
High-Density Genetic Map Construction and QTL Detection for Cotyledon Color in Faba Bean Based on Double Digest Restriction-Site Associated DNA Sequencing (ddRAD-Seq)
Abstract
:1. Introduction
2. Materials and Methods
2.1. Construction of Populations and Phenotypic Data Analysis
2.2. The ddRAD-Seq Library Construction and High-Throughput Sequencing
2.3. Development of Polymorphic ddRAD Markers and Construction of High-Density Genetic Map
2.4. QTL Detection for Cotyledon Color and Candidate Gene Annotation
3. Results
3.1. Analysis of the Heredity of Green-Cotyledon Trait in Faba Bean
3.2. Analysis of Sequencing Data
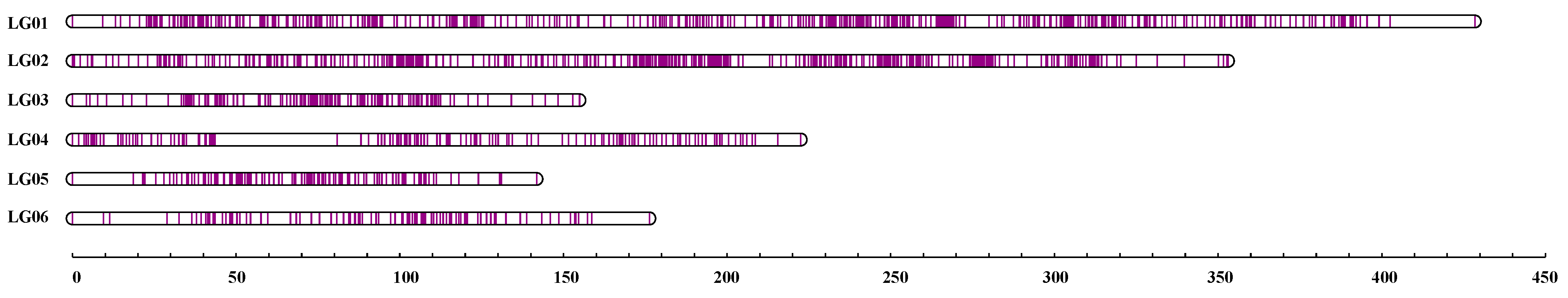
3.3. Development of SNP Markers and Construction of a High-Density Genetic Map
3.4. QTL Mapping for Cotyledon Color of Faba Bean
3.5. Candidate Regions and Genes for Cotyledon Color
4. Discussion
4.1. Construction of a High-Density Linkage Map for Faba Bean
4.2. Identification of QTL for Green-Cotyledon Trait in Faba Bean
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Luo, Y.W.; Li, J. Soluble sugars and myo-inositol phosphates during germination and seedling growth of green and white faba bean (Vicia faba L.). Legume Res. 2017, 40, 47–54. [Google Scholar] [CrossRef]
- Landry, E.J.; Fuchs, S.J.; Hu, J.G. Carbohydrate composition of mature and immature faba bean seeds. J. Food Compos. Anal. 2016, 50, 55–60. [Google Scholar] [CrossRef]
- Chen, X.Z. Variety Annals of Food Legumes in China, 2nd ed.; Science Press: Beijing, China, 2023; pp. 294–353. ISBN 9787030740120. [Google Scholar]
- Ma, J.R.; Jia, X.L.; Guo, Y.P.; Li, L.; Zhang, L.S. A new green cotyledon variety of spring faba bean—‘Lincan No.11’. China Seed Ind. 2016, 2, 71. (In Chinese) [Google Scholar]
- Guo, X.L.; Liu, Y.J.; Duo, X.L. A green cotyledon and suitable for processing faba bean variety—‘Qingcan No.19’. China Seed Ind. 2022, 1, 102–103. (In Chinese) [Google Scholar]
- Sha, W.C. Study on SSR Markers of Cotyledon Colour Traits in Faba Bean. Master’s Thesis, Qinghai University, Xining, China, 2017. (In Chinese). [Google Scholar]
- Ronning, C.M.; Bouwkamp, J.C.; Solomos, T. Observations on the senescence of a mutant non-yellowing genotype of Phaseolus vulgaris L. J. Exp. Bot. 1991, 42, 235–241. [Google Scholar] [CrossRef]
- Thomas, H.; Schellenberg, M.; Vicentini, F.; Matile, P. Gregor Mendel’s green and yellow pea seeds. Bot. Acta 1996, 109, 3–4. [Google Scholar] [CrossRef]
- Zhou, C.; Han, L.; Pislariu, C.; Nakashima, J.; Fu, C.; Jiang, Q.; Quan, L.; Blancaflor, E.B.; Tang, Y.; Bouton, J.H.; et al. From model to crop: Functional analysis of a STAY-GREEN gene in the model legume Medicago truncatula and effective use of the gene for alfalfa improvement. Plant Physiol. 2011, 157, 1483–1496. [Google Scholar] [CrossRef]
- Guiamét, J.J.; Schwartz, E.; Pichersky, E.; Noodén, L.D. Characterization of cytoplasmic and nuclear mutations affecting chlorophyll and chlorophyll-binding proteins during senescence in soybean. Plant Physiol. 1991, 96, 227–231. [Google Scholar] [CrossRef]
