Qi, X.; Li, F.; Li, M.; Fan, L.; Zhang, H.; Zhang, X.; Yang, X. Bcl-2 Enhances Chimeric Antigen Receptor T Cell Persistence by Reducing Activation-Induced Apoptosis. Cancers 2021, 13, 197. [Google Scholar] [CrossRef] [PubMed]
- Fischer, U.; Forster, M.; Rinaldi, A.; Risch, T.; Sungalee, S.; Warnatz, H.-J.; Bornhauser, B.; Gombert, M.; Kratsch, C.; Stütz, A.M.; et al. Genomics and drug profiling of fatal TCF3-HLF-positive acute lymphoblastic leukemia identifies recurrent mutation patterns and therapeutic options. Nat. Genet. 2015, 47, 1020–1029. [Google Scholar] [CrossRef] [PubMed]
- Slayton, W.B.; Schultz, K.R.; Kairalla, J.A.; Devidas, M.; Mi, X.; Pulsipher, M.A.; Chang, B.H.; Mullighan, C.; Iacobucci, I.; Silverman, L.B.; et al. Dasatinib Plus Intensive Chemotherapy in Children, Adolescents, and Young Adults With Philadelphia Chromosome–Positive Acute Lymphoblastic Leukemia: Results of Children’s Oncology Group Trial AALL0622. J. Clin. Oncol. 2018, 36, 2306–2314. [Google Scholar] [CrossRef] [PubMed]
- Harrison, C.J.; Moorman, A.V.; Schwab, C.; Carroll, A.J.; Raetz, E.A.; Devidas, M.; Strehl, S.; Nebral, K.; Harbott, J. An international study of intrachromosomal amplification of chromosome 21 (iAMP21): Cytogenetic characterization and outcome. Leukemia 2013, 28, 1015–1021. [Google Scholar] [CrossRef] [PubMed]
- Tasian, S.K.; Loh, M.L.; Hunger, S.P. Philadelphia chromosome–like acute lymphoblastic leukemia. Blood 2017, 130, 2064–2072. [Google Scholar] [CrossRef]
- Jain, S.; Abraham, A. BCR-ABL1–like B-Acute Lymphoblastic Leukemia/Lymphoma: A Comprehensive Review. Arch. Pathol. Lab. Med. 2019, 144, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Boer, M.L.D.; Cario, G.; Moorman, A.V.; Boer, J.M.; Groot-Kruseman, H.A.D.; Fiocco, M.; Escherich, G.; Imamura, T.; Yeoh, A.; Sutton, R.; et al. Outcomes of paediatric patients with B-cell acute lymphocytic leukaemia with ABL-class fusion in the pre-tyrosine-kinase inhibitor era: A multicentre, retrospective, cohort study. Lancet Haematol. 2021, 8, e55–e66. [Google Scholar] [CrossRef]
- Liu, Y.; Easton, J.; Shao, Y.; Maciaszek, J.; Wang, Z.; Wilkinson, M.R.; Mccastlain, K.; Edmonson, M.; Pounds, S.B.; Shi, L.; et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat. Genet. 2017, 49, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- Rooij, J.D.; Zwaan, C.; Heuvel-Eibrink, M.V.D. Pediatric AML: From Biology to Clinical Management. J. Clin. Med. 2015, 4, 127–149. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Beau, M.M.L.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Lonetti, A.; Pession, A.; Masetti, R. Targeted Therapies for Pediatric AML: Gaps and Perspective. Front. Pediatrics 2019, 7, 463. [Google Scholar] [CrossRef]
- Creutzig, U.; Heuvel-Eibrink, M.M.V.D.; Gibson, B.; Dworzak, M.N.; Adachi, S.; Bont, E.D.; Harbott, J.; Hasle, H.; Johnston, D.; Kinoshita, A.; et al. Diagnosis and management of acute myeloid leukemia in children and adolescents: Recommendations from an international expert panel. Blood 2012, 120, 3187–3205. [Google Scholar] [CrossRef] [PubMed]
- Quessada, J.; Cuccuini, W.; Saultier, P.; Loosveld, M.; Harrison, C.J.; Lafage-Pochitaloff, M. Cytogenetics of Pediatric Acute Myeloid Leukemia: A Review of the Current Knowledge. Genes 2021, 12, 924. [Google Scholar] [CrossRef] [PubMed]
- Balgobind, B.V.; Hollink, I.H.I.M.; Arentsen-Peters, S.T.C.J.M.; Zimmermann, M.; Harbott, J.; Beverloo, H.B.; Bergh, A.R.M.V.; Cloos, J.; Kaspers, G.J.L.; Haas, V.D.; et al. Integrative analysis of type-I and type-II aberrations underscores the genetic heterogeneity of pediatric acute myeloid leukemia. Haematologica 2011, 96, 1478–1487. [Google Scholar] [CrossRef] [PubMed]
- Testi, A.M.; Pession, A.; Diverio, D.; Grimwade, D.; Gibson, B.; Azevedo, A.C.D.; Moran, L.; Leverger, G.; Elitzur, S.; Hasle, H.; et al. Risk-adapted treatment of acute promyelocytic leukemia: Results from the International Consortium for Childhood APL. Blood 2018, 132, 405–412. [Google Scholar] [CrossRef]
- Kutny, M.A.; Geyer, S.; Laumann, K.M.; Gregory, J.; Willman, C.L.; Stock, W.; Larson, R.A.; Powell, B.L.; Feusner, J.H. Outcome for pediatric acute promyelocytic leukemia patients at Childrens Oncology Group sites on the Leukemia Intergroup Study CALGB 9710 (Alliance). Pediatric Blood Cancer 2018, 66, e27542. [Google Scholar] [CrossRef]
- Balgobind, B.V.; Raimondi, S.C.; Harbott, J.; Zimmermann, M.; Alonzo, T.A.; Auvrignon, A.; Beverloo, H.B.; Chang, M.; Creutzig, U.; Dworzak, M.N.; et al. Novel prognostic subgroups in childhood 11q23/MLL-rearranged acute myeloid leukemia: Results of an international retrospective study. Blood 2009, 114, 2489–2496. [Google Scholar] [CrossRef]
- Lonetti, A.; Indio, V.; Laginestra, M.A.; Tarantino, G.; Chiarini, F.; Astolfi, A.; Bertuccio, S.N.; Martelli, A.M.; Locatelli, F.; Pession, A.; et al. Inhibition of Methyltransferase DOT1L Sensitizes to Sorafenib Treatment AML Cells Irrespective of MLL-Rearrangements: A Novel Therapeutic Strategy for Pediatric AML. Cancers 2020, 12, 1972. [Google Scholar] [CrossRef]
- Casillas, J.N.; Woods, W.G.; Hunger, S.P.; Mcgavran, L.; Alonzo, T.A.; Feig, S.A. Prognostic Implications of t(10;11) Translocations in Childhood Acute Myelogenous Leukemia: A Report From the Childrens Cancer Group. J. Pediatric Hematol. Oncol. 2003, 25, 594–600. [Google Scholar] [CrossRef][Green Version]
- Lopez, C.K.; Malinge, S.; Gaudry, M.; Bernard, O.A.; Mercher, T. Pediatric Acute Megakaryoblastic Leukemia: Multitasking Fusion Proteins and Oncogenic Cooperations. Trends Cancer 2017, 3, 631–642. [Google Scholar] [CrossRef] [PubMed]
- Rooij, J.D.E.D.; Branstetter, C.; Ma, J.; Li, Y.; Walsh, M.P.; Cheng, J.; Obulkasim, A.; Dang, J.; Easton, J.; Verboon, L.J.; et al. Pediatric non–Down syndrome acute megakaryoblastic leukemia is characterized by distinct genomic subsets with varying outcomes. Nat. Genet. 2017, 49, 451–456. [Google Scholar] [CrossRef]
- Hollink, I.H.I.M.; Heuvel-Eibrink, M.M.V.D.; Arentsen-Peters, S.T.; Pratcorona, M.; Abbas, S.; Kuipers, J.E.; Galen, J.F.V.; Beverloo, H.B.; Sonneveld, E.; Kaspers, G.-J.J.L.; et al. NUP98/NSD1 characterizes a novel poor prognostic group in acute myeloid leukemia with a distinct HOX gene expression pattern. Blood 2011, 118, 3645–3656. [Google Scholar] [CrossRef]
- Haferlach, T.; Kohlmann, A.; Klein, H.-U.; Ruckert, C.; Dugas, M.; Williams, P.M.; Kern, W.; Schnittger, S.; Bacher, U.; Löffler, H.; et al. AML with translocation t(8;16)(p11;p13) demonstrates unique cytomorphological, cytogenetic, molecular and prognostic features. Leukemia 2009, 23, 934–943. [Google Scholar] [CrossRef] [PubMed]
- Falini, B.; Nicoletti, I.; Bolli, N.; Martelli, M.P.; Liso, A.; Gorello, P.; Mandelli, F.; Mecucci, C.; Martelli, M.F. Translocations and mutations involving the nucleophosmin (NPM1) gene in lymphomas and leukemias. Haematologica 2007, 92, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Noort, S.; Zimmermann, M.; Reinhardt, D.; Cuccuini, W.; Pigazzi, M.; Smith, J.; Ries, R.E.; Alonzo, T.A.; Hirsch, B.; Tomizawa, D.; et al. Prognostic impact of t(16;21)(p11;q22) and t(16;21)(q24;q22) in pediatric AML: A retrospective study by the I-BFM Study Group. Blood 2018, 132, 1584–1592. [Google Scholar] [CrossRef] [PubMed]
- Wolman, S.R.; Gundacker, H.; Appelbaum, F.R.; Slovak, M.L. Impact of trisomy 8 (8) on clinical presentation, treatment response, and survival in acute myeloid leukemia: A Southwest Oncology Group study. Blood 2002, 100, 29–35. [Google Scholar] [CrossRef]
- Sandahl, J.D.; Kjeldsen, E.; Abrahamsson, J.; Ha, S.-Y.; Heldrup, J.; Jahnukainen, K.; Jónsson, Ó.G.; Lausen, B.; Palle, J.; Zeller, B.; et al. Ploidy and clinical characteristics of childhood acute myeloid leukemia: A NOPHO-AML study. Genes Chromosomes Cancer 2014, 53, 667–675. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, J.; Zhong, J.F.; Zhang, X. Engineering CAR-T cells. Biomark. Res. 2017, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Baldo, B.A. Chimeric Fusion Proteins Used for Therapy: Indications, Mechanisms, and Safety. Drug Saf. 2015, 38, 455–479. [Google Scholar] [CrossRef] [PubMed]
- Chandran, S.S.; Klebanoff, C.A. T cell receptor-based cancer immunotherapy: Emerging efficacy and pathways of resistance. Immunol. Rev. 2019, 290, 127–147. [Google Scholar] [CrossRef]
- Qin, L.; Lai, Y.; Zhao, R.; Wei, X.; Weng, J.; Lai, P.; Li, B.; Lin, S.; Wang, S.; Wu, Q.; et al. Incorporation of a hinge domain improves the expansion of chimeric antigen receptor T cells. J. Hematol. Oncol. 2017, 10, 68. [Google Scholar] [CrossRef]
- Dotti, G.; Gottschalk, S.; Savoldo, B.; Brenner, M.K. Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunol. Rev. 2013, 257, 107–126. [Google Scholar] [CrossRef]
- Hartmann, J.; Schüßler-Lenz, M.; Bondanza, A.; Buchholz, C.J. Clinical development of CAR T cells—challenges and opportunities in translating innovative treatment concepts. EMBO Mol. Med. 2017, 9, 1183–1197. [Google Scholar] [CrossRef]
- Sermer, D.; Brentjens, R. CAR T-cell therapy: Full speed ahead. Hematol. Oncol. 2019, 37, 95–100. [Google Scholar] [CrossRef]
- Srivastava, S.; Riddell, S.R. Chimeric Antigen Receptor T Cell Therapy: Challenges to Bench-to-Bedside Efficacy. J. Immunol. 2018, 200, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Iwakuma, T.; Cui, Y.; Chang, L.J. Self-Inactivating Lentiviral Vectors with U3 and U5 Modifications. Virology 1999, 261, 120–132. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.-P.; Harbottle, R.P. Genetic modification of dividing cells using episomally maintained S/MAR DNA vectors. Mol. Ther. Nucleic Acids 2013, 2, e115. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Fotaki, G.; Ramachandran, M.; Nilsson, B.; Essand, M.; Yu, D. Safe engineering of CAR T cells for adoptive cell therapy of cancer using long-term episomal gene transfer. EMBO Mol. Med. 2016, 8, 702–711. [Google Scholar] [CrossRef]
- Schmidt-Wolf, I.G.; Negrin, R.S.; Kiem, H.P.; Blume, K.G.; Weissman, I.L. Use of a SCID mouse/human lymphoma model to evaluate cytokine-induced killer cells with potent antitumor cell activity. J. Exp. Med. 1991, 174, 139–149. [Google Scholar] [CrossRef]
- Ivics, Z.; Hackett, P.B.; Plasterk, R.H.; Izsvák, Z. Molecular Reconstruction of Sleeping Beauty, a Tc1-like Transposon from Fish, and Its Transposition in Human Cells. Cell 1997, 91, 501–510. [Google Scholar] [CrossRef]
- Magnani, C.F.; Gaipa, G.; Lussana, F.; Belotti, D.; Gritti, G.; Napolitano, S.; Matera, G.; Cabiati, B.; Buracchi, C.; Borleri, G.; et al. Sleeping Beauty–engineered CAR T cells achieve antileukemic activity without severe toxicities. J. Clin. Investig. 2020, 130, 6021–6033. [Google Scholar] [CrossRef]
- Yakoub-Agha, I.; Chabannon, C.; Bader, P.; Basak, G.W.; Bonig, H.; Ciceri, F.; Corbacioglu, S.; Duarte, R.F.; Einsele, H.; Hudecek, M.; et al. Management of adults and children undergoing chimeric antigen receptor T-cell therapy: Best practice recommendations of the European Society for Blood and Marrow Transplantation (EBMT) and the Joint Accreditation Committee of ISCT and EBMT (JACIE). Haematologica 2019, 105, 297–316. [Google Scholar] [CrossRef] [PubMed]
- Turtle, C.J.; Hanafi, L.-A.; Berger, C.; Gooley, T.A.; Cherian, S.; Hudecek, M.; Sommermeyer, D.; Melville, K.; Pender, B.; Budiarto, T.M.; et al. CD19 CAR–T cells of defined CD4: CD8 composition in adult B cell ALL patients. J. Clin. Investig. 2016, 126, 2123–2138. [Google Scholar] [CrossRef]
- Marks, P. The FDA’s Regulatory Framework for Chimeric Antigen Receptor-T Cell Therapies. Clin. Transl. Sci. 2019, 12, 428–430. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Teachey, D.T.; Rheingold, S.R.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Barker, C.S.; Callahan, C.; Frey, N.V.; Nazimuddin, F.; et al. Sustained remissions with CD19-specific chimeric antigen receptor (CAR)-modified T cells in children with relapsed/refractory ALL. J. Clin. Oncol. 2016, 34, 3011. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Wang, J.; Hu, Y.; Huang, H. Acute lymphoblastic leukemia relapse after CD19-targeted chimeric antigen receptor T cell therapy. J. Leukoc. Biol. 2017, 102, 1347–1356. [Google Scholar] [CrossRef]
- Zhang, L.-N.; Song, Y.; Liu, D. CD19 CAR-T cell therapy for relapsed/refractory acute lymphoblastic leukemia: Factors affecting toxicities and long-term efficacies. J. Hematol. Oncol. 2018, 11, 41. [Google Scholar] [CrossRef]
- Davila, M.L.; Riviere, I.; Wang, X.; Bartido, S.; Park, J.; Curran, K.; Chung, S.S.; Stefanski, J.; Borquez-Ojeda, O.; Olszewska, M.; et al. Efficacy and Toxicity Management of 19-28z CAR T Cell Therapy in B Cell Acute Lymphoblastic Leukemia. Sci. Transl. Med. 2014, 6, 224ra25. [Google Scholar] [CrossRef]
- Lee, D.W.; Kochenderfer, J.N.; Stetler-Stevenson, M.; Cui, Y.K.; Delbrook, C.; Feldman, S.A.; Fry, T.J.; Orentas, R.; Sabatino, M.; Shah, N.N.; et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial. Lancet 2015, 385, 517–528. [Google Scholar] [CrossRef]
- Pan, J.; Niu, Q.; Deng, B.; Liu, S.; Wu, T.; Gao, Z.; Liu, Z.; Zhang, Y.; Qu, X.; Zhang, Y.; et al. CD22 CAR T-cell therapy in refractory or relapsed B acute lymphoblastic leukemia. Leukemia 2019, 33, 2854–2866. [Google Scholar] [CrossRef]
- Schultz, L.; Mackall, C. Driving CAR T cell translation forward. Sci. Transl. Med. 2019, 11, eaaw2127. [Google Scholar] [CrossRef]
- Sotillo, E.; Barrett, D.M.; Black, K.L.; Bagashev, A.; Oldridge, D.; Wu, G.; Sussman, R.; Lanauze, C.; Ruella, M.; Gazzara, M.R.; et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov. 2015, 5, 1282–1295. [Google Scholar] [CrossRef]
- Klesmith, J.R.; Wu, L.; Lobb, R.R.; Rennert, P.D.; Hackel, B.J. Fine Epitope Mapping of the CD19 Extracellular Domain Promotes. Design. Biochem. 2019, 58, 4869–4881. [Google Scholar] [CrossRef] [PubMed]
- Teachey, D.T.; Lacey, S.F.; Shaw, P.A.; Melenhorst, J.J.; Maude, S.L.; Frey, N.; Pequignot, E.; Gonzalez, V.E.; Chen, F.; Finklestein, J.; et al. Identification of Predictive Biomarkers for Cytokine Release Syndrome after Chimeric Antigen Receptor T-cell Therapy for Acute Lymphoblastic Leukemia. Cancer Discov. 2016, 6, 664–679. [Google Scholar] [CrossRef]
- Bonifant, C.L.; Jackson, H.J.; Brentjens, R.J.; Curran, K.J. Toxicity and management in CAR T-cell therapy. Mol. Ther. Oncolytics 2016, 3, 16011. [Google Scholar] [CrossRef]
- Gofshteyn, J.S.; Shaw, P.A.; Teachey, D.T.; Grupp, S.A.; Maude, S.; Banwell, B.; Chen, F.; Lacey, S.F.; Melenhorst, J.J.; Edmonson, M.J.; et al. Neurotoxicity after CTL019 in a pediatric and young adult cohort. Ann. Neurol. 2018, 84, 537–546. [Google Scholar] [CrossRef]
- Trede, N.; Juno, T. Study Evaluating the Efficacy and Safety of JCAR015 in Adult B-Cell Acute Lymphoblastic Leukemia (B-ALL) (ROCKET). Available online: https://clinicaltrials.gov/ct2/show/NCT02535364 (accessed on 12 October 2021).
