Virus-Heat Shock Protein Interaction and a Novel Axis for Innate Antiviral Immunity

Abstract
:1. Introduction
2. The 70 kDa Heat Shock Protein Family (HSP70)
2.1. Viral Induction of hsp70 (the Major Inducible HSP70 Family Member)
2.2. The Role of the Major Inducible 70 kDa HSP (hsp70) in Support of Viral Replication

{kind=link}
| Virus | Viral Target of Hsp70 Interaction | Effect of Virus-Hsp70 Interaction | Reference |
|---|---|---|---|
| human papillomavirus | E1, E2 | increased genome replication | [42] |
| herpes simplex virus type 1 | UL9 | increased genome replication | [41] |
| canine distemper virus, measles virus | nucleocapsid | increased nucleocapsid transcriptional activity and genome replication | [43,44,45,46,47] |
| respiratory syncytial virus | nucleocapsid | increased nucleocapsid transcriptional activity | [51] |
| rabies virus | nucleocapsid | support formation of replication factories (Negri bodies) | [52] |
| tomato bush stunt virus | P33 | support replicase function | [53,54] |
| simian virus 40 | Tag | Genome replication | [55] |
| human cytomegalovirus | Hap46 | stimulate viral transcriptional activity | [56] |
2.3. Hsp70 Levels and the Virus Infection Phenotype
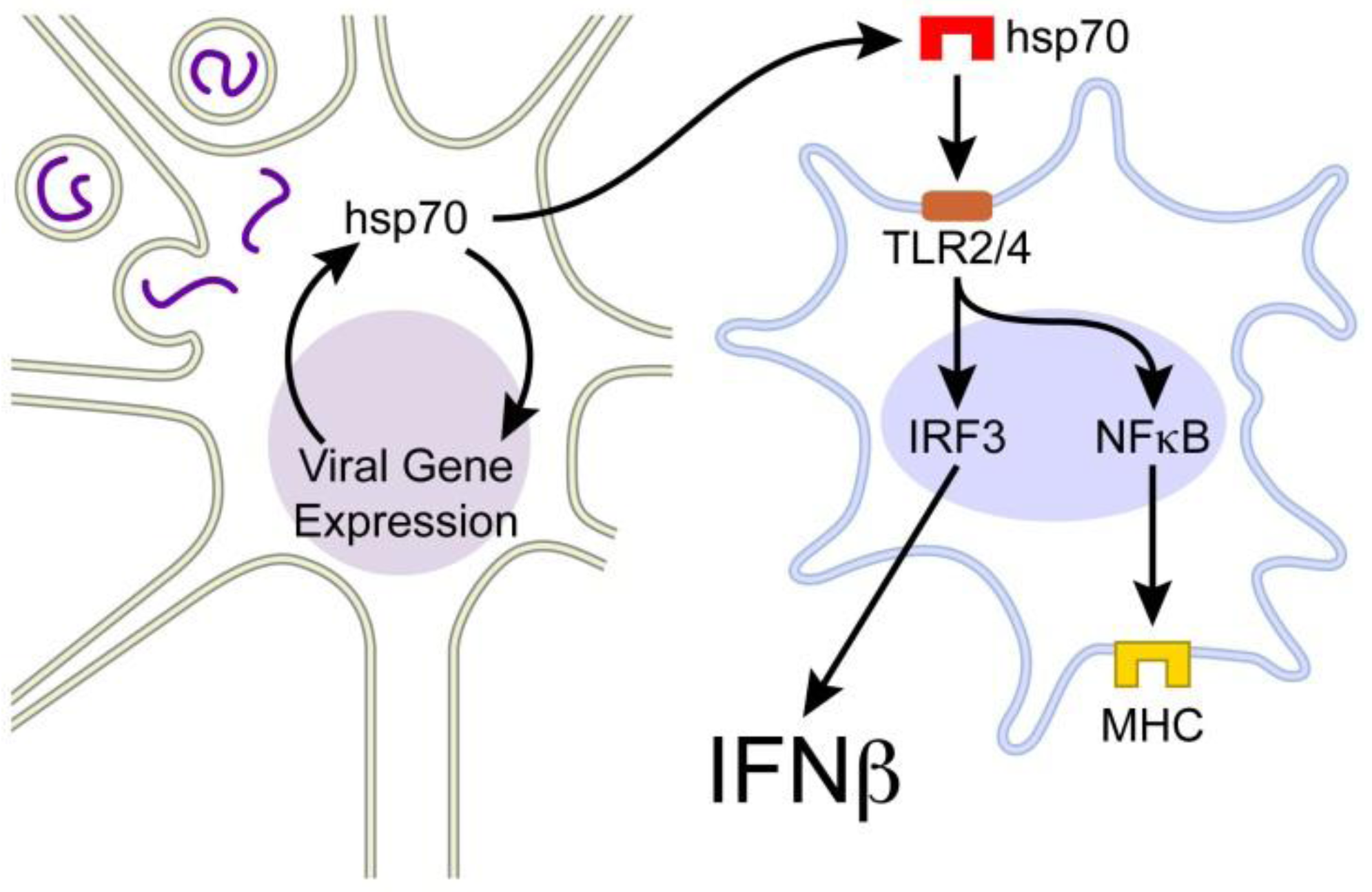
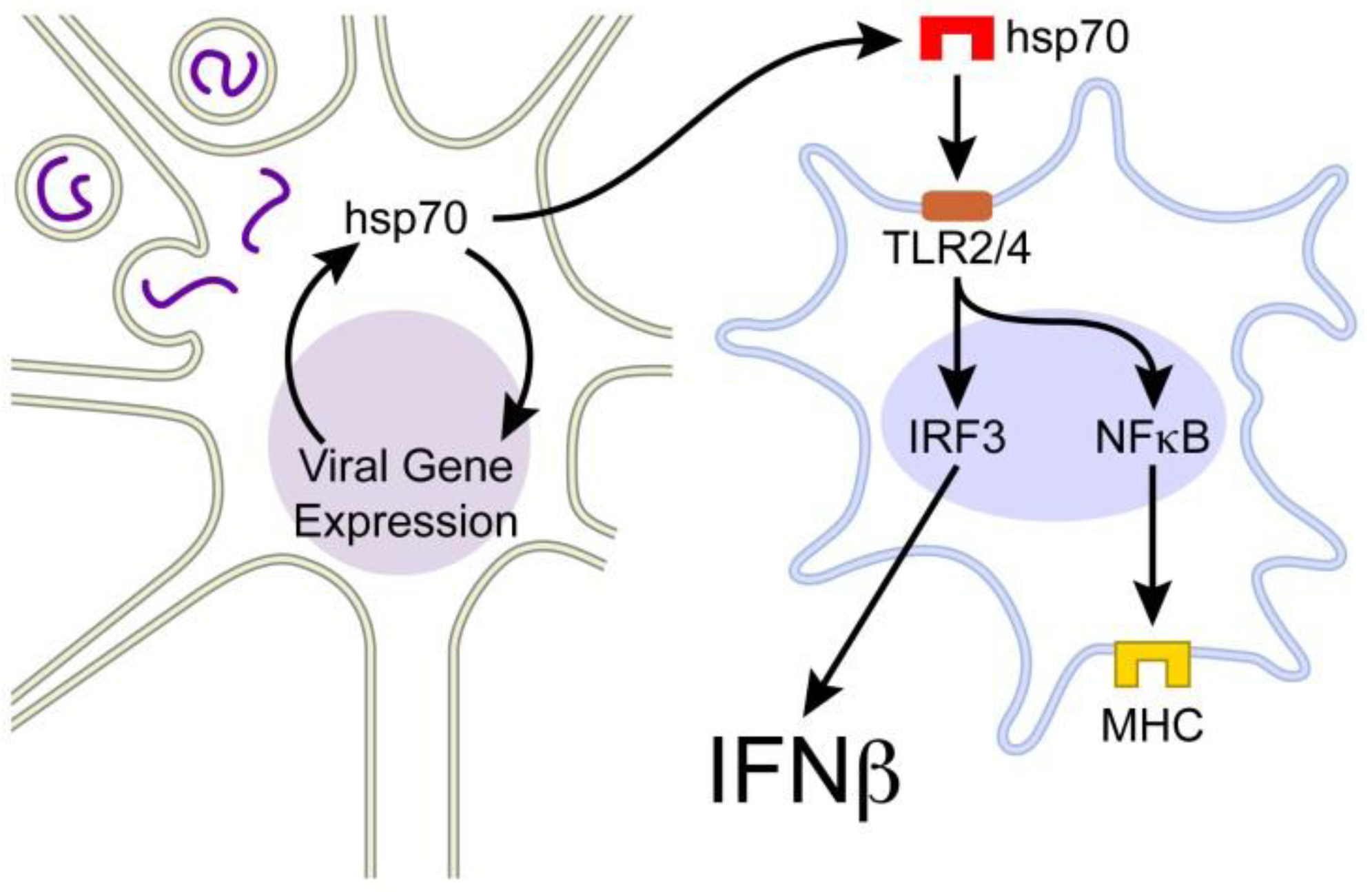
3. Extracellular hsp70
3.1. Extracellular hsp70 and Immunity
4. Hsp70-Mediated Host Protection in the Mouse Model of MeV Brain Infection
5. Future Directions
6. Conclusion
Acknowledgements
References
- Hartl, F.U. Molecular chaperones in cellular protein folding. Nature 1996, 381, 571–579. [Google Scholar] [CrossRef]
- Lindquist, S.; Craig, E.A. The heat-shock proteins. Annu. Rev. Genet. 1988, 22, 631–677. [Google Scholar] [CrossRef]
- Srivastava, P. Roles of heat-shock proteins in innate and adaptive immunity. Nat. Rev. Immunol. 2002, 2, 185–194. [Google Scholar] [CrossRef]
- Gamer, J.; Multhaup, G.; Tomoyasu, T.; McCarty, J.S.; Rudiger, S.; Schonfeld, H.J.; Schirra, C.; Bujard, H.; Bukau, B. A cycle of binding and release of the dnak, dnaj and grpe chaperones regulates activity of the escherichia coli heat shock transcription factor sigma32. EMBO J. 1996, 15, 607–617. [Google Scholar]
- Hunt, C.; Morimoto, R.I. Conserved features of eukaryotic hsp70 genes revealed by comparison with the nucleotide sequence of human hsp70. Proc. Natl. Acad. Sci. USA 1985, 82, 6455–6459. [Google Scholar] [CrossRef]
- Milner, C.M.; Campbell, R.D. Structure and expression of the three mhc-linked hsp70 genes. Immunogenetics 1990, 32, 242–251. [Google Scholar]
- Radford, N.B.; Fina, M.; Benjamin, I.J.; Moreadith, R.W.; Graves, K.H.; Zhao, P.; Gavva, S.; Wiethoff, A.; Sherry, A.D.; Malloy, C.R.; et al. Cardioprotective effects of 70-kDa heat shock protein in transgenic mice. Proc. Natl. Acad. Sci. USA 1996, 93, 2339–2342. [Google Scholar]
- Plumier, J.C.; Ross, B.M.; Currie, R.W.; Angelidis, C.E.; Kazlaris, H.; Kollias, G.; Pagoulatos, G.N. Transgenic mice expressing the human heat shock protein 70 have improved post-ischemic myocardial recovery. J. Clin. Invest. 1995, 95, 1854–1860. [Google Scholar] [CrossRef]
- Mestril, R.; Chi, S.H.; Sayen, M.R.; O'Reilly, K.; Dillmann, W.H. Expression of inducible stress protein 70 in rat heart myogenic cells confers protection against simulated ischemia-induced injury. J. Clin. Invest. 1994, 93, 759–767. [Google Scholar] [CrossRef]
- Marber, M.S.; Mestril, R.; Chi, S.H.; Sayen, M.R.; Yellon, D.M.; Dillmann, W.H. Overexpression of the rat inducible 70-kD heat stress protein in a transgenic mouse increases the resistance of the heart to ischemic injury. J. Clin. Invest. 1995, 95, 1446–1456. [Google Scholar] [CrossRef]
- Wissing, D.; Jaattela, M. HSP27 and Hsp70 increase the survival of WEHI-S cells exposed to hyperthermia. Int. J. Hyperthermia 1996, 12, 125–138. [Google Scholar] [CrossRef]