- Guiamet, J.J.; Giannibelli, M.C. Nuclear and cytoplasmic “stay-green” mutations of soybean alter the loss of leaf soluble proteins during senescence. Physiol. Plant. 1996, 96, 655–661. [Google Scholar] [CrossRef]
- Kohzuma, K.; Sato, Y.; Ito, H.; Okuzaki, A.; Watanabe, M.; Kobayashi, H.; Nakano, M.; Yamatani, H.; Masuda, Y.; Nagashima, Y.; et al. The non-Mendelian green cotyledon gene in soybean encodes a small subunit of photosystem II. Plant Physiol. 2017, 173, 2138–2147. [Google Scholar] [CrossRef]
- Zhang, H.S.; Cheng, Y.X.; Wang, D.Q.; Xiang, S.H.; Gao, M.; Wang, X.F.; Niu, J.G. Correlation analysis of variation in chlorophyll content and yield in pod period of soybean. Soybean Sci. 2011, 4, 275–279. (In Chinese) [Google Scholar]
- Hörtensteiner, S. Update on the biochemistry of chlorophyll breakdown. Plant Mol. Biol. 2012, 82, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.Y.; Liu, Z.X.; Jin, W.W. Biosynthesis, catabolism and related signal regulations of plant chlorophyll. Hereditas 2009, 31, 698–704. [Google Scholar] [CrossRef]
- Sato, Y.S.; Morita, R.; Katsuma, S.; Nishimura, M.; Tanaka, A.; Kusaba, M. Two short-chain dehydrogenase/reductases, NON-YELLOW COLORING 1 and NYC1-LIKE, are required for chlorophyll b and light-harvesting complex II degradation during senescence in rice. Plant J. 2009, 57, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Morita, R.; Nishimura, M.; Yamaguchi, H.; Kusaba, M. Mendel’s green cotyledon gene encodes a positive regulator of the chlorophyll-degrading pathway. Proc. Natl. Acad. Sci. USA 2007, 104, 14169–14174. [Google Scholar] [CrossRef] [PubMed]
- Yadava, Y.K.; Chaudhary, P.; Yadav, S.; Rizvi, A.H.; Kumar, T.; Srivastava, R.; Soren, K.R.; Bharadwaj, C.; Srinivasan, R.; Singh, N.K.; et al. Genetic mapping of quantitative trait loci associated with drought tolerance in chickpea (Cicer arietinum L.). Sci. Rep. 2023, 13, 17623. [Google Scholar] [CrossRef] [PubMed]
- Li, J.S.; Gong, D.; Wang, S.H.; Chen, H.L.; Chen, X.Z.; Xiong, T.; Wang, L.X. Construction of SNP high-density genetic map and QTL analysis of agronomic traits in cowpea (Vigna unguiculata (L.) Walp.). Acta Agron. Sin. 2022, 48, 2475–2482. (In Chinese) [Google Scholar]
- Chen, T.X.; Hu, L.L.; Wang, S.H.; Wang, L.X.; Cheng, X.Z.; Chen, H.L. Construction of high-density genetic map and identification of a bruchid resistance locus in mung bean (Vigna radiata L.). Front. Genet. 2022, 13, 903267. [Google Scholar] [CrossRef] [PubMed]
- Li, X.M.; Tang, Y.S.; Wang, L.F.; Chang, Y.J.; Wu, J.; Wang, S.M. QTL mapping and identification of genes associated with the resistance to Acanthoscelides obtectus in cultivated common bean using a high-density genetic linkage map. BMC Plant Biol. 2022, 22, 260–274. [Google Scholar] [CrossRef]
- Johnston, J.S.; Bennett, M.D.; Rayburn, A.L.; Galbraith, D.W.; Price, H.J. Reference standards for determination of DNA content of plant nuclei. Am. J. Bot. 1999, 86, 609–613. [Google Scholar] [CrossRef] [PubMed]