- Sterner, R.M.; Sakemura, R.; Cox, M.J.; Yang, N.; Khadka, R.H.; Forsman, C.L.; Hansen, M.J.; Jin, F.; Ayasoufi, K.; Hefazi, M.; et al. GM-CSF inhibition reduces cytokine release syndrome and neuroinflammation but enhances CAR-T cell function in xenografts. Blood 2019, 133, 697–709. [Google Scholar] [CrossRef]
- Cooper, M.L.; Dipersio, J.F. Chimeric antigen receptor T cells (CAR-T) for the treatment of T-cell malignancies. Best Pract. Res. Clin. Haematol. 2019, 32, 101097. [Google Scholar] [CrossRef]
- Li, S.; Wang, X.; Yuan, Z.; Liu, L.; Luo, L.; Li, Y.; Wu, K.; Liu, J.; Yang, C.; Li, Z.; et al. Eradication of T-ALL Cells by CD7-targeted Universal CAR-T Cells and Initial Test of Ruxolitinib-based CRS Management. Clin. Cancer Res. 2020, 27, 1242–1246. [Google Scholar] [CrossRef]
- Sánchez-Martínez, D.; Baroni, M.L.; Gutierrez-Agüera, F.; Roca-Ho, H.; Blanch-Lombarte, O.; González-García, S.; Torrebadell, M.; Junca, J.; Ramírez-Orellana, M.; Velasco-Hernández, T.; et al. Fratricide-resistant CD1a-specific CAR T cells for the treatment of cortical T-cell acute lymphoblastic leukemia. Blood 2019, 133, 2291–2304. [Google Scholar] [CrossRef]
- Neeson, P.; Shin, A.; Tainton, K.M.; Guru, P.; Prince, H.M.; Harrison, S.J.; Peinert, S.; Smyth, M.J.; Trapani, J.A.; Kershaw, M.H.; et al. Ex vivo culture of chimeric antigen receptor T cells generates functional CD8 T cells with effector and central memory-like phenotype. Gene Ther. 2010, 17, 1105–1116. [Google Scholar] [CrossRef]
- Ritchie, D.S.; Neeson, P.J.; Khot, A.; Peinert, S.; Tai, T.; Tainton, K.; Chen, K.; Shin, M.; Wall, D.M.; Hönemann, D.; et al. Persistence and Efficacy of Second-Generation CAR T Cell Against the LeY Antigen in Acute Myeloid Leukemia. Mol. Ther. 2013, 21, 2122–2129. [Google Scholar] [CrossRef]
- Ehninger, A.; Kramer, M.; Röllig, C.; Thiede, C.; Bornhäuser, M.; Bonin, M.V.; Wermke, M.; Feldmann, A.; Bachmann, M.; Ehninger, G.; et al. Distribution and levels of cell surface expression of CD33 and CD123 in acute myeloid leukemia. Blood Cancer J. 2014, 4, e218. [Google Scholar] [CrossRef]
- Bordignon, C.; Bonini, C.; Verzeletti, S.; Nobili, N.; Maggioni, D.; Traversari, C.; Giavazzi, R.; Servida, P.; Zappone, E.; Benazzi, E.; et al. Transfer of the HSV-tk Gene into Donor Peripheral Blood Lymphocytes for In Vivo Modulation of Donor Anti-Tumor Immunity after Allogeneic Bone Marrow Transplantation. The San Raffaele Hospital, Milan, Italy. Hum. Gene Ther. 1995, 6, 813–819. [Google Scholar] [CrossRef] [PubMed]
- Gargett, T.; Brown, M.P. The inducible caspase-9 suicide gene system as a “safety switch” to limit on-target, off-tumor toxicities of chimeric antigen receptor T cells. Front. Pharmacol. 2014, 5, 235. [Google Scholar] [CrossRef]
- Straathof, K.C.; Pulè Martin, A.; Yotnda, P.; Dotti, G.; Vanin, E.F.; Brenner, M.K.; Heslop, H.E.; Spencer, D.M.; Rooney, C.M. An inducible caspase 9 safety switch for T-cell therapy. Blood 2005, 105, 4247–4254. [Google Scholar] [CrossRef]
- Hoyos, V.; Savoldo, B.; Quintarelli, C.; Mahendravada, A.; Zhang, M.; Vera, J.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; Dotti, G. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia 2010, 24, 1160–1170. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef]
- Goswami, M.; Hourigan, C. Novel Antigen Targets for Immunotherapy of Acute Myeloid Leukemia. Curr. Drug Targets 2017, 18, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Rafiq, S.; Purdon, T.J.; Daniyan, A.F.; Koneru, M.; Dao, T.; Liu, C.; Scheinberg, D.A.; Brentjens, R.J. Optimized T-cell receptor-mimic chimeric antigen receptor T cells directed toward the intracellular Wilms Tumor 1 antigen. Leukemia 2016, 31, 1788–1797. [Google Scholar] [CrossRef]
- Hoffmann, P.; Hofmeister, R.; Brischwein, K.; Brandl, C.; Crommer, S.; Bargou, R.; Itin, C.; Prang, N.; Baeuerle, P.A. Serial killing of tumor cells by cytotoxic T cells redirected with a CD19-/CD3-bispecific single-chain antibody construct. Int. J. Cancer 2005, 115, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, U.; Kontermann, R.E. The making of bispecific antibodies. Mabs 2017, 9, 182–212. [Google Scholar] [CrossRef] [PubMed]
- Viardot, A.; Goebeler, M.; Scheele, J.S.; Zugmaier, G.; Noppeney, R.; Knop, S.; Topp, M.S.; Nagorsen, D.; Klinger, M.; Schmidt, M.; et al. Treatment of Patients with Non-Hodgkin Lymphoma (NHL) with CD19/CD3 Bispecific Antibody Blinatumomab (MT103): Double-Step Dose Increase to Continuous Infusion of 60 μg/m2/d Is Tolerable and Highly Effective. Blood 2010, 116, 2880. [Google Scholar] [CrossRef]
- Topp, M.S.; Kufer, P.; Gökbuget, N.; Goebeler, M.; Klinger, M.; Neumann, S.; Horst, H.-A.; Raff, T.; Viardot, A.; Schmid, M.; et al. Targeted Therapy With the T-Cell–Engaging Antibody Blinatumomab of Chemotherapy-Refractory Minimal Residual Disease in B-Lineage Acute Lymphoblastic Leukemia Patients Results in High Response Rate and Prolonged Leukemia-Free Survival. J. Clin. Oncol. 2011, 29, 2493–2498. [Google Scholar] [CrossRef]
- Przepiorka, D.; Ko, C.-W.; Deisseroth, A.; Yancey, C.L.; Candau-Chacon, R.; Chiu, H.-J.; Gehrke, B.J.; Gomez-Broughton, C.; Kane, R.C.; Kirshner, S.; et al. FDA Approval: Blinatumomab. Clin. Cancer Res. 2015, 21, 4035–4039. [Google Scholar] [CrossRef]
- Stackelberg, A.V.; Locatelli, F.; Zugmaier, G.; Handgretinger, R.; Trippett, T.M.; Rizzari, C.; Bader, P.; O’Brien, M.M.; Brethon, B.; Bhojwani, D.; et al. Phase I/Phase II Study of Blinatumomab in Pediatric Patients With Relapsed/Refractory Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2016, 34, 4381–4389. [Google Scholar] [CrossRef]
- Locatelli, F.; Zugmaier, G.; Mergen, N.; Bader, P.; Jeha, S.; Schlegel, P.-G.; Bourquin, J.-P.; Handgretinger, R.; Brethon, B.; Rossig, C.; et al. Blinatumomab in pediatric patients with relapsed/refractory acute lymphoblastic leukemia: Results of the RIALTO trial, an expanded access study. Blood Cancer J. 2020, 10, 77. [Google Scholar] [CrossRef] [PubMed]
- Queudeville, M.; Schlegel, P.; Heinz, A.T.; Lenz, T.; Döring, M.; Holzer, U.; Hartmann, U.; Kreyenberg, H.; Stackelberg, A.; Schrappe, M.; et al. Blinatumomab in pediatric patients with relapsed/refractory B-cell precursor acute lymphoblastic leukemia. Eur. J. Haematol. 2021, 106, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, F.; Zugmaier, G.; Rizzari, C.; Morris, J.D.; Gruhn, B.; Klingebiel, T.; Parasole, R.; Linderkamp, C.; Flotho, C.; Petit, A.; et al. Effect of Blinatumomab vs Chemotherapy on Event-Free Survival Among Children With High-risk First-Relapse B-Cell Acute Lymphoblastic Leukemia. JAMA 2021, 325, 843. [Google Scholar] [CrossRef]
- Brown, P.A.; Ji, L.; Xu, X.; Devidas, M.; Hogan, L.E.; Borowitz, M.J.; Raetz, E.A.; Zugmaier, G.; Sharon, E.; Bernhardt, M.B.; et al. Effect of Postreinduction Therapy Consolidation With Blinatumomab vs Chemotherapy on Disease-Free Survival in Children, Adolescents, and Young Adults With First Relapse of B-Cell Acute Lymphoblastic Leukemia. JAMA 2021, 325, 833. [Google Scholar] [CrossRef]
- Karachunskiy, A.I. Federal Research Institute of Pediatric Hematology, Oncology and Immunology. Acute Lymphoblastic Leukemia Treatment Protocol Moscow-Berlin 2019 Pilot. Available online: https://clinicaltrials.gov/ct2/show/NCT04723342 (accessed on 12 October 2021).
- Schrappe, M.; University Hospital of Schleswig-Holstein. Treatment Protocol for Children and Adolescents With Acute Lymphoblastic Leukemia AIEOP-BFM ALL 2017. Available online: https://clinicaltrials.gov/ct2/show/NCT03643276 (accessed on 12 October 2021).
- A Pilot Study to Test the Feasibility, Safety and Efficacy of the Addition of the BiTE Antibody Blinatumomab to the Interfant-06 Backbone in Infants with MLL-Rearranged Acute Lymphoblastic Leukemia. A Collaborative Study of the Interfant Network. Available online: https://www.clinicaltrialsregister.eu/ctr-search/trial/2016-004674-17/AT (accessed on 12 October 2021).
- Burke, M. Medical College of Wisconsin. Blinatumomab Bridging Therapy. Available online: https://clinicaltrials.gov/ct2/show/NCT04556084 (accessed on 12 October 2021).
- Gupta, S.; Children’s Oncology Group. A Study to Investigate Blinatumomab in Combination with Chemotherapy in Patients With Newly Diagnosed B-Lymphoblastic Leukemia. Available online: https://clinicaltrials.gov/ct2/show/NCT03914625 (accessed on 12 October 2021).
- Gojo, I.; JHU Sidney Kimmel Comprehensive Cancer Center LAO. Blinatumomab and Nivolumab with or Without Ipilimumab in Treating Patients with Poor-Risk Relapsed or Refractory CD19+ Precursor B-Lymphoblastic Leukemia. Available online: https://clinicaltrials.gov/ct2/show/NCT02879695 (accessed on 12 October 2021).
