Applied Genetics and Genomics in Alfalfa Breeding

Abstract
:1. Introduction
2. Alfalfa Origins and Genetic Diversity
3. Alfalfa Breeding
3.1. Breeding Goals and Traits of Interest
3.2. Biomass Yield
3.3. Hybrid Alfalfa
4. Genetic Dissection of Quantitative Traits Using Molecular Markers
4.1. Marker Development
4.2. Genetic Linkage Map Construction

{kind=link}
| Reference | Mapping population | Ploidy | Population size | Markers a | No. markers | Linkage groups | Map length | Seg. Dist.b | Traits of QTL identified | |
|---|---|---|---|---|---|---|---|---|---|---|
| a RFLP = restriction fragment length polymorphism; RAPD = random amplified polymorphic DNA; I = isozyme; M = morphological marker; AFLP = amplified fragment length polymorphism; SSR = simple sequence repeat. b Seg.Dist. means percentage of markers that showed segregation distortion. c means composite map derived from both parents. | ||||||||||
| (cM) | (%) | |||||||||
| Brummer et al. [61] | F2 | 2x | 86 | RFLP | 108 | 10 | 468 | 50 | ||
| Kiss et al. [63] | F2 | 2x | 137 | RFLP, RAPD, I, M | 89 | 8 | 659 | -- | ||
| Kalo et al. [64] | F2 | 2x | 137 | RFLP, RAPD, I, M | 868 | 8 | 754 | 63 | ||
| Echt et al. [62] | BC | 2x | 87 | RFLP, RAPD | 86/61 | 10/7 | 553/603 | 34 | ||
| Narasimhamoorthy et al. [57] | BC | 2x | 130 | RFLP, SSR | 132 | 10 | 764 | 43 | Aluminum tolerance [57] | |
| Tavoletti et al. [65] | F1 | 2x | 55 | RFLP | 50/55 | 10/8 | 234/261 | 8.8 | ||
| Li et al. [53] | F1 | 2x | 190 | SSR | 99 | 8 | 528 | 24 | ||
| Li et al. [53] | F1 | 2x | 183 | SSR | 99 | 10 | 547 | 34 | ||
| Li et al. [53] | F2 | 2x | 152 | SSR | 90 | 13 | 391 | 68 | Viability [53] | |
| Brouwer and Osborn [69] | BC | 4x | 101/101 | RFLP | 88 c | 7 | 443 | 4–9 | Fall dormancy and winter hardiness [69] | |
| Sledge et al. [55] | BC | 4x | 93/93 | SSR | 286 c | 8 | 624 | 10 | ||
| Julier et al. [54] | F1 | 4x | 168 | AFLP, SSR | 107 c | 8 | 709 | 25-SSR | ||
| Robins et al. [37] | F1 | 4x | 200 | RFLP, SSR | 172 c | 8 | 546 | 32 | Biomass [37], morphological traits [38], winter hardiness [70], persistence [71], and self-fertility [72] | |
| Musial et al. [68] | BC | 4x | 145 | AFLP, SSR | 203 c | 8 | 794 | 14.3 | Stagonospora root and crown rot resistance [68] and Anthracnose resistance [73] |
4.3. Characteristics of Alfalfa Genetic Linkage Maps
4.4. Association Mapping
5. Using Markers in Selection
5.1. Simple Uses—Diversity Selection and Paternity Testing
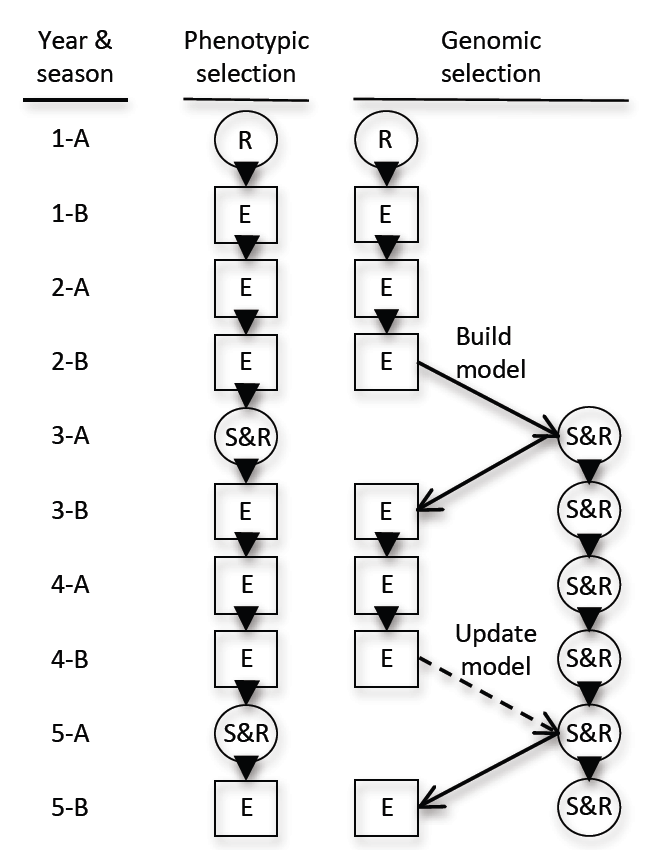
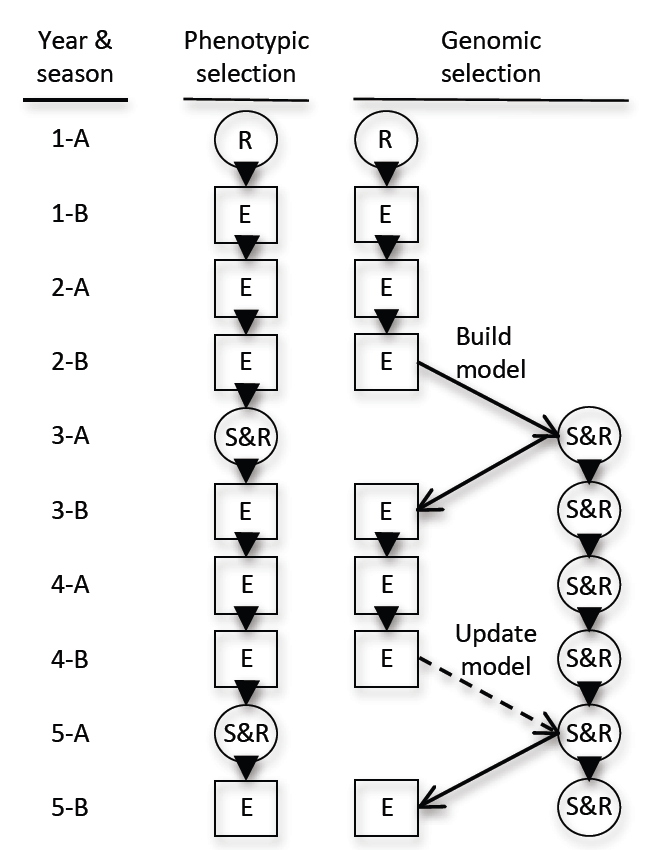
5.3. Marker-Only Selection and Genomic Selection
6. Conclusions
Acknowledgements
References
- Small, E. Alfalfa and Relatives: Evolution and Classification of Medicago; 2011; NRC Research Press/CAB International: Ottawa, Canada. [Google Scholar]
- Russelle, M. After an 8,000-year journey, the “Queen of forages” stands poised to enjoy renewed popularity. Am. Sci. 2001, 89, 252–261. [Google Scholar]
- Kondorosi, A.; Brown, S.; Marie, D.; Blondon, F. Genome size and base composition in Medicago sativa and M. truncatula species. Genome 1994, 37, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Bauchan, G.R.; Quiros, C.F. The Genus Medicago and the origin of the Medicago sativa complex. In Alfalfa and Alfalfa Improvement; Ed., Hanson, A.A., Eds.; 1988; American Society of Agronomy,Inc.,Crop Science Society of America,Inc.,Soil Science Society of America,Inc.: Madison, WI, USA. [Google Scholar]
- Sprague, E.W. Cytological and fertility relationships of Medicago sativa, Medicago falcata, and Medicago gaetula. Agron. J. 1959, 51, 249–252. [Google Scholar] [CrossRef]
- Brummer, E.C.; Li, X.H. Inbreeding depression for fertility and biomass in advanced generations of inter- and intrasubspecific hybrids of tetraploid alfalfa. Crop Sci. 2009, 49, 13–19. [Google Scholar] [CrossRef]
