
Cell-Based Therapy for the Treatment of Glioblastoma: An Update from Preclinical to Clinical Studies

 ,
,  , , ,
, , ,
Abstract
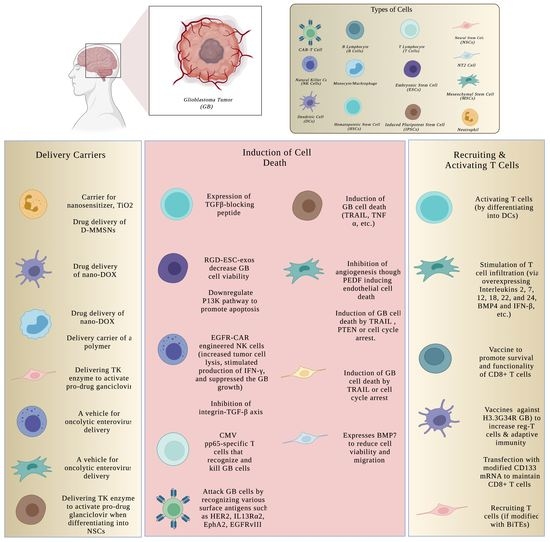
1. Introduction
2. Cell Therapy for Glioblastoma
2.1. Immune Cell Therapy
2.1.1. Lymphocytes/CAR-T
2.1.2. Natural Killer (NK) Cells
2.1.3. Dendritic Cells (DCs)
2.1.4. Monocytes/Macrophages
2.1.5. Neutrophils
2.2. Stem Cell Therapy
2.2.1. Mesenchymal Stem Cells (MSCs)
Antitumor Effect of MSCs
Cytokine-Based Therapy
Oncolytic Virus Therapy
Anti-Angiogenesis Therapy
Induction of Tumor Cell Death
Protumor Effects of MSCs
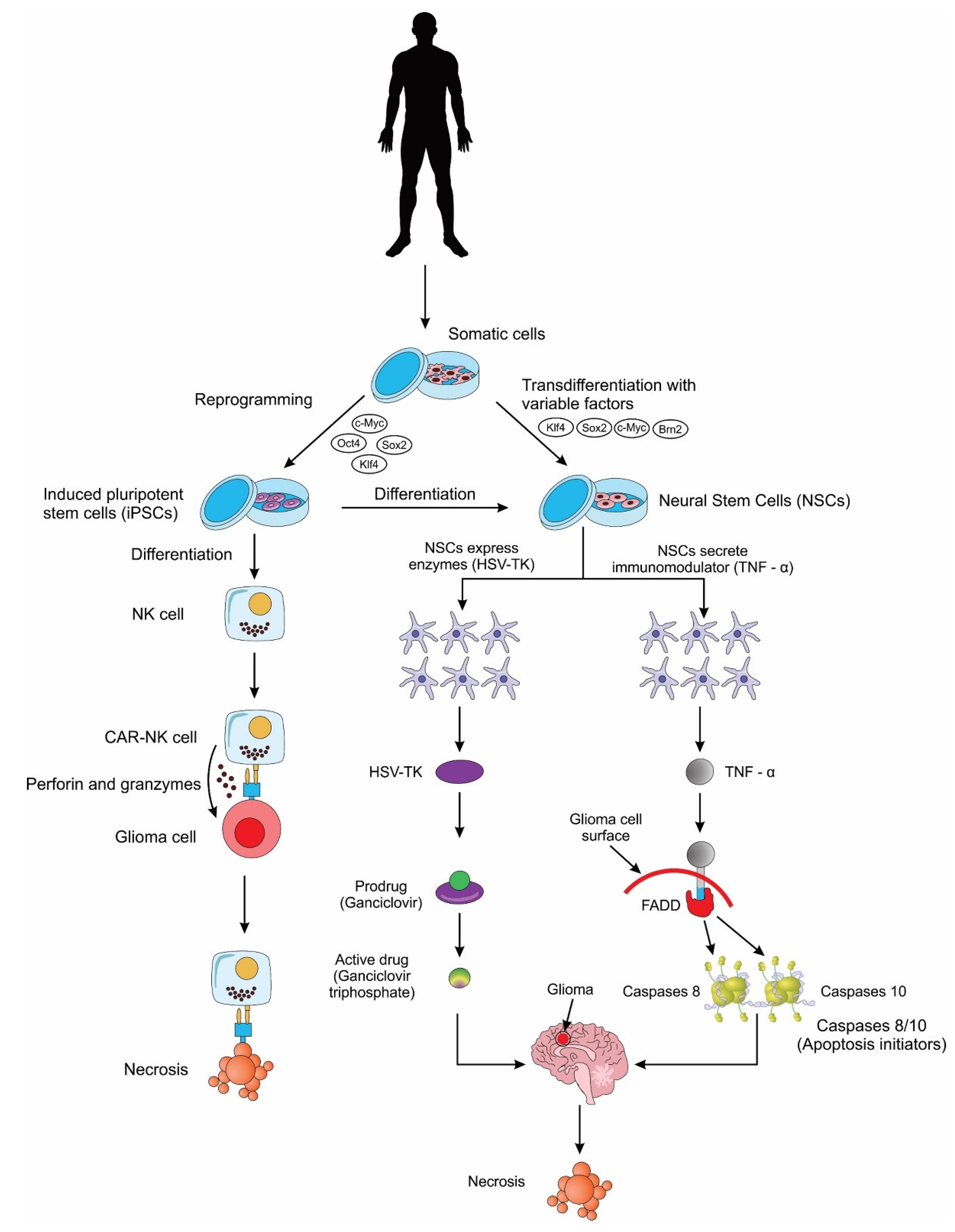
2.2.2. Neural Stem Cells (NSCs)
2.2.3. Induced Pluripotent Stem Cells (iPSCs)
2.2.4. Other Stem Cells
3. Clinical Trials on Cell-Based Treatment for Glioblastoma
4. Future Perspectives
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ALE | Autologous lymphoid effector cells |
| ALECSAT | Autologous lymphoid effector cells specific against tumor |
| AD | Adipose tissue |
| CAR-T- | Chimeric antigen receptor T cells |
| BM | Bone marrow |
| CD | Cluster of differentiation |
| CMV | Cytomegalovirus |
| CMV-ALT | CMV-autologous lymphocyte transfer |
| CNS | Central nervous system |
| CSCs | Cancer stem cells |
| CYNK-001 | Human placental hematopoietic stem cell-derived natural killer cells |
| DC | Dendritic cells |
| EGFRvIII | Epidermal growth factor receptor variant III |
| GB | Glioblastoma |
| GPX4 | Glutathione peroxidase 4 |
| GSC | Glioma stem cell |
| HER2 | Human epidermal growth factor receptor 2 |
| ID | Intradermal |
| IL | Interleukin |
| IN | Intranodal |
| IT | Intratumoral |
| IV | Intravenous |
| MSC | Mesenchymal stem cell |
| NK cells | Natural killer cells |
| NSCs | Neural stem cells |
| PBMCs | Peripheral blood mononuclear cells |
| PBSCs | Peripheral blood stem cells |
| PDT | Photodynamic therapy |
| SC | Subcutaneous |
| UC | Umbilical cord |
| UCB | Umbilical cord blood |
| WBCs | White blood cells |
References
- Nizamutdinov, D.; Stock, E.M.; Dandashi, J.A.; Vasquez, E.A.; Mao, Y.; Dayawansa, S.; Zhang, J.; Wu, E.; Fonkem, E.; Huang, J.H. Prognostication of survival outcomes in patients diagnosed with glioblastoma. World Neurosurg. 2018, 109, e67–e74. [Google Scholar] [CrossRef]
- Tan, A.C.; Ashley, D.M.; López, G.Y.; Malinzak, M.; Friedman, H.S.; Khasraw, M. Management of glioblastoma: State of the art and future directions. CA Cancer J. Clin. 2020, 70, 299–312. [Google Scholar] [CrossRef]
- Ghosh, D.; Nandi, S.; Bhattacharjee, S. Combination therapy to checkmate glioblastoma: Clinical challenges and advances. Clin. Transl. Med. 2018, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Da Ros, M.; De Gregorio, V.; Iorio, A.L.; Giunti, L.; Guidi, M.; De Martino, M.; Genitori, L.; Sardi, I. Glioblastoma chemoresistance: The double play by microenvironment and blood-brain barrier. Int. J. Mol. Sci. 2018, 19, 2879. [Google Scholar] [CrossRef]
- Kim, G.B.; Aragon-Sanabria, V.; Randolph, L.; Jiang, H.; Reynolds, J.A.; Webb, B.S.; Madhankumar, A.; Lian, X.; Connor, J.R.; Yang, J.; et al. High-affinity mutant Interleukin-13 targeted CAR T cells enhance delivery of clickable biodegradable fluorescent nanoparticles to glioblastoma. Bioact. Mater. 2020, 5, 624–635. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.K.; De Leon, G.; Boczkowski, D.; Schmittling, R.; Xie, W.; Staats, J.; Liu, R.; Johnson, L.A.; Weinhold, K.; Archer, G.E.; et al. Recognition and killing of autologous, primary glioblastoma tumor cells by human cytomegalovirus pp65-specific cytotoxic T cells. Clin. Cancer Res. 2014, 20, 2684–2694. [Google Scholar] [CrossRef] [PubMed]
- Lee-Chang, C.; Miska, J.; Hou, D.; Rashidi, A.; Zhang, P.; Burga, R.A.; Jusué-Torres, I.; Xiao, T.; Arrieta, V.A.; Zhang, D.Y.; et al. Activation of 4-1BBL+B cells with CD40 agonism and IFNγ elicits potent immunity against glioblastoma. J. Exp. Med. 2020, 218, e20200913. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Aguilar, B.; Starr, R.; Alizadeh, D.; Brito, A.; Sarkissian, A.; Ostberg, J.R.; Forman, S.J.; Brown, C.E. Glioblastoma-targeted CD4+CAR T cells mediate superior antitumor activity. JCI Insight 2018, 3, 99048. [Google Scholar] [CrossRef] [PubMed]
- Krenciute, G.; Prinzing, B.L.; Yi, Z.; Wu, M.-F.; Liu, H.; Dotti, G.; Balyasnikova, I.V.; Gottschalk, S. Transgenic expression of IL15 improves antiglioma activity of IL13Rα2-CAR T cells but results in antigen loss variants. Cancer Immunol. Res. 2017, 5, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Pituch, K.C.; Miska, J.; Krenciute, G.; Panek, W.K.; DeFelice, G.; Rodriguez-Cruz, T.; Wu, M.; Han, Y.; Lesniak, M.S.; Gottschalk, S.; et al. Adoptive transfer of IL13Rα2-specific chimeric antigen receptor T cells creates a Pro-inflammatory environment in glioblastoma. Mol. Ther. 2018, 26, 986–995. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Sun, B.; Dai, H.; Li, W.; Shi, L.; Zhang, P.; Li, S.; Zhao, X. T cells expressing NKG2D chimeric antigen receptors efficiently eliminate glioblastoma and cancer stem cells. J. Immunother. Cancer 2019, 7, 171. [Google Scholar] [CrossRef]
- Wang, D.; Starr, R.; Chang, W.-C.; Aguilar, B.; Alizadeh, D.; Wright, S.L.; Yang, X.; Brito, A.; Sarkissian, A.; Ostberg, J.R.; et al. Chlorotoxin-directed CAR T cells for specific and effective targeting of glioblastoma. Sci. Transl. Med. 2020, 12, 2672. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Li, H.; Bin, S.; Li, P.; Chen, J.; Gu, H.; Yuan, W. The efficacy of third generation anti-HER2 chimeric antigen receptor T cells in combination with PD1 blockade against malignant glioblastoma cells. Oncol. Rep. 2019, 42, 1549–1557. [Google Scholar] [CrossRef] [PubMed]
- Bielamowicz, K.; Fousek, K.; Byrd, T.T.; Samaha, H.; Mukherjee, M.; Aware, N.; Wu, M.-F.; Orange, J.S.; Sumazin, P.; Man, T.-K.; et al. Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro-Oncology 2018, 20, 506–518. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Sun, R.; Shi, B.; Wang, Y.; Di, S.; Luo, H.; Sun, Y.; Li, Z.; Zhou, M.; Jiang, H. Antitumor efficacy of chimeric antigen receptor T cells against EGFRvIII-expressing glioblastoma in C57BL/6 mice. Biomed. Pharmacother. 2019, 113, 108734. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.D.; Yu, X.; Castano, A.P.; Darr, H.; Henderson, D.B.; Bouffard, A.A.; Larson, R.C.; Scarfò, I.; Bailey, S.R.; Gerhard, G.M.; et al. CRISPR-Cas9 disruption of PD-1 enhances activity of universal EGFRvIII CAR T cells in a preclinical model of human glioblastoma. J. Immunother. Cancer 2019, 7, 304. [Google Scholar] [CrossRef] [PubMed]
- Ravanpay, A.C.; Gust, J.; Johnson, A.J.; Rolczynski, L.S.; Cecchini, M.; Chang, C.A.; Hoglund, V.J.; Mukherjee, R.; Vitanza, N.A.; Orentas, R.J.; et al. EGFR806-CAR T cells selectively target a tumor-restricted EGFR epitope in glioblastoma. Oncotarget 2019, 10, 7080–7095. [Google Scholar] [CrossRef] [PubMed]
- Hegde, M.; Corder, A.; Chow, K.K.; Mukherjee, M.; Ashoori, A.; Kew, Y.; Zhang, Y.J.; Baskin, D.S.; Merchant, F.; Brawley, V.S.; et al. Combinational targeting offsets antigen escape and enhances effector functions of adoptively transferred T cells in glioblastoma. Mol. Ther. 2013, 21, 2087–2101. [Google Scholar] [CrossRef]
- Hegde, M.; Mukherjee, M.; Grada, Z.; Pignata, A.; Landi, D.; Navai, S.; Wakefield, A.; Fousek, K.; Bielamowicz, K.; Chow, K.K.; et al. Tandem CAR T cells targeting HER2 and IL13Rα2 mitigate tumor antigen escape. J. Clin. Investig. 2016, 126, 3036–3052. [Google Scholar] [CrossRef]