- Clark, M.C.; Stein, A. CD33 directed bispecific antibodies in acute myeloid leukemia. Best Pract. Res. Clin. Haematol. 2020, 33, 101224. [Google Scholar] [CrossRef] [PubMed]
- Kuo, S.-R.; Wong, L.; Liu, J.-S. Engineering a CD123xCD3 bispecific scFv immunofusion for the treatment of leukemia and elimination of leukemia stem cells. Protein Eng. Des. Sel. 2012, 25, 561–570. [Google Scholar] [CrossRef]
- Liu, L.; Lam, C.-Y.K.; Long, V.; Widjaja, L.; Yang, Y.; Li, H.; Jin, L.; Burke, S.; Gorlatov, S.; Brown, J.; et al. MGD011, A CD19 x CD3 Dual-Affinity Retargeting Bi-specific Molecule Incorporating Extended Circulating Half-life for the Treatment of B-Cell Malignancies. Clin. Cancer Res. 2016, 23, 1506–1518. [Google Scholar] [CrossRef]
- Rader, C. DARTs take aim at BiTEs. Blood 2011, 117, 4403–4404. [Google Scholar] [CrossRef]
- Shi, M.; Su, R.J.; Parmar, K.-P.; Chaudhry, R.; Sun, K.; Rao, J.; Chen, M. CD123: A Novel Biomarker for Diagnosis and Treatment of Leukemia. Cardiovasc. Hematol. Disord. Drug Targets 2019, 19, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, L.; Nomdedéu, J.F.; López, O.; Carnicer, M.J.; Bellido, M.; Aventin, A.; Brunet, B.; Sierra, J. Interleukin-3 receptor alpha chain (CD123) is widely expressed in hematologic malignancies. Haematologica 2001, 86, 1261–1269. [Google Scholar]
- Liu, K.; Zhu, M.; Huang, Y.; Wei, S.; Xie, J.; Xiao, Y. CD123 and its potential clinical application in leukemias. Life Sci. 2015, 122, 59–64. [Google Scholar] [CrossRef]
- Uy, G.L.; Aldoss, I.; Foster, M.C.; Sayre, P.H.; Wieduwilt, M.J.; Advani, A.S.; Godwin, J.E.; Arellano, M.L.; Sweet, K.L.; Emadi, A.; et al. Flotetuzumab as salvage immunotherapy for refractory acute myeloid leukemia. Blood 2021, 137, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Lamble, A.L. Pediatric Early Phase Clinical Trial Network. Flotetuzumab for the Treatment of Pediatric Recurrent or Refractory Acute Myeloid Leukemia. Available online: https://clinicaltrials.gov/ct2/show/NCT04158739 (accessed on 12 October 2021).
- Aldoss, I.T.; City of Hope Medical Center. Flotetuzumab for the Treatment of Relapsed or Refractory Advanced CD123-Positive Hematological Malignancies. Available online: https://clinicaltrials.gov/ct2/show/NCT04681105 (accessed on 12 October 2021).
- Ward, A. MacroGenics. Flotetuzumab Expanded Access Program. Available online: https://clinicaltrials.gov/ct2/show/NCT04678466 (accessed on 12 October 2021).
- Martens, A.W.J.; Janssen, S.R.; Derks, I.A.M.; III, H.C.A.; Izhak, L.; Kampen, R.V.; Tonino, S.H.; Eldering, E.; van der Windt, G.J.W.; Kater, A.P. CD3xCD19 DART molecule treatment induces non-apoptotic killing and is efficient against high-risk chemotherapy and venetoclax-resistant chronic lymphocytic leukemia cells. J. Immunother. Cancer 2020, 8, e000218. [Google Scholar] [CrossRef]
- Circosta, P.; Elia, A.R.; Landra, I.; Machiorlatti, R.; Todaro, M.; Aliberti, S.; Brusa, D.; Deaglio, S.; Chiaretti, S.; Bruna, R.; et al. Tailoring CD19xCD3-DART exposure enhances T-cells to eradication of B-cell neoplasms. OncoImmunology 2017, 7, e1341032. [Google Scholar] [CrossRef] [PubMed]
- Lorenczewski, G.; Friedrich, M.; Kischel, R.; Dahlhoff, C.; Anlahr, J.; Balazs, M.; Rock, D.; Boyle, M.C.; Goldstein, R.; Coxon, A.; et al. Generation of a Half-Life Extended Anti-CD19 BiTE® Antibody Construct Compatible with Once-Weekly Dosing for Treatment of CD19-Positive Malignancies. Blood 2017, 130, 2815. [Google Scholar]
- Ravandi, F.; Walter, R.B.; Subklewe, M.; Buecklein, V.; Jongen-Lavrencic, M.; Paschka, P.; Ossenkoppele, G.J.; Kantarjian, H.M.; Hindoyan, A.; Agarwal, S.K.; et al. Updated results from phase I dose-escalation study of AMG 330, a bispecific T-cell engager molecule, in patients with relapsed/refractory acute myeloid leukemia (R/R AML). J. Clin. Oncol. 2020, 38, 7508. [Google Scholar] [CrossRef]
- Subklewe, M.; Stein, A.; Walter, R.B.; Bhatia, R.; Wei, A.H.; Ritchie, D.; Bücklein, V.; Vachhani, P.; Dai, T.; Hindoyan, A.; et al. Preliminary Results from a Phase 1 First-in-Human Study of AMG 673, a Novel Half-Life Extended (HLE) Anti-CD33/CD3 BiTE® (Bispecific T-Cell Engager) in Patients with Relapsed/Refractory (R/R) Acute Myeloid Leukemia (AML). Blood 2019, 134, 833. [Google Scholar] [CrossRef]
- Yurkiewicz, I.R.; Muffly, L.; Liedtke, M. Inotuzumab ozogamicin: A CD22 mAb–drug conjugate for adult relapsed or refractory B-cell precursor acute lymphoblastic leukemia. Drug Des. Dev. Ther. 2018, 12, 2293–2300. [Google Scholar] [CrossRef]
- Hafeez, U.; Parakh, S.; Gan, H.K.; Scott, A.M. Antibody–Drug Conjugates for Cancer Therapy. Molecules 2020, 25, 4764. [Google Scholar] [CrossRef]
- Inaba, H.; Pui, C.-H. Immunotherapy in pediatric acute lymphoblastic leukemia. Cancer Metastasis Rev. 2019, 38, 595–610. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; Deangelo, D.J.; Stelljes, M.; Martinelli, G.; Liedtke, M.; Stock, W.; Gökbuget, N.; O’Brien, S.; Wang, K.; Wang, T.; et al. Inotuzumab Ozogamicin versus Standard Therapy for Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2016, 375, 740–753. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.M.; Deangelo, D.J.; Stelljes, M.; Liedtke, M.; Stock, W.; Gökbuget, N.; O’Brien, S.M.; Jabbour, E.; Wang, T.; White, J.L.; et al. Inotuzumab ozogamicin versus standard of care in relapsed or refractory acute lymphoblastic leukemia: Final report and long-term survival follow-up from the randomized, phase 3 INO-VATE study. Cancer 2019, 125, 2474–2487. [Google Scholar] [CrossRef] [PubMed]
- Bhojwani, D.; Sposto, R.; Shah, N.N.; Rodriguez, V.; Yuan, C.; Stetler-Stevenson, M.; O’Brien, M.M.; Mcneer, J.L.; Quereshi, A.; Cabannes, A.; et al. Inotuzumab ozogamicin in pediatric patients with relapsed/refractory acute lymphoblastic leukemia. Leukemia 2018, 33, 884–892. [Google Scholar] [CrossRef]
- Calvo, C.; Cabannes-Hamy, A.; Adjaoud, D.; Bruno, B.; Blanc, L.; Boissel, N.; Tabone, M.D.; Willson-Plat, G.; Villemonteix, J.; Baruchel, A.; et al. Inotuzumab ozogamicin compassionate use for French paediatric patients with relapsed or refractory CD22-positive B-cell acute lymphoblastic leukaemia. Br. J. Haematol. 2020, 190, e53–e56. [Google Scholar] [CrossRef]
- Brivio, E.; Locatelli, F.; Lopez-Yurda, M.; Malone, A.; Díaz-De-Heredia, C.; Bielorai, B.; Rossig, C.; van der Velden, V.H.J.; Ammerlaan, A.C.J.; Thano, A. A phase 1 study of inotuzumab ozogamicin in pediatric relapsed/refractory acute lymphoblastic leukemia (ITCC-059 study). Blood 2021, 137, 1582–1590. [Google Scholar] [CrossRef] [PubMed]
- Duong, H.; Sekeres, M. Targeted treatment of acute myeloid leukemia in older adults: Role of gemtuzumab ozogamicin. Clin. Interv. Aging 2009, 4, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Hammood, M.; Craig, A.; Leyton, J.V. Impact of Endocytosis Mechanisms for the Receptors Targeted by the Currently Approved Antibody-Drug Conjugates (ADCs)—A Necessity for Future ADC Research and Development. Pharmaceuticals 2021, 14, 674. [Google Scholar] [CrossRef]
- Zein, N.; Poncin, M.; Nilakantan, R.; Ellestad, G.A. Calicheamicin gamma 1I and DNA: Molecular recognition process responsible for site-specificity. Science 1989, 244, 697–699. [Google Scholar] [CrossRef] [PubMed]
- Bross, P.F.; Beitz, J.; Chen, G.; Chen, X.H.; Duffy, E.; Kieffer, L.; Roy, S.; Sridhara, R.; Rahman, A.; Williams, G.; et al. Approval summary: Gemtuzumab ozogamicin in relapsed acute myeloid leukemia? Clin. Cancer Res. 2001, 7, 1490–1496. [Google Scholar]
- Larson, R.A.; Sievers, E.L.; Stadtmauer, E.A.; Löwenberg, B.; Estey, E.H.; Dombret, H.; Theobald, M.; Voliotis, D.; Bennett, J.M.; Richie, M.; et al. Final report of the efficacy and safety of gemtuzumab ozogamicin (Mylotarg) in patients with CD33-positive acute myeloid leukemia in first recurrence. Cancer 2005, 104, 1442–1452. [Google Scholar] [CrossRef] [PubMed]
- Arceci, R.J. Safety and efficacy of gemtuzumab ozogamicin in pediatric patients with advanced CD33 acute myeloid leukemia. Blood 2005, 106, 1183–1188. [Google Scholar] [CrossRef]
- Petersdorf, S.H.; Kopecky, K.J.; Slovak, M.; Willman, C.; Nevill, T.; Brandwein, J.; Larson, R.A.; Erba, H.P.; Stiff, P.J.; Stuart, R.K.; et al. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood 2013, 121, 4854–4860. [Google Scholar] [CrossRef] [PubMed]
- Gamis, A.S.; Alonzo, T.A.; Meshinchi, S.; Sung, L.; Gerbing, R.B.; Raimondi, S.C.; Hirsch, B.A.; Kahwash, S.B.; Heerema-Mckenney, A.; Winter, L.; et al. Gemtuzumab Ozogamicin in Children and Adolescents With De Novo Acute Myeloid Leukemia Improves Event-Free Survival by Reducing Relapse Risk: Results From the Randomized Phase III Childrens Oncology Group Trial AAML0531. J. Clin. Oncol. 2014, 32, 3021–3032. [Google Scholar] [CrossRef]
- Niktoreh, N.; Lerius, B.; Zimmermann, M.; Gruhn, B.; Escherich, G.; Bourquin, J.-P.; Dworzak, M.; Sramkova, L.; Rossig, C.; Creutzig, U.; et al. Gemtuzumab ozogamicin in children with relapsed or refractory acute myeloid leukemia: A report by Berlin-Frankfurt-Münster study group. Haematologica 2018, 104, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Pollard, J.A.; Guest, E.; Alonzo, T.A.; Gerbing, R.B.; Loken, M.R.; Brodersen, L.E.; Kolb, E.A.; Aplenc, R.; Meshinchi, S.; Raimondi, S.C.; et al. Gemtuzumab Ozogamicin Improves Event-Free Survival and Reduces Relapse in Pediatric KMT2A-Rearranged AML: Results From the Phase III Childrens Oncology Group Trial AAML0531. J. Clin. Oncol. 2021, 39, 3149–3160. [Google Scholar] [CrossRef] [PubMed]
- Raedler, L.A. Darzalex (Daratumumab): First Anti-CD38 Monoclonal Antibody Approved for Patients with Relapsed Multiple Myeloma. Am. Health Drug Benefits 2016, 9, 70–73. [Google Scholar] [PubMed]
- Overdijk, M.B.; Verploegen, S.; Bögels, M.; Egmond, M.V.; Bueren, J.J.L.V.; Mutis, T.; Groen, R.W.; Breij, E.; Martens, A.C. Antibody-mediated phagocytosis contributes to the anti-tumor activity of the therapeutic antibody daratumumab in lymphoma and multiple myeloma. MAbs 2015, 7, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Morandi, F.; Horenstein, A.L.; Costa, F.; Giuliani, N.; Pistoia, V.; Malavasi, F. CD38: A Target for Immunotherapeutic Approaches in Multiple Myeloma. Front. Immunol. 2018, 9, 2722. [Google Scholar] [CrossRef] [PubMed]
- Roccatello, D.; Fenoglio, R.; Sciascia, S.; Naretto, C.; Rossi, D.; Ferro, M.; Barreca, A.; Malavasi, F.; Baldovino, S. CD38 and Anti-CD38 Monoclonal Antibodies in AL Amyloidosis: Targeting Plasma Cells and beyond. Int. J. Mol. Sci. 2020, 21, 4129. [Google Scholar] [CrossRef]
- Deaglio, S.; Vaisitti, T.; Billington, R.; Bergui, L.; Omede, P.; Genazzani, A.A.; Malavasi, F. CD38/CD19: A lipid raft–dependent signaling complex in human B cells. Blood 2007, 109, 5390–5398. [Google Scholar] [CrossRef]
- Konopleva, M.; Estrov, Z.; Zhao, S.; Andreeff, M.; Mehta, K. Ligation of Cell Surface CD38 Protein with Agonistic Monoclonal Antibody Induces a Cell Growth Signal in Myeloid Leukemia Cells. J. Immunol. 1998, 161, 4702–4708. [Google Scholar]
- Lokhorst, H.M.; Plesner, T.; Laubach, J.P.; Nahi, H.; Gimsing, P.; Hansson, M.; Minnema, M.C.; Lassen, U.; Krejcik, J.; Palumbo, A.; et al. Targeting CD38 with Daratumumab Monotherapy in Multiple Myeloma. N. Engl. J. Med. 2015, 373, 1207–1219. [Google Scholar] [CrossRef]
- Darzalex Full Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/761036s000lbl.pdf (accessed on 12 October 2021).
- Bride, K.L.; Vincent, T.L.; Im, S.-Y.; Aplenc, R.; Barrett, D.M.; Carroll, W.L.; Carson, R.; Dai, Y.; Devidas, M.; Dunsmore, K.P.; et al. Preclinical efficacy of daratumumab in T-cell acute lymphoblastic leukemia. Blood 2018, 131, 995–999. [Google Scholar] [CrossRef]
- Vogiatzi, F.; Winterberg, D.; Lenk, L.; Buchmann, S.; Cario, G.; Schrappe, M.; Peipp, M.; Richter-Pechanska, P.; Kulozik, A.E.; Lentes, J.; et al. Daratumumab eradicates minimal residual disease in a preclinical model of pediatric T-cell acute lymphoblastic leukemia. Blood 2019, 134, 713–716. [Google Scholar] [CrossRef]
- Janssen Research & Development. Study to Evaluate the Efficacy and Safety of Daratumumab in Pediatric and Young Adult Participants Greater Than or Equal to (>=)1 and Less Than or Equal to (<=)30 Years of Age with Relapsed/Refractory Precursor B-Cell or T-Cell Acute Lymphoblastic Leukemia or Lymphoblastic Lymphoma. Available online: https://clinicaltrials.gov/ct2/show/NCT03384654 (accessed on 12 October 2021).