- Hendry, G.W. Alfalfa in history. J. Am. Soc. Agron. 1923, 15, 171–176. [Google Scholar] [CrossRef]
- Teuber, L.R.; Skrdla, W.H.; Beard, D.F.; Hunt, O.J.; Murphy, R.P.; Bingham, E.T.; Barnes, D.K. Alfalfa Germplasm in the United States: Genetic Vulnerability,Use,Improvement,and Maintenance; 1977; USDA Tech. Bull. No. 1571; U.S. Gov. Print. Office: Washington, DC, USA. [Google Scholar]
- Brummer, E.C. Genomics research in alfalfa, Medicago sativa L. In Legume Crop Genomics; Eds., Brummer, E.C., Stalker, H.T., Wilson, R.F., Eds.; 2004; AOCS Press: Champaign, IL, USA. [Google Scholar]
- Brummer, E.C.; Doyle, J.J.; Sakiroglu, M. The population genetic structure of diploid Medicago sativa L. germplasm. In Sustainable Use of Genetic Diversity in Forage and Turf Breeding; Ed., Huyghe, C., Eds.; 2010; Springer: Berlin, Germany. [Google Scholar]
- Charles Brummer, E.; Doyle, J.J.; Sakiroglu, M. Inferring population structure and genetic diversity of broad range of wild diploid alfalfa (Medicago sativa L.) accessions using SSR markers. Theor. Appl. Genet. 2010, 121, 403–415. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J.J.; Maureira-Butler, I.J.; Brummer, E.C.; Havananda, T. Relationships among diploid members of the Medicago sativa (Fabaceae) species complex based on chloroplast and mitochondrial DNA sequences. Syst. Bot. 2010, 35, 140–150. [Google Scholar] [CrossRef]
- Doyle, J.J.; Brummer, E.C.; Havananda, T. Complex patterns of autopolyploid evolution in alfalfa and allies (Medicago sativa; Leguminosae). Am. J. Bot. 2011, 98, 1633–1646. [Google Scholar] [CrossRef] [PubMed]
- Ronfort, J.; Santoni, S.; Prosperi, J.M.; Muller, M.H. Inferences from mitochondrial DNA patterns on the domestication history of alfalfa (Medicago sativa). Mol. Ecol. 2003, 12, 2187–2199. [Google Scholar] [CrossRef] [PubMed]
- Ronfort, J.; Santoni, S.; Prosperi, J.M.; Poncet, C.; Muller, M.H. Domestication history in the Medicago sativa species complex: Inferences from nuclear sequence polymorphism. Mol. Ecol. 2006, 15, 1589–1602. [Google Scholar] [CrossRef] [PubMed]
- Jenczewski, E.; Prosperi, J.M.; Ronfort, J. Evidence for gene flow between wild and cultivatedMedicago sativa (Leguminosae) based on allozyme markers andquantitative traits. Am. J. Bot. 1999, 86, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Dzyubenko, N.I.; Kisha, T.J.; Greene, S.L. Conserving alfalfa wild relatives: is past introgression with Russian varieties evident today? Crop Sci. 2008, 48, 1853–1864. [Google Scholar] [CrossRef]
- Van Acker, R.C.; Gulden, R.H.; Barre, P.; Julier, B.; Bagavathiannan, M.V. Genetic diversity of feral alfalfa (Medicago sativa L.) populations occurring in Manitoba, Canada and comparison with alfalfa cultivars: An analysis using SSR markers and phenotypic traits. Euphytica 2010, 173, 419–432. [Google Scholar] [CrossRef]
- Julier, B.; Huyghe, C.; Huguet, T.; Barre, P.; Baudouin, P.; Ronfort, J.; Flajoulot, S. Genetic diversity among alfalfa (Medicago sativa) cultivars coming from a breeding program, using SSR markers. Theor. Appl. Genet. 2005, 111, 1420–1429. [Google Scholar] [CrossRef] [PubMed]
- Barnes, R.F.; Shenk, J.S.; R.R. Hill, J. Breeding for yield and quality. In Alfalfa and Alfalfa Improvement; Ed., Hanson, A.A., Eds.; 1988; American Society of Agronomy,Inc.,Crop Science Society of America,Inc.,Soil Science Society of America,Inc.: Madison, WI, USA. [Google Scholar]
- Brummer, E.C. Capturing heterosis in forage crop cultivar development. Crop Sci. 1999, 39, 943–954. [Google Scholar] [CrossRef]