- van de Ven, W.T.G.; Waugh, R.; Duncan, N.; Ramsay, G.; Dow, N.; Powell, W. Development of a genetic linkage map in Vicia faba using molecular and biochemical techniques. Asp. Appl. Biol. 1991, 27, 49–54. [Google Scholar]
- Satovic, Z.; Avila, C.M.; Cruz-Izquierdo, S.; Díaz-Ruíz, R.; García-Ruíz, G.M.; Palomino, C.; Gutiérrez, N.; Vitale, S.; Ocaña-Moral, S.; Gutiérrez, M.V.; et al. A reference consensus genetic map for molecular markers and economically important traits in faba bean (Vicia faba L.). BMC Genom. 2013, 14, 932–946. [Google Scholar] [CrossRef] [PubMed]
- Webb, A.; Cottage, A.; Wood, T.A.; Khamassi, K.; Hobbs, D.H.; Gostkiewicz, K.; White, M.P.; Khazaei, H.; Ali, M.; Street, D.; et al. A SNP-based consensus genetic map for synteny-based trait targeting in faba bean (Vicia faba L.). Plant Biotechnol. J. 2015, 14, 177–185. [Google Scholar] [CrossRef]
- Hou, W.W.; Zhang, X.J.; Yan, Q.B.; Li, P.; Sha, W.C.; Tian, Y.Y.; Liu, Y.J. Linkage map of a gene controlling zero tannins (zt-1) in faba bean (Vicia faba L.) with SSR and ISSR markers. Agronomy 2018, 8, 80. [Google Scholar] [CrossRef]
- El-Rodeny, W.M.; Kimura, M.; Hirakawa, H.; Sabah, A.; Shirasawa, K.; Sato, S.; Tabata, S.; Sasamoto, S.; Watanabe, A.; Kawashima, K.; et al. Development of EST-SSR markers and construction of a linkage map in faba bean (Vicia faba). Breed. Sci. 2014, 64, 252–263. [Google Scholar] [CrossRef]
- Zhao, N.; Xue, D.; Miao, Y.M.; Wang, Y.Q.; Zhou, E.Q.; Zhou, Y.; Yao, M.N.; Gu, C.Y.; Wang, K.H.; Li, B.; et al. Construction of a high-density genetic map for faba bean (Vicia faba L.) and quantitative trait loci mapping of seed-related traits. Front. Plant Sci. 2023, 14, 1201103–1201116. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.Q.; Liao, L.; Peng, Q.; Fang, T.; Zhou, H.; Korban, S.S.; Han, Y.P. Reduced representation genome sequencing reveals patterns of genetic diversity and selection in apple. J. Integr. Plant Biol. 2017, 59, 190–204. [Google Scholar] [CrossRef] [PubMed]
- Peterson, B.K.; Weber, J.N.; Kay, E.H.; Fisher, H.S.; Hoekstra, H.E. Double digest RADseq: An inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS ONE 2012, 7, e37135. [Google Scholar] [CrossRef]
- Yol, E.; Basak, M.; Kızıl, S.; Lucas, S.J.; Uzun, B. A high-density SNP genetic map construction using ddRAD-Seq and mapping of capsule shattering trait in sesame. Front. Plant Sci. 2021, 12, 679659–679671. [Google Scholar] [CrossRef]
- Wang, G.P.; Lai, H.; Bi, S.; Guo, D.L.; Zhao, X.P.; Chen, X.L.; Liu, S.; Liu, X.G.; Su, Y.Q.; Yi, H.D.; et al. ddRAD-Seq reveals evolutionary insights into population differentiation and the cryptic phylogeography of Hyporhamphus intermedius in Mainland China. Ecol. Evol. 2022, 12, e9053. [Google Scholar] [CrossRef]