- Li, Z.; Richards, S.; Surks, H.K.; Jacobs, A.; Panzara, M.A. Clinical pharmacology of alemtuzumab, an anti-CD52 immunomodulator, in multiple sclerosis. Clin. Exp. Immunol. 2018, 194, 295–314. [Google Scholar] [CrossRef] [PubMed]
- Frota, N.F.; Rebouças, A.D.S.; Fuzo, C.A.; Lourenzoni, M.R. Alemtuzumab scFv fragments and CD52 interaction study through molecular dynamics simulation and binding free energy. J. Mol. Graph. Model. 2021, 107, 107949. [Google Scholar] [CrossRef]
- Möhn, N.; Pfeuffer, S.; Ruck, T.; Gross, C.C.; Skripuletz, T.; Klotz, L.; Wiendl, H.; Stangel, M.; Meuth, S.G. Alemtuzumab therapy changes immunoglobulin levels in peripheral blood and CSF. Neurol. Neuroimmunol. Neuroinflamm. 2019, 7, e654. [Google Scholar] [CrossRef] [PubMed]
- Dumont, F.J. CAMPATH (alemtuzumab) for the treatment of chronic lymphocytic leukemia and beyond. Expert Rev. Anticancer Ther. 2002, 2, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Demko, S.; Summers, J.; Keegan, P.; Pazdur, R. FDA Drug Approval Summary: Alemtuzumab as Single-Agent Treatment for B-Cell Chronic Lymphocytic Leukemia. Oncologist 2008, 13, 167–174. [Google Scholar] [CrossRef]
- Lemtrada (Alemtuzumab) a New Treatment Option Approved by the FDA for the Treatment of Relapsing Forms of Multiple Sclerosis. Available online: https://www.ahdbonline.com/web-exclusives/1943-lemtrada-alemtuzumab-a-new-treatment-option-approved-by-the-fda-for-the-treatment-of-relapsing-forms-of-multiple-sclerosis (accessed on 12 October 2021).
- FDA Approves Lemtrada™ (Alemtuzumab) for Relapsing MS-UPDATE. Available online: https://www.nationalmssociety.org/About-the-Society/News/FDA-Approves-Lemtrada%E2%84%A2-(alemtuzumab)-for-Relapsing (accessed on 12 October 2021).
- Tibes, R.; Keating, M.J.; Ferrajoli, A.; Wierda, W.; Ravandi, F.; Garcia-Manero, G.; Obrien, S.; Cortes, J.; Verstovsek, S.; Browning, M.L.; et al. Activity of alemtuzumab in patients with CD52-positive acute leukemia. Cancer 2006, 106, 2645–2651. [Google Scholar] [CrossRef]
- Angiolillo, A.L.; Yu, A.L.; Reaman, G.; Ingle, A.M.; Secola, R.; Adamson, P.C. A phase II study of Campath-1H in children with relapsed or refractory acute lymphoblastic leukemia: A Childrens Oncology Group report. Pediatric Blood Cancer 2009, 53, 978–983. [Google Scholar] [CrossRef]
- Kennedy-Nasser, A.A.; Bollard, C.M.; Myers, G.D.; Leung, K.S.; Gottschalk, S.; Zhang, Y.; Liu, H.; Heslop, H.E.; Brenner, M.K.; Krance, R.A. Comparable Outcome of Alternative Donor and Matched Sibling Donor Hematopoietic Stem Cell Transplant for Children with Acute Lymphoblastic Leukemia in First or Second Remission Using Alemtuzumab in a Myeloablative Conditioning Regimen. Biol. Blood Marrow Transplant. 2008, 14, 1245–1252. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.A.N.; Kumar, R.; Altaf, S.; Gourde, J.A.; Rodriguez, V.; Khan, S.P. Pretransplant Conditioning With Campath-1H (Alemtuzumab) in Pediatric Matched Unrelated Hematopoietic Stem Cell Transplants. J. Pediatric Hematol. Oncol. 2012, 34, 96–100. [Google Scholar]
- Lindsay, J.; Kerridge, I.; Wilcox, L.; Tran, S.; Obrien, T.A.; Greenwood, M.; Chen, S.C.-A.; Kong, D.C.; Pergam, S.A.; Liu, C.; et al. Infection-Related Mortality in Adults and Children Undergoing Allogeneic Hematopoietic Cell Transplantation: An Australian Registry Report. Transplant. Cell. Ther. 2021, 27, 798.e1–798.e10. [Google Scholar] [CrossRef]
- Sikaria, S.; Aldoss, I.; Akhtari, M. Monoclonal antibodies and immune therapies for adult precursor B-acute lymphoblastic leukemia. Immunol. Lett. 2016, 172, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Cerny, T.; Borisch, B.; Introna, M.; Johnson, P.; Rose, A.L. Mechanism of action of rituximab. Anti Cancer Drugs 2002, 13, S3–S10. [Google Scholar] [CrossRef] [PubMed]
- James, J.S.; Dubs, G. FDA approves new kind of lymphoma treatment. Food and Drug Administration. AIDS Treat. News 1997, 284, 2–3. [Google Scholar]
- Jaglowski, S.M.; Byrd, J.C. Rituximab in Chronic Lymphocytic Leukemia. Semin. Hematol. 2010, 47, 156–169. [Google Scholar] [CrossRef]
- Mok, C.C. Rituximab for the treatment of rheumatoid arthritis: An update. Drug Des. Dev. Ther. 2013, 8, 87–100. [Google Scholar] [CrossRef]
- Ho, C.; Adcock, L. Rituximab Maintenance Therapy for the Management of Granulomatosis with Polyangiitis or Microscopic Polyangiitis: A Review of Clinical Effectiveness, Cost-Effectiveness, and Guidelines; Canadian Agency for Drugs and Technologies in Health: Ottawa, ON, Canada, 2018; pp. 1–21.
- Merkel, P.A.; Niles, J.L.; Mertz, L.E.; Lehane, P.B.; Pordeli, P.; Erblang, F.; Allen, N.; Block, J.A.; Cartin-Ceba, R.; Koening, C.; et al. Long-Term Safety of Rituximab in Granulomatosis With Polyangiitis and in Microscopic Polyangiitis. Arthritis Care Res. 2021, 73, 1372–1378. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Odueyungbo, A.; Csinady, E.; Gearhart, L.; Lehane, P.; Cheu, M.; Maho-Vaillant, M.; Prost-Squarcioni, C.; Hebert, V.; Houivet, E.; et al. Rituximab is an effective treatment in patients with pemphigus vulgaris and demonstrates a steroid-sparing effect. Br. J. Dermatol. 2019, 182, 1111–1119. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.A. Treatment of Philadelphia chromosome-positive acute lymphocytic leukemia with hyper-CVAD and imatinib mesylate. Blood 2004, 103, 4396–4407. [Google Scholar] [CrossRef]
- Jeha, S.; Behm, F.; Pei, D.; Sandlund, J.T.; Ribeiro, R.C.; Razzouk, B.I.; Rubnitz, J.E.; Hijiya, N.; Howard, S.C.; Cheng, C.; et al. Prognostic significance of CD20 expression in childhood B-cell precursor acute lymphoblastic leukemia. Blood 2006, 108, 3302–3304. [Google Scholar] [CrossRef] [PubMed]
- Griffin, T.C.; Weitzman, S.; Weinstein, H.; Chang, M.; Cairo, M.; Hutchison, R.; Shiramizu, B.; Wiley, J.; Woods, D.; Barnich, M.; et al. A study of rituximab and ifosfamide, carboplatin, and etoposide chemotherapy in children with recurrent/refractory B-cell (CD20 ) non-Hodgkin lymphoma and mature B-cell acute lymphoblastic leukemia: A report from the Childrens Oncology Group. Pediatric Blood Cancer 2009, 52, 177–181. [Google Scholar] [CrossRef]
- Rigaud, C.; Auperin, A.; Jourdain, A.; Haouy, S.; Couec, M.L.; Aladjidi, N.; Gandemer, V.; Lambliotte, A.; Plat, G.; Landman-Parker, J.; et al. Outcome of relapse in children and adolescents with B-cell non-Hodgkin lymphoma and mature acute leukemia: A report from the French LMB study. Pediatric Blood Cancer 2019, 66, e27873. [Google Scholar] [CrossRef]
- Dworzak, M.N.; Schumich, A.; Printz, D.; Pötschger, U.; Husak, Z.; Attarbaschi, A.; Basso, G.; Gaipa, G.; Ratei, R.; Mann, G.; et al. CD20 up-regulation in pediatric B-cell precursor acute lymphoblastic leukemia during induction treatment: Setting the stage for anti-CD20 directed immunotherapy. Blood 2008, 112, 3982–3988. [Google Scholar] [CrossRef] [PubMed]
- Pavlasova, G.; Mraz, M. The regulation and function of CD20: An “enigma” of B-cell biology and targeted therapy. Haematologica 2020, 105, 1494–1506. [Google Scholar] [CrossRef]
- Teeling, J.L.; Mackus, W.J.M.; Wiegman, L.J.J.M.; van den Brakel, J.H.; Beers, S.A.; French, R.R.; Meerten, T.V.; Ebeling, S.; Vink, T.; Slootstra, J.W.; et al. The Biological Activity of Human CD20 Monoclonal Antibodies Is Linked to Unique Epitopes on CD20. J. Immunol. 2006, 177, 362–371. [Google Scholar] [CrossRef]
- Teeling, J.L. Characterization of new human CD20 monoclonal antibodies with potent cytolytic activity against non-Hodgkin lymphomas. Blood 2004, 104, 1793–1800. [Google Scholar] [CrossRef]
- Lemery, S.J.; Zhang, J.; Rothmann, M.D.; Yang, J.; Earp, J.; Zhao, H.; Mcdougal, A.; Pilaro, A.; Chiang, R.; Gootenberg, J.E.U.S. Food and Drug Administration Approval: Ofatumumab for the Treatment of Patients with Chronic Lymphocytic Leukemia Refractory to Fludarabine and Alemtuzumab. Clin. Cancer Res. 2010, 16, 4331–4338. [Google Scholar] [CrossRef]
- Jabbour, E.; Anderson Cancer Center. Combination Chemotherapy and Ofatumumab in Treating Patients With Acute Lymphoblastic Leukemia or Lymphoblastic Lymphoma. Available online: https://clinicaltrials.gov/ct2/show/NCT01363128 (accessed on 12 October 2021).
- Ohanian, M.; Anderson Cancer Center. Combination Chemotherapy in Treating Patients With Relapsed or Refractory Acute Lymphoblastic Leukemia, Lymphoblastic Lymphoma, Burkitt Lymphoma/Leukemia, or Double-Hit Lymphoma/Leukemia. Available online: https://clinicaltrials.gov/ct2/show/NCT03136146 (accessed on 12 October 2021).
- Jabbour, E.; Anderson Cancer Center. Blinatumomab, Inotuzumab Ozogamicin, and Combination Chemotherapy as Frontline Therapy in Treating Patients With B Acute Lymphoblastic Leukemia. Available online: https://clinicaltrials.gov/ct2/show/NCT02877303 (accessed on 12 October 2021).
- Dhillon, S. Moxetumomab Pasudotox: First Global Approval. Drugs 2018, 78, 1763–1767. [Google Scholar] [CrossRef] [PubMed]
- Alderson, R.F.; Kreitman, R.J.; Chen, T.; Yeung, P.; Herbst, R.; Fox, J.A.; Pastan, I. CAT-8015: A Second-Generation Pseudomonas Exotoxin A–Based Immunotherapy Targeting CD22-Expressing Hematologic Malignancies. Clin. Cancer Res. 2009, 15, 832–839. [Google Scholar] [CrossRef]
- Bokori-Brown, M.; Metz, J.; Petrov, P.G.; Mussai, F.; Santo, C.D.; Smart, N.J.; Saunders, S.; Knight, B.; Pastan, I.; Titball, R.W.; et al. Interactions Between Pseudomonas Immunotoxins and the Plasma Membrane: Implications for CAT-8015 Immunotoxin Therapy. Front. Oncol. 2018, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- FDA Approves Moxetumomab Pasudotox-Tdfk for Hairy Cell Leukemia. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-moxetumomab-pasudotox-tdfk-hairy-cell-leukemia (accessed on 12 October 2021).
- Mussai, F.; Campana, D.; Bhojwani, D.; Stetler-Stevenson, M.; Steinberg, S.M.; Wayne, A.S.; Pastan, I. Cytotoxicity of the anti-CD22 immunotoxin HA22 (CAT-8015) against paediatric acute lymphoblastic leukaemia. Br. J. Haematol. 2010, 150, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Wayne, A.S.; Shah, N.N.; Bhojwani, D.; Silverman, L.B.; Whitlock, J.A.; Stetler-Stevenson, M.; Sun, W.; Liang, M.; Yang, J.; Kreitman, R.J.; et al. Phase 1 study of the anti-CD22 immunotoxin moxetumomab pasudotox for childhood acute lymphoblastic leukemia. Blood 2017, 130, 1620–1627. [Google Scholar] [CrossRef]
- MedImmune, L.L.C. CAT-8015 in Children, Adolescents and Young Adults With Acute Lymphoblastic Leukemia or Non-Hodgkin’s Lymphoma. Available online: https://clinicaltrials.gov/ct2/show/NCT00659425 (accessed on 12 October 2021).
- Shah, N.N.; Bhojwani, D.; August, K.; Baruchel, A.; Bertrand, Y.; Boklan, J.; Dalla-Pozza, L.; Dennis, R.; Hijiya, N.; Locatelli, F.; et al. Results from an international phase 2 study of the anti-CD22 immunotoxin moxetumomab pasudotox in relapsed or refractory childhood B-lineage acute lymphoblastic leukemia. Pediatric Blood Cancer 2020, 67, e2811. [Google Scholar] [CrossRef] [PubMed]
- Herrera, L.; Bostrom, B.; Gore, L.; Sandler, E.; Lew, G.; Schlegel, P.G.; Aquino, V.; Ghetie, V.; Vitetta, E.S.; Schindler, J.A.; et al. Phase 1 Study of Combotox in Pediatric Patients With Refractory B-lineage Acute Lymphoblastic Leukemia. J. Pediatric Hematol. Oncol. 2009, 31, 936–941. [Google Scholar] [CrossRef]
- Jegalian, A.G.; Wayne, A.S.; Kreitman, R.J.; Mussai, F.J.; Pastan, I.; Yuan, C.M.; Stetler-Stevenson, M. CD22 Expression in Pediatric B-Lineage Acute Lymphoblastic Leukemia. Blood 2009, 114, 4119. [Google Scholar] [CrossRef]
- Barta, S.K.; Zou, Y.; Schindler, J.; Shenoy, N.; Bhagat, T.D.; Steidl, U.; Verma, A. Synergy of sequential administration of a deglycosylated ricin A chain-containing combined anti-CD19 and anti-CD22 immunotoxin (Combotox) and cytarabine in a murine model of advanced acute lymphoblastic leukemia. Leuk. Lymphoma 2012, 53, 1999–2003. [Google Scholar] [CrossRef] [PubMed]
- Herrera, L.; Farah, R.; Pellegrini, V.; Aquino, D.; Sandler, E.; Buchanan, G.; Vitetta, E. Immunotoxins against CD19 and CD22 are effective in killing precursor-B acute lymphoblastic leukemia cells in vitro. Leukemia 2000, 14, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Ghetie, M.-A.; Podar, E.M.; Gordon, B.E.; Pantazis, P.; Uhr, J.W.; Vitetta, E.S. Combination immunotoxin treatment and chemotherapy in SCID mice with advanced, disseminated Daudi lymphoma. Int. J. Cancer 1996, 68, 93–96. [Google Scholar] [CrossRef]
- Herrera, L.; Stanciu-Herrera, C.; Morgan, C.; Ghetie, V.; Vitetta, E.S. Anti-CD19 immunotoxin enhances the activity of chemotherapy in severe combined immunodeficient mice with human pre-B acute lymphoblastic leukemia. Leuk. Lymphoma 2006, 47, 2380–2387. [Google Scholar] [CrossRef]
- Immunotoxin Therapy and Cytarabine in Treating Patients With Relapsed or Refractory B-Cell Acute Lymphoblastic Leukemia. Available online: https://clinicaltrials.gov/ct2/show/study/NCT01408160 (accessed on 12 October 2021).