- Bellmann, K.; Jaattela, M.; Wissing, D.; Burkart, V.; Kolb, H. Heat shock protein hsp70 overexpression confers resistance against nitric oxide. FEBS Lett. 1996, 391, 185–188. [Google Scholar] [CrossRef]
- Amin, V.; Cumming, D.V.; Latchman, D.S. Over-expression of heat shock protein 70 protects neuronal cells against both thermal and ischaemic stress but with different efficiencies. Neurosci. Lett. 1996, 206, 45–48. [Google Scholar] [CrossRef]
- Beckmann, R.P.; Mizzen, L.E.; Welch, W.J. Interaction of hsp 70 with newly synthesized proteins: Implications for protein folding and assembly. Science 1990, 248, 850–854. [Google Scholar]
- Chirico, W.J.; Waters, M.G.; Blobel, G. 70k heat shock related proteins stimulate protein translocation into microsomes. Nature 1988, 332, 805–810. [Google Scholar]
- Deshaies, R.J.; Koch, B.D.; Werner-Washburne, M.; Craig, E.A.; Schekman, R. A subfamily of stress proteins facilitates translocation of secretory and mitochondrial precursor polypeptides. Nature 1988, 332, 800–805. [Google Scholar]
- Kang, P.J.; Ostermann, J.; Shilling, J.; Neupert, W.; Craig, E.A.; Pfanner, N. Requirement for hsp70 in the mitochondrial matrix for translocation and folding of precursor proteins. Nature 1990, 348, 137–143. [Google Scholar]
- Jaattela, M. Heat shock proteins as cellular lifeguards. Ann. Med. 1999, 31, 261–271. [Google Scholar]
- Arya, R.; Mallik, M.; Lakhotia, S.C. Heat shock genes—Integrating cell survival and death. J. Biosci. 2007, 32, 595–610. [Google Scholar] [CrossRef]
- Nylandsted, J.; Rohde, M.; Brand, K.; Bastholm, L.; Elling, F.; Jaattela, M. Selective depletion of heat shock protein 70 (hsp70) activates a tumor-specific death program that is independent of caspases and bypasses bcl-2. Proc. Natl. Acad. Sci. USA 2000, 97, 7871–7876. [Google Scholar] [CrossRef]
- Theodorakis, N.G.; Morimoto, R.I. Posttranscriptional regulation of hsp70 expression in human cells: Effects of heat shock, inhibition of protein synthesis, and adenovirus infection on translation and mrna stability. Mol. Cell. Biol. 1987, 7, 4357–4368. [Google Scholar]
- Mosser, D.D.; Duchaine, J.; Massie, B. The DNA-binding activity of the human heat shock transcription factor is regulated in vivo by hsp70. Mol. Cell. Biol. 1993, 13, 5427–5438. [Google Scholar]
- Snoeckx, L.H.; Cornelussen, R.N.; Van Nieuwenhoven, F.A.; Reneman, R.S.; Van Der Vusse, G.J. Heat shock proteins and cardiovascular pathophysiology. Physiol. Rev. 2001, 81, 1461–1497. [Google Scholar]
- Mandrekar, P.; Catalano, D.; Jeliazkova, V.; Kodys, K. Alcohol exposure regulates heat shock transcription factor binding and heat shock proteins 70 and 90 in monocytes and macrophages: Implication for tnf-alpha regulation. J. Leukoc. Biol. 2008, 84, 1335–1345. [Google Scholar] [CrossRef]
- Lum, L.S.; Hsu, S.; Vaewhongs, M.; Wu, B. The hsp70 gene ccaat-binding factor mediates transcriptional activation by the adenovirus e1a protein. Mol. Cell. Biol. 1992, 12, 2599–2605. [Google Scholar]
- Su, C.Y.; Chong, K.Y.; Chen, J.; Ryter, S.; Khardori, R.; Lai, C.C. A physiologically relevant hyperthermia selectively activates constitutive hsp70 in h9c2 cardiac myoblasts and confers oxidative protection. J. Mol. Cell. Cardiol. 1999, 31, 845–855. [Google Scholar] [CrossRef]
- Morrison-Bogorad, M.; Zimmerman, A.L.; Pardue, S. Heat-shock 70 messenger rna levels in human brain: Correlation with agonal fever. J. Neurochem. 1995, 64, 235–246. [Google Scholar] [CrossRef]
- Mayer, M.P. Recruitment of hsp70 chaperones: A crucial part of viral survival strategies. Rev. Physiol. Biochem. Pharmacol. 2005, 153, 1–46. [Google Scholar] [CrossRef]
- Oglesbee, M.; Krakowka, S. Cellular stress response induces selective intranuclear trafficking and accumulation of morbillivirus major core protein. Lab. Invest. 1993, 68, 109–117. [Google Scholar]