- Zhu, H.; Liu, J.; Gao, M.R.; Yue, C.L.; Li, H.P. Population genetic assessment of Viburnum japonicum in China using ddRAD-seq. Front. Genet. 2023, 14, 1150437–1150444. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, X.M.; Yan, M.; Wang, P.F.; Wang, H.; Xin, Q.; Yang, L.Y.; Hong, D.F.; Yang, G.S. Fine mapping and candidate gene analysis of a seed glucosinolate content QTL, qGSL-C2, in rapeseed (Brassica napus L.). Theor. Appl. Genet. 2019, 133, 479–490. [Google Scholar] [CrossRef]
- Luo, H.Y.; Guo, J.B.; Yu, B.; Chen, W.G.; Zhang, H.; Zhou, X.J.; Chen, Y.N.; Huang, L.; Liu, N.; Ren, X.P.; et al. Construction of ddRADseq-based high-density genetic map and identification of quantitative trait loci for trans-resveratrol content in peanut seeds. Front. Plant Sci. 2021, 12, 644402–644412. [Google Scholar] [CrossRef] [PubMed]
- Armstead, I.; Donnison, I.; Aubry, S.; Harper, J.; Hortensteiner, S.; James, C.; Mani, J.; Moffet, M.; Ougham, H.; Roberts, L.; et al. Cross-species identification of Mendel’s I locus. Science 2007, 315, 73. [Google Scholar] [CrossRef]
- Jayakodi, M.; Golicz, A.A.; Kreplak, J.; Fechete, L.I.; Angra, D.; Bednář, P.; Bornhofen, E.; Zhang, H.L.; Boussageon, R.; Kaur, S.; et al. The giant diploid faba genome unlocks variation in a global protein crop. Nature 2023, 615, 652–659. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.Y.; Ma, C.X.; Hong, W.G.; Huang, L.; Liu, M.; Liu, H.; Zeng, H.P.; Deng, D.J.; Xin, H.G.; Song, J.; et al. Construction and analysis of high-density linkage map using high-throughput sequencing data. PLoS ONE 2014, 9, e98855. [Google Scholar] [CrossRef]
- Jansen, J.; de Jong, A.G.; van Ooijen, J.W. Constructing dense genetic linkage maps. Theor. Appl. Genet. 2001, 102, 1113–1122. [Google Scholar] [CrossRef]
- van Ooijen, J.W. Multipoint maximum likelihood mapping in a full-sib family of an outbreeding species. Genet. Res. 2011, 93, 343–349. [Google Scholar] [CrossRef] [PubMed]
- van Os, H.; Stam, P.; Visser, R.G.F.; van Eck, H.J. SMOOTH: A statistical method for successful removal of genotyping errors from high-density genetic linkage data. Theor. Appl. Genet. 2005, 112, 187–194. [Google Scholar] [CrossRef]
- Huang, X.H.; Zhao, Y.; Wei, X.H.; Li, C.Y.; Wang, A.H.; Zhao, Q.; Li, W.J.; Guo, Y.L.; Deng, L.W.; Zhu, C.R.; et al. Genome-wide association study of flowering time and grain yield traits in a worldwide collection of rice germplasm. Nat. Genet. 2012, 44, 32–39. [Google Scholar] [CrossRef]
- Kosambi, D.D. The estimation of map distances from recombination values. Ann. Eugen. 1943, 12, 172–175. [Google Scholar] [CrossRef]
- Schwacke, R.; Ponce-Soto, G.Y.; Krause, K.; Bolger, A.M.; Arsova, B.; Hallab, A.; Gruden, K.; Stitt, M.; Bolger, M.E.; Usadel, B. MapMan4: A refined protein classification and annotation framework applicable to multi-omics data analysis. Mol. Plant 2019, 12, 879–892. [Google Scholar] [CrossRef] [PubMed]
- Bolger, M.; Schwacke, R.; Usadel, B. MapMan visualization of RNA-seq data using Mercator4 functional annotations. In Solanum Tuberosum: Methods in Molecular Biology; Humana: New York, NY, USA, 2021; pp. 195–212. [Google Scholar] [CrossRef]