- Doronina, S.O.; Mendelsohn, B.A.; Bovee, T.D.; Cerveny, C.G.; Alley, S.C.; Meyer, D.L.; Oflazoglu, E.; Toki, B.E.; Sanderson, R.J.; Zabinski, R.F.; et al. Enhanced Activity of Monomethylauristatin F through Monoclonal Antibody Delivery: Effects of Linker Technology on Efficacy and Toxicity. Bioconjug. Chem. 2005, 17, 114–124. [Google Scholar] [CrossRef]
- Fathi, A.T.; Borate, U.; Deangelo, D.J.; Obrien, M.M.; Trippett, T.; Shah, B.D.; Hale, G.A.; Foran, J.M.; Silverman, L.B.; Tibes, R.; et al. A Phase 1 Study of Denintuzumab Mafodotin (SGN-CD19A) in Adults with Relapsed or Refractory B-Lineage Acute Leukemia (B-ALL) and Highly Aggressive Lymphoma. Blood 2015, 126, 1328. [Google Scholar] [CrossRef]
- Feldman, E.; Seagen Inc. A Safety Study of SGN-CD19A for Leukemia and Lymphoma. Available online: https://clinicaltrials.gov/ct2/show/NCT01786096 (accessed on 12 October 2021).
- Kostic, A.; Seagen Inc. A Safety Study of SGN-CD19A for B-Cell Lymphoma. Available online: https://clinicaltrials.gov/ct2/show/NCT01786135 (accessed on 12 October 2021).
- Jones, L.; Mccalmont, H.; Evans, K.; Mayoh, C.; Kurmasheva, R.T.; Billups, C.A.; Houghton, P.J.; Smith, M.A.; Lock, R.B. Preclinical activity of the antibody-drug conjugate denintuzumab mafodotin (SGN-CD19A) against pediatric acute lymphoblastic leukemia xenografts. Pediatric Blood Cancer 2019, 66, e27765. [Google Scholar] [CrossRef]
- Kahl, B.S.; Hamadani, M.; Radford, J.; Carlo-Stella, C.; Caimi, P.; Reid, E.; Feingold, J.M.; Ardeshna, K.M.; Solh, M.; Heffner, L.T.A.; et al. Phase I Study of ADCT-402 (Loncastuximab Tesirine), a Novel Pyrrolobenzodiazepine-Based Antibody–Drug Conjugate, in Relapsed/Refractory B-Cell Non-Hodgkin Lymphoma. Clin. Cancer Res. 2019, 25, 6986–6994. [Google Scholar] [CrossRef] [PubMed]
- Hartley, J.A.; Flynn, M.J.; Bingham, J.P.; Corbett, S.; Reinert, H.; Tiberghien, A.; Masterson, L.A.; Antonow, D.; Adams, L.; Chowdhury, S.; et al. Pre-clinical pharmacology and mechanism of action of SG3199, the pyrrolobenzodiazepine (PBD) dimer warhead component of antibody-drug conjugate (ADC) payload tesirine. Sci. Rep. 2018, 8, 10479. [Google Scholar] [CrossRef]
- Jain, N.; Stock, W.; Zeidan, A.; Atallah, E.; Mccloskey, J.; Heffner, L.; Tomlinson, B.; Bhatnagar, B.; Feingold, J.; Ungar, D.; et al. Loncastuximab tesirine, an anti-CD19 antibody-drug conjugate, in relapsed/refractory B-cell acute lymphoblastic leukemia. Blood Adv. 2020, 4, 449–457. [Google Scholar] [CrossRef]
- Nakase, K.; Kita, K.; Miwa, H.; Nishii, K.; Shikami, M.; Tanaka, I.; Tsutani, H.; Ueda, T.; Nasu, K.; Kyo, T.; et al. Clinical and prognostic significance of cytokine receptor expression in adult acute lymphoblastic leukemia: Interleukin-2 receptor α-chain predicts a poor prognosis. Leukemia 2007, 21, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.D.; Atallah, E.; Rizzieri, D.; Walter, R.B.; Chung, K.-Y.; Spira, A.; Stock, W.; Tallman, M.S.; Cruz, H.G.; Boni, J.; et al. Camidanlumab tesirine, an antibody-drug conjugate, in relapsed/refractory CD25-positive acute myeloid leukemia or acute lymphoblastic leukemia: A phase I study. Leuk. Res. 2020, 95, 106385. [Google Scholar] [CrossRef]
- Study to Evaluate the Efficacy and Safety of Camidanlumab Tesirine (ADCT-301) in Patients with Relapsed or Refractory Hodgkin Lymphoma. Available online: https://clinicaltrials.gov/ct2/show/NCT04052997 (accessed on 12 October 2021).
- Rizzieri, D. ADCT-301 in Patients with R/R AML, MDS, or MDS/MPN. Available online: https://clinicaltrials.gov/ct2/show/NCT04639024 (accessed on 12 October 2021).
- Hong, E.E.; Erickson, H.; Lutz, R.J.; Whiteman, K.R.; Jones, G.; Kovtun, Y.; Blanc, V.; Lambert, J.M. Design of Coltuximab Ravtansine, a CD19-Targeting Antibody–Drug Conjugate (ADC) for the Treatment of B-Cell Malignancies: Structure–Activity Relationships and Preclinical Evaluation. Mol. Pharm. 2015, 12, 1703–1716. [Google Scholar] [CrossRef] [PubMed]
- Widdison, W.C.; Wilhelm, S.D.; Cavanagh, E.E.; Whiteman, K.R.; Leece, B.A.; Kovtun, Y.; Goldmacher, V.S.; Xie, H.; Steeves, R.M.; Lutz, R.J.; et al. Semisynthetic Maytansine Analogues for the Targeted Treatment of Cancer. J. Med. Chem. 2006, 49, 4392–4408. [Google Scholar] [CrossRef]
- SAR3419 in Acute Lymphoblastic Leukemia (MYRALL). Available online: https://clinicaltrials.gov/ct2/show/NCT01440179 (accessed on 12 October 2021).
- Carol, H.; Szymanska, B.; Evans, K.; Boehm, I.; Houghton, P.J.; Smith, M.A.; Lock, R.B. The Anti-CD19 Antibody–Drug Conjugate SAR3419 Prevents Hematolymphoid Relapse Postinduction Therapy in Preclinical Models of Pediatric Acute Lymphoblastic Leukemia. Clin. Cancer Res. 2013, 19, 1795–1805. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; Lioure, B.; Kim, S.K.; Atallah, E.; Leguay, T.; Kelly, K.; Marolleau, J.-P.; Escoffre-Barbe, M.; Thomas, X.G.; Cortes, J.; et al. A Phase II Study of Coltuximab Ravtansine (SAR3419) Monotherapy in Patients With Relapsed or Refractory Acute Lymphoblastic Leukemia. Clin. Lymphoma Myeloma Leuk. 2016, 16, 139–145. [Google Scholar] [CrossRef]
- Hicks, S.W.; Tarantelli, C.; Wilhem, A.; Gaudio, E.; Li, M.; Arribas, A.J.; Spriano, F.; Bordone, R.; Cascione, L.; Lai, K.C.; et al. The novel CD19-targeting antibody-drug conjugate huB4-DGN462 shows improved anti-tumor activity compared to SAR3419 in CD19-positive lymphoma and leukemia models. Haematologica 2019, 104, 1633–1639. [Google Scholar] [CrossRef]
- Touma, Z.; Rayes, H.A. Profile of epratuzumab and its potential in the treatment of systemic lupus erythematosus. Drug Des. Dev. Ther. 2014, 8, 2303. [Google Scholar] [CrossRef]
- Raetz, E.A.; Cairo, M.S.; Borowitz, M.J.; Blaney, S.M.; Krailo, M.D.; Leil, T.A.; Reid, J.M.; Goldenberg, D.M.; Wegener, W.A.; Carroll, W.L.; et al. Chemoimmunotherapy Reinduction with Epratuzumab in Children With Acute Lymphoblastic Leukemia in Marrow Relapse: A Childrens Oncology Group Pilot Study. J. Clin. Oncol. 2008, 26, 3756–3762. [Google Scholar] [CrossRef] [PubMed]
- Raetz, E.A.; Cairo, M.S.; Borowitz, M.J.; Lu, X.; Devidas, M.; Reid, J.M.; Goldenberg, D.M.; Wegener, W.A.; Whitlock, J.A.; Adamson, P.C.; et al. Reinduction Chemoimmunotherapy with Epratuzumab in Relapsed Acute Lymphoblastic Leukemia (ALL) in Children, Adolescents and Young Adults: Results From Childrens Oncology Group (COG) Study ADVL04P2. Blood 2011, 118, 573. [Google Scholar] [CrossRef]
- Raetz, E.A.; Borowitz, M.J.; Devidas, M.; Hunger, S.P.; Winick, N.; Camitta, B.; Gaynon, P.S.; Carroll, W.L. Outcomes of Children with First Marrow Relapse: Results from Children’s Oncology Group (COG) Study AALL01P2. Blood 2006, 108, 1871. [Google Scholar] [CrossRef]
- Brunet, J.-F.; Denizot, F.; Luciani, M.-F.; Roux-Dosseto, M.; Suzan, M.; Mattei, M.-G.; Golstein, P. A new member of the immunoglobulin superfamily—CTLA-4. Nature 1987, 328, 267–270. [Google Scholar] [CrossRef]
- Schwartz, J.-C.D.; Zhang, X.; Fedorov, A.A.; Nathenson, S.G.; Almo, S.C. Structural basis for co-stimulation by the human CTLA-4/B7-2 complex. Nature 2001, 410, 604–608. [Google Scholar] [CrossRef]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef]
- Saudemont, A.; Quesnel, B. In a model of tumor dormancy, long-term persistent leukemic cells have increased B7-H1 and B7.1 expression and resist CTL-mediated lysis. Blood 2004, 104, 2124–2133. [Google Scholar] [CrossRef]
- Allison, J.P. Immune Checkpoint Blockade in Cancer Therapy. JAMA 2015, 314, 1113. [Google Scholar] [CrossRef]
- Oday, S.J.; Hamid, O.; Urba, W.J. Targeting cytotoxic T-lymphocyte antigen-4 (CTLA-4). Cancer 2007, 110, 2614–2627. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Neyns, B.; Linette, G.; Negrier, S.; Lutzky, J.; Thomas, L.; Waterfield, W.; Schadendorf, D.; Smylie, M.; Guthrie, T.; et al. Ipilimumab monotherapy in patients with pretreated advanced melanoma: A randomised, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncol. 2010, 11, 155–164. [Google Scholar] [CrossRef]
- Bashey, A.; Medina, B.; Corringham, S.; Pasek, M.; Carrier, E.; Vrooman, L.; Lowy, I.; Solomon, S.R.; Morris, L.E.; Holland, H.K. CTLA4 blockade with ipilimumab to treat relapse of malignancy after allogeneic hematopoietic cell transplantation. Blood 2009, 113, 1581–1588. [Google Scholar] [CrossRef]
- Davids, M.S.; Kim, H.T.; Bachireddy, P.; Costello, C.; Liguori, R.; Savell, A.; Lukez, A.P.; Avigan, D.; Chen, Y.-B.; McSweeney, P.; et al. Ipilimumab for Patients with Relapse after Allogeneic Transplantation. N. Engl. J. Med. 2016, 375, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Penter, L.; Zhang, Y.; Savell, A.; Huang, T.; Cieri, N.; Thrash, E.M.; Kim-Schulze, S.; Jhaveri, A.; Fu, J.; Ranasinghe, S.; et al. Molecular and cellular features of CTLA-4 blockade for relapsed myeloid malignancies after transplantation. Blood 2021, 137, 3212–3217. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.S.; Farber, D.; Harvard Cancer Center LAO. Ipilimumab and Decitabine in Treating Patients With Relapsed or Refractory Myelodysplastic Syndrome or Acute Myeloid Leukemia. Available online: https://clinicaltrials.gov/ct2/show/NCT02890329 (accessed on 12 October 2021).