- Brummer, E.C.; Undersander, D.J.; Sulc, R.M.; Rhodes, L.H.; Sheaffer, C.C.; Lamb, J.F.S. Five decades of alfalfa cultivar improvement: Impact on forage yield, persistence, and nutritive value. Crop Sci. 2006, 46, 902–909. [Google Scholar] [CrossRef]
- Bingham, E.T.; Holland, J.B. Genetic improvement for yield and fertility of alfalfa cultivars representing different eras of breeding. Crop Sci. 1994, 34, 953–957. [Google Scholar] [CrossRef]
- Noël, D.; Ghesquière, M.; Galbrun, C.; Deneufbourg, F.; Chosson, J.F.; Bourdon, P.; Béguier, V.; Bayle, B.; Baudouin, P.; Sampoux, J.P. Breeding perennial grasses for forage usage: An experimental assessment of trait changes in diploid perennial ryegrass (Lolium perenne L.) cultivars released in the last four decades. Field Crops Res. 2011, 123, 117–129. [Google Scholar] [CrossRef]
- Wilsie, C.P.; Busbice, T.H. Inbreeding depression and heterosis in autotetraploids with application to Medicago sativa L. Euphytica 1966, 15, 52–67. [Google Scholar]
- Bingham, E.T.; Dunbier, M.W. Maximizing heterozygosity in alfalfa: results using haploid-derived autotetraploid. Crop Sci. 1975, 15, 527–531. [Google Scholar]
- Kidwell, K.K.; Woodfield, D.R.; Groose, R.W.; Bingham, E.T. Complementary gene interaction in alfalfa is greater in autotetraploids than diploid. Crop Sci. 1994, 34, 823–829. [Google Scholar] [CrossRef]
- Bingham, E.T.; Woodfield, D.R. Improvement in two-allele autotetraploid populations of alfalfa explained by accumulation of favorable alleles. Crop Sci. 1995, 35, 988–994. [Google Scholar] [CrossRef]
- Brummer, E.C. Thoughts on breeding for increased forage yield. In 20th International Grassland Congress; Eds., Boland, T.M., Rogers, P.A.M., Lovett, D.K., Mannetje, L.T., Wilkins, R.J., O’Mara, F.P., Eds.; 2005; p. 63. Wangeningen Academic Publishers: Dublin, Ireland. [Google Scholar]
- Wilsie, C.P.; Sriwatanapongse, S. Intra- and intervariety crosses of Medicago sativa L. and Medicago falcata L. Crop Sci. 1968, 8, 465–466. [Google Scholar] [CrossRef]
- Rawlings, J.O.; Busbice, T.H. Combining ability in crosses within and between diverse groups of alfalfa introductions. Euphytica 1974, 23, 86–94. [Google Scholar] [CrossRef]
- Bingham, E.T.; Jones, J.S. breeding depression in alfalfa and cross-pollinated crops. In Plant Breeding Reviews; Ed., Janick, J., Eds.; 1995; John Wiley : New York, NY, USA. [Google Scholar]
- Brummer, E.C.; Riday, H. Forage yield heterosis in alfalfa. Crop Sci. 2002, 42, 716–723. [Google Scholar] [CrossRef]
- Brummer, E.C.; Riday, H. Heterosis of agronomic traits in alfalfa. Crop Sci. 2002, 42, 1081–1087. [Google Scholar] [CrossRef]
- Moore, K.J.; Brummer, E.C.; Riday, H. Heterosis of forage quality in alfalfa. Crop Sci. 2002, 42, 1088–1093. [Google Scholar] [CrossRef]
- Brummer, E.C.; Riday, H. Heterosis in a broad range of alfalfa germplasm. Crop Sci. 2005, 45, 8–17. [Google Scholar]
- Brummer, E.C.; Hansen, J.L.; Viands, D.R.; He, C.L.; Bauchan, G.R.; Campbell, I.A.; Luth, D.; Robins, J.G. Genetic mapping of biomass production in tetraploid alfalfa. Crop Sci. 2007, 47, 1–10. [Google Scholar] [CrossRef]
- Brummer, E.C.; Bauchan, G.R.; Robins, J.G. Genetic mapping forage yield, plant height, and regrowth at multiple harvests in tetraploid alfalfa (Medicago sativa L.). Crop Sci. 2007, 47, 11–18. [Google Scholar] [CrossRef]
- Ray, I.M.; Townsend, M.S.; Murray, L.W.; Segovia-Lerma, A. Population-based diallel analyses among nine historically recognized alfalfa germplasms. Theor. Appl. Genet. 2004, 109, 1568–1575. [Google Scholar] [CrossRef] [PubMed]
- Osborn, T.C.; Campos, H.; Ortega, F.; Maureira, I.J. Population structure and combining ability of diverse Medicago sativa germplasms. Theor. Appl. Genet. 2004, 109, 775–782. [Google Scholar] [CrossRef] [PubMed]