- Brown, C.E.; Aguilar, B.; Starr, R.; Yang, X.; Chang, W.-C.; Weng, L.; Chang, B.; Sarkissian, A.; Brito, A.; Sanchez, J.F.; et al. Optimization of IL13Rα2-targeted chimeric antigen receptor T cells for improved anti-tumor efficacy against glioblastoma. Mol. Ther. 2018, 26, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Zhao, S.; Zhang, Y.; Wang, Y.; Zhang, Z.; Yang, M.; Zhu, Y.; Zhang, G.; Guo, G.; Tong, A.; et al. B7-H3 as a novel CAR-T therapeutic target for glioblastoma. Mol. Ther. Oncolytics 2019, 14, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Nehama, D.; Di Ianni, N.; Musio, S.; Du, H.; Patanè, M.; Pollo, B.; Finocchiaro, G.; Park, J.H.; Dunn, D.E.; Edwards, D.; et al. B7-H3-redirected chimeric antigen receptor T cells target glioblastoma and neurospheres. EBioMedicine 2019, 47, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Murty, S.; Haile, S.T.; Beinat, C.; Aalipour, A.; Alam, I.S.; Murty, T.; Shaffer, T.M.; Patel, C.B.; Graves, E.E.; Mackall, C.L.; et al. Intravital imaging reveals synergistic effect of CAR T-cells and radiation therapy in a preclinical immunocompetent glioblastoma model. OncoImmunology 2020, 9, 1757360. [Google Scholar] [CrossRef] [PubMed]
- Navarro, A.G.; Kmiecik, J.; Leiss, L.; Zelkowski, M.; Engelsen, A.; Bruserud, Ø.; Zimmer, J.; Enger, P.Ø.; Chekenya, M. NK Cells with KIR2DS2 immunogenotype have a functional activation advantage to efficiently kill glioblastoma and prolong animal survival. J. Immunol. 2014, 193, 6192–6206. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Kang, W.Y.; Yoon, Y.; Jin, J.Y.; Song, H.J.; Her, J.H.; Kang, S.M.; Hwang, Y.K.; Kang, K.J.; Joo, K.M.; et al. Natural killer (NK) cells inhibit systemic metastasis of glioblastoma cells and have therapeutic effects against glioblastomas in the brain. BMC Cancer 2015, 15, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bi, J.; Zheng, X.; Chen, Y.; Wang, H.; Wu, W.; Wang, Z.; Wu, Q.; Peng, H.; Wei, H.; et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat. Immunol. 2018, 19, 723–732. [Google Scholar] [CrossRef]
- Hung, A.L.; Maxwell, R.; Theodros, D.; Belcaid, Z.; Mathios, D.; Luksik, A.S.; Kim, E.; Wu, A.; Xia, Y.; Garzon-Muvdi, T.; et al. TIGIT and PD-1 dual checkpoint blockade enhances antitumor immunity and survival in GBM. OncoImmunology 2018, 7, e1466769. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Chu, J.; Chan, W.K.; Zhang, J.; Wang, Y.; Cohen, J.; Victor, A.; Meisen, W.H.; Kim, S.-H.; Grandi, P.; et al. CAR-engineered NK cells targeting wild-type EGFR and EGFRvIII enhance killing of glioblastoma and patient-derived glioblastoma stem cells. Sci. Rep. 2015, 5, 11483. [Google Scholar] [CrossRef]
- Genßler, S.; Burger, M.C.; Zhang, C.; Oelsner, S.; Mildenberger, I.; Wagner, M.; Steinbach, J.P.; Wels, W.S. Dual targeting of glioblastoma with chimeric antigen receptor-engineered natural killer cells overcomes heterogeneity of target antigen expression and enhances antitumor activity and survival. OncoImmunology 2016, 5, e1119354. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Nakazawa, T.; Nakamura, M.; Nishimura, F.; Matsuda, R.; Omoto, K.; Shida, Y.; Murakami, T.; Nakagawa, I.; Motoyama, Y.; et al. Ex vivo-expanded highly purified natural killer cells in combination with temozolomide induce antitumor effects in human glioblastoma cells in vitro. PLoS ONE 2019, 14, e0212455. [Google Scholar] [CrossRef]
- Navarro, A.G.; Espedal, H.; Joseph, J.V.; Trachsel-Moncho, L.; Bahador, M.; Gjertsen, B.T.; Kristoffersen, E.K.; Simonsen, A.; Miletic, H.; Enger, P.Ø.; et al. Pretreatment of glioblastoma with Bortezomib potentiates natural killer cell cytotoxicity through TRAIL/DR5 mediated apoptosis and prolongs animal survival. Cancers 2019, 11, 996. [Google Scholar] [CrossRef]
- Sharifzad, F.; Mardpour, S.; Mardpour, S.; Fakharian, E.; Taghikhani, A.; Sharifzad, A.; Kiani, S.; Heydarian, Y.; Los, M.J.; Ebrahimi, M.; et al. HSP70/IL-2 treated NK cells effectively cross the blood brain barrier and target tumor cells in a rat model of induced glioblastoma multiforme (GBM). Int. J. Mol. Sci. 2020, 21, 2263. [Google Scholar] [CrossRef] [PubMed]
- Vichchatorn, P.; Wongkajornsilp, A.; Petvises, S.; Tangpradabkul, S.; Pakakasama, S.; Hongeng, S. Dendritic cells pulsed with total tumor RNA for activation NK-like T cells against glioblastoma multiforme. J. Neuro-Oncology 2005, 75, 111–118. [Google Scholar] [CrossRef]
- Podshivalova, E.S.; Semkina, A.S.; Kravchenko, D.S.; Frolova, E.I.; Chumakov, S.P. Efficient delivery of oncolytic enterovirus by carrier cell line NK-92. Mol. Ther. Oncolytics 2021, 21, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Rich, J.N. Cancer stem cells in radiation resistance. Cancer Res. 2007, 67, 8980–8984. [Google Scholar] [CrossRef] [PubMed]
- Shaim, H.; Shanley, M.; Basar, R.; Daher, M.; Gumin, J.; Zamler, D.B.; Uprety, N.; Wang, F.; Huang, Y.; Gabrusiewicz, K.; et al. Targeting the αv integrin/TGF-β axis improves natural killer cell function against glioblastoma stem cells. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef] [PubMed]
- Li, T.-F.; Li, K.; Zhang, Q.; Wang, C.; Yue, Y.; Chen, Z.; Yuan, S.-J.; Liu, X.; Wen, Y.; Han, M.; et al. Dendritic cell-mediated delivery of doxorubicin-polyglycerol-nanodiamond composites elicits enhanced anti-cancer immune response in glioblastoma. Biomaterials 2018, 181, 35–52. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Lv, X.; Zhang, X.; Li, T.; Zang, G.; Yang, N.; Wang, X.; Wu, J.; Chen, W.; Liu, Y.-J.; et al. An effective dendritic cell-based vaccine containing glioma stem-like cell lysate and CpG adjuvant for an orthotopic mouse model of glioma. Int. J. Cancer 2018, 144, 2867–2879. [Google Scholar] [CrossRef]
- Peeters, S.; Owens, G.; Sun, M.Z.; Lee, A.; Kienzler, J.C.; Orpilla, J.; Contreras, E.; Treger, J.; Odesa, S.; Wang, A.C.; et al. Dendritic cell vaccination is effective against H3. 3G34R mutant glioblastoma in a novel syngeneic genetically engineered mouse model. Neurosurgery 2020, 67, nyaa447_795. [Google Scholar] [CrossRef]
- Shao, G.; Zhou, C.; Ma, K.; Zhao, W.; Feng, G.; Xiong, Q.; Yang, L.; Yang, Z. Dendritic cells transduced with glioma-expressed antigen 2 recombinant adenovirus induces specific cytotoxic lymphocyte response and anti-tumor effect in mice. J. Inflamm. 2020, 17, 1–7. [Google Scholar] [CrossRef]
- Do, A.S.-M.S.; Amano, T.; Edwards, L.A.; Zhang, L.; De Peralta-Venturina, M.; John, S.Y. CD133 mRNA-loaded dendritic cell vaccination abrogates glioma stem cell propagation in humanized glioblastoma mouse model. Mol. Ther.-Oncolytics 2020, 18, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Li, K.; Li, T.; Chen, Z.; Wen, Y.; Liu, X.; Jia, X.; Zhang, Y.; Xu, Y.; Han, M.; et al. Monocyte-mediated chemotherapy drug delivery in glioblastoma. Nanomedicine 2018, 13, 157–178. [Google Scholar] [CrossRef] [PubMed]
- Ibarra, L.; Beaugé, L.; Arias-Ramos, N.; Rivarola, V.; Chesta, C.; López-Larrubia, P.; Palacios, R. Trojan horse monocyte-mediated delivery of conjugated polymer nanoparticles for improved photodynamic therapy of glioblastoma. Nanomedicine 2020, 15, 1687–1707. [Google Scholar] [CrossRef] [PubMed]
- Gattas, M.; Estecho, I.; Huvelle, M.L.; Errasti, A.; Silva, E.C.; Simian, M. A heterotypic tridimensional model to study the interaction of macrophages and glioblastoma in vitro. Int. J. Mol. Sci. 2021, 22, 5105. [Google Scholar] [CrossRef]
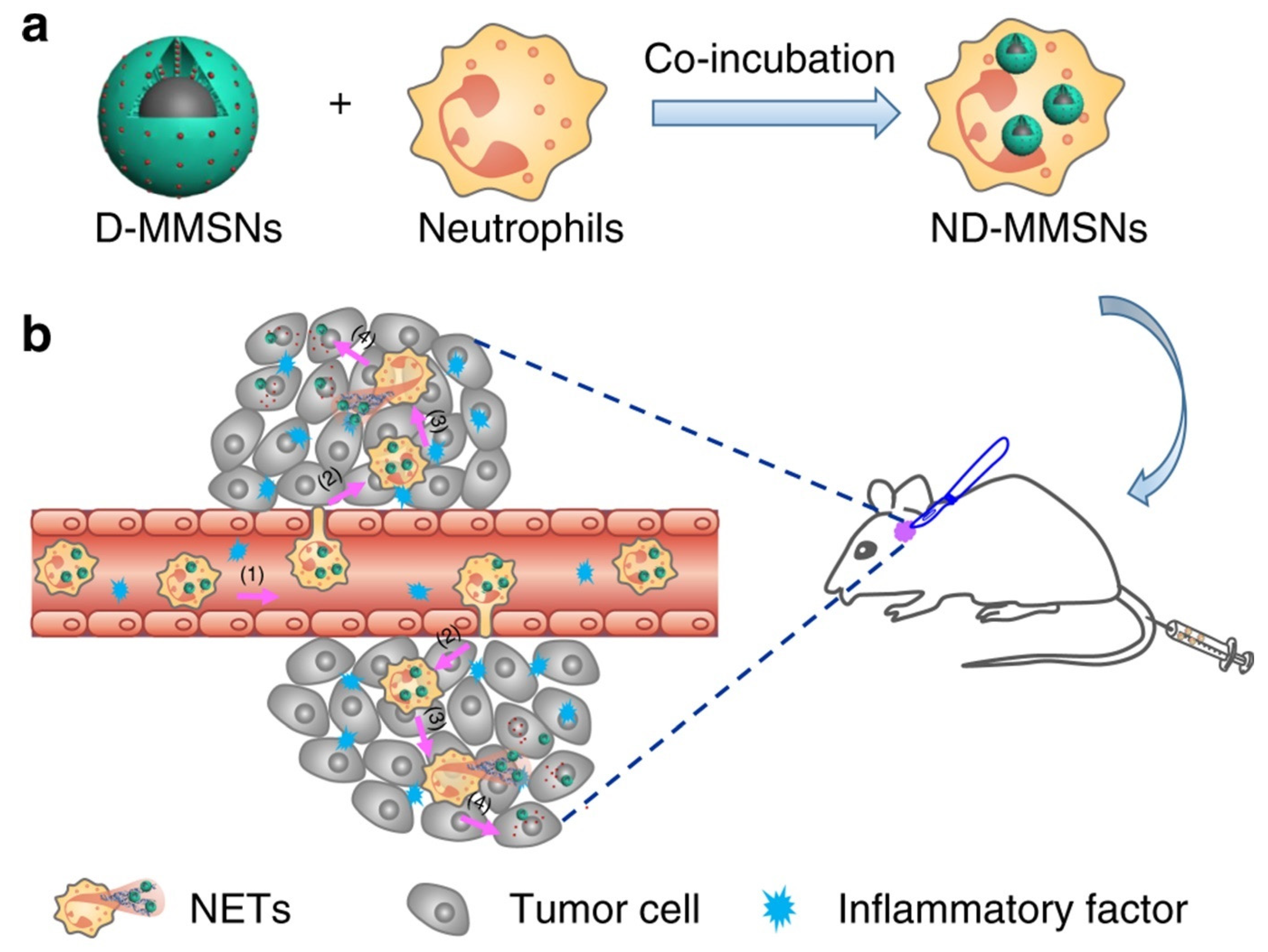
- Li, Y.; Teng, X.; Wang, Y.; Yang, C.; Yan, X.; Li, J. Neutrophil delivered hollow titania covered persistent luminescent nanosensitizer for ultrosound augmented chemo/immuno glioblastoma therapy. Adv. Sci. 2021, 8, 2004381. [Google Scholar] [CrossRef]