- Ramos-Casals, M.; Brahmer, J.R.; Callahan, M.K.; Flores-Chávez, A.; Keegan, N.; Khamashta, M.A.; Lambotte, O.; Mariette, X.; Prat, A.; Suárez-Almazor, M.E.; et al. Immune-related adverse events of checkpoint inhibitors. Nat. Rev. Dis. Primers 2020, 6, 38. [Google Scholar] [CrossRef]
- Coutzac, C.; Adam, J.; Soularue, E.; Collins, M.; Racine, A.; Mussini, C.; Boselli, L.; Kamsukom, N.; Mateus, C.; Charrier, M.; et al. Colon Immune-Related Adverse Events: Anti-CTLA-4 and Anti-PD-1 Blockade Induce Distinct Immunopathological Entities. J. Crohns Colitis 2017, 11, 1238–1246. [Google Scholar] [CrossRef]
- Duan, J.; Cui, L.; Zhao, X.; Bai, H.; Cai, S.; Wang, G.; Zhao, Z.; Zhao, J.; Chen, S.; Song, J.; et al. Use of Immunotherapy With Programmed Cell Death 1 vs Programmed Cell Death Ligand 1 Inhibitors in Patients With Cancer. JAMA Oncol. 2020, 6, 375. [Google Scholar] [CrossRef]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef]
- Zhang, J.; Medeiros, L.J.; Young, K.H. Cancer Immunotherapy in Diffuse Large B-Cell Lymphoma. Front. Oncol. 2018, 8, 351. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Erratum: Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 1039. [Google Scholar] [CrossRef]
- Chen, X.; Liu, S.; Wang, L.; Zhang, W.-G.; Ji, Y.; Ma, X. Clinical significance of B7-H1(PD-L1) expression in human acute leukemia. Cancer Biol. Ther. 2008, 7, 622–627. [Google Scholar] [CrossRef]
- Dong, Y.; Han, Y.; Huang, Y.; Jiang, S.; Huang, Z.; Chen, R.; Yu, Z.; Yu, K.; Zhang, S. PD-L1 Is Expressed and Promotes the Expansion of Regulatory T Cells in Acute Myeloid Leukemia. Front. Immunol. 2020, 11, 1710. [Google Scholar] [CrossRef] [PubMed]
- Hamid, O.; Robert, C.; Daud, A.; Hodi, F.S.; Hwu, W.-J.; Kefford, R.; Wolchok, J.D.; Hersey, P.; Joseph, R.W.; Weber, J.S.; et al. Safety and Tumor Responses with Lambrolizumab (Anti–PD-1) in Melanoma. N. Engl. J. Med. 2013, 369, 134–144. [Google Scholar] [CrossRef]
- Robert, C.; Ribas, A.; Wolchok, J.D.; Hodi, F.S.; Hamid, O.; Kefford, R.; Weber, J.S.; Joshua, A.M.; Hwu, W.-J.; Gangadhar, T.C.; et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: A randomised dose-comparison cohort of a phase 1 trial. Lancet 2014, 384, 1109–1117. [Google Scholar] [CrossRef]
- Cassaday, R.D.; Garcia, K.-L.A.; Fromm, J.R.; Percival, M.-E.M.; Turtle, C.J.; Nghiem, P.T.; Stevenson, P.A.; Estey, E.H. Phase 2 study of pembrolizumab for measurable residual disease in adults with acute lymphoblastic leukemia. Blood Adv. 2020, 4, 3239–3245. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Drake, C.G.; Wollner, I.; Powderly, J.D.; Picus, J.; Sharfman, W.H.; Stankevich, E.; Pons, A.; Salay, T.M.; Mcmiller, T.L.; et al. Phase I Study of Single-Agent Anti–Programmed Death-1 (MDX-1106) in Refractory Solid Tumors: Safety, Clinical Activity, Pharmacodynamics, and Immunologic Correlates. J. Clin. Oncol. 2010, 28, 3167–3175. [Google Scholar] [CrossRef]
- Felip, E.; Ardizzoni, A.; Ciuleanu, T.; Cobo, M.; Laktionov, K.; Szilasi, M.; Califano, R.; Carcereny, E.; Griffiths, R.; Paz-Ares, L.; et al. CheckMate 171: A phase 2 trial of nivolumab in patients with previously treated advanced squamous non-small cell lung cancer, including ECOG PS 2 and elderly populations. Eur. J. Cancer 2020, 127, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.S.; Dangelo, S.P.; Minor, D.; Hodi, F.S.; Gutzmer, R.; Neyns, B.; Hoeller, C.; Khushalani, N.I.; Miller, W.H.; Lao, C.D.; et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): A randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015, 16, 375–384. [Google Scholar] [CrossRef]
- Kadia, T.M.; Cortes, J.E.; Ghorab, A.; Ravandi, F.; Jabbour, E.; Daver, N.G.; Alvarado, Y.; Ohanian, M.; Konopleva, M.; Kantarjian, H.M. Nivolumab (Nivo) maintenance (maint) in high-risk (HR) acute myeloid leukemia (AML) patients. J. Clin. Oncol. 2018, 36, 7014. [Google Scholar] [CrossRef]
- Cooper, S.L.; Children’s Oncology Group. A Study to Compare Blinatumomab Alone to Blinatumomab with Nivolumab in Patients Diagnosed With First Relapse B-Cell Acute Lymphoblastic Leukemia (B-ALL). Available online: https://clinicaltrials.gov/ct2/show/NCT04546399 (accessed on 12 October 2021).
- Breese, E.; Children’s Hospital Medical Center: Cincinnati. Pembro + Blina Combination in Pediatric and Young Adult Patients with Relapsed/Refractory Acute Leukemia or Lymphoma. Available online: https://clinicaltrials.gov/ct2/show/NCT03605589 (accessed on 12 October 2021).
- Kamphorst, A.O.; Wieland, A.; Nasti, T.; Yang, S.; Zhang, R.; Barber, D.L.; Konieczny, B.T.; Daugherty, C.Z.; Koenig, L.; Yu, K.; et al. Rescue of exhausted CD8 T cells by PD-1–targeted therapies is CD28-dependent. Science 2017, 355, 1423–1427. [Google Scholar] [CrossRef]
- Park, S.H.; You, E.; Park, C.J.; Cho, Y.U.; Jang, S.; Im, H.J.; Seo, J.J.; Park, H.S.; Lee, J.H. Increased expression of immune checkpoint programmed cell death protein-1 (PD-1) on T cell subsets of bone marrow aspirates in patients with B-Lymphoblastic leukemia, especially in relapse and at diagnosis. Cytom. Part B Clin. Cytom. 2020, 98, 336–347. [Google Scholar] [CrossRef]
- Tan, J.; Yu, Z.; Huang, J.; Chen, Y.; Huang, S.; Yao, D.; Xu, L.; Lu, Y.; Chen, S.; Li, Y. Increased PD-1 Tim-3 exhausted T cells in bone marrow may influence the clinical outcome of patients with AML. Biomark. Res. 2020, 8, 6. [Google Scholar] [CrossRef] [PubMed]
- Martins, F.; Sofiya, L.; Sykiotis, G.P.; Lamine, F.; Maillard, M.; Fraga, M.; Shabafrouz, K.; Ribi, C.; Cairoli, A.; Guex-Crosier, Y.; et al. Adverse effects of immune-checkpoint inhibitors: Epidemiology, management and surveillance. Nat. Rev. Clin. Oncol. 2019, 16, 563–580. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, J.E.; Hoffman-Censits, J.; Powles, T.; Heijden, M.S.V.D.; Balar, A.V.; Necchi, A.; Dawson, N.; Odonnell, P.H.; Balmanoukian, A.; Loriot, Y.; et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: A single-arm, multicentre, phase 2 trial. Lancet 2016, 387, 1909–1920. [Google Scholar] [CrossRef]
- Herbst, R.S.; Soria, J.-C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; Mcdermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef]
- Hoffmann-La Roche. A Study Evaluating the Safety and Pharmacology of Atezolizumab Administered in Combination with Immunomodulatory Agents in Participants With Acute Myeloid Leukemia (AML). Available online: https://clinicaltrials.gov/ct2/show/NCT02892318 (accessed on 12 October 2021).
- Powles, T.; Odonnell, P.H.; Massard, C.; Arkenau, H.-T.; Friedlander, T.W.; Hoimes, C.J.; Lee, J.L.; Ong, M.; Sridhar, S.S.; Vogelzang, N.J.; et al. Efficacy and Safety of Durvalumab in Locally Advanced or Metastatic Urothelial Carcinoma. JAMA Oncol. 2017, 3, e172411. [Google Scholar] [CrossRef]
- Lutzky, J.; Antonia, S.J.; Blake-Haskins, A.; Li, X.; Robbins, P.B.; Shalabi, A.M.; Vasselli, J.; Ibrahim, R.A.; Khleif, S.; Segal, N.H. A phase 1 study of MEDI4736, an anti–PD-L1 antibody, in patients with advanced solid tumors. J. Clin. Oncol. 2014, 32, 3001. [Google Scholar] [CrossRef]
- Massard, C.; Gordon, M.S.; Sharma, S.; Rafii, S.; Wainberg, Z.A.; Luke, J.; Curiel, T.J.; Colon-Otero, G.; Hamid, O.; Sanborn, R.E.; et al. Safety and Efficacy of Durvalumab (MEDI4736), an Anti–Programmed Cell Death Ligand-1 Immune Checkpoint Inhibitor, in Patients With Advanced Urothelial Bladder Cancer. J. Clin. Oncol. 2016, 34, 3119–3125. [Google Scholar] [CrossRef] [PubMed]
- Zeidan, A.M.; Cavenagh, J.; Voso, M.T.; Taussig, D.; Tormo, M.; Boss, I.; Copeland, W.B.; Gray, V.E.; Previtali, A.; Oconnor, T.; et al. Efficacy and Safety of Azacitidine (AZA) in Combination with the Anti-PD-L1 Durvalumab (durva) for the Front-Line Treatment of Older Patients (pts) with Acute Myeloid Leukemia (AML) Who Are Unfit for Intensive Chemotherapy (IC) and Pts with Higher-Risk Myelodysplastic Syndromes (HR-MDS): Results from a Large, International, Randomized Phase 2 Study. Blood 2019, 134, 829. [Google Scholar]
- Soto-Pantoja, D.R.; Kaur, S.; Roberts, D.D. CD47 signaling pathways controlling cellular differentiation and responses to stress. Crit. Rev. Biochem. Mol. Biol. 2015, 50, 212–230. [Google Scholar] [CrossRef] [PubMed]
- Willingham, S.B.; Volkmer, J.-P.; Gentles, A.J.; Sahoo, D.; Dalerba, P.; Mitra, S.S.; Wang, J.; Contreras-Trujillo, H.; Martin, R.; Cohen, J.D.; et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 6662–6667. [Google Scholar] [CrossRef] [PubMed]
- Majeti, R.; Chao, M.P.; Alizadeh, A.A.; Pang, W.W.; Jaiswal, S.; Gibbs, K.D.; Rooijen, N.V.; Weissman, I.L. CD47 Is an Adverse Prognostic Factor and Therapeutic Antibody Target on Human Acute Myeloid Leukemia Stem Cells. Cell 2009, 138, 286–299. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, L.; Zhao, F.; Tseng, S.; Narayanan, C.; Shura, L.; Willingham, S.; Howard, M.; Prohaska, S.; Volkmer, J.; et al. Pre-Clinical Development of a Humanized Anti-CD47 Antibody with Anti-Cancer Therapeutic Potential. PLoS ONE 2015, 10, e0137345. [Google Scholar]
- Brierley, C.; Staves, J.; Roberts, C.; Johnson, H.; Vyas, P.; Goodnough, L.; Murphy, M. The effects of monoclonal anti-CD47 on RBCs, compatibility testing, and transfusion requirements in refractory acute myeloid leukemia. Transfusion 2019, 59, 2248–2254. [Google Scholar] [CrossRef]
- Sallman, D.A.; Asch, A.S.; Malki, M.M.A.; Lee, D.J.; Donnellan, W.B.; Marcucci, G.; Kambhampati, S.; Daver, N.G.; Garcia-Manero, G.; Komrokji, R.S.; et al. The First-in-Class Anti-CD47 Antibody Magrolimab (5F9) in Combination with Azacitidine Is Effective in MDS and AML Patients: Ongoing Phase 1b Results. Blood 2019, 134, 569. [Google Scholar] [CrossRef]
- Fisher, G.A.; Lakhani, N.J.; Eng, C.; Hecht, J.R.; Bendell, J.C.; Philip, P.A.; Odwyer, P.J.; Johnson, B.; Kardosh, A.; Ippolito, T.M.; et al. A phase Ib/II study of the anti-CD47 antibody magrolimab with cetuximab in solid tumor and colorectal cancer patients. J. Clin. Oncol. 2020, 38, 114. [Google Scholar] [CrossRef]
- Chiba, S.; Baghdadi, M.; Akiba, H.; Yoshiyama, H.; Kinoshita, I.; Dosaka-Akita, H.; Fujioka, Y.; Ohba, Y.; Gorman, J.V.; Colgan, J.D.; et al. Tumor-infiltrating DCs suppress nucleic acid–mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat. Immunol. 2012, 13, 832–842. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Fueyo, A.; Tian, J.; Picarella, D.; Domenig, C.; Zheng, X.X.; Sabatos, C.A.; Manlongat, N.; Bender, O.; Kamradt, T.; Kuchroo, V.K.; et al. Tim-3 inhibits T helper type 1–mediated auto- and alloimmune responses and promotes immunological tolerance. Nat. Immunol. 2003, 4, 1093–1101. [Google Scholar] [CrossRef]
- Brunner, A.M.; Esteve, J.; Porkka, K.; Knapper, S.; Vey, N.; Scholl, S.; Garcia-Manero, G.; Wermke, M.; Janssen, J.; Traer, E.; et al. Efficacy and Safety of Sabatolimab (MBG453) in Combination with Hypomethylating Agents (HMAs) in Patients with Acute Myeloid Leukemia (AML) and High-Risk Myelodysplastic Syndrome (HR-MDS): Updated Results from a Phase 1b Study. Blood 2020, 136, 1–2. [Google Scholar] [CrossRef]
- Wolf, Y.; Anderson, A.C.; Kuchroo, V.K. TIM3 comes of age as an inhibitory receptor. Nat. Rev. Immunol. 2019, 20, 173–185. [Google Scholar] [CrossRef]
- Dama, P.; Tang, M.; Fulton, N.; Kline, J.; Liu, H. Gal9/Tim-3 expression level is higher in AML patients who fail chemotherapy. J. Immunother. Cancer 2019, 7, 175. [Google Scholar] [CrossRef] [PubMed]
- Novartis Pharmaceuticals. Sabatolimab as a Treatment for Patients with Acute Myeloid Leukemia and Presence of Measurable Residual Disease after Allogeneic Stem Cell Transplantation. Available online: https://clinicaltrials.gov/ct2/show/NCT04623216 (accessed on 12 October 2021).
- Novartis Pharmaceuticals. Study of PDR001 and/or MBG453 in Combination with Decitabine in Patients with AML or High Risk MDS. Available online: https://clinicaltrials.gov/ct2/show/NCT03066648 (accessed on 12 October 2021).