- Hutchins, S.S.; Papania, M.J.; Amler, R.; Maes, E.F.; Grabowsky, M.; Bromberg, K.; Glasglow, V.; Speed, T.; Bellini, W.J.; Orenstein, W.A. Evaluation of the measles clinical case definition. J. Infect. Dis. 2004, 189, S153–S159. [Google Scholar] [CrossRef]
- Morimoto, R.I.; Tissières, A.; Georgopoulos, C. The Biology of Heat Shock Proteins and Molecular Chaperones; Cold Spring Harbor Laboratory Press: Plainview, NY, USA, 1994; Volume vii, p. 610. [Google Scholar]
- Jockusch, H.; Wiegand, C.; Mersch, B.; Rajes, D. Mutants of tobacco mosaic virus with temperature-sensitive coat proteins induce heat shock response in tobacco leaves. Mol. Plant. Microbe. Interact. 2001, 14, 914–917. [Google Scholar] [CrossRef]
- Aparicio, F.; Thomas, C.L.; Lederer, C.; Niu, Y.; Wang, D.; Maule, A.J. Virus induction of heat shock protein 70 reflects a general response to protein accumulation in the plant cytosol. Plant Physiol. 2005, 138, 529–536. [Google Scholar] [CrossRef]
- Liberman, E.; Fong, Y.L.; Selby, M.J.; Choo, Q.L.; Cousens, L.; Houghton, M.; Yen, T.S. Activation of the grp78 and grp94 promoters by hepatitis c virus e2 envelope protein. J. Virol. 1999, 73, 3718–3722. [Google Scholar]
- Phillips, B.; Abravaya, K.; Morimoto, R.I. Analysis of the specificity and mechanism of transcriptional activation of the human hsp70 gene during infection by DNA viruses. J. Virol. 1991, 65, 5680–5692. [Google Scholar]
- Yang, U.C.; Huang, W.; Flint, S.J. Mrna export correlates with activation of transcription in human subgroup c adenovirus-infected cells. J. Virol. 1996, 70, 4071–4080. [Google Scholar]
- Damania, B.; Lieberman, P.; Alwine, J.C. Simian virus 40 large t antigen stabilizes the tata-binding protein-tfiia complex on the tata element. Mol. Cell. Biol 1998, 18, 3926–3935. [Google Scholar]
- Hagemeier, C.; Walker, S.; Caswell, R.; Kouzarides, T.; Sinclair, J. The human cytomegalovirus 80-kilodalton but not the 72-kilodalton immediate-early protein transactivates heterologous promoters in a tata box-dependent mechanism and interacts directly with tfiid. J. Virol. 1992, 66, 4452–4456. [Google Scholar]
- Erbse, A.; Mayer, M.P.; Bukau, B. Mechanism of substrate recognition by hsp70 chaperones. Biochem. Soc. Trans. 2004, 32, 617–621. [Google Scholar] [CrossRef]
- Perez-Vargas, J.; Romero, P.; Lopez, S.; Arias, C.F. The peptide-binding and atpase domains of recombinant hsc70 are required to interact with rotavirus and reduce its infectivity. J. Virol. 2006, 80, 3322–3331. [Google Scholar] [CrossRef]
- Tanguy Le Gac, N.; Boehmer, P.E. Activation of the herpes simplex virus type-1 origin-binding protein (ul9) by heat shock proteins. J. Biol. Chem. 2002, 277, 5660–5666. [Google Scholar] [CrossRef]
- Liu, J.S.; Kuo, S.R.; Makhov, A.M.; Cyr, D.M.; Griffith, J.D.; Broker, T.R.; Chow, L.T. Human hsp70 and hsp40 chaperone proteins facilitate human papillomavirus-11 e1 protein binding to the origin and stimulate cell-free DNA replication. J. Biol. Chem. 1998, 273, 30704–30712. [Google Scholar]
- Carsillo, T.; Zhang, X.; Vasconcelos, D.; Niewiesk, S.; Oglesbee, M. A single codon in the nucleocapsid protein c terminus contributes to in vitro and in vivo fitness of edmonston measles virus. J. Virol. 2006, 80, 2904–2912. [Google Scholar] [CrossRef]
- Oglesbee, M.; Ringler, S.; Krakowka, S. Interaction of canine distemper virus nucleocapsid variants with 70k heat-shock proteins. J. Gen. Virol. 1990, 71, 1585–1590. [Google Scholar] [CrossRef]
- Oglesbee, M.J.; Kenney, H.; Kenney, T.; Krakowka, S. Enhanced production of morbillivirus gene-specific rnas following induction of the cellular stress response in stable persistent infection. Virology 1993, 192, 556–567. [Google Scholar] [CrossRef]