- Brummer, E.C.; Sakiroglu, M. Little heterosis between alfalfa populations derived from the Midwestern and Southwestern United States. Crop Sci. 2007, 47, 2364–2371. [Google Scholar] [CrossRef]
- Ray, I.M.; Murray, L.W.; Pierce, C.A.; Bhandari, H.S. Combining abilities and heterosis for forage yield among high-yielding accessions of the alfalfa core collection. Crop Sci. 2007, 47, 665–673. [Google Scholar] [CrossRef]
- Lowe, K.F.; Mackie, J.M.; Armour, D.; Pepper, P.M.; Irwin, J.A.G. Use of single cross hybrids to measure heterosis for yield in diverse lucerne genotypes growing in a subtropical environment. Aust. J. Agric. Res. 2008, 59, 999–1009. [Google Scholar] [CrossRef]
- Chandler, M.A.; Tracy, W.F. The historical and biological basis of the concept of heterotic patterns in corn belt dent maize. In Plant breeding: The Arnel R. Hallauer International Symposium; Eds., Lee, M., Lamkey, K.R., Eds.; 2006; Blackwell: Ames, IA, USA. [Google Scholar]
- Brummer, E.C.; Scotti, C. Creation of heterotic groups and hybrid varieties. In Sustainable use of Genetic Diversity in Forage and Turf Breeding; Ed., Huyghe, C., Eds.; 2010; Springer: Berlin, Germany. [Google Scholar]
- Yamada, T.; Fujii, H.; Ashikaga, K.; Tamaki, H.; Tanaka, T. Proposal for shift to reciprocal recurrent selections in “Clone and Strain Synthesis” timothy breeding using molecular marker diversity. Crop Sci. 2011, 51, 2589–2596. [Google Scholar] [CrossRef]
- Kolliker, R.; ShojaieFar, S.; Majidi, M.M.; Mirlohi, A.; Amini, F. Improved polycross breeding of tall fescue through marker-based parental selection. Plant Breeding 2011, 130, 701–707. [Google Scholar] [CrossRef]
- Woodman, W.L.; Lee, M.; Lamkey, K.R.; Labate, J.A. Molecular genetic diversity after reciprocal recurrent selection in BSSS and BSCB1 maize populations. Crop Sci. 1997, 37, 416–423. [Google Scholar] [CrossRef]
- Botstein, D.; Lander, E.S. Mapping mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics 1989, 121, 185–199. [Google Scholar] [PubMed]
- Hospital, F.; Dekkers, J.C.M. The use of molecular genetics in the improvement of agricultural populations. Nat. Rev. Genet. 2002, 3, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Mackill, D.J.; Collard, B.C. Marker-assisted selection: an approach for precision plant breeding in the twenty-first century. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2008, 363, 557–572. [Google Scholar] [CrossRef] [PubMed]
- Mian, M.A.R.; Zwonitzer, J.C.; Chekhovskiy, K.; May, G.D.; Wang, L.; Sledge, M.K.; Eujayl, I. Medicago truncatula EST-SSRs reveal cross-species genetic markers for Medicago spp. Theor. Appl. Genet. 2004, 108, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Brummer, E.C.; Wei, Y.; Wang, X.; Li, X. Prevalence of segregation distortion in diploid alfalfa and its implications for genetics and breeding applications. Theor. Appl. Genet. 2011, 123, 667–679. [Google Scholar] [CrossRef] [PubMed]
- Huyghe, C.; Huguet, T.; Santoni, S.; Cardinet, G.; Barre, P.; Flajoulot, S.; Julier, B. Construction of two genetic linkage maps in cultivated tetraploid alfalfa (Medicago sativa) using microsatellite and AFLP markers. BMC Plant Biol. 2003, 3, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.; Ray, I.M.; Sledge, M.K. An expressed sequence tag SSR map of tetraploid alfalfa (Medicago sativa L.). Theor. Appl. Genet. 2005, 111, 980–992. [Google Scholar] [CrossRef] [PubMed]
- Bouton, J.H.; Sledge, M.K.; Zhang, Y. Genome mapping of white clover (Trifolium repens L.) and comparative analysis within the Trifolieae using cross-species SSR markers. Theor. Appl. Genet. 2007, 114, 1367–1378. [Google Scholar] [CrossRef] [PubMed]
- Sledge, M.K.; Olsen, K.M.; Bouton, J.H.; Narasimhamoorthy, B. Quantitative trait loci and candidate gene mapping of aluminum tolerance in diploid alfalfa. Theor. Appl. Genet. 2007, 114, 901–913. [Google Scholar] [CrossRef] [PubMed]