- Curigliano, G.; Gelderblom, H.; Mach, N.; Doi, T.; Tai, D.; Forde, P.M.; Sarantopoulos, J.; Bedard, P.L.; Lin, C.-C.; Hodi, F.S.; et al. Phase I/Ib Clinical Trial of Sabatolimab, an Anti–TIM-3 Antibody, Alone and in Combination with Spartalizumab, an Anti–PD-1 Antibody, in Advanced Solid Tumors. Clin. Cancer Res. 2021, 27, 3620–3629. [Google Scholar] [CrossRef] [PubMed]
- Amarante-Mendes, G.P.; Adjemian, S.; Branco, L.M.; Zanetti, L.C.; Weinlich, R.; Bortoluci, K.R. Pattern Recognition Receptors and the Host Cell Death Molecular Machinery. Front. Immunol. 2018, 9, 2379. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The Inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Pattern Recognition Receptors and Inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Shin, J.-S.; Nahm, M.H. NOD-Like Receptors in Infection, Immunity, and Diseases. Yonsei Med. J. 2016, 57, 5. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Yamamoto, M.; Takeda, K. Role of adapters in Toll-like receptor signalling. Biochem. Soc. Trans. 2003, 31, 637–642. [Google Scholar] [CrossRef]
- Takeda, K.; Akira, S. Toll-like receptors in innate immunity. Int. Immunol. 2004, 17, 1–14. [Google Scholar] [CrossRef]
- Burns, K.; Janssens, S.; Brissoni, B.; Olivos, N.; Beyaert, R.; Tschopp, J. Inhibition of Interleukin 1 Receptor/Toll-like Receptor Signaling through the Alternatively Spliced, Short Form of MyD88 Is Due to Its Failure to Recruit IRAK-4. J. Exp. Med. 2003, 197, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Landström, M. The TAK1–TRAF6 signalling pathway. Int. J. Biochem. Cell Biol. 2010, 42, 585–589. [Google Scholar] [CrossRef]
- Oshiumi, H.; Matsumoto, M.; Funami, K.; Akazawa, T.; Seya, T. TICAM-1, an adaptor molecule that participates in Toll-like receptor 3–mediated interferon-β induction. Nat. Immunol. 2003, 4, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Monlish, D.A.; Bhatt, S.T.; Schuettpelz, L.G. The Role of Toll-Like Receptors in Hematopoietic Malignancies. Front. Immunol. 2016, 7, 390. [Google Scholar] [CrossRef]
- Sato, S.; Sugiyama, M.; Yamamoto, M.; Watanabe, Y.; Kawai, T.; Takeda, K.; Akira, S. Toll/IL-1 Receptor Domain-Containing Adaptor Inducing IFN-β (TRIF) Associates with TNF Receptor-Associated Factor 6 and TANK-Binding Kinase 1, and Activates Two Distinct Transcription Factors, NF-κB and IFN-Regulatory Factor-3, in the Toll-Like Receptor Signaling. J. Immunol. 2003, 171, 4304–4310. [Google Scholar] [PubMed]
- Hoebe, K.; Du, X.; Georgel, P.; Janssen, E.; Tabeta, K.; Kim, S.O.; Goode, J.; Lin, P.; Mann, N.; Mudd, S.; et al. Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature 2003, 424, 743–748. [Google Scholar] [CrossRef]
- Okamoto, M.; Hirai, H.; Taniguchi, K.; Shimura, K.; Inaba, T.; Shimazaki, C.; Taniwaki, M.; Imanishi, J. Toll-like Receptors (TLRs) are expressed by myeloid leukaemia cell lines, but fail to trigger differentiation in response to the respective TLR ligands. Br. J. Haematol. 2009, 147, 585–587. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.; Singh, S.; Anang, V.; Bhatt, A.N.; Natarajan, K.; Dwarakanath, B.S. Pattern Recognition Receptors in Cancer Progression and Metastasis. Cancer Growth Metastasis 2015, 8, 25–34. [Google Scholar] [CrossRef]
- Akira, S.; Uematsu, S.; Takechi, O. Pathogen Recognition and Innate Immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef]
- Guiducci, C.; Gong, M.; Cepika, A.-M.; Xu, Z.; Tripodo, C.; Bennett, L.; Crain, C.; Quartier, P.; Cush, J.J.; Pascual, V.; et al. RNA recognition by human TLR8 can lead to autoimmune inflammation. J. Exp. Med. 2013, 210, 2903–2919. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.M.Y.; Kok, K.-H.; Jaume, M.; Cheung, T.K.W.; Yip, T.-F.; Lai, J.C.C.; Guan, Y.; Webster, R.G.; Jin, D.-Y.; Peiris, J.S.M. Toll-like receptor 10 is involved in induction of innate immune responses to influenza virus infection. Proc. Natl. Acad. Sci. USA 2014, 111, 3793–3798. [Google Scholar] [CrossRef]
- Peana, M.; Zdyb, K.; Medici, S.; Pelucelli, A.; Simula, G.; Gumienna-Kontecka, E.; Zoroddu, M.A. Ni(II) interaction with a peptide model of the human TLR4 ectodomain. J. Trace Elem. Med. Biol. 2017, 44, 151–160. [Google Scholar] [CrossRef]
- Tsujimoto, H.; Ono, S.; Efron, P.A.; Scumpia, P.O.; Moldawer, L.L.; Mochizuki, H. Role Of Toll-Like Receptors In The Development Of Sepsis. Shock 2008, 29, 315–321. [Google Scholar] [CrossRef]
- Nagai, Y.; Garrett, K.P.; Ohta, S.; Bahrun, U.; Kouro, T.; Akira, S.; Takatsu, K.; Kincade, P.W. Toll-like Receptors on Hematopoietic Progenitor Cells Stimulate Innate Immune System Replenishment. Immunity 2006, 24, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Sioud, M.; Fløisand, Y.; Forfang, L.; Lund-Johansen, F. Signaling through Toll-like Receptor 7/8 Induces the Differentiation of Human Bone Marrow CD34 Progenitor Cells along the Myeloid Lineage. J. Mol. Biol. 2006, 364, 945–954. [Google Scholar] [CrossRef] [PubMed]
- Dorantes-Acosta, E.; Pelayo, R. Lineage Switching in Acute Leukemias: A Consequence of Stem Cell Plasticity? Bone Marrow Res. 2012, 2012, 406796. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, M.; Peña, P.; Chapellier, M.; Högberg, C.; Fioretos, T.; Ebert, B.L.; Järås, M. Toll-like Receptor 1 Is a Candidate Therapeutic Target in Acute Myeloid Leukemia. Blood 2014, 124, 5782. [Google Scholar] [CrossRef]
- Wei, Y.; Dimicoli, S.; Bueso-Ramos, C.; Chen, R.; Yang, H.; Neuberg, D.; Pierce, S.; Jia, Y.; Zheng, H.; Wang, H.; et al. Toll-like receptor alterations in myelodysplastic syndrome. Leukemia 2013, 27, 1832–1840. [Google Scholar] [CrossRef]
- Baakhlagh, S.; Kashani, B.; Zandi, Z.; Bashash, D.; Moradkhani, M.; Nasrollahzadeh, A.; Yaghmaei, M.; Mousavi, S.A.; Ghaffari, S.H. Toll-like receptor 4 signaling pathway is correlated with pathophysiological characteristics of AML patients and its inhibition using TAK-242 suppresses AML cell proliferation. Int. Immunopharmacol. 2021, 90, 107202. [Google Scholar] [CrossRef]
- Aref, S.; Elmaksoud, A.S.A.; Elaziz, S.A.; Mabed, M.; Ayed, M. Clinical Implication of Toll-Like Receptors (TLR2 and TLR4) in Acute Myeloid Leukemia Patients. Asian Pac. J. Cancer Prev. 2020, 21, 3177–3183. [Google Scholar] [CrossRef]
- Schnetzke, U.; Spies-Weisshart, B.; Yomade, O.; Fischer, M.; Rachow, T.; Schrenk, K.; Glaser, A.; Lilienfeld-Toal, M.V.; Hochhaus, A.; Scholl, S. Polymorphisms of Toll-like receptors (TLR2 and TLR4) are associated with the risk of infectious complications in acute myeloid leukemia. Genes Immun. 2014, 16, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Cuaxospa, M.; Contreras-Ramos, A.; Pérez-Figueroa, E.; Medina-Sansón, A.; Jiménez-Hernández, E.; Torres-Nava, J.R.; Rojas-Castillo, E.; Maldonado-Bernal, C. Low expression of Toll-like receptors in peripheral blood mononuclear cells of pediatric patients with acute lymphoblastic leukemia. Int. J. Oncol. 2016, 49, 675–681. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Krieg, A.M. Therapeutic potential of Toll-like receptor 9 activation. Nat. Rev. Drug Discov. 2006, 5, 471–484. [Google Scholar] [CrossRef]
- Reid, G.S.D.; She, K.; Terrett, L.; Food, M.R.; Trudeau, J.D.; Schultz, K.R. CpG stimulation of precursor B-lineage acute lymphoblastic leukemia induces a distinct change in costimulatory molecule expression and shifts allogeneic T cells toward a Th1 response. Blood 2005, 105, 3641–3647. [Google Scholar] [CrossRef][Green Version]
- Ronsley, R.; Kariminia, A.; Ng, B.; Mostafavi, S.; Reid, G.; Subrt, P.; Hijiya, N.; Schultz, K.R. The TLR9 agonist (GNKG168) induces a unique immune activation pattern in vivo in children with minimal residual disease positive acute leukemia: Results of the TACL T2009-008 phase I study. Pediatric Hematol. Oncol. 2019, 36, 468–481. [Google Scholar] [CrossRef] [PubMed]
- Lichtenegger, F.S.; Schnorfeil, F.M.; Rothe, M.; Deiser, K.; Altmann, T.; Bücklein, V.L.; Köhnke, T.; Augsberger, C.; Konstandin, N.P.; Spiekermann, K.; et al. Toll-like receptor 7/8-matured RNA-transduced dendritic cells as post-remission therapy in acute myeloid leukaemia: Results of a phase I trial. Clin. Transl. Immunol. 2020, 9, e1117. [Google Scholar] [CrossRef] [PubMed]
- Creagh, E.M.; O’Neill, L.A. TLRs, NLRs and RLRs: A trinity of pathogen sensors that co-operate in innate immunity. Trends Immunol. 2006, 27, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Velloso, F.J.; Trombetta-Lima, M.; Anschau, V.; Sogayar, M.C.; Correa, R.G. NOD-like receptors: Major players (and targets) in the interface between innate immunity and cancer. Biosci. Rep. 2019, 39, BSR20181709. [Google Scholar] [CrossRef]
- Kanneganti, T.-D.; Lamkanfi, M.; Núñez, G. Intracellular NOD-like Receptors in Host Defense and Disease. Immunity 2007, 27, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Motta, V.; Soares, F.; Sun, T.; Philpott, D.J. NOD-Like Receptors: Versatile Cytosolic Sentinels. Physiol. Rev. 2015, 95, 149–178. [Google Scholar] [CrossRef]
- Stutz, A.; Golenbock, D.T.; Latz, E. Inflammasomes: Too big to miss. J. Clin. Investig. 2009, 119, 3502–3511. [Google Scholar] [CrossRef]
- Hasegawa, M.; Yang, K.; Hashimoto, M.; Park, J.-H.; Kim, Y.-G.; Fujimoto, Y.; Nuñez, G.; Fukase, K.; Inohara, N. Differential Release and Distribution of Nod1 and Nod2 Immunostimulatory Molecules among Bacterial Species and Environments. J. Biol. Chem. 2006, 281, 29054–29063. [Google Scholar] [CrossRef] [PubMed]
- Sioud, M.; Fløisand, Y. NOD2/CARD15 on bone marrow CD34 hematopoietic cells mediates induction of cytokines and cell differentiation. J. Leukoc. Biol. 2009, 85, 939–946. [Google Scholar] [CrossRef]
- Buteyn, N.J.; Santhanam, R.; Merchand-Reyes, G.; Murugesan, R.A.; Dettorre, G.M.; Byrd, J.C.; Sarkar, A.; Vasu, S.; Mundy-Bosse, B.L.; Butchar, J.P.; et al. Activation of the Intracellular Pattern Recognition Receptor NOD2 Promotes Acute Myeloid Leukemia (AML) Cell Apoptosis and Provides a Survival Advantage in an Animal Model of AML. J. Immunol. 2020, 204, 1988–1997. [Google Scholar] [CrossRef] [PubMed]
- Damon, L.E.; Rugo, H.S.; Ries, C.A.; Linker, C.A. Post-remission cytopenias following intense induction chemotherapy for acute myeloid leukemia. Leukemia 1994, 8, 535–541. [Google Scholar]