- Xue, J.; Zhao, Z.; Zhang, L.; Xue, L.; Shen, S.; Wen, Y.; Wei, Z.; Wang, L.; Kong, L.; Sun, H.; et al. Neutrophil-mediated anticancer drug delivery for suppression of postoperative malignant glioma recurrence. Nat. Nanotechnol. 2017, 12, 692–700. [Google Scholar] [CrossRef]
- Wu, M.; Zhang, H.; Tie, C.; Yan, C.; Deng, Z.; Wan, Q.; Liu, X.; Yan, F.; Zheng, H. MR imaging tracking of inflammation-activatable engineered neutrophils for targeted therapy of surgically treated glioma. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Goenka, A.; Tiek, D.; Song, X.; Huang, T.; Hu, B.; Cheng, S.-Y. The many facets of therapy resistance and tumor recurrence in glioblastoma. Cells 2021, 10, 484. [Google Scholar] [CrossRef]
- Lim, M.; Xia, Y.; Bettegowda, C.; Weller, M. Current state of immunotherapy for glioblastoma. Nat. Rev. Clin. Oncol. 2018, 15, 422–442. [Google Scholar] [CrossRef]
- Wang, X.; Lu, J.; Guo, G.; Yu, J. Immunotherapy for recurrent glioblastoma: Practical insights and challenging prospects. Cell Death Dis. 2021, 12, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Friedenstein, A.J.; Piatetzky, S., II; Petrakova, K.V. Osteogenesis in transplants of bone marrow cells. J. Embryol. Exp. Morphol. 1966, 16, 381–390. [Google Scholar] [CrossRef]
- Caplan, A.I. Mesenchymal stem cells. J. Orthop. Res. 1991, 9, 641–650. [Google Scholar] [CrossRef]
- Rogers, I.; Casper, R.F. Umbilical cord blood stem cells. Best Pract. Res. Clin. Obs. Gynaecol. 2004, 18, 893–908. [Google Scholar] [CrossRef] [PubMed]
- Bussolati, B.; Bruno, S.; Grange, C.; Buttiglieri, S.; Deregibus, M.C.; Cantino, D.; Camussi, G. Isolation of renal progenitor cells from adult human kidney. Am. J. Pathol. 2005, 166, 545–555. [Google Scholar] [CrossRef]
- Wang, X.-J.; Xiang, B.-Y.; Ding, Y.-H.; Chen, L.; Zou, H.; Mou, X.-Z.; Xiang, C. Human menstrual blood-derived mesenchymal stem cells as a cellular vehicle for malignant glioma gene therapy. Oncotarget 2017, 8, 58309–58321. [Google Scholar] [CrossRef] [PubMed]
- Wexler, S.A.; Donaldson, C.; Denning-Kendall, P.; Rice, C.; Bradley, B.; Hows, J.M. Adult bone marrow is a rich source of human mesenchymal ‘stem’ cells but umbilical cord and mobilized adult blood are not. Br. J. Haematol. 2003, 121, 368–374. [Google Scholar] [CrossRef]
- Chen, L.-B.; Jiang, X.-B.; Yang, L. Differentiation of rat marrow mesenchymal stem cells into pancreatic islet beta-cells. World J. Gastroenterology 2004, 10, 3016–3020. [Google Scholar] [CrossRef]
- Lee, K.-D.; Kuo, T.K.-C.; Whang-Peng, J.; Chung, Y.-F.; Lin, C.-T.; Chou, S.-H.; Chen, J.-R.; Chen, Y.-P.; Lee, O.K.-S. In vitro hepatic differentiation of human mesenchymal stem cells. Hepatology 2004, 40, 1275–1284. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.-Y.; Jung, J.-H.; Kim, A.A.; Marasini, S.; Lee, Y.J.; Paek, S.H.; Kim, S.-S.; Suh-Kim, H. Combined effects of mesenchymal stem cells carrying cytosine deaminase gene with 5-fluorocytosine and temozolomide in orthotopic glioma model. Am. J. Cancer Res. 2020, 10, 1429–1441. [Google Scholar] [PubMed]
- Pittenger, M.F.; Mackay, A.M.; Beck, S.C.; Jaiswal, R.K.; Douglas, R.; Mosca, J.D.; Moorman, M.A.; Simonetti, D.W.; Craig, S.; Marshak, D.R. Multilineage potential of adult human mesenchymal stem cells. Science 1999, 284, 143–147. [Google Scholar] [CrossRef]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.C.; Krause, D.S.; Deans, R.J.; Keating, A.; Prockop, D.J.; Horwitz, E.M. Minimal criteria for defining multipotent mesenchymal stromal cells. The international society for cellular therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Attia, N.; Mashal, M. Mesenchymal stem cells: The past present and future. Adv. Exp. Med. Biol. 2021, 1312, 107–129. [Google Scholar] [PubMed]
- Mushahary, D.; Spittler, A.; Kasper, C.; Weber, V.; Charwat, V. Isolation, cultivation, and characterization of human mesenchymal stem cells. Cytom. Part A 2018, 93, 19–31. [Google Scholar] [CrossRef]
- Yi, D.; Xiang, W.; Zhang, Q.; Cen, Y.; Su, Q.; Zhang, F.; Lu, Y.; Zhao, H.; Fu, P. Human Glioblastoma-derived Mesenchymal stem cell to Pericytes transition and Angiogenic capacity in Glioblastoma microenvironment. Cell. Physiol. Biochem. 2018, 46, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Xiang, W.; Yi, D.-Y.; Xue, B.-Z.; Wen, W.-W.; Abdelmaksoud, A.; Xiong, N.-X.; Jiang, X.-B.; Zhao, H.-Y.; Fu, P. Current status and potential challenges of mesenchymal stem cell-based therapy for malignant gliomas. Stem Cell Res. Ther. 2018, 9, 228. [Google Scholar] [CrossRef] [PubMed]
- Attia, N.; Mashal, M.; Puras, G.; Pedraz, J. Mesenchymal stem cells as a gene delivery tool: Promise, problems, and prospects. Pharmaceutics 2021, 13, 843. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Du, L.; Lin, L.; Wang, Y. Tumour-associated mesenchymal stem/stromal cells: Emerging therapeutic targets. Nat. Rev. Drug Discov. 2017, 16, 35–52. [Google Scholar] [CrossRef]
- Langroudi, L.; Hassan, Z.M.; Soleimani, M.; Hashemi, S.M. Tumor associated Mesenchymal stromal cells show higher immunosuppressive and angiogenic properties compared to adipose derived MSCs. Iran. J. Immunol. IJI 2015, 12, 226–239. [Google Scholar]
- Schichor, C.; Birnbaum, T.; Etminan, N.; Schnell, O.; Grau, S.; Miebach, S.; Aboody, K.; Padovan, C.; Straube, A.; Tonn, J.-C.; et al. Vascular endothelial growth factor A contributes to glioma-induced migration of human marrow stromal cells (hMSC). Exp. Neurol. 2006, 199, 301–310. [Google Scholar] [CrossRef]
- Kim, D.-S.; Kim, J.H.; Lee, J.K.; Choi, S.J.; Kim, J.-S.; Jeun, S.-S.; Oh, W.; Yang, Y.S.; Chang, J.W. Overexpression of CXC Chemokine receptors is required for the superior glioma-tracking property of umbilical cord blood-derived mesenchymal stem cells. Stem Cells Dev. 2009, 18, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Shi, J.; Yu, B.; Ni, W.; Wu, X.; Gu, Z. Chemokines mediate mesenchymal stem cell migration toward gliomas in vitro. Oncol. Rep. 2010, 23, 1561–1567. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Ito, Y.; Kawano, Y.; Kurozumi, K.; Kobune, M.; Tsuda, H.; Bizen, A.; Honmou, O.; Niitsu, Y.; Hamada, H. Antitumor effect of genetically engineered mesenchymal stem cells in a rat glioma model. Gene Ther. 2004, 11, 1155–1164. [Google Scholar] [CrossRef]
- Xu, G.; Jiang, X.-D.; Xu, Y.; Zhang, J.; Huang, F.-H.; Chen, Z.-Z.; Zhou, D.-X.; Shang, J.-H.; Zou, Y.-X.; Cai, Y.-Q. Adenoviral-mediated interleukin-18 expression in mesenchymal stem cells effectively suppresses the growth of glioma in rats. Cell Biol. Int. 2009, 33, 466–474. [Google Scholar] [CrossRef]
- Mohme, M.; Maire, C.L.; Geumann, U.; Schliffke, S.; Dührsen, L.; Fita, K.D.; Akyüz, N.; Binder, M.; Westphal, M.; Guenther, C.; et al. Local intracerebral immunomodulation using interleukin-expressing mesenchymal stem cells in glioblastoma. Clin. Cancer Res. 2020, 26, 2626–2639. [Google Scholar] [CrossRef] [PubMed]
- Ryu, C.H.; Park, S.-H.; Park, S.; Kim, S.M.; Lim, J.Y.; Jeong, C.H.; Yoon, W.-S.; Oh, W.-I.; Sung, Y.C.; Jeun, S.-S. Gene therapy of intracranial glioma using interleukin 12–secreting human umbilical cord blood–derived mesenchymal stem cells. Hum. Gene Ther. 2011, 22, 733–743. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Xiang, W.; Tian, J.; Wang, H.; Hu, S.; Wang, K.; Chen, L.; Huang, C.; Zhou, J. Bone marrow-derived mesenchymal stem cells differentially affect glioblastoma cell proliferation, migration, and invasion: A 2D-DIGE proteomic analysis. BioMed Res. Int. 2021, 2021, 4952876. [Google Scholar] [CrossRef]