- Chen, J.B.; Zhou, H.M.; Yuan, X.X.; He, Y.M.; Yan, Q.; Lin, Y.; Wu, R.R.; Liu, J.; Xue, C.C.; Chen, X. Homolog of pea SGR controls Stay-Green in faba bean (Vicia faba L.). Genes 2023, 14, 1030. [Google Scholar] [CrossRef]
- Torres, A.M.; Weeden, N.F.; Martín, A. Linkage among isozyme, RFLP and RAPD markers in Vicia faba. Theor. Appl. Genet. 1993, 85, 937–945. [Google Scholar] [CrossRef] [PubMed]
- Vaz Patto, M.C.V.; Torres, A.M.; Koblizkova, A.; Macas, J.; Cubero, J.I. Development of a genetic composite map of Vicia faba using F2 populations derived from trisomic plants. Theor. Appl. Genet. 1999, 98, 736–743. [Google Scholar] [CrossRef]
- Román, B.; Torres, A.M.; Rubiales, D.; Cubero, J.I.; Satovic, Z. Mapping of quantitative trait loci controlling broomrape (Orobanche crenata Forsk.) resistance in faba bean (Vicia faba L.). Genome 2002, 45, 1057–1063. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Bao, S.Y.; Yang, T.; Hu, J.G.; Guan, J.P.; He, Y.H.; Wang, X.J.; Wan, Y.L.; Sun, X.L.; Jiang, J.Y.; et al. Genetic linkage map of Chinese native variety faba bean (Vicia faba L.) based on simple sequence repeat markers. Plant Breed. 2013, 132, 397–400. [Google Scholar] [CrossRef]
- Yang, T.; Jiang, J.Y.; Zhang, H.Y.; Liu, R.; Strelkov, S.; Hwang, S.F.; Chang, K.F.; Yang, F.; Miao, Y.M.; He, Y.H.; et al. Density enhancement of a faba bean (Vicia faba) genetic linkage map based on simple sequence repeats markers. Plant Breed. 2019, 138, 207–215. [Google Scholar] [CrossRef]
- Kaur, S.; Kimber, R.B.E.; Cogan, N.O.I.; Materne, M.; Forster, J.W.; Paull, J.G. SNP discovery and high-density genetic mapping in faba bean (Vicia faba L.) permits identification of QTLs for ascochyta blight resistance. Plant Sci. 2014, 217–218, 47–55. [Google Scholar] [CrossRef]
- Sudheesh, S.; Kimber, R.B.E.; Braich, S.; Forster, J.W.; Paull, J.G.; Kaur, S. Construction of an integrated genetic linkage map and detection of quantitative trait loci for ascochyta blight resistance in faba bean (Vicia faba L.). Euphytica 2019, 215, 42–57. [Google Scholar] [CrossRef]
- Carrillo-Perdomo, E.; Vidal, A.; Kreplak, J.; Duborjal, H.; Leveugle, M.; Duarte, J.; Desmetz, C.; Deulvot, C.; Raffiot, B.; Marget, P.; et al. Development of new genetic resources for faba bean (Vicia faba L.) breeding through the discovery of gene-based SNP markers and the construction of a high-density consensus map. Sci. Rep. 2020, 10, 6790–6803. [Google Scholar] [CrossRef] [PubMed]
- Li, M.W.; He, Y.H.; Liu, R.; Li, G.; Wang, D.; Ji, Y.S.; Yan, X.; Huang, S.X.; Wang, C.Y.; Ma, Y.; et al. Construction of SNP genetic maps based on targeted next-generation sequencing and QTL mapping of vital agronomic traits in faba bean (Vicia faba L.). J. Integr. Agric. 2023, 22, 2648–2659. [Google Scholar] [CrossRef]
- Lou, S.B.; Teng, C.C.; Hou, W.W.; Zhou, X.L.; Zhang, H.Y.; Liu, Y.J. Quantitative trait loci mapping of first pod height in faba bean based on genotyping-by-sequencing (GBS). Agronomy 2024, 14, 2013. [Google Scholar] [CrossRef]