- Rafalski, A. Applications of single nucleotide polymorphisms in crop genetics. Curr. Opin. Plant Biol. 2002, 5, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Monteros, M.J.; Udvardi, M.K.; Zhao, P.X.; Town, C.D.; Cheung, F.; Torres-Jerez, I.; Kang, Y.; Han, Y. Genome-wide SNP discovery in tetraploid alfalfa using 454 sequencing and high resolution melting analysis. BMC Genomics 2011, 12, 350. [Google Scholar] [CrossRef]
- Monteros, M.J.; Khu, D.-M.; Han, Y. High-resolution melting analysis for SNP genotyping and mapping in tetraploid alfalfa (Medicago sativa L.). Mol. Breeding 2012, 29, 489–501. [Google Scholar] [CrossRef]
- Kochert, G.; Bouton, J.H.; Brummer, E.C. Development of an RFLP map in diploid alfalfa. Theor. Appl. Genet. 1993, 86, 329–332. [Google Scholar]
- Mccoy, T.J.; Osborn, T.C.; Knapp, S.J.; Kidwell, K.K.; Echt, C.S. Linkage mapping in diploid alfalfa (Medicago-Sativa). Genome 1994, 37, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Okresz, L.; Kalo, P.; Kalman, K.; Csanadi, G.; Kiss, G.B. Construction of a basic genetic-map for alfalfa using RFLP, RAPD, isozyme and morphological markers. Mol. Gen. Genet. 1993, 238, 129–137. [Google Scholar] [PubMed]
- Kiss, G.B.; Csanádi, G.; Zimányi, L.; Endre, G.; Kalo, P. Construction of an improved linkage map of diploid alfalfa (Medicago sativa). Theor. Appl. Genet. 2000, 100, 641–657. [Google Scholar] [CrossRef]
- Osborn, T.C.; Veronesi, F.; Tavoletti, S. RFLP linkage map of an alfalfa meiotic mutant based on an F-1 population. J. Hered. 1996, 87, 167–170. [Google Scholar] [CrossRef]
- Bingham, E.T.; McCoy, T.J. Cytology and cytogenetics of alfalfa. In Alfalfa and Alfalfa Improvement; Ed., Hanson, A.A., Eds.; 1988; American Society of Agronomy,Inc.,Crop Science Society of America,Inc.,Soil Science Society of America,Inc.: Madison, WI, USA. [Google Scholar]
- Luo, Z.; Bradshaw, J.E.; Milne, I.; Hackett, C.A. TetraploidMap for Windows: Linkage map construction and QTL mapping in autotetraploid species. J. Hered. 2007, 98, 727–729. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.A.; Aitken, K.S.; Ellwood, S.E.; Phan, H.T.; Armour, D.J.; Mackie, J.M.; Musial, J.M. Identification of QTL for resistance and susceptibility to Stagonospora meliloti in autotetraploid lucerne. Theor. Appl. Genet. 2007, 114, 1427–1435. [Google Scholar] [CrossRef] [PubMed]
- Osborn, T.C.; Duke, S.H.; Brouwer, D.J. Mapping genetic factors associated with winter hardiness, fail growth, and freezing injury in autotetraploid alfalfa. Crop Sci. 2000, 40, 1387–1396. [Google Scholar] [CrossRef]
- Brummer, E.; Luth, D.; Moore, K.; Scott, P.; Alarcon Zuniga, B. Quantitative trait locus mapping of winter hardiness metabolites in autotetraploid alfalfa (M. sativa). In Molecular Breeding of Forage and Turf; Eds., Barker, R.E., Sledge, M., Mian, R., Wang, Z.Y., Hopkins, A., Eds.; 2004; Kluwer: Dordrecht, the Netherlands. [Google Scholar]
- Brummer, E.C.; Viands, D.R.; Hansen, J.L.; Robins, J.G. Genetic mapping of persistence in tetraploid alfalfa. Crop Sci. 2008, 48, 1780–1786. [Google Scholar] [CrossRef]
- Brummer, E.C.; Robins, J.G. QTL underlying self-fertility in tetraploid alfalfa. Crop Sci. 2010, 50, 143–149. [Google Scholar] [CrossRef]
- Irwin, J.A.; Aitken, K.S.; Ellwood, S.E.; Phan, H.T.; Armour, D.J.; Musial, J.M.; Mackie, J.M. Identification of QTL for reaction to three races of Colletotrichum trifolii and further analysis of inheritance of resistance in autotetraploid lucerne. Theor. Appl. Genet. 2007, 114, 1417–1426. [Google Scholar] [CrossRef] [PubMed]