- Vasconcelos, D.; Norrby, E.; Oglesbee, M. The cellular stress response increases measles virus-induced cytopathic effect. J. Gen. Virol. 1998, 79, 1769–1773. [Google Scholar]
- Vasconcelos, D.Y.; Cai, X.H.; Oglesbee, M.J. Constitutive overexpression of the major inducible 70 kDa heat shock protein mediates large plaque formation by measles virus. J. Gen. Virol. 1998, 79, 2239–2247. [Google Scholar]
- Oglesbee, M.J.; Liu, Z.; Kenney, H.; Brooks, C.L. The highly inducible member of the 70 kda family of heat shock proteins increases canine distemper virus polymerase activity. J. Gen. Virol. 1996, 77, 2125–2135. [Google Scholar] [CrossRef]
- Longhi, S. Measles Virus Nucleoprotein; Nova Biomedical Books: New York, NY, USA, 2007; Volume ix, p. 163. [Google Scholar]
- Bourhis, J.M.; Canard, B.; Longhi, S. Structural disorder within the replicative complex of measles virus: Functional implications. Virology 2006, 344, 94–110. [Google Scholar] [CrossRef]
- Brown, G.; Rixon, H.W.; Steel, J.; McDonald, T.P.; Pitt, A.R.; Graham, S.; Sugrue, R.J. Evidence for an association between heat shock protein 70 and the respiratory syncytial virus polymerase complex within lipid-raft membranes during virus infection. Virology 2005, 338, 69–80. [Google Scholar] [CrossRef]
- Menager, P.; Roux, P.; Megret, F.; Bourgeois, J.P.; Le Sourd, A.M.; Danckaert, A.; Lafage, M.; Prehaud, C.; Lafon, M. Toll-like receptor 3 (tlr3) plays a major role in the formation of rabies virus negri bodies. PLoS Pathog. 2009, 5, e1000315. [Google Scholar] [CrossRef]
- Jiang, Y.; Serviene, E.; Gal, J.; Panavas, T.; Nagy, P.D. Identification of essential host factors affecting tombusvirus rna replication based on the yeast tet promoters hughes collection. J. Virol. 2006, 80, 7394–7404. [Google Scholar] [CrossRef]
- Nagy, P.D. Yeast as a model host to explore plant virus-host interactions. Annu. Rev. Phytopathol. 2008, 46, 217–242. [Google Scholar] [CrossRef]
- Campbell, K.S.; Mullane, K.P.; Aksoy, I.A.; Stubdal, H.; Zalvide, J.; Pipas, J.M.; Silver, P.A.; Roberts, T.M.; Schaffhausen, B.S.; DeCaprio, J.A. Dnaj/hsp40 chaperone domain of sv40 large t antigen promotes efficient viral DNA replication. Genes. Dev. 1997, 11, 1098–1110. [Google Scholar] [CrossRef]
- Niyaz, Y.; Frenz, I.; Petersen, G.; Gehring, U. Transcriptional stimulation by the DNA binding protein hap46/bag-1m involves hsp70/hsc70 molecular chaperones. Nucleic. Acids. Res. 2003, 31, 2209–2216. [Google Scholar] [CrossRef]
- Heller, M.; Vasconcelos, D.; Cummins, J.; Oglesbee, M. Interferon-alpha inhibits the emergence of cellular stress response-dependent morbillivirus large plaque variants. Antiviral Res. 1998, 38, 195–207. [Google Scholar] [CrossRef]
- Carsillo, T.; Carsillo, M.; Niewiesk, S.; Vasconcelos, D.; Oglesbee, M. Hyperthermic pre-conditioning promotes measles virus clearance from brain in a mouse model of persistent infection. Brain Res. 2004, 1004, 73–82. [Google Scholar]
- Oglesbee, M.; Niewiesk, S. Measles virus neurovirulence and host immunity. Future Virol 2011, 6, 85–99. [Google Scholar] [CrossRef]
- Manchester, M.; Rall, G.F. Model systems: Transgenic mouse models for measles pathogenesis. Trends Microbiol. 2001, 9, 19–23. [Google Scholar] [CrossRef]
- Carsillo, T.; Traylor, Z.; Choi, C.; Niewiesk, S.; Oglesbee, M. Hsp72, a host determinant of measles virus neurovirulence. J. Virol. 2006, 80, 11031–11039. [Google Scholar] [CrossRef]
- Basu, S.; Binder, R.J.; Suto, R.; Anderson, K.M.; Srivastava, P.K. Necrotic but not apoptotic cell death releases heat shock proteins, which deliver a partial maturation signal to dendritic cells and activate the nf-kappa b pathway. Int. Immunol. 2000, 12, 1539–1546. [Google Scholar] [CrossRef]
- Hightower, L.E.; Guidon, P.T., Jr. Selective release from cultured mammalian cells of heat-shock (stress) proteins that resemble glia-axon transfer proteins. J. Cell. Physiol. 1989, 138, 257–266. [Google Scholar] [CrossRef]