- Mao, J.; Cao, M.; Zhang, F.; Zhang, J.; Duan, X.; Lu, L.; Yang, Z.; Zhang, X.; Zhu, W.; Zhang, Q.; et al. Peritumoral administration of IFNbeta upregulated mesenchymal stem cells inhibits tumor growth in an orthotopic, immunocompetent rat glioma model. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Jing, X.; Yi, Q.; Xiang, Z.; Tian, J.; Tan, B.; Zhu, J. IL22 furthers malignant transformation of rat mesenchymal stem cells, possibly in association with IL22RA1/STAT3 signaling. Oncol. Rep. 2019, 41, 2148–2158. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Gao, H.; Ji, W.; Zhu, F.; Sun, L.; Liu, Y.; Zhang, S.; Xu, Y.; Yan, Y.; Gao, Y. Umbilical cord-derived mesenchymal stromal/stem cells expressing IL-24 induce apoptosis in gliomas. J. Cell. Physiol. 2020, 235, 1769–1779. [Google Scholar] [CrossRef]
- Mangraviti, A.; Tzeng, S.Y.; Gullotti, D.; Kozielski, K.L.; Kim, J.E.; Seng, M.; Abbadi, S.; Schiapparelli, P.; Sarabia-Estrada, R.; Vescovi, A.; et al. Non-virally engineered human adipose mesenchymal stem cells produce BMP4, target brain tumors, and extend survival. Biomaterials 2016, 100, 53–66. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Guo, Y.; Seng, Z.; Cui, G.; Qu, J. Bone marrow-derived mesenchymal stem cells co-expressing interleukin-18 and interferon-beta exhibit potent antitumor effect against intracranial glioma in rats. Oncol. Rep. 2015, 34, 1915–1922. [Google Scholar] [CrossRef] [PubMed]
- Motaln, H.; Gruden, K.; Hren, M.; Schichor, C.; Primon, M.; Rotter, A.; Lah, T.T. Human Mesenchymal stem cells exploit the immune response mediating chemokines to impact the phenotype of glioblastoma. Cell Transplant. 2012, 21, 1529–1545. [Google Scholar] [CrossRef]
- Hong, X.; Miller, C.; Savant-Bhonsale, S.; Kalkanis, S.N. Antitumor treatment using interleukin-12-secreting marrow stromal cells in an invasive glioma model. Neurosurgery 2009, 64, 1139–1147. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Chen, G.; Yang, J.; Ma, Z.; Yang, Y.; Hu, Y.; Lu, Y.; Cao, Z.; Wang, Y.; Wang, X. Bone marrow mesenchymal stem cells suppress growth and promote the apoptosis of glioma U251 cells through downregulation of the PI3K/AKT signaling pathway. Biomed. Pharmacother. 2019, 112, 108625. [Google Scholar] [CrossRef]
- Choi, S.H.; Stuckey, D.W.; Pignatta, S.; Reinshagen, C.; Khalsa, J.K.; Roozendaal, N.; Martinez-Quintanilla, J.; Tamura, K.; Keles, E.; Shah, K. Tumor resection recruits effector T cells and boosts therapeutic efficacy of encapsulated stem cells expressing IFNbeta in glioblastomas. Clin. Cancer Res. 2017, 23, 7047–7058. [Google Scholar] [CrossRef]
- Chastkofsky, M.I.; Pituch, K.C.; Katagi, H.; Zannikou, M.; Ilut, L.; Xiao, T.; Han, Y.; Sonabend, A.M.; Curiel, D.T.; Bonner, E.R.; et al. Mesenchymal stem cells successfully deliver oncolytic virotherapy to diffuse intrinsic pontine glioma. Clin. Cancer Res. 2021, 27, 1766–1777. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, H.; Chen, C.; Liu, H.; He, Y.; Zhao, J.; Yang, P.; Mao, Q.; Xia, H. Systemic administration of mesenchymal stem cells loaded with a novel oncolytic adenovirus carrying IL-24/endostatin enhances glioma therapy. Cancer Lett. 2021, 509, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Quintanilla, J.; He, D.; Wakimoto, H.; Alemany, R.; Shah, K. Encapsulated stem cells loaded with hyaluronidase-expressing oncolytic virus for brain tumor therapy. Mol. Ther. 2015, 23, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Duebgen, M.; Martinez-Quintanilla, J.; Tamura, K.; Hingtgen, S.; Redjal, N.; Wakimoto, H.; Shah, K. Stem cells loaded with multimechanistic oncolytic herpes simplex virus variants for brain tumor therapy. J. Natl. Cancer Inst. 2014, 106, dju090. [Google Scholar] [CrossRef] [PubMed]
- Josiah, D.T.; Zhu, D.; Dreher, F.; Olson, J.; McFadden, G.; Caldas, H. Adipose-derived stem cells as therapeutic delivery vehicles of an oncolytic virus for glioblastoma. Mol. Ther. 2010, 18, 377–385. [Google Scholar] [CrossRef]
- Sonabend, A.M.; Ulasov, I.V.; Tyler, M.A.; Rivera, A.A.; Mathis, J.M.; Lesniak, M.S. Mesenchymal stem cells effectively deliver an oncolytic adenovirus to intracranial glioma. Stem Cells 2008, 26, 831–841. [Google Scholar] [CrossRef] [PubMed]
- Yong, R.L.; Shinojima, N.; Fueyo, J.; Gumin, J.; Vecil, G.G.; Marini, F.C.; Bogler, O.; Andreeff, M.; Lang, F.F. Human bone marrow-derived mesenchymal stem cells for intravascular delivery of oncolytic adenovirus Delta24-RGD to human gliomas. Cancer Res. 2009, 69, 8932–8940. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, Z.; Ding, T.; Chen, Z.; Zhang, T. Mesenchymal stem cells overexpressing PEDF decrease the angiogenesis of gliomas. Biosci. Rep. 2013, 33, e00019. [Google Scholar] [CrossRef] [PubMed]
- Ho, I.A.; Toh, H.C.; Ng, W.H.; Teo, Y.L.; Guo, C.M.; Hui, K.M.; Lam, P.Y. Human bone marrow-derived mesenchymal stem cells suppress human glioma growth through inhibition of angiogenesis. Stem Cells 2012, 31, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Aslam, N.; Abusharieh, E.; Abuarqoub, D.; Alhattab, D.; Jafar, H.; Alshaer, W.; Masad, R.J.; Awidi, A.S. An in vitro comparison of anti-tumoral potential of Wharton’s jelly and bone marrow mesenchymal stem cells exhibited by cell cycle arrest in glioma cells (U87MG). Pathol. Oncol. Res. 2021, 27, 584710. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.-J.; Lu, J.-T.; Tu, H.-J.; Huang, K.-M.; Fu, R.; Cao, G.; Huang, M.; Cheng, L.-H.; Dai, L.-J.; Zhang, L. TRAIL-engineered bone marrow-derived mesenchymal stem cells: TRAIL expression and cytotoxic effects on C6 glioma cells. Anticancer Res. 2014, 34, 729–734. [Google Scholar]
- Park, S.A.; Han, H.R.; Ahn, S.; Ryu, C.H.; Jeun, S. Combination treatment with VPA and MSCs-TRAIL could increase anti-tumor effects against intracranial glioma. Oncol. Rep. 2021, 45, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Baik, S.K.; Yoon, Y.; Hwang, S.; Sohn, J.H.; Jo, M.; Kim, W.-S.; Rhee, K.-J.; Whang, K.; Eom, Y.W. DNA-binding cell-penetrating peptide-based TRAIL over-expression in Adipose tissue-derived mesenchymal stem cells inhibits glioma U251MG growth. Anticancer Res. 2021, 41, 2859–2866. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Fitch, S.; Wang, C.; Wilson, C.; Li, J.; Grant, G.A.; Yang, F. Nanoparticle engineered TRAIL-overexpressing adipose-derived stem cells target and eradicate glioblastoma via intracranial delivery. Proc. Natl. Acad. Sci. USA 2016, 113, 13857–13862. [Google Scholar] [CrossRef]
- Akimoto, K.; Kimura, K.; Nagano, M.; Takano, S.; Salazar, G.; Yamashita, T.; Ohneda, O. Umbilical cord blood-derived mesenchymal stem cells inhibit, but adipose tissue-derived mesenchymal stem cells promote, glioblastoma multiforme proliferation. Stem Cells Dev. 2013, 22, 1370–1386. [Google Scholar] [CrossRef] [PubMed]
- Sasportas, L.S.; Kasmieh, R.; Wakimoto, H.; Hingtgen, S.; van de Water, J.A.; Mohapatra, G.; Figueiredo, J.L.; Martuza, R.L.; Weissleder, R.; Shah, K. Assessment of therapeutic efficacy and fate of engineered human mesenchymal stem cells for cancer therapy. Proc. Natl. Acad. Sci. USA 2009, 106, 4822–4827. [Google Scholar] [CrossRef] [PubMed]
- Malik, Y.S.; Sheikh, M.A.; Xing, Z.; Guo, Z.; Zhu, X.; Tian, H.; Chen, X. Polylysine-modified polyethylenimine polymer can generate genetically engineered mesenchymal stem cells for combinational suicidal gene therapy in glioblastoma. Acta Biomater. 2018, 80, 144–153. [Google Scholar] [CrossRef]
- Chung, T.; Na, J.; Kim, Y.-I.; Chang, D.-Y.; Kim, H.; Moon, H.E.; Kang, K.W.; Lee, D.S.; Chung, J.-K.; Kim, S.-S.; et al. Dihydropyrimidine dehydrogenase is a prognostic marker for mesenchymal stem cell-mediated cytosine deaminase gene and 5-fluorocytosine prodrug therapy for the treatment of recurrent gliomas. Theranostics 2016, 6, 1477–1490. [Google Scholar] [CrossRef]