- Monteros, M.J.; Bouton, J.H.; Brummer, E.C.; Reyno, R.; Khu, D.M. QTL mapping of aluminum tolerance in tetraploid alfalfa. In Sustainable Use of Genetic Diversity in Forage and Turf Breeding; Ed., Huyghe, C., Eds.; 2010; Springer: Berlin, Germany. [Google Scholar]
- Lelievre, F.; Huguet, T.; Gibelin, C.; Bernard, K.; Julier, B. QTL for water use efficiency in alfalfa. In Sustainable Use of Genetic Diversity in Forage and Turf Breeding; Ed., Huyghe, C., Eds.; 2010; Springer: Berlin, Germany. [Google Scholar]
- Beavis, W.D. QTL analyses: powver, precision, and accuracy. In Molecular Dissection of Complex Traits; Ed., Paterson, A.H., Eds.; 1998; pp. 145–162. CGC Press: New York, NY, USA. [Google Scholar]
- Xu, S. Theoretical basis of the Beavis effect. Genetics 2003, 165, 2259–2268. [Google Scholar] [PubMed]
- Kiss, G.B.; Kereszt, A.; Szakal, B.; Mihacea, S.; Kiss, P.; Kevei, Z.; Kalo, P.; Endre, G. Genetic mapping of the non-nodulation phenotype of the mutant MN-1008 in tetraploid alfalfa (Medicago sativa). Mol. Genet. Genomics 2002, 266, 1012–1019. [Google Scholar] [CrossRef] [PubMed]
- Kiss, G.B.; Kalo, P.; Mihacea, S.; Kevei, Z.; Kereszt, A.; Endre, G. A receptor kinase gene regulating symbiotic nodule development. Nature 2002, 417, 962–966. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Roe, B.A.; Lin, S.; Deshpande, S.; Gao, J.; Xu, C.; Gao, M.; Yang, S. Alfalfa benefits from Medicago truncatula: the RCT1 gene from M. truncatula confers broad-spectrum resistance to anthracnose in alfalfa. Proc. Natl. Acad. Sci. USA 2008, 105, 12164–12169. [Google Scholar] [CrossRef] [PubMed]
- Huguet, T.; Delalande, M.; Deniot, G.; Hervé, M.; Le Goff, I.; Lecointe, R.; Lesné, A.; Hamon, C.; Prospéri, J.M.; Pilet-Nayel, M.L. AER1, a major gene conferring resistance to Aphanomyces euteiches in Medicago truncatula. Phytopathology 2009, 99, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Baranger, A.; Pilet-Nayel, M.L.; Cazaux, M.; Bottin, A.; Mathé, C.; Jaulneau, V.; Chardon, F.; Ameline-Torregrosa, C.; Jauneau, A.; Djébali, N. Partial resistance of Medicago truncatula to Aphanomyces euteiches is associated with protection of the root stele and is controlled by a major QTL rich in proteasome-related genes. Mol. Plant Microbe In. 2009, 22, 1043–1055. [Google Scholar] [CrossRef]
- Ellwood, S.; Oliver, R.; Lichtenzveig, J.; Kamphuis, L. Two alternative recessive quantitative trait loci influence resistance to spring black stem and leaf spot in Medicago truncatula. BMC Plant Biol. 2008, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Munier-Jolain, N.; Salon, C.; Prosperi, J.M.; Ben, C.; Huguet, T.; Aubert, G.; Burstin, J.; Moreau, D. Using a physiological framework for improving the detection of quantitative trait loci related to nitrogen nutrition in Medicago truncatula. Theor. Appl. Genet. 2012, 124, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Julier, B.; Huyghe, C.; Barre, P.; Huguet, T.; Pierre, J.B. Detection of QTLs for flowering date in three mapping populations of the model legume species Medicago truncatula. Theor. Appl. Genet. 2008, 117, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Huyghe, C.; Barre, P.; Prosperi, J.M.; Pierre, J.B.; Ayadi, R.; Chardon, F.; Huguet, T.; Julier, B. Identification of quantitative trait loci influencing aerial morphogenesis in the model legume Medicago truncatula. Theor. Appl. Genet. 2007, 114, 1391–1406. [Google Scholar] [CrossRef] [PubMed]
- Kulikova, O.; Seres, A.; Penmetsa, R.V.; Kalo, P.; Mun, J.H.; Lim, H.; Limpens, E.; Uhm, T.; Kim, D.; Choi, H.K.; et al. A sequence-based genetic map of Medicago truncatula and comparison of marker colinearity with M. sativa. Genetics 2004, 166, 1463–1502. [Google Scholar] [CrossRef] [PubMed]