- Hunter-Lavin, C.; Davies, E.L.; Bacelar, M.M.; Marshall, M.J.; Andrew, S.M.; Williams, J.H. Hsp70 release from peripheral blood mononuclear cells. Biochem. Biophys. Res. Commun. 2004, 324, 511–517. [Google Scholar] [CrossRef]
- Moehler, M.; Zeidler, M.; Schede, J.; Rommelaere, J.; Galle, P.R.; Cornelis, J.J.; Heike, M. Oncolytic parvovirus h1 induces release of heat-shock protein hsp72 in susceptible human tumor cells but may not affect primary immune cells. Cancer. Gene. Ther. 2003, 10, 477–480. [Google Scholar] [CrossRef]
- Meckes, D.G., Jr.; Raab-Traub, N. Microvesicles and viral infection. J. Virol. 2011, 85, 12844–12854. [Google Scholar] [CrossRef]
- Simpson, R.J.; Lim, J.W.; Moritz, R.L.; Mathivanan, S. Exosomes: Proteomic insights and diagnostic potential. Expert Rev. Proteomics 2009, 6, 267–283. [Google Scholar] [CrossRef]
- Faure, J.; Lachenal, G.; Court, M.; Hirrlinger, J.; Chatellard-Causse, C.; Blot, B.; Grange, J.; Schoehn, G.; Goldberg, Y.; Boyer, V.; et al. Exosomes are released by cultured cortical neurones. Mol. Cell. Neurosci. 2006, 31, 642–648. [Google Scholar] [CrossRef]
- Clayton, A.; Turkes, A.; Navabi, H.; Mason, M.D.; Tabi, Z. Induction of heat shock proteins in b-cell exosomes. J. Cell. Sci. 2005, 118, 3631–3638. [Google Scholar] [CrossRef]
- De Maio, A. Extracellular heat shock proteins, cellular export vesicles, and the stress observation system: A form of communication during injury, infection, and cell damage. It is never known how far a controversial finding will go! Dedicated to ferruccio ritossa. Cell Stress Chaperones 2011, 16, 235–249. [Google Scholar] [CrossRef]
- Anand, P.K.; Anand, E.; Bleck, C.K.; Anes, E.; Griffiths, G. Exosomal hsp70 induces a pro-inflammatory response to foreign particles including mycobacteria. PLoS One 2010, 5, e10136. [Google Scholar]
- Wheeler, D.S.; Chase, M.A.; Senft, A.P.; Poynter, S.E.; Wong, H.R.; Page, K. Extracellular hsp72, an endogenous damp, is released by virally infected airway epithelial cells and activates neutrophils via toll-like receptor (tlr)-4. Respir. Res. 2009, 10, 31. [Google Scholar] [CrossRef]
- Njemini, R.; Lambert, M.; Demanet, C.; Mets, T. Elevated serum heat-shock protein 70 levels in patients with acute infection: Use of an optimized enzyme-linked immunosorbent assay. Scand. J. Immunol. 2003, 58, 664–669. [Google Scholar] [CrossRef]
- Kono, H.; Rock, K.L. How dying cells alert the immune system to danger. Nat. Rev. Immunol. 2008, 8, 279–289. [Google Scholar] [CrossRef]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef]
- Vabulas, R.M.; Wagner, H.; Schild, H. Heat shock proteins as ligands of toll-like receptors. Curr. Top. Microbiol. Immunol. 2002, 270, 169–184. [Google Scholar]
- Asea, A.; Rehli, M.; Kabingu, E.; Boch, J.A.; Bare, O.; Auron, P.E.; Stevenson, M.A.; Calderwood, S.K. Novel signal transduction pathway utilized by extracellular hsp70: Role of toll-like receptor (tlr) 2 and tlr4. J. Biol. Chem. 2002, 277, 15028–15034. [Google Scholar]
- Kakimura, J.; Kitamura, Y.; Takata, K.; Umeki, M.; Suzuki, S.; Shibagaki, K.; Taniguchi, T.; Nomura, Y.; Gebicke-Haerter, P.J.; Smith, M.A.; et al. Microglial activation and amyloid-beta clearance induced by exogenous heat-shock proteins. Faseb. J. 2002, 16, 601–603. [Google Scholar]
- Toshchakov, V.; Jones, B.W.; Perera, P.Y.; Thomas, K.; Cody, M.J.; Zhang, S.; Williams, B.R.; Major, J.; Hamilton, T.A.; Fenton, M.J.; et al. Tlr4, but not tlr2, mediates ifn-beta-induced stat1alpha/beta-dependent gene expression in macrophages. Nat. Immunol. 2002, 3, 392–398. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, Z.Y.; Wu, Y.; Schluesener, H.J. Immunolocalization of toll-like receptors 2 and 4 as well as their endogenous ligand, heat shock protein 70, in rat traumatic brain injury. Neuroimmunomodulation 2012, 19, 10–19. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Murshid, A.; Gong, J.; Calderwood, S.K. The role of heat shock proteins in antigen cross presentation. Front. Immunol. 2012, 3, 63. [Google Scholar]