- Chang, D.-Y.; Yoo, S.-W.; Hong, Y.; Kim, S.; Kim, S.J.; Yoon, S.-H.; Cho, K.-G.; Paek, S.H.; Lee, Y.-D.; Kim, S.-S.; et al. The growth of brain tumors can be suppressed by multiple transplantation of mesenchymal stem cells expressing cytosine deaminase. Int. J. Cancer 2010, 127, 1975–1983. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Gao, J.-Q.; Ouyang, X.; Wang, J.; Sun, X.; Lv, Y. Mesenchymal stem cells loaded with paclitaxel–poly(lactic-co-glycolic acid) nanoparticles for glioma-targeting therapy. Int. J. Nanomed. 2018, 13, 5231–5248. [Google Scholar] [CrossRef]
- Bonomi, A.; Ghezzi, E.; Pascucci, L.; Aralla, M.; Ceserani, V.; Pettinari, L.; Coccè, V.; Guercio, A.; Alessandri, G.; Parati, E.; et al. Effect of canine mesenchymal stromal cells loaded with paclitaxel on growth of canine glioma and human glioblastoma cell lines. Vet. J. 2017, 223, 41–47. [Google Scholar] [CrossRef]
- Goodarzi, A.; Khanmohammadi, M.; Ai, A.; Khodayari, H.; Ai, A.; Farahani, M.S.; Khodayari, S.; Ebrahimi-Barough, S.; Mohandesnezhad, S.; Ai, J. Simultaneous impact of atorvastatin and mesenchymal stem cells for glioblastoma multiform suppression in rat glioblastoma multiform model. Mol. Biol. Rep. 2020, 47, 7783–7795. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.R.; Hu, Q.Y.; Yuan, Y.H.; Tang, X.J.; Yang, Z.S.; Zou, D.D.; Bian, L.J.; Dai, L.J.; Li, D.S. PTEN-mRNA engineered mesenchymal stem cell-mediated cytotoxic effects on U251 glioma cells. Oncol. Lett. 2016, 11, 2733–2740. [Google Scholar] [CrossRef] [PubMed]
- Mahjoor, M.; Afkhami, H.; Mollaei, M.; Nasr, A.; Shahriary, S.; Khorrami, S. MicroRNA-30c delivered by bone marrow-mesenchymal stem cells induced apoptosis and diminished cell invasion in U-251 glioblastoma cell line. Life Sci. 2021, 279, 119643. [Google Scholar] [CrossRef] [PubMed]
- Allahverdi, A.; Arefian, E.; Soleimani, M.; Ai, J.; Nahanmoghaddam, N.; Yousefi-Ahmadipour, A.; Ebrahimi-Barough, S. MicroRNA-4731-5p delivered by AD-mesenchymal stem cells induces cell cycle arrest and apoptosis in glioblastoma. J. Cell. Physiol. 2020, 235, 8167–8175. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, C.; Cai, L.; Lu, J.; Zhu, Z.; Wang, C.; Su, Z.; Lu, X. miR-34a derived from mesenchymal stem cells stimulates senescence in glioma cells by inducing DNA damage. Mol. Med. Rep. 2018, 19, 1849–1857. [Google Scholar] [CrossRef] [PubMed]
- Yan, T.; Wu, M.; Lv, S.; Hu, Q.; Xu, W.; Zeng, A.; Huang, K.; Zhu, X. Exosomes derived from microRNA-512-5p-transfected bone mesenchymal stem cells inhibit glioblastoma progression by targeting JAG1. Aging 2021, 13, 9911–9926. [Google Scholar] [CrossRef] [PubMed]
- Hao, S.C.; Ma, H.; Niu, Z.F.; Sun, S.Y.; Zou, Y.R.; Xia, H.C. hUC-MSCs secreted exosomes inhibit the glioma cell progression through PTENP1/miR-10a-5p/PTEN pathway. Eur. Rev. Med. Pharm. Sci. 2019, 23, 10013–10023. [Google Scholar]
- Yu, L.; Gui, S.; Liu, Y.; Qiu, X.; Zhang, G.; Zhang, X.; Pan, J.; Fan, J.; Qi, S.; Qiu, B. Exosomes derived from microRNA-199a-overexpressing mesenchymal stem cells inhibit glioma progression by down-regulating AGAP2. Aging 2019, 11, 5300–5318. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.; Hossain, A.; Gumin, J.; Momin, E.N.; Shimizu, Y.; Ledbetter, D.; Shahar, T.; Yamashita, S.; Kerrigan, B.P.; Fueyo, J.; et al. Mesenchymal stem cells as natural biofactories for exosomes carrying miR-124a in the treatment of gliomas. Neuro-Oncology 2018, 20, 380–390. [Google Scholar] [CrossRef] [PubMed]
- Pavon, L.F.; Sibov, T.T.; De Souza, A.V.; da Cruz, E.F.; Malheiros, S.M.F.; Cabral, F.R.; De Souza, J.G.; Boufleur, P.; Oliveira, D.; De Toledo, S.R.C.; et al. Tropism of mesenchymal stem cell toward CD133+ stem cell of glioblastoma in vitro and promote tumor proliferation in vivo. Stem Cell Res. Ther. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Zhao, D.; Dai, X.; Chen, J.; Rong, X.; Wang, H.; Wang, A.; Li, M.; Dong, J.; Huang, Q.; et al. Fusion of cancer stem cells and mesenchymal stem cells contributes to glioma neovascularization. Oncol. Rep. 2015, 34, 2022–2030. [Google Scholar] [CrossRef]
- Rodini, C.O.; Da Silva, P.B.G.; Assoni, A.F.; Carvalho, V.M.; Okamoto, O.K. Mesenchymal stem cells enhance tumorigenic properties of human glioblastoma through independent cell-cell communication mechanisms. Oncotarget 2018, 9, 24766–24777. [Google Scholar] [CrossRef]
- Figueroa, J.; Phillips, L.M.; Shahar, T.; Hossain, A.; Gumin, J.; Kim, H.; Bean, A.J.; Calin, G.; Fueyo, J.; Walters, E.T.; et al. Exosomes from glioma-associated mesenchymal stem cells increase the tumorigenicity of glioma stem-like cells via transfer of miR-1587. Cancer Res. 2017, 77, 5808–5819. [Google Scholar] [CrossRef]
- Iser, I.C.; Beckenkamp, L.R.; Azambuja, J.H.; Rahmeier, F.L.; Bracco, P.A.; Bertoni, A.P.S.; de Cássia Sant Anna Alves, R.; Braganhol, E.; Xavier, L.L.; Fernandes, M.D.C.; et al. Rat Adipose-Derived Stromal Cells (ADSCs) increases the glioblastoma growth and decreases the animal survival. Stem Cell Rev. Rep. 2021, 1–15. [Google Scholar] [CrossRef]
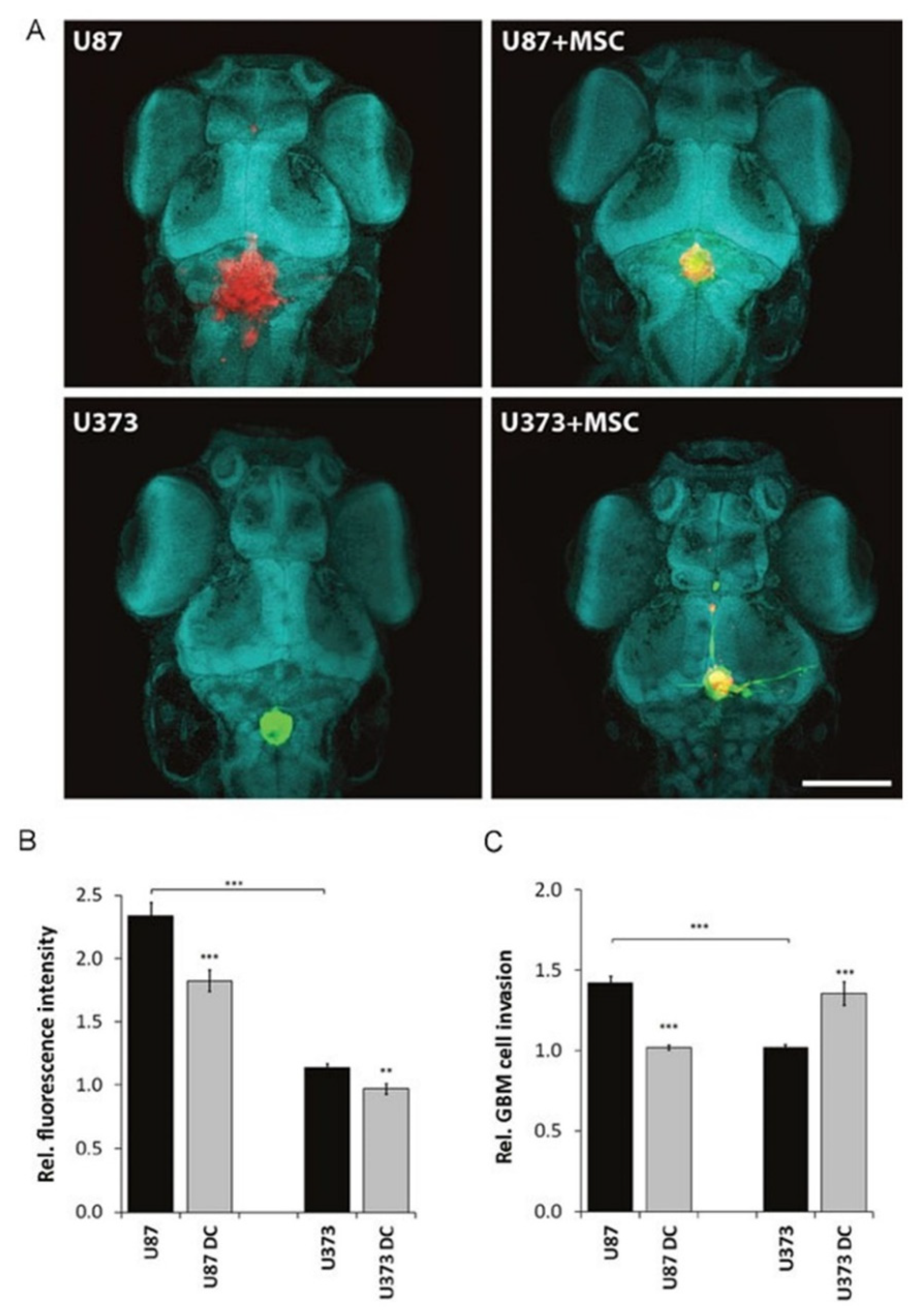
- Breznik, B.; Motaln, H.; Vittori, M.; Rotter, A.; Turnšek, T.L. Mesenchymal stem cells differentially affect the invasion of distinct glioblastoma cell lines. Oncotarget 2017, 8, 25482–25499. [Google Scholar] [CrossRef] [PubMed]
- Völker, J.; Engert, J.; Völker, C.; Bieniussa, L.; Schendzielorz, P.; Hagen, R.; Rak, K. Isolation and characterization of neural stem cells from the rat inferior colliculus. Stem Cells Int. 2019, 2019, 12. [Google Scholar] [CrossRef]
- Zhang, G.-L.; Wang, C.-F.; Qian, C.; Ji, Y.-X.; Wang, Y.-Z. Role and mechanism of neural stem cells of the subventricular zone in glioblastoma. World J. Stem Cells 2021, 13, 877–893. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhou, R.; Xiong, Y.; Zhou, L.; Yan, X.; Wang, M.; Fan, L.; Xie, C.; Zhang, Y.; Wang, Y.; et al. Sequential fate-switches in stem-like cells drive the tumorigenic trajectory from human neural stem cells to malignant glioma. Cell Res. 2021, 31, 684–702. [Google Scholar] [CrossRef] [PubMed]
- Satterlee, A.B.; Dunn, D.; Lo, D.C.; Khagi, S.; Hingtgen, S. Tumoricidal stem cell therapy enables killing in novel hybrid models of heterogeneous glioblastoma. Neuro-Oncology 2019, 21, 1552–1564. [Google Scholar] [CrossRef] [PubMed]
- Tamura, R.; Miyoshi, H.; Morimoto, Y.; Oishi, Y.; Sampetrean, O.; Iwasawa, C.; Mine, Y.; Saya, H.; Yoshida, K.; Okano, H.; et al. Gene therapy using neural stem/progenitor cells derived from human induced pluripotent stem cells: Visualization of migration and bystander killing effect. Hum. Gene Ther. 2020, 31, 352–366. [Google Scholar] [CrossRef] [PubMed]
- Bomba, H.N.; Sheets, K.T.; Valdivia, A.; Khagi, S.; Ruterbories, L.; Mariani, C.L.; Borst, L.; Tokarz, D.A.; Hingtgen, S.D. Personalized-induced neural stem cell therapy: Generation, transplant, and safety in a large animal model. Bioeng. Transl. Med. 2021, 6, 1. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Tokuyama, T.; Yamamoto, J.; Koide, M.; Yokota, N.; Namba, H. Bystander effect-mediated gene therapy of gliomas using genetically engineered neural stem cells. Cancer Gene Ther. 2005, 12, 600–607. [Google Scholar] [CrossRef]
- Pituch, K.C.; Zannikou, M.; Ilut, L.; Xiao, T.; Chastkofsky, M.; Sukhanova, M.; Bertolino, N.; Procissi, D.; Amidei, C.; Balyasnikova, I.V. Neural stem cells secreting bispecific T cell engager to induce selective antiglioma activity. Proc. Natl. Acad. Sci. USA 2021, 118, 9. [Google Scholar] [CrossRef] [PubMed]
- Carey-Ewend, A.G.; Hagler, S.B.; Bomba, H.N.; Goetz, M.J.; Bago, J.R.; Hingtgen, S.D. Developing bioinspired three-dimensional models of brain cancer to evaluate tumor-homing neural stem cell therapy. Tissue Eng. Part A 2020, 10, 1089. [Google Scholar] [CrossRef]