- Kiss, G.B.; Ellis, T.H.; Endre, G.; Kereszt, A.; Kevei, Z.; Jakab, J.; Taylor, S.A.; Seres, A.; Kalo, P. Comparative mapping between Medicago sativa and Pisum sativum. Mol. Genet. Genomics 2004, 272, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Schoof, H.; Gouzy, J.; Mayer, K.F.X.; Benedito, V.A.; Udvardi, M.K.; Cannon, S.B.; Geurts, R.; Oldroyd, G.E.D.; Debellé, F.; Young, N.D. The Medicago genome provides insight into the evolution of rhizobial symbioses. Nature 2011, 480, 520–524. [Google Scholar] [PubMed]
- Walsh, B.; Jannink, J.L. Association mapping in plant populations. In Quantitative Genetics,Genomics and Plant Breeding; Ed., Kang, M.S., Eds.; 2002; CAB International: New York, NY, USA. [Google Scholar]
- Weigel, D.; Nordborg, M. Next-generation genetics in plants. Nature 2008, 456, 720–723. [Google Scholar] [CrossRef] [PubMed]
- Brummer, E.C.; Moore, K.J.; Doyle, J.J.; Story, A.; Sherman-Broyles, S.; Sakiroglu, M. Paterns of linkage diequilibium and association mapping in diploid alfalfa (M. sativa L.). Theor. Appl. Genet. 2012. [Google Scholar]
- Julier, B.; Santoni, S.; Barre, P.; Herrmann, D. Association of a CONSTANS-LIKE gene to flowering and height in autotetraploid alfalfa. Theor. Appl. Genet. 2010, 121, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Brummer, E.C.; Acharya, A.; Hansen, J.L.; Viands, D.R.; Michaud, R.; Moore, K.J.; Wei, Y.; Li, X. Association mapping of biomass yield and stem composition in a tetraploid alfalfa breeding population. Plant Genome 2011, 4, 24–35. [Google Scholar] [CrossRef]
- Hamilton, N.R.S.; Chorlton, K.H.; Thomas, I.D.; Sanderson, R.; SkØt, K.P.; Heywood, S.; Armstead, I.; Humphreys, M.O.; SkØt, L. An association mapping approach to identify flowering time genes in natural populations of Lolium perenne (L.). Mol. Breeding 2005, 15, 233–245. [Google Scholar] [CrossRef]
- Barre, P.; Julier, B.; Huyghe, C.; Auzanneau, J. Linkage disequilibrium in synthetic varieties of perennial ryegrass. Theor. Appl. Genet. 2007, 115, 837–847. [Google Scholar] [CrossRef] [PubMed]
- Julier, B. A program to test linkage disequilibrium between loci in autotetraploid species. Mol. Ecol. Resour. 2009, 9, 746–748. [Google Scholar] [CrossRef] [PubMed]
- Thomas, I.D.; Armstead, I.P.; Sanderson, R.; Gallagher, J.; Thorogood, D.; Humphreys, M.O.; Humphreys, J.; SkØt, L. Association of candidate genes with flowering time and water-soluble carbohydrate content in Lolium perenne (L.). Genetics 2007, 177, 535–547. [Google Scholar] [CrossRef] [PubMed]
- Brummer, E.C.; Nettleton, D.; Wei, Y.; Li, X. Comparative gene expression profiles between heterotic and non-heterotic hybrids of tetraploid Medicago sativa. BMC Plant Biol. 2009, 9, 107. [Google Scholar] [CrossRef] [PubMed]
- Snyder, M.; Gerstein, M.; Wang, Z. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Gilad, Y.; Stephens, M.; Mane, S.M.; Mason, C.E.; Marioni, J.C. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008, 18, 1509–1517. [Google Scholar] [CrossRef] [PubMed]
- Gronwald, J.W.; Vance, C.P.; Jung, H.J.; Lamb, J.F.; Xu, W.W.; Cheung, F.; Tu, Z.J.; Yang, S.S. Using RNA-Seq for gene identification, polymorphism detection and transcript profiling in two alfalfa genotypes with divergent cell wall composition in stems. BMC genomics 2011, 12, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Widmer, F.; Boller, B.; Kölliker, R. Marker assisted polycross breeding to increase diversity and yield in perennial ryegrass (Lolium perenne L.). Euphytica 2005, 146, 55–65. [Google Scholar] [CrossRef]
- Osborn, T.C.; Moutray, J.; Brummer, J.E.; Crump, P.M.; Yandell, B.S.; Hartweck, L.M.; Kidwell, K.K. Forage yields of alfalfa populations derived from parents selected on the basis of molecular marker diversity. Crop Sci. 1999, 39, 223–227. [Google Scholar] [CrossRef]