- Zhang, H.L.; Fechete, L.I.; Himmelbach, A.; Poehlein, A.; Lohwasser, U.; Börner, A.; Maalouf, F.; Kumar, S.; Khazaei, H.; Stein, N.; et al. Optimization of genotyping-by-sequencing (GBS) for germplasm fingerprinting and trait mapping in faba bean. Legume Sci. 2024, 6, e254. [Google Scholar] [CrossRef]
- Yang, Y.Z.; Gu, Z.C.; Yang, L.M. Cultivation techniques of ‘Touxinlv’ broad bean in Baoshan. Rural. Pract. Sci. Technol. 2006, 6, 16. (In Chinese) [Google Scholar]
- Xu, J.; Liu, G.; Ou, Q.H.; Zhao, S.Q.; Ren, J.; Hu, X.F. Infrared spectrum analysis and elements determination of broad bean of different colors. Hubei Agric. Sci. 2015, 54, 3244–3247. (In Chinese) [Google Scholar]
- Duc, G.; Moussy, F.; Zong, X.X.; Ding, G. Single gene mutation for green cotyledons as a marker for the embryonic genotype in faba bean, Vicia faba. Plant Breed. 1999, 118, 577–578. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Generation | Total of Plants | Cotyledon Color | Expected Ratio | Observed Ratio | ||
|---|---|---|---|---|---|---|
| Yellow | Green | |||||
| P1 | 25 | 25 | 0 | - | ||
| P2 | 20 | 0 | 20 | - | ||
| F1 | 32 | 32 | 0 | - | ||
| F2 | 180 | 137 | 43 | 3:1 | 137:43 | 0.1185 |
| BC1P1 | 19 | 19 | 0 | - | ||
| BC1P2 | 42 | 23 | 19 | 1:1 | 23:19 | 0.381 |
| LG | No. of Markers | Distance (cM) | Average Distance (cM) | No. of Gaps (<5 cM) | Max. Gap (cM) |
|---|---|---|---|---|---|
| LG01 | 729 | 428.45 | 0.59 | 4 | 25.91 |
| LG02 | 613 | 353.05 | 0.58 | 4 | 10.4 |
| LG03 | 213 | 154.91 | 0.73 | 3 | 7.16 |
| LG04 | 189 | 222.5 | 1.18 | 5 | 37.25 |
| LG05 | 138 | 141.77 | 1.03 | 4 | 18.62 |
| LG06 | 109 | 176.28 | 1.63 | 4 | 17.65 |
| Total | 1991 | 1476.95 | 0.74 | 24 |
| LG | QTL Clusters | No. of SNPs | 2-LOD Confidence Interval | LOD | Add | Dom | Expl. (%) | |
|---|---|---|---|---|---|---|---|---|
| Left-Side Marker Name (Position/cM) | Right-Side Marker Name (Position/cM) | |||||||
| LG02 | QTL-cluster1 | 79 | un_106165709 (86.132) | un_7144751 (130.042) | 3.20~26.30 | 0.2629~0.7278 | −0.6547~−0.2154 | 7.90–49.40 |
| LG04 | QTL-cluster2 | 4 | un_29953217 (24.058) | un_109876029 (27.159) | 3.16~3.64 | 0.2914~0.3383 | −0.5922~−0.5348 | 7.90–9.00 |
| LG04 | QTL-cluster3 | 3 | un_2266011 (38.537) | un_109876101 (38.838) | 3.51 | 0.3317 | −0.4985 | 8.7 |
| LG04 | QTL-cluster4 | 22 | un_51389317 (88.124) | un_102388622 (103.219) | 3.19~5.60 | 0.3095~0.3642 | −0.3207~−0.1196 | 7.90–13.50 |
| Database | Functional Genes | Unknown Genes |
|---|---|---|
| Prot-Scriber | 1797 | 224 |
| Swiss-Prot | 1285 | 736 |
| Mercator4v6.0 | 1037 | 984 |
| Prot-Scriber ∪ Swiss-Prot ∪ Mercator4v6.0 | 1804 | 217 |
| Chromosome | Percentage of Identical Matches | Alignment Length | Start | End | E-Value | Bit Score |
|---|---|---|---|---|---|---|
| Chr1L | 61.983 | 242 | 1,196,354,963 | 1,196,355,688 | 3.95 × 10−78 | 264 |
| Chr1L | 98.077 | 104 | 1,196,354,406 | 1,196,354,717 | 3.79 × 10−62 | 218 |
| Chr1L | 48.276 | 58 | 1,237,744,660 | 1,237,744,824 | 1.15 × 10−6 | 57 |