- Wang, J.; Liu, J.; Meng, H.; Guan, Y.; Yin, Y.; Zhao, Z.; Sun, G.; Wu, A.; Chen, L.; Yu, X. Neural stem cells promote glioblastoma formation in nude mice. Clin. Transl. Oncol. 2019, 21, 1551–1560. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Omole, A.E.; Fakoya, A.O.J. Ten years of progress and promise of induced pluripotent stem cells: Historical origins, characteristics, mechanisms, limitations, and potential applications. PeerJ 2018, 6, e4370. [Google Scholar] [CrossRef]
- Yamazoe, T.; Koizumi, S.; Yamasaki, T.; Amano, S.; Tokuyama, T.; Namba, H. Potent tumor tropism of induced pluripotent stem cells and induced pluripotent stem cell-derived neural stem cells in the mouse intracerebral glioma model. Int. J. Oncol. 2014, 46, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Bago, J.R.; Alfonso-Pecchio, A.; Okolie, O.; Dumitru, R.; Rinkenbaugh, A.; Baldwin, A.S.; Miller, C.; Magness, S.T.; Hingtgen, S.D. Therapeutically engineered induced neural stem cells are tumour-homing and inhibit progression of glioblastoma. Nat. Commun. 2016, 7, 10593. [Google Scholar] [CrossRef]
- Bagó, J.R.; Okolie, O.; Dumitru, R.; Ewend, M.G.; Parker, J.S.; Werff, R.V.; Underhill, T.M.; Schmid, R.S.; Miller, C.R.; Hingtgen, S.D. Tumor-homing cytotoxic human induced neural stem cells for cancer therapy. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Pitti, R.M.; Marsters, S.A.; Ruppert, S.; Donahue, C.J.; Moore, A.; Ashkenazi, A. Induction of apoptosis by apo-2 ligand, a new member of the tumor necrosis factor Cytokine family. J. Biol. Chem. 1996, 271, 12687–12690. [Google Scholar] [CrossRef] [PubMed]
- Oberst, A.; Pop, C.; Tremblay, A.G.; Blais, V.; Denault, J.-B.; Salvesen, G.S.; Green, D.R. Inducible dimerization and inducible cleavage reveal a requirement for both processes in caspase-8 activation. J. Biol. Chem. 2010, 285, 16632–16642. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chun, H.; Wong, W.; Spencer, D.M.; Lenardo, M.J. Caspase-10 is an initiator caspase in death receptor signaling. Proc. Natl. Acad. Sci. USA 2001, 98, 13884–13888. [Google Scholar] [CrossRef] [PubMed]
- Hingtgen, S.; Ren, X.; Terwilliger, E.; Classon, M.; Weissleder, R.; Shah, K. Targeting multiple pathways in gliomas with stem cell and viral delivered S-TRAIL and Temozolomide. Mol. Cancer Ther. 2008, 7, 3575–3585. [Google Scholar] [CrossRef] [PubMed]
- Kock, N.; Kasmieh, R.; Weissleder, R.; Shah, K. Tumor therapy mediated by lentiviral expression of shBcl-2 and S-TRAIL. Neoplasia 2007, 9, 435–442. [Google Scholar] [CrossRef][Green Version]
- Balyasnikova, I.V.; Ferguson, S.D.; Han, Y.; Liu, F.; Lesniak, M.S. Therapeutic effect of neural stem cells expressing TRAIL and bortezomib in mice with glioma xenografts. Cancer Lett. 2011, 310, 148–159. [Google Scholar] [CrossRef]
- Calinescu, A.-A.; Kauss, M.C.; Sultan, Z.; Al-Holou, W.N.; O’Shea, S.K. Stem cells for the treatment of glioblastoma: A 20-year perspective. CNS Oncol. 2021, 10, CNS73. [Google Scholar] [CrossRef] [PubMed]
- Bhere, D.; Khajuria, R.K.; Hendriks, W.T.; Bandyopadhyay, A.; Bagci-Onder, T.; Shah, K. Stem cells engineered during different stages of reprogramming reveal varying therapeutic efficacies. Stem Cells 2018, 36, 932–942. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.; Cichocki, F.; Ning, J.; Bjordahl, R.; Davis, Z.; Tuininga, K.; Wang, H.; Rogers, P.; He, M.; Chen, C. 155 iPSC-derived NK cells mediate robust anti-tumor activity against glioblastoma. J. ImmunoTher. Cancer 2020, 8, A93–A94. [Google Scholar] [CrossRef]
- Yu, M.; Mansour, A.G.; Teng, K.Y.; Sun, G.; Shi, Y.; Caligiuri, M.A. Abstract 3313: iPSC-derived natural killer cells expressing EGFR-CAR against glioblastoma. Cancer Res. 2020, 80, 3313. [Google Scholar]
- Yamanaka, S. Pluripotent stem cell-based cell therapy—Promise and challenges. Cell Stem Cell 2020, 27, 523–531. [Google Scholar] [CrossRef]
- Hayashi, Y.; Ohnuma, K.; Furue, M.K. Pluripotent stem cell heterogeneity. Adv. Exp. Med. Biol. 2019, 1123, 71–94. [Google Scholar] [CrossRef]
- Bravery, C.A. Do human Leukocyte antigen-typed cellular therapeutics based on induced pluripotent stem cells make commercial sense? Stem Cells Dev. 2015, 24, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Sugita, S.; Iwasaki, Y.; Makabe, K.; Kamao, H.; Mandai, M.; Shiina, T.; Ogasawara, K.; Hirami, Y.; Kurimoto, Y.; Takahashi, M. Successful transplantation of retinal pigment epithelial cells from MHC homozygote iPSCs in MHC-matched models. Stem Cell Rep. 2016, 7, 635–648. [Google Scholar] [CrossRef] [PubMed]
- Meissner, T.; Strominger, J.; Cowan, C. The universal donor stem cell: Removing the immune barrier to transplantation using CRISPR/Cas9 (TRAN1P.946). J. Immunol. 2015, 194, 140. [Google Scholar]
- Gornalusse, G.G.; Hirata, R.K.; Funk, S.E.; Riolobos, L.; Lopes, V.S.; Manske, G.; Prunkard, D.; Colunga, G.A.; Hanafi, L.-A.; Clegg, D.O.; et al. HLA-E-expressing pluripotent stem cells escape allogeneic responses and lysis by NK cells. Nat. Biotechnol. 2017, 35, 765–772. [Google Scholar] [CrossRef]
- Deuse, T.; Hu, X.; Gravina, A.; Wang, D.; Tediashvili, G.; De, C.; Thayer, W.O.; Wahl, A.; Garcia, J.V.; Reichenspurner, H.; et al. Hypoimmunogenic derivatives of induced pluripotent stem cells evade immune rejection in fully immunocompetent allogeneic recipients. Nat. Biotechnol. 2019, 37, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Wang, B.; Ono, M.; Kagita, A.; Fujii, K.; Sasakawa, N.; Ueda, T.; Gee, P.; Nishikawa, M.; Nomura, M.; et al. Targeted disruption of HLA genes via CRISPR-Cas9 generates iPSCs with enhanced immune compatibility. Cell Stem Cell 2019, 24, 566–578.e7. [Google Scholar] [CrossRef] [PubMed]
- Okita, K.; Matsumura, Y.; Sato, Y.; Okada, A.; Morizane, A.; Okamoto, S.; Hong, H.; Nakagawa, M.; Tanabe, K.; Tezuka, K.-I.; et al. A more efficient method to generate integration-free human iPS cells. Nat. Methods 2011, 8, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Umekage, M.; Sato, Y.; Takasu, N. Overview: An iPS cell stock at CiRA. Inflamm. Regen. 2019, 39, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.J.; Peacock, S.; Chaudhry, A.N.; Bradley, J.A.; Bolton, E. Generating an iPSC Bank for HLA-matched tissue transplantation based on known donor and recipient HLA types. Cell Stem Cell 2012, 11, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Ling, X.; Yang, Y.; Zhang, J.; Li, Q.; Niu, X.; Hu, G.; Chen, B.; Li, H.; Wang, Y.; et al. Embryonic stem cells-derived exosomes endowed with targeting properties as chemotherapeutics delivery vehicles for glioblastoma therapy. Adv. Sci. 2019, 6, 1801899. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Liu, J.; Wang, X.; Liu, T.; Yang, L.; Li, C.; Wang, C.; Liu, Y.; Sang, X.; Wang, Z.; et al. The embryonic stem cell microenvironment inhibits mouse glioma cell proliferation by regulating the PI3K/AKT pathway. Transl. Cancer Res. 2021, 10, 487–498. [Google Scholar] [CrossRef]
- Milkina, E.; Ponomarenko, A.; Korneyko, M.; Lyakhova, I.; Zayats, Y.; Zaitsev, S.; Mischenko, P.; Eliseikina, M.; Khotimchenko, Y.; Shevchenko, V.; et al. Interaction of hematopoietic CD34+ CD45+ stem cells and cancer cells stimulated by TGF-β1 in a model of glioblastoma in vitro. Oncol. Rep. 2018, 40, 2595–2607. [Google Scholar] [CrossRef] [PubMed]
- Andreou, T.; Williams, J.; Brownlie, R.J.; Salmond, R.J.; Watson, E.; Shaw, G.; Melcher, A.; Wurdak, H.; Short, S.C.; Lorger, M. Hematopoietic stem cell gene therapy targeting TGFβ enhances the efficacy of irradiation therapy in a preclinical glioblastoma model. J. Immunother. Cancer 2021, 9, e001143. [Google Scholar] [CrossRef]