- Riday, H. Paternity Testing: A non-linkage based marker-assisted selection scheme for outbred forage species. Crop Sci. 2011, 51, 631–641. [Google Scholar] [CrossRef]
- Brummer, E.C.; Casler, M.D. Theoretical expected genetic gains for among-and-within-family selection methods in perennial forage crops. Crop Sci. 2008, 48, 890–902. [Google Scholar] [CrossRef]
- Elgin, J.H.; Elden, T.C. Recurrent seedling and individual-plant selection for potato leafhopper (Homoptera: Cicadellidae) resistance in alfalfa. J. Econ. Entomol. 1987, 80, 690–695. [Google Scholar]
- Elgin, J.H., Jr.; Elden, T.C. Registration of B16-PLH alfalfa germplasm resistant to the potato leafhopper. Crop Sci. 1989, 29, 1577–1578. [Google Scholar]
- Stuteville, D.L.; Horber, E.K.; Sorensen, E.L. Registration of KS108GH5 glandular-haired alfalfa germplasm with multiple pest resistance. Crop Sci. 1985, 25, 1132. [Google Scholar]
- Jannink, J.L. Dynamics of long-term genomic selection. Genet. Sel. Evol. 2010, 42, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Jannink, J.L.; Sorrells, M.E.; Heffner, E.L. Genomic selection for crop improvement. Crop Sci. 2009, 49, 1–12. [Google Scholar] [CrossRef]
- Iwata, H.; Lorenz, A.J.; Jannink, J.L. Genomic selection in plant breeding: from theory to practice. Brief. Funct. Genomics 2010, 9, 166–177. [Google Scholar] [CrossRef] [PubMed]
- Jannink, J.L.; Sorrells, M.E.; Smith, K.P.; Iwata, H.; Hayashi, T.; Heffner, E.L.; Asoro, F.G.; Chao, S.; Lorenz, A.J. Genomic selection in plant breeding: Knowledge and prospects. Adv. Agron. 2011, 110, 77–123. [Google Scholar]
- Sorrels, M.E.; Jannink, J.L.; Lorenz, A.J.; Heffner, E.L. Plant breeding with genomic selection: gain per unit time and cost. Crop Sci. 2010, 50, 1681–1690. [Google Scholar] [CrossRef]
- Goddard, M.E.; Chamberlain, A.J.; Bowman, P.J.; Hayes, B.J. Invited review: Genomic selection in dairy cattle: progress and challenges. J. Dairy Sci. 2009, 92, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Schenkel, F.S.; Taylor, J.F.; Schnabel, R.D.; Sonstegard, T.S.; Wiggans, G.R.; Van Tassell, C.P.; VanRaden, P.M. Invited review: reliability of genomic predictions for north American Holstein bulls. J. Dairy Sci. 2009, 92, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Sorrells, M.E.; Heffner, E.L.; Rutkoski, J.E. Genomic selection for durable stem rust resistance in wheat. Euphytica 2011, 179, 161–173. [Google Scholar] [CrossRef]
- Mitchell, S.E.; Buckler, E.S.; Kawamoto, K.; Poland, J.A.; Sun, Q.; Glaubitz, J.C.; Elshire, R.J. A Robust, Simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One 2011, 6, e1–e10. [Google Scholar]
- Zhang, Z.; Lu, T.; Zhu, C.; Li, C.; Zhao, Y.; Feng, Q.; Zhao, Q.; Sang, T.; Wei, X.; Huang, X.; et al. Genome-wide association studies of 14 agronomic traits in rice landraces. Nat. Genet. 2010, 42, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, R. Molecular markers and selection for complex traits in plants: Learning from the last 20 years. Crop Sci. 2008, 48, 1649–1664. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Li, X.; Brummer, E.C. Applied Genetics and Genomics in Alfalfa Breeding. Agronomy 2012, 2, 40-61. https://doi.org/10.3390/agronomy2010040
Li X, Brummer EC. Applied Genetics and Genomics in Alfalfa Breeding. Agronomy. 2012; 2(1):40-61. https://doi.org/10.3390/agronomy2010040
Chicago/Turabian StyleLi, Xuehui, and E. Charles Brummer. 2012. "Applied Genetics and Genomics in Alfalfa Breeding" Agronomy 2, no. 1: 40-61. https://doi.org/10.3390/agronomy2010040
APA StyleLi, X., & Brummer, E. C. (2012). Applied Genetics and Genomics in Alfalfa Breeding. Agronomy, 2(1), 40-61. https://doi.org/10.3390/agronomy2010040