- Suto, R.; Srivastava, P.K. A mechanism for the specific immunogenicity of heat shock protein-chaperoned peptides. Science 1995, 269, 1585–1588. [Google Scholar]
- Carbone, F.R.; Kurts, C.; Bennett, S.R.; Miller, J.F.; Heath, W.R. Cross-presentation: A general mechanism for ctl immunity and tolerance. Immunol. Today 1998, 19, 368–373. [Google Scholar] [CrossRef]
- Ciupitu, A.M.; Petersson, M.; O'Donnell, C.L.; Williams, K.; Jindal, S.; Kiessling, R.; Welsh, R.M. Immunization with a lymphocytic choriomeningitis virus peptide mixed with heat shock protein 70 results in protective antiviral immunity and specific cytotoxic t lymphocytes. J. Exp. Med. 1998, 187, 685–691. [Google Scholar] [CrossRef]
- Chen, T.; Guo, J.; Han, C.; Yang, M.; Cao, X. Heat shock protein 70, released from heat-stressed tumor cells, initiates antitumor immunity by inducing tumor cell chemokine production and activating dendritic cells via tlr4 pathway. J. Immunol. 2009, 182, 1449–1459. [Google Scholar]
- Campisi, J.; Leem, T.H.; Fleshner, M. Stress-induced extracellular hsp72 is a functionally significant danger signal to the immune system. Cell Stress Chaperones 2003, 8, 272–286. [Google Scholar] [CrossRef]
- Wright, B.H.; Corton, J.M.; El-Nahas, A.M.; Wood, R.F.; Pockley, A.G. Elevated levels of circulating heat shock protein 70 (hsp70) in peripheral and renal vascular disease. Heart Vessels 2000, 15, 18–22. [Google Scholar] [CrossRef]
- Dybdahl, B.; Wahba, A.; Lien, E.; Flo, T.H.; Waage, A.; Qureshi, N.; Sellevold, O.F.; Espevik, T.; Sundan, A. Inflammatory response after open heart surgery: Release of heat-shock protein 70 and signaling through toll-like receptor-4. Circulation 2002, 105, 685–690. [Google Scholar] [CrossRef]
- Pockley, A.G.; Georgiades, A.; Thulin, T.; de Faire, U.; Frostegard, J. Serum heat shock protein 70 levels predict the development of atherosclerosis in subjects with established hypertension. Hypertension 2003, 42, 235–238. [Google Scholar] [CrossRef]
- Moore, S.A.; Kim, M.Y.; Maiolini, A.; Tipold, A.; Oglesbee, M.J. Extracellular hsp70 release in canine steroid responsive meningitis-arteritis. Vet. Immunol. Immunopathol. 2012, 145, 129–133. [Google Scholar] [CrossRef]
- Awad, H.; Suntres, Z.; Heijmans, J.; Smeak, D.; Bergdall-Costell, V.; Christofi, F.L.; Magro, C.; Oglesbee, M. Intracellular and extracellular expression of the major inducible 70 kDa heat shock protein in experimental ischemia-reperfusion injury of the spinal cord. Exp. Neurol. 2008, 212, 275–284. [Google Scholar] [CrossRef]
- Hecker, J.G.; Sundram, H.; Zou, S.; Praestgaard, A.; Bavaria, J.E.; Ramchandren, S.; McGarvey, M. Heat shock proteins hsp70 and hsp27 in the cerebral spinal fluid of patients undergoing thoracic aneurysm repair correlate with the probability of postoperative paralysis. Cell Stress Chaperones 2008, 13, 435–446. [Google Scholar] [CrossRef]
- Vega, V.L.; Rodriguez-Silva, M.; Frey, T.; Gehrmann, M.; Diaz, J.C.; Steinem, C.; Multhoff, G.; Arispe, N.; De Maio, A. Hsp70 translocates into the plasma membrane after stress and is released into the extracellular environment in a membrane-associated form that activates macrophages. J. Immunol. 2008, 180, 4299–4307. [Google Scholar]
- Calderwood, S.K.; Murshid, A.; Gong, J. Heat shock proteins: Conditional mediators of inflammation in tumor immunity. Front. Immunol. 2012, 3, 75. [Google Scholar]
- Gong, J.; Zhu, B.; Murshid, A.; Adachi, H.; Song, B.; Lee, A.; Liu, C.; Calderwood, S.K. T cell activation by heat shock protein 70 vaccine requires tlr signaling and scavenger receptor expressed by endothelial cells-1. J. Immunol. 2009, 183, 3092–3098. [Google Scholar] [CrossRef]