- Wildes, T.J.; Grippin, A.; Dyson, K.A.; Wummer, B.M.; Damiani, D.J.; Abraham, R.S.; Flores, C.T.; Mitchell, D.A. Cross-talk between T cells and hematopoietic stem cells during adoptive cellular therapy for malignant glioma. Clin. Cancer Res. 2018, 24, 3955–3966. [Google Scholar] [CrossRef]
- Marzouk, S.; Attia, N.; Mashal, M. Insights into the potential role of alpha1-antitrypsin in COVID-19 patients: Mechanisms, current update, and future perspectives. Clin. Respir. J. 2021, 15, 1019–1024. [Google Scholar] [CrossRef]
- Attia, N.; Mashal, M.; Grijalvo, S.; Eritja, R.; Puras, G.; Pedraz, J.L. Cationic niosome-based hBMP7 gene transfection of neuronal precursor NT2 cells to reduce the migration of glioma cells in vitro. J. Drug Deliv. Sci. Technol. 2019, 53, 101219. [Google Scholar] [CrossRef]
- Moore, K.M.; Murthy, A.B.; Graham-Gurysh, E.G.; Hingtgen, S.D.; Bachelder, E.M.; Ainslie, K. Polymeric biomaterial scaffolds for tumoricidal stem cell glioblastoma therapy. ACS Biomater. Sci. Eng. 2020, 6, 3762–3777. [Google Scholar] [CrossRef] [PubMed]
- Catoira, M.C.; Fusaro, L.; Di Francesco, D.; Ramella, M.; Boccafoschi, F. Overview of natural hydrogels for regenerative medicine applications. J. Mater. Sci. Mater. Med. 2019, 30, 1–10. [Google Scholar] [CrossRef]
- Echave, C.M.; Burgo, S.L.; Pedraz, L.J.; Orive, G. Gelatin as biomaterial for tissue engineering. Curr. Pharm. Des. 2017, 23, 3567–3584. [Google Scholar] [CrossRef]
- Moore, K.M.; Graham-Gurysh, E.G.; Bomba, H.N.; Murthy, A.B.; Bachelder, E.M.; Hingtgen, S.D.; Ainslie, K.M. Impact of composite scaffold degradation rate on neural stem cell persistence in the glioblastoma surgical resection cavity. Mater. Sci. Eng. C 2020, 111, 110846. [Google Scholar] [CrossRef] [PubMed]
- George, N.; Geller, H.M. Extracellular matrix and traumatic brain injury. J. Neurosci. Res. 2018, 96, 573–588. [Google Scholar] [CrossRef] [PubMed]
- Bagó, J.R.; Pegna, G.J.; Okolie, O.; Mohiti-Asli, M.; Loboa, E.G.; Hingtgen, S.D. Electrospun nanofibrous scaffolds increase the efficacy of stem cell-mediated therapy of surgically resected glioblastoma. Biomaterials 2016, 90, 116–125. [Google Scholar] [CrossRef]
- Akbar, U.; Jones, T.; Winestone, J.; Michael, M.; Shukla, A.; Sun, Y.; Duntsch, C. Delivery of temozolomide to the tumor bed via biodegradable gel matrices in a novel model of intracranial glioma with resection. J. Neuro-Oncol. 2009, 94, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Ning, W.; Wang, J.; Choi, A.; Lee, P.-Y.; Tyagi, P.; Huang, L. Controlled gene delivery system based on thermosensitive biodegradable hydrogel. Pharm. Res. 2003, 20, 884–888. [Google Scholar] [CrossRef]
- Hoare, T.R.; Kohane, D.S. Hydrogels in drug delivery: Progress and challenges. Polymer 2008, 49, 1993–2007. [Google Scholar] [CrossRef]
- Guo, W.; Li, A.; Jia, Z.; Yuan, Y.; Dai, H.; Li, H. Transferrin modified PEG-PLA-resveratrol conjugates: In vitro and in vivo studies for glioma. Eur. J. Pharmacol. 2013, 718, 41–47. [Google Scholar] [CrossRef]
- Kefayat, A.; Ghahremani, F.; Motaghi, H.; Amouheidari, A. Ultra-small but ultra-effective: Folic acid-targeted gold nanoclusters for enhancement of intracranial glioma tumors’ radiation therapy efficacy. Nanomed. Nanotechnol. Biol. Med. 2019, 16, 173–184. [Google Scholar] [CrossRef]
- Ruiz-Garcia, H.; Alvarado-Estrada, K.; Krishnan, S.; Quinones-Hinojosa, A.; Trifiletti, D.M. Nanoparticles for stem cell therapy bioengineering in glioma. Front. Bioeng. Biotechnol. 2020, 8, 558375. [Google Scholar] [CrossRef]
- Chung, T.-H.; Hsiao, J.-K.; Hsu, S.-C.; Yao, M.; Chen, Y.-C.; Wang, S.-W.; Kuo, M.Y.-P.; Yang, C.-S.; Huang, D.-M. Iron oxide nanoparticle-induced epidermal growth factor receptor expression in human stem cells for tumor therapy. ACS Nano 2011, 5, 9807–9816. [Google Scholar] [CrossRef]
- Caban-Toktas, S.; Sahin, A.; Lule, S.; Esendagli, G.; Vural, I.; Oguz, K.K.; Soylemezoglu, F.; Mut, M.; Dalkara, T.; Khan, M.; et al. Combination of Paclitaxel and R-flurbiprofen loaded PLGA nanoparticles suppresses glioblastoma growth on systemic administration. Int. J. Pharm. 2020, 578, 119076. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.; Ullah, I.; Kim, N.; Lim, J.; Shin, J.; Lee, S.C.; Jeon, S.; Kim, S.H.; Kumar, P.; Lee, S.-K. Intranasal delivery of cancer-targeting doxorubicin-loaded PLGA nanoparticles arrests glioblastoma growth. J. Drug Target. 2020, 28, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Ullah, I.; Chung, K.; Bae, S.; Li, Y.; Kim, C.; Choi, B.; Nam, H.Y.; Kim, S.H.; Yun, C.-O.; Lee, K.Y.; et al. Nose-to-brain delivery of cancer-targeting paclitaxel-loaded nanoparticles potentiates antitumor effects in malignant glioblastoma. Mol. Pharm. 2020, 17, 1193–1204. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Rovira, I.; Seksek, O.; Dokic, I.; Brons, S.; Abdollahi, A.; Yousef, I. Study of the intracellular nanoparticle-based radiosensitization mechanisms in F98 glioma cells treated with charged particle therapy through synchrotron-based infrared microspectroscopy. Analyst 2020, 145, 2345–2356. [Google Scholar] [CrossRef] [PubMed]
- Gomes, E.; de Castro, J.V.; Costa, B.; Salgado, A. The impact of Mesenchymal Stem Cells and their secretome as a treatment for gliomas. Biochimie 2018, 155, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Suryaprakash, S.; Lao, Y.-H.; Cho, H.-Y.; Li, M.; Ji, H.Y.; Shao, D.; Hu, H.; Quek, C.H.; Huang, D.; Mintz, R.L.; et al. Engineered mesenchymal stem cell/nanomedicine spheroid as an active drug delivery platform for combinational glioblastoma therapy. Nano Lett. 2019, 19, 1701–1705. [Google Scholar] [CrossRef] [PubMed]
- Song, R.; Li, T.; Ye, J.; Sun, F.; Hou, B.; Saeed, M.; Gao, J.; Wang, Y.; Zhu, Q.; Xu, Z.; et al. Acidity-activatable dynamic nanoparticles boosting ferroptotic cell death for immunotherapy of cancer. Adv. Mater. 2021, 33, 2101155. [Google Scholar] [CrossRef]
- Yang, T.; Kong, Z.; Ma, W. PD-1/PD-L1 immune checkpoint inhibitors in glioblastoma: Clinical studies, challenges and potential. Hum. Vaccines Immunother. 2021, 17, 546–553. [Google Scholar] [CrossRef]
- Sun, J.; Lu, Q.; Sanmanmed, M.F.; Wang, J. Siglec-15 as an emerging target for next-generation cancer immunotherapy. Clin. Cancer Res. 2021, 27, 680–688. [Google Scholar] [CrossRef]
- Kugeratski, F.G.; Kalluri, R. Exosomes as mediators of immune regulation and immunotherapy in cancer. FEBS J. 2021, 288, 10–35. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study ID | Start Date | End Date | Cellular Intervention | Phase | Participants | Cell Type | Cell Dose | Cell Delivery Route | Status |
|---|---|---|---|---|---|---|---|---|---|
| NCT00005813 | Mar 1997 | Jan 2003 | Lymphokine-activated killer cells | I | 13 | Monocytes/ WBCs | Not specified | IT | Completed |
| NCT00003185 | Aug 1997 | Jul 1998 | Tumor-draining lymph node lymphocyte therapy | II | 40 | Lymphocytes | 9 × 108 to 1.5 × 1011 | IV Infusion | Completed |
| NCT00576537 | Mar 2001 | Oct 2011 | Dendritic cell immunotherapy | II | 50 | DCs | Not specified | SC | Completed |
| NCT00068510 | Jun 2003 | Sept 2012 | Therapeutic autologous dendritic cells | I | 28 | DCs | 1, 5, or 10 × 106 DCs | ID | Completed |
| NCT00107185 | Jan 2005 | Mar 2010 | Biological: therapeutic autologous dendritic cells | I | 7 | DCs | Not specified | ID | Completed |
| NCT00639639 | Jan 2006 | Est. Jan 2022 | Therapeutic autologous dendritic cells/ therapeutic autologous lymphocytes | I | 42 | DCs/Lymphocytes | DCs: 2 × 107 Lymphocytes: 3 × 107 | IV/ID | Active |
| NCT00323115 | May 2006 | Jul 2013 | Dendritic cell vaccine | II | 11 | DCs | 1 × 107 DCs | IN vaccine | Completed |