| Chr2 | 53.125 | 64 | 792,668,091 | 792,668,273 | 8.69 × 10−12 | 72.4 |
| Chr2 | 46.988 | 83 | 74,072,565 | 74,072,804 | 1.46 × 10−9 | 65.9 |
| Chr2 | 49.296 | 71 | 74,137,277 | 74,137,480 | 1.98 × 10−9 | 65.5 |
| Chr2 | 51.02 | 49 | 792,667,442 | 792,667,588 | 5.85 × 10−7 | 58.2 |
| Chr2 | 48.333 | 60 | 792,668,407 | 792,668,586 | 1.54 × 10−6 | 56.6 |
| Chr5 | 47.887 | 71 | 181,755,951 | 181,756,154 | 3.12 × 10−9 | 64.7 |
| Chr5 | 46.479 | 71 | 181,777,262 | 181,777,465 | 5.22 × 10−8 | 61.2 |
| Chr5 | 41.772 | 79 | 531,003,825 | 531,004,052 | 7.14 × 10−8 | 60.8 |
| Chr4 | 46.154 | 78 | 1,390,849,993 | 1,390,850,217 | 3.59 × 10−9 | 64.7 |
| Chr4 | 47.887 | 71 | 1,390,761,333 | 1,390,761,536 | 3.72 × 10−9 | 64.7 |
| Chr4 | 43.038 | 79 | 969,946,743 | 969,946,970 | 1.22 × 10−8 | 63.2 |
| Chr4 | 47.143 | 70 | 970,018,899 | 970,019,099 | 2.56 × 10−7 | 59.3 |
| Chr4 | 47.143 | 70 | 970,038,677 | 970,038,877 | 2.56 × 10−7 | 59.3 |
| Chr4 | 40.506 | 79 | 1,225,324,609 | 1,225,324,836 | 4.32 × 10−7 | 58.5 |
| Chr4 | 40.506 | 79 | 1,225,303,984 | 1,225,304,211 | 2.01 × 10−6 | 56.6 |
| Chr6 | 47.887 | 71 | 1,140,361,221 | 1,140,361,424 | 1.12 × 10−8 | 63.2 |
| Chr6 | 47.887 | 71 | 1,140,342,993 | 1,140,343,196 | 1.63 × 10−8 | 62.8 |
| Chr6 | 47.059 | 51 | 1,445,004,990 | 1,445,005,142 | 3.71 × 10−5 | 52.8 |
| Chr3 | 46.479 | 71 | 920,577,449 | 920,577,652 | 7.75 × 10−8 | 60.8 |
| Chr3 | 42.254 | 71 | 920,556,707 | 920,556,919 | 1.11 × 10−6 | 57.4 |
| Chr3 | 41.772 | 79 | 764,135,754 | 764,135,981 | 1.15 × 10−7 | 60.1 |
| Chr2 | 41.772 | 79 | 1,209,841,431 | 1,209,841,658 | 1.60 × 10−7 | 59.7 |
| Chr5 | 52.083 | 48 | 1,237,512,370 | 1,237,512,513 | 4.90 × 10−4 | 49.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teng, C.; Zhang, H.; Hou, W.; Li, P.; Zhou, X.; Liu, Y. High-Density Genetic Map Construction and QTL Detection for Cotyledon Color in Faba Bean Based on Double Digest Restriction-Site Associated DNA Sequencing (ddRAD-Seq). Agronomy 2025, 15, 193. https://doi.org/10.3390/agronomy15010193
Teng C, Zhang H, Hou W, Li P, Zhou X, Liu Y. High-Density Genetic Map Construction and QTL Detection for Cotyledon Color in Faba Bean Based on Double Digest Restriction-Site Associated DNA Sequencing (ddRAD-Seq). Agronomy. 2025; 15(1):193. https://doi.org/10.3390/agronomy15010193
Chicago/Turabian StyleTeng, Changcai, Hongyan Zhang, Wanwei Hou, Ping Li, Xianli Zhou, and Yujiao Liu. 2025. "High-Density Genetic Map Construction and QTL Detection for Cotyledon Color in Faba Bean Based on Double Digest Restriction-Site Associated DNA Sequencing (ddRAD-Seq)" Agronomy 15, no. 1: 193. https://doi.org/10.3390/agronomy15010193
APA StyleTeng, C., Zhang, H., Hou, W., Li, P., Zhou, X., & Liu, Y. (2025). High-Density Genetic Map Construction and QTL Detection for Cotyledon Color in Faba Bean Based on Double Digest Restriction-Site Associated DNA Sequencing (ddRAD-Seq). Agronomy, 15(1), 193. https://doi.org/10.3390/agronomy15010193