- Pulido, J.; Kottke, T.; Thompson, J.; Galivo, F.; Wongthida, P.; Diaz, R.M.; Rommelfanger, D.; Ilett, E.; Pease, L.; Pandha, H.; et al. Using virally expressed melanoma cdna libraries to identify tumor-associated antigens that cure melanoma. Nat. Biotechnol. 2012, 30, 337–343. [Google Scholar] [CrossRef]
- Pack, C.D.; Kumaraguru, U.; Suvas, S.; Rouse, B.T. Heat-shock protein 70 acts as an effective adjuvant in neonatal mice and confers protection against challenge with herpes simplex virus. Vaccine 2005, 23, 3526–3534. [Google Scholar] [CrossRef]
- Chen, W.; Lin, Y.; Liao, C.; Hsieh, S. Modulatory effects of the human heat shock protein 70 on DNA vaccination. J. Biomed. Sci. 2000, 7, 412–419. [Google Scholar] [CrossRef]
- Pack, C.D.; Gierynska, M.; Rouse, B.T. An intranasal heat shock protein based vaccination strategy confers protection against mucosal challenge with herpes simplex virus. Hum. Vaccin. 2008, 4, 360–364. [Google Scholar] [CrossRef]
- Pardue, S.; Wang, S.; Miller, M.M.; Morrison-Bogorad, M. Elevated levels of inducible heat shock 70 proteins in human brain. Neurobiol. Aging 2007, 28, 314–324. [Google Scholar] [CrossRef]
- Niewiesk, S.; Brinckmann, U.; Bankamp, B.; Sirak, S.; Liebert, U.G.; ter Meulen, V. Susceptibility to measles virus-induced encephalitis in mice correlates with impaired antigen presentation to cytotoxic t lymphocytes. J. Virol. 1993, 67, 75–81. [Google Scholar]
- Lawrence, D.M.; Vaughn, M.M.; Belman, A.R.; Cole, J.S.; Rall, G.F. Immune response-mediated protection of adult but not neonatal mice from neuron-restricted measles virus infection and central nervous system disease. J. Virol. 1999, 73, 1795–1801. [Google Scholar]
- Neighbour, P.A.; Rager-Zisman, B.; Bloom, B.R. Susceptibility of mice to acute and persistent measles infection. Infect. Immun. 1978, 21, 764–770. [Google Scholar]
- Griffin, D.E.; Mullinix, J.; Narayan, O.; Johnson, R.T. Age dependence of viral expression: Comparative pathogenesis of two rodent-adapted strains of measles virus in mice. Infect. Immun. 1974, 9, 690–695. [Google Scholar]
- Finke, D.; Liebert, U.G. CD4+ T cells are essential in overcoming experimental murine measles encephalitis. Immunology 1994, 83, 184–189. [Google Scholar]
- Rall, G.F.; Manchester, M.; Daniels, L.R.; Callahan, E.M.; Belman, A.R.; Oldstone, M.B. A transgenic mouse model for measles virus infection of the brain. Proc. Natl. Acad. Sci. U.SA 1997, 94, 4659–4663. [Google Scholar]
- Mrkic, B.; Pavlovic, J.; Rulicke, T.; Volpe, P.; Buchholz, C.J.; Hourcade, D.; Atkinson, J.P.; Aguzzi, A.; Cattaneo, R. Measles virus spread and pathogenesis in genetically modified mice. J. Virol. 1998, 72, 7420–7427. [Google Scholar]
- Lawrence, D.M.; Patterson, C.E.; Gales, T.L.; D'Orazio, J.L.; Vaughn, M.M.; Rall, G.F. Measles virus spread between neurons requires cell contact but not cd46 expression, syncytium formation, or extracellular virus production. J. Virol. 2000, 74, 1908–1918. [Google Scholar] [CrossRef]
- Duprex, W.P.; Duffy, I.; McQuaid, S.; Hamill, L.; Cosby, S.L.; Billeter, M.A.; Schneider-Schaulies, J.; ter Meulen, V.; Rima, B.K. The h gene of rodent brain-adapted measles virus confers neurovirulence to the edmonston vaccine strain. J. Virol. 1999, 73, 6916–6922. [Google Scholar]
- Weidinger, G.; Czub, S.; Neumeister, C.; Harriott, P.; ter Meulen, V.; Niewiesk, S. Role of CD4(+) and CD8(+) t cells in the prevention of measles virus-induced encephalitis in mice. J. Gen. Virol. 2000, 81, 2707–2713. [Google Scholar]
- Neumeister, C.; Niewiesk, S. Recognition of measles virus-infected cells by CD8+ t cells depends on the h-2 molecule. J. Gen. Virol. 1998, 79, 2583–2591. [Google Scholar]