| NCT00626483 | Apr 2007 | Jul 2016 | RNA-loaded dendritic cell vaccine | I | 34 | DCs | 2 × 107 DCs | ID | Completed |
| NCT00576641 | May 2007 | Apr 2012 | Autologous dendritic cells | I | 22 | DCs | Not specified | SC | Completed |
| NCT00693095 | Sept 2008 | Apr 2015 | CMV-ALT + CMV-DC | I | 23 | CMV-ALT +/− CMV-DCs | 3 × 107/Kg CMV-ALT +/− 2 × 107 CMV-DCs | SC | Completed |
| NCT00846456 | Jan 2009 | Feb 2013 | Dendritic cell vaccine with mRNA from tumor stem cells | I/II | 20 | DCs | 1 × 107 DCs | ID | Completed |
| NCT00890032 | Sept 2009 | Feb 2016 | Brain tumor stem cell mRNA-loaded DCs | I | 50 | DCs | 2 × 106, 5 × 106, or 2 × 107 | ID | Completed |
| NCT01006044 | Oct 2009 | Aug 2014 | Autologous DCs | II | 26 | DCs | Not specified | SC | Completed |
| NCT01081223 | Apr 2010 | Mar 2011 | Activated white blood cells + cancer vaccine+ immune adjuvant activated WBCs | I/II | 14 | Activated WBCs | N/A | IV infusion | Completed |
| NCT01171469 | Sept 2010 | Jun 2012 | DCs | I | 8 | DCs | 5, 10 or 15 × 106 DCs | ID | Completed |
| NCT01109095 | Oct 2010 | Mar 2018 | HER.CAR CMV-specific CTLs | I | 16 | T-lymphocytes/ DCs | -CMV-ALT (3 × 107) - CMV-DCs (2 × 107) | SC | Completed |
| NCT01204684 | Oct 2010 | Jan 2023 | Autologous tumor lysate-pulsed DC vaccination | II | 60 | DCs | 1, 5, or 10 × 106 DCs | ID | Active |
| NCT01280552 | Jan 2011 | Dec 2015 | Autologous dendritic cells pulsed with immunogenic antigens (ICT-107) | II | 124 | DCs | Not specified | ID | Completed |
| NCT01588769 | Aug 2011 | Apr 2013 | ALECSAT cell-based immunotherapy | I | 23 | NK/ Cytotoxic T-lymphocytes | 10 × 106 to 1 billion cytotoxic T cells and NK cells | IV Infusion | Completed |
| NCT01454596 | May 2012 | Jan 2019 | (EGFRv) III CAR transduced PBL | I/II | 18 | WBCs | Not specified | IV Infusion | Completed |
| NCT01808820 | Aug 2013 | est. Nov 2023 | DC vaccine | I | 20 | DCs | 1.2 to 12 × 106 DCs per dose | ID | Active |
| NCT01957956 | Nov 2013 | est. Nov 2021 | Malignant glioma tumor lysate-pulsed autologous DC vaccine | I | 21 | DCs | Not specified | ID | Active |
| NCT02049489 | Dec 2013 | Mar 2017 | Autologous vaccine of DC pulsed with purified peptides from CD133 cancer cells (ICT-12) | I | 20 | DCs | Not specified | ID | Completed |
| NCT02010606 | Jan 2014 | est. Jan 2022 | DC vaccination | I | 39 | DCs | Not specified | SC | Active |
| NCT02366728 | Oct 2015 | Aug 2020 | Unpulsed DCs, Td, human CMV pp65-LAMP mRNA-pulsed autologous DCs, 111In-labeled DCs | II | 100 | DCs | -1 × 106 autologous unpulsed DCs -2 × 107 hCMV pp65-LAMP mRNA-pulsed autologous DCs | ID | Active |
| NCT02529072 | Jan 2016 | Dec 2019 | DCs | I | 6 | DCs | Not specified | ID | Completed |
| NCT02799238 | Mar 2016 | Feb 2020 | ALECSAT | II | 62 | ALE Cells | Not Specified | IV | Completed |
| NCT02661282 | Jun 2016 | est. Jun 2021 | Autologous cytomegalovirus-specific cytotoxic T-lymphocytes | I/ II | 65 | T-lymphocytes | Not specified | IV Infusion | Active |
| NCT02820584 | Sept 2016 | Jun 2017 | GSC-loaded autologous dendritic cells | I | 20 | GSC-DCs | -1st vaccine: 20 × 106 DCs -2nd and 3rd: 10 × 106 DCs -4–6th vaccine: 5 × 106 DCs | Vaccine | Completed |
| NCT03400917 | Jun 2018 | est. Feb 2023 | AV-GBM-1 | II | 55 | DCs | Not specified | SC | Active |
| NCT03615404 | Oct 2018 | Jul 2020 | CMV-DCs with GM-CSF | I | 11 | CMV-DCs | Not specified | Vaccine | Completed |
| NCT03726515 | Mar 2019 | Feb 2021 | CART-EGFRvIII T cells | I | 7 | T-lymphocytes | Not specified | IV Infusion | Completed |
| NCT03360708 | Jun 2019 | Dec 2022 | Malignant glioma tumor lysate-pulsed autologous DC vaccine | I | 20 | DCs | Not specified | ID | Active |
| NCT04489420 | Oct 2020 | Feb 2024 | Cryopreserved, allogeneic, off-the-shelf, NK cells (CYNK001) | I | 36 | NK cells | For IV 1.2 × 109 cells/dose For IT 2 × 108 +/− 5 × 107 cells | IV Infusion/ IT | Active |
| Study ID | Start Date | End Date | Cellular Intervention | Phase | Participants | Cell Type | Cell Dose | Cell Delivery Route | Status |
|---|---|---|---|---|---|---|---|---|---|
| NCT00002619 | Sept1994 | Apr 2000 | Autologous peripheral blood stem cell transplantation | II | 60 | PBSCs | Not specified | IV Infusion | Completed |
| NCT00008008 | Sept1997 | May 2008 | Transplantation of autologous PBSCs | II | 40 | PBSCs | Not specified | IV Infusion | Completed |
| NCT00014573 | Aug 1998 | Oct 2004 | Transplantation of PBSCs or BM stem cells | II | 30 | PBSCs/BMSCs | Not specified | IV Infusion | Completed |
| NCT00179803 | Mar 1998 | Sept 2009 | Transplantation of autologous PBSCs | II | 24 | PBSCs | Not specified | IV Infusion | Completed |
| NCT00003141 | Mar 1998 | Oct 2011 | Transplantation of autologous PBSCs | I | 94 | PBSCs | Not specified | IV Infusion | Completed |
| NCT00005796 | Feb 2000 | Fibronectin-assisted, retroviral-mediated modification of CD34+ peripheral blood cells with O6-methylguanine DNA methyltransferase | I | 10 | CD34+ PBSCs | Not specified | IV Infusion | Completed | |
| NCT00005952 | Aug 2000 | Nov 2005 | Transplantation of autologous PBSCs | II | 30 | PBSCs | Not specified | IV Infusion | Completed |
| NCT00025558 | Oct 2000 | May 2007 | Transplantation of autologous peripheral blood stem cells | I | 30 | PBSCs | Not specified | IV Infusion | Completed |
| NCT00078988 | Oct 2004 | Sept 2006 | Transplantation of autologous PBSCs | III | 1 | PBSCs | Not specified | IV Infusion | Completed |
| NCT00253487 | Aug 2005 | Aug 2012 | Transplantation of peripheral blood or bone marrow CD34-positive stem cells transduced with the MGMT gene | N/A | 12 | CD34-positive PBSCs/ CD34-positive BMSCs | 5 × 106 | IV Infusion | Completed |
| NCT00669669 | Feb 2009 | Jan 2021 | In vitro-transfected (Phoenix-RD114 pseudotype vector) peripheral blood stem cell transplant | I/II | 12 | CD34+ HSCs | Not specified | IV Infusion | Completed |
| NCT01172964 | Aug 2010 | Feb 2015 | E. coli CD-expressing genetically modified neural stem cells | I | 15 | NSCs | Not specified | Brain injection | Completed |
| NCT03072134 | Apr 2017 | Dec 2021 | Neural stem cells loaded with an oncolytic adenovirus | I | 13 | NSCs | First cohort 5 × 107 NSCs, second cohort 1 × 108 NSCs, third cohort 1.5 × 106 NSCs | Injected into the walls of the resection cavity | Active |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Attia, N.; Mashal, M.; Pemminati, S.; Omole, A.; Edmondson, C.; Jones, W.; Priyadarshini, P.; Mughal, T.; Aziz, P.; Zenick, B.; et al. Cell-Based Therapy for the Treatment of Glioblastoma: An Update from Preclinical to Clinical Studies. Cells 2022, 11, 116. https://doi.org/10.3390/cells11010116
Attia N, Mashal M, Pemminati S, Omole A, Edmondson C, Jones W, Priyadarshini P, Mughal T, Aziz P, Zenick B, et al. Cell-Based Therapy for the Treatment of Glioblastoma: An Update from Preclinical to Clinical Studies. Cells. 2022; 11(1):116. https://doi.org/10.3390/cells11010116
Chicago/Turabian StyleAttia, Noha, Mohamed Mashal, Sudhakar Pemminati, Adekunle Omole, Carolyn Edmondson, Will Jones, Priyanka Priyadarshini, Temoria Mughal, Pauline Aziz, Blesing Zenick, and et al. 2022. "Cell-Based Therapy for the Treatment of Glioblastoma: An Update from Preclinical to Clinical Studies" Cells 11, no. 1: 116. https://doi.org/10.3390/cells11010116
APA StyleAttia, N., Mashal, M., Pemminati, S., Omole, A., Edmondson, C., Jones, W., Priyadarshini, P., Mughal, T., Aziz, P., Zenick, B., Perez, A., & Lacken, M. (2022). Cell-Based Therapy for the Treatment of Glioblastoma: An Update from Preclinical to Clinical Studies. Cells, 11(1), 116. https://doi.org/10.3390/cells11010116