- Ekman, A.-K.; Cardell, L.O. The expression and function of Nod-like receptors in neutrophils. Immunology 2010, 130, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Yomade, O.; Spies-Weisshart, B.; Glaser, A.; Schnetzke, U.; Hochhaus, A.; Scholl, S. Impact of NOD2 polymorphisms on infectious complications following chemotherapy in patients with acute myeloid leukaemia. Ann. Hematol. 2013, 92, 1071–1077. [Google Scholar] [CrossRef] [PubMed]



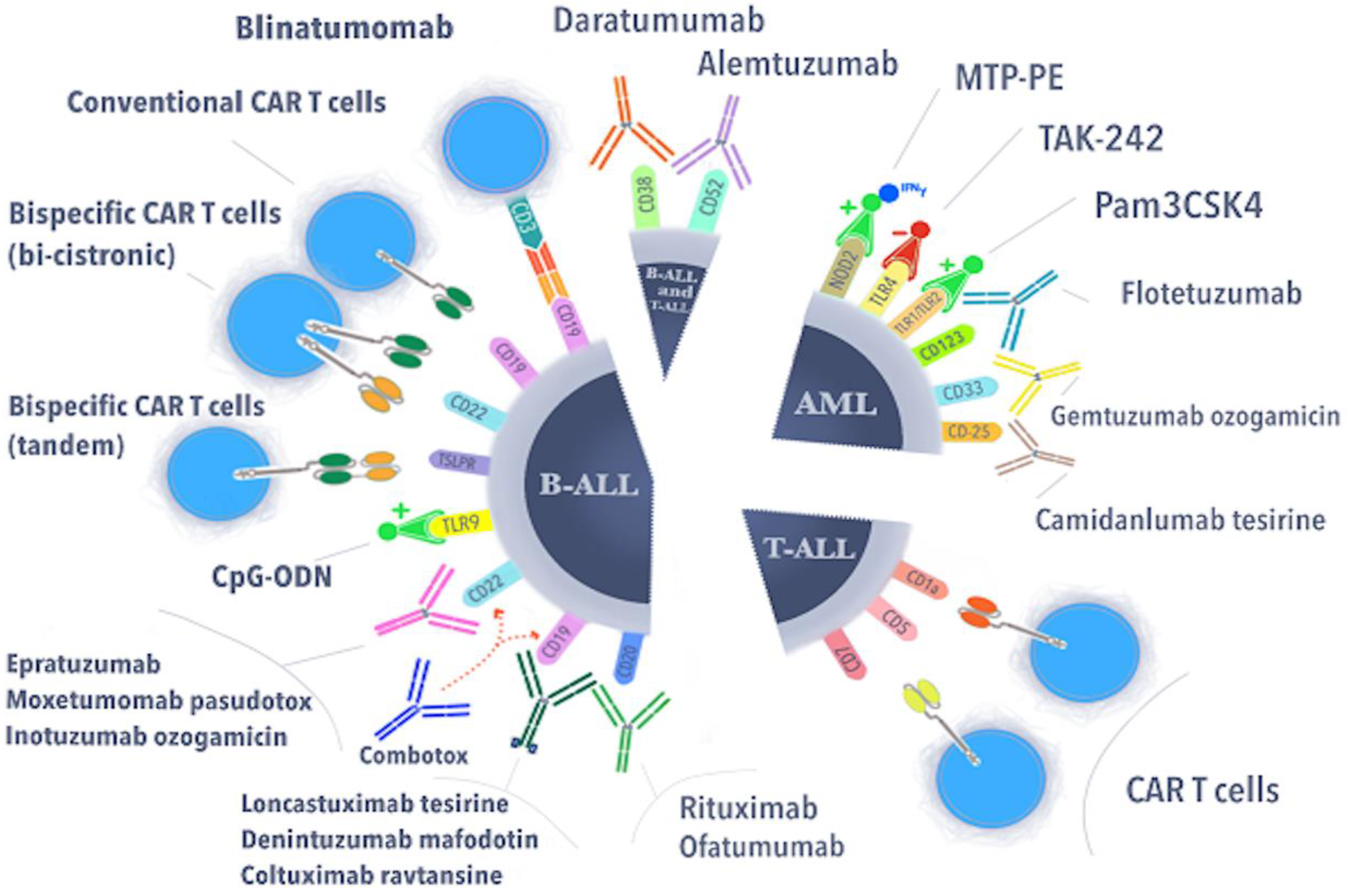{kind=link}
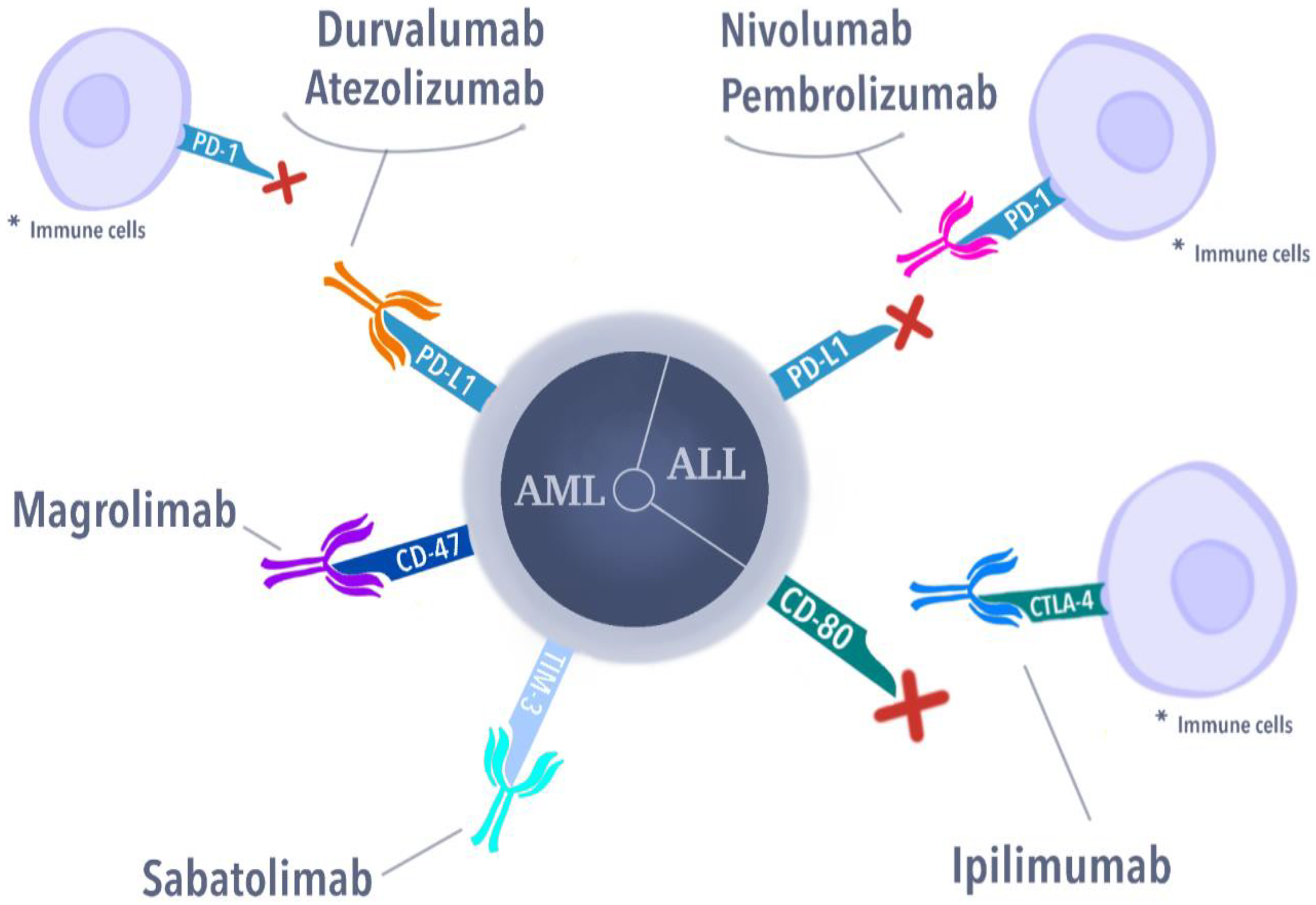{kind=link}
| Genetic Subgroup | Frequency in BCP-ALL | Characteristics | Prognosis | Therapeutic Approach | References |
|---|---|---|---|---|---|
| Hyperdiploidy | 25–30% | 51–67 chromosomes; 3 to 5 years old | excellent | MRD-based reduction in intensity treatment; condensin-complex members, AURKB, or the spindle assembly checkpoint | [4,5] |
| ETV6::RUNX1 t(12;21)(p13.2;q22.1) | 20–25% | Commonly occurring with the deletion of non-rearranged ETV6 allele, the deletion of PAX5, and the deletion of 6q | excellent | Conventional chemotherapy with reduced intensity | [6] |
| High hypodiploidy | 2–3% | 40–44 chromosomes, often occurring with dic(9;20) or ETV6::RUNX1 | poor | MRD risk-stratified therapy | [7] |
| Near haploidy | <1% | 24–31 chromosomes; associated with mutations of NF1, FLT3, NRAS, KRAS, MAPK1, and PTPN11 and the deletion/mutation of IKZF3 | poor | BCL-2 inhibitors, immunotherapy, phosphoinositide 3-kinase (PI3K) inhibitors | [8] |
| Low haploidy | 2% | 32–32 chromosomes; the deletion of IKZF2, RB1, CDKN2A/CDKN2B, and TP53 mutations | poor | BCL-2 inhibitors, immunotherapy, phosphoinositide 3-kinase (PI3K) inhibitors | [8] |
| TCF3::PBX1 t(1;19)(q23;p13) | 5–6% | higher WBC at diagnosis; increased CNS relapse cases | poor | dasatinib and ponatinib | [9] |
| KMT2A::AFF1 t(4;11)(q21;q23) | 4% | pro-B (CD10-) immunophenotype, expression of myeloid markers | very poor | Intensification of treatment; DOT1L inhibitors, menin inhibitors, proteasome inhibitors, histone deacetylase inhibitors, BCL-2 inhibitors; CAR-T therapy | [10,11,12] |
| KMT2A-rearranged (11q23) | 2–3% | common in infant ALL (80%); <100 different partner genes | poor | DOT1L inhibitors, menin inhibitors, proteasome inhibitors, histone deacetylase inhibitors, BCL-2 inhibitors; CAR-T therapy | [12] |
| TCF3::HLF t(17;19)( | <0.7% | associated with the expression of stem cells and myeloid markers, alterations of PAX5, VPREB1 and the Ras signaling pathway | very poor | BCL-2 inhibitors (venetoclax), immunologic therapies, Aurora A kinase inhibitors | [13] |
| BCR::ABL1 t(9;22)(q34.1;q11.2) | 2–3% | increases with age; associated with IKZF1, PAX5, CDKN2A/B deletions, and hypodiploidy | poor | improved outcome when chemotherapy combined with TKI: imatinib, dasatinib, ponatinib | [14] |
| iAMP21 | 1–2% | additional copies of a region of chromosome 21 that includes RUNX1, and it can be associated with older age (median, 9 years) and low white blood cell counts | poor | intensive treatment improves outcome | [15] |
| Ph-like or BCR-ABL1-like ALL | 10–15% | increases with age; frequently harbor alterations of IKZF1; alterations include: JAK-STAT (CRLF2 rearrangement (P2RY8::CRLF2 and IGH::CRLF2; CRLF2 F232C mutation) ABL1 class fusions (ABL1, ABL2, PDGFRB, CSF1R, PDGFRA, and LYN) fewer common fusions (FLT3, FGFR1, NTRK3, and PTK2B) | poor | ABL1 inhibitors, BCL-2 inhibitors, blinatumomab, inotuzumab, and CAR-T cells | [16,17,18] |
| Genetic Subgroup | Frequency | Characteristics | Prognosis | References |
|---|---|---|---|---|
| RUNX1::RUNX1T1 t(8;21)(q22;q22) | 10–12% | FAB M2, blasts with single and thin Auer rods, median age 8 years; CBF AML; standard risk group; almost 90% of patients achieve complete remission with chemotherapy alone; dasatinib (targeting KIT kinase); GO for relapsed patients | very good | [20,21,22,23,24,25] |
| CBFB::MYH11 inv(16)(p13.1q22) or t(16;16)(p13.1;q22) | 8–10% | FAB M4eo, median age 9 years; core binding factor (CBF) AML; standard risk group; almost 90% of patients achieve complete remission with chemotherapy alone; dasatinib (targeting KIT kinase); gemtuzumab ozogamicin (GO) for relapsed patients | very good | [20,21,22,23,24,25] |
| PML::RARA t(15;17)(q24.1;q21.2) | 5–10% | FAB M3, median age 7 years (1–18 years); acute promyelocytic leukemia (APL); standard risk group; ATRA, ATO treatment | very good | [20,21,22,23,24,26,27] |
| KMT2A- rearranged (11q23) | 16–21% | FAB M4 and M5, infant, median age 7 years (1–18 years) KMT2A with multiple partners; hypomethylating agents, DOT1L inhibitors, Menin-KMT2A protein–protein interaction inhibitors, protein interaction inhibitors | poor or intermediate | [10,11,20,21,22,23,24,25,28,29,30] |
| KMT2A::MLLT3 t(9;11)(p22;q23) | 6–9% | identified in 40% of KMT2Ar AML cases | intermediate | |
| KMT2A::MLLT1 t(11;19)(q23;p13.3) | 1% | identified in 7% of KMT2Ar AML cases | intermediate | |
| KMT2A::ELL t(11;19)(q23;p13.1) | 1–2% | identified in 7% of KMT2Ar AML cases | poor | |
| KMT2A::MLLT10 t(10;11)(p12;q23) | 2–3% | identified in 6% of KMT2Ar AML cases | poor | |
| KMT2A::MLLT4 t(6;11)(q27;q23) | 1–2% | identified in 8% of KMT2Ar AML cases | poor | |
| NUP98::NSD1 t(5;11)(q35;p15) | 3–4% | FAB M4 and M5; median age 10.4 years; 10% strong association with FLT3-ITD | poor | [20,21,22,23,24,25,31,32,33] |
| NUP98::KMD5A t(11;12)(p15;p13) | 1–2% | 30% of FAB M7 (AMKL); median age 3.2 years | ||
| MNX1::ETV6 t(7;12)(q36;p13) | 1% | Only infants (4% of infants); 3-year EFS below 24%; KAT inhibitors, C646, I-CBP112, CCS1477 | poor | [20,21,22,23,24,25] |
| DEK::NUP214 t(6;9)(p22;q34) | 1–4% | FAB M2 and M4; median age 12 years, no infant; association with FLT3-ITD; benefit from HSCT in first CR | poor | [20,21,22,23,24,25] |
| CBFA2T3::GLIS2 inv(16)(p13.3;q24.3) | 2–3% | FAB M7 (AMKL); infants; median age 1.5 years; 20% of non-DS-AMKL; high rates of relapse, and dismal survival; GLI inhibitors (GANT61); the AURKA inhibitor alisertib (MLN8237) | very poor | [20,21,22,23,24,25,31,32] |
| BCR::ABL1 t(9;22)(q34;q11) | 1% | sensitivity to TKI | poor | [20,21,22,23,24,25] |
| KAT6A::CREBBP t(8;16)(p11;p13) | <1% | FAB M4 and M5; infants; spontaneous remission has been observed | intermediate | [20,21,22,23,24,25,34] |
| RBM15::MKL1 t(1;22)(p13;q13) | <1% | FAB M7 (AMKL); median age 0.7 years; 14% of non-DS-AMKL | intermediate | [20,21,22,23,24,25,31,32] |
| PICALM::MLLT10 t(10;11)(p12;q14) | <1% | Extramedullary disease, CD7+, older children | intermediate | [20,21,22,23,24,25] |
| EVI1(MECOM) inv(3)(q21q26.2)/ t(3;3)(q21;q26.2) | <2% | Median age 3 years; secondary abnormality monosomy 7 | poor | [20,21,22,23,24,25] |
| NPM1::MLF1 t(3;5)(q25;q35) | <0.5% | FAB M2, M4, and M5; median age 3.5 years | intermediate | [35] |
| FUS::ERG t(16;21)(p11;q22) | <0.4% | Median age 8.5 years | poor | [36] |
| RUNX1::CBFA2T3 t(16;21)(q24;q22) | <0.2% | FAB M1/M2, t-AML; median age 6.8 years | unknown | [36] |
| Monosomy 7/del(7q) | 3% | median age 7 years; are considered candidates for allo-HSCT in first complete remission (CR) | poor | [20,21,22,23,24,25] |
| Monosomy 5/del(5q) | 1–2% | FAB M0; median age 12.5 years; 10-year OS 30–40% 10-year EFS 30%; are considered candidates for allo-HSCT in first CR | poor | [20,21,22,23,24,25] |
| Trisomy 8 | 10–14% | median age 10 years | unknown | [37] |
| Hyperdiploidy (48–65 chromosomes) | 11% | FAB M7 (AMKL); infants; median age 2 years | no significance | [38] |
| Targeted Molecule | Drug Name (Symbol) | Mechanism of Action | Pediatric Hematological Malignancy |
|---|---|---|---|
| CD19 | Denintuzumab mafodotin (SGN-CD19A) | Inhibition of cell division in CD19+ cells | ALL/B-ALL |
| Coltuximab Ravtansine | Disruption of microtubules, impeding cell cycle | B-ALL | |
| Loncastuximab tesirine (ADCT-402) | Creating cytotoxic interstrand DNA cross-links in CD19+ cells | B-ALL | |
| CD25 | Camidanlumab tesirine (ADCT-301) | Creating cytotoxic interstrand DNA cross-links in CD25+ cells | ALL/AML |
| CD20 | Rituximab | Complement-dependent cytotoxicity, antibody-dependent cell-mediated cytotoxicity | B-ALL |
| Ofatumumab | Complement-dependent cytotoxicity | B-ALL | |
| CD22 | Epratuzumab | Modulate the activation of B lymphocytes | relapse B-ALL |
| Inotuzumab ozogamicin | Breaking double-stranded DNA, hampering the cell cycle (at the G2/M phase) | B-ALL | |
| Moxetumomab pasudotox | Targeting in CD22 causes cytotoxicity relative to B-ALL blasts | B-ALL | |
| CD33 | Gemtuzumab ozogamicin | Blocking cell cycle resulting in tumor cell death | AML |
| CD52 | Alemtuzumab | Binding to CD52 leads to cell death by ADCC and through CDC | risk reduction in GVHD in patients with ALL after an alternative donor transplantation |
| CD38 | Daratumumab | Its role is to bind to a specific epitope on CD38-expressing cells, thus leading to their apoptosis. | T-ALL |
| Toll-Like Receptor | Dependent Pathway | Ligand | Localization |
|---|---|---|---|
| TLR1 | MyD88 | Lipopeptides | Cell surface |
| TLR2 | MyD88, TIRAP, MAL | Lipopeptides | Cell surface |
| TLR3 | TRIF | Nucelic acid fragments | Inner cell compartments |
| TLR4 | MyD88, TIRAP, TRAM | Lipopolisaccharydes | Cell surface |
| TLR5 | MyD88 | Flagellin | Cell surface |
| TLR6 | MyD88 | Lipopeptides | Cell surface |
| TLR7 | MyD88 | Nucelic acid fragments | Inner cell compartments |
| TLR8 | MyD88 | Nucelic acid fragments | Inner cell compartments |
| TLR9 | MyD88 | Nucelic acid fragments | Inner cell compartments |
| TLR10 | MyD88 | Lipopeptides | Cell surface |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panuciak, K.; Margas, M.; Makowska, K.; Lejman, M. Insights into Modern Therapeutic Approaches in Pediatric Acute Leukemias. Cells 2022, 11, 139. https://doi.org/10.3390/cells11010139
Panuciak K, Margas M, Makowska K, Lejman M. Insights into Modern Therapeutic Approaches in Pediatric Acute Leukemias. Cells. 2022; 11(1):139. https://doi.org/10.3390/cells11010139
Chicago/Turabian StylePanuciak, Kinga, Mikołaj Margas, Karolina Makowska, and Monika Lejman. 2022. "Insights into Modern Therapeutic Approaches in Pediatric Acute Leukemias" Cells 11, no. 1: 139. https://doi.org/10.3390/cells11010139
APA StylePanuciak, K., Margas, M., Makowska, K., & Lejman, M. (2022). Insights into Modern Therapeutic Approaches in Pediatric Acute Leukemias. Cells, 11(1), 139. https://doi.org/10.3390/cells11010139