- Finke, D.; Brinckmann, U.G.; ter Meulen, V.; Liebert, U.G. Gamma interferon is a major mediator of antiviral defense in experimental measles virus-induced encephalitis. J. Virol. 1995, 69, 5469–5474. [Google Scholar]
- Matthews, V.B.; Christiansen, F.T.; Price, P. Lymphocytes from h2 mice produce lower levels of several cytokines than congenic h2 or h2 mice. Immunol. Cell. Biol. 2000, 78, 247–253. [Google Scholar] [CrossRef]
- Patterson, C.E.; Lawrence, D.M.; Echols, L.A.; Rall, G.F. Immune-mediated protection from measles virus-induced central nervous system disease is noncytolytic and gamma interferon dependent. J. Virol. 2002, 76, 4497–4506. [Google Scholar]
- Beauvillain, C.; Donnou, S.; Jarry, U.; Scotet, M.; Gascan, H.; Delneste, Y.; Guermonprez, P.; Jeannin, P.; Couez, D. Neonatal and adult microglia cross-present exogenous antigens. Glia 2008, 56, 69–77. [Google Scholar] [CrossRef]
- Delhaye, S.; Paul, S.; Blakqori, G.; Minet, M.; Weber, F.; Staeheli, P.; Michiels, T. Neurons produce type I interferon during viral encephalitis. Proc. Natl. Acad. Sci. USA 2006, 103, 7835–7840. [Google Scholar]
- Dhib-Jalbut, S.; Xia, J.; Rangaviggula, H.; Fang, Y.Y.; Lee, T. Failure of measles virus to activate nuclear factor-kappa b in neuronal cells: Implications on the immune response to viral infections in the central nervous system. J. Immunol. 1999, 162, 4024–4029. [Google Scholar]
- Robinzon, S.; Dafa-Berger, A.; Dyer, M.D.; Paeper, B.; Proll, S.C.; Teal, T.H.; Rom, S.; Fishman, D.; Rager-Zisman, B.; Katze, M.G. Impaired cholesterol biosynthesis in a neuronal cell line persistently infected with measles virus. J. Virol. 2009, 83, 5495–5504. [Google Scholar]
- Manchester, M.; Eto, D.S.; Oldstone, M.B. Characterization of the inflammatory response during acute measles encephalitis in nse-cd46 transgenic mice. J. Neuroimmunol. 1999, 96, 207–217. [Google Scholar] [CrossRef]
- Faul, E.J.; Wanjalla, C.N.; McGettigan, J.P.; Schnell, M.J. Interferon-beta expressed by a rabies virus-based hiv-1 vaccine vector serves as a molecular adjuvant and decreases pathogenicity. Virology 2008, 382, 226–238. [Google Scholar] [CrossRef]
- Faul, E.J.; Wanjalla, C.N.; Suthar, M.S.; Gale, M.; Wirblich, C.; Schnell, M.J. Rabies virus infection induces type i interferon production in an ips-1 dependent manner while dendritic cell activation relies on ifnar signaling. PLoS Pathog 2010, 6, e1001016. [Google Scholar] [CrossRef]
- Slifka, M.K.; Rodriguez, F.; Whitton, J.L. Rapid on/off cycling of cytokine production by virus-specific cd8+ t cells. Nature 1999, 401, 76–79. [Google Scholar] [CrossRef]
- Pien, G.C.; Nguyen, K.B.; Malmgaard, L.; Satoskar, A.R.; Biron, C.A. A unique mechanism for innate cytokine promotion of t cell responses to viral infections. J. Immunol. 2002, 169, 5827–5837. [Google Scholar]
- Carsillo, T.; Carsillo, M.; Traylor, Z.; Rajala-Schultz, P.; Popovich, P.; Niewiesk, S.; Oglesbee, M. Major histocompatibility complex haplotype determines hsp70-dependent protection against measles virus neurovirulence. J. Virol. 2009, 83, 5544–5555. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kim, M.Y.; Oglesbee, M. Virus-Heat Shock Protein Interaction and a Novel Axis for Innate Antiviral Immunity. Cells 2012, 1, 646-666. https://doi.org/10.3390/cells1030646
Kim MY, Oglesbee M. Virus-Heat Shock Protein Interaction and a Novel Axis for Innate Antiviral Immunity. Cells. 2012; 1(3):646-666. https://doi.org/10.3390/cells1030646
Chicago/Turabian StyleKim, Mi Young, and Michael Oglesbee. 2012. "Virus-Heat Shock Protein Interaction and a Novel Axis for Innate Antiviral Immunity" Cells 1, no. 3: 646-666. https://doi.org/10.3390/cells1030646
APA StyleKim, M. Y., & Oglesbee, M. (2012). Virus-Heat Shock Protein Interaction and a Novel Axis for Innate Antiviral Immunity. Cells, 1(3), 646-666. https://doi.org/10.3390/cells1030646