
Optimizing Genomic Selection Methods to Improve Prediction Accuracy of Sugarcane Single-Stalk Weight

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. DNA Extraction and Genotyping
2.2. Determination of Stem Heaviness
2.3. Broad-Sense Heritability and the Coefficient of Variation
2.4. Genomic Relationship Matrices
2.5. Scoring of Genomic Regions
2.6. Estimation of GEBVs
3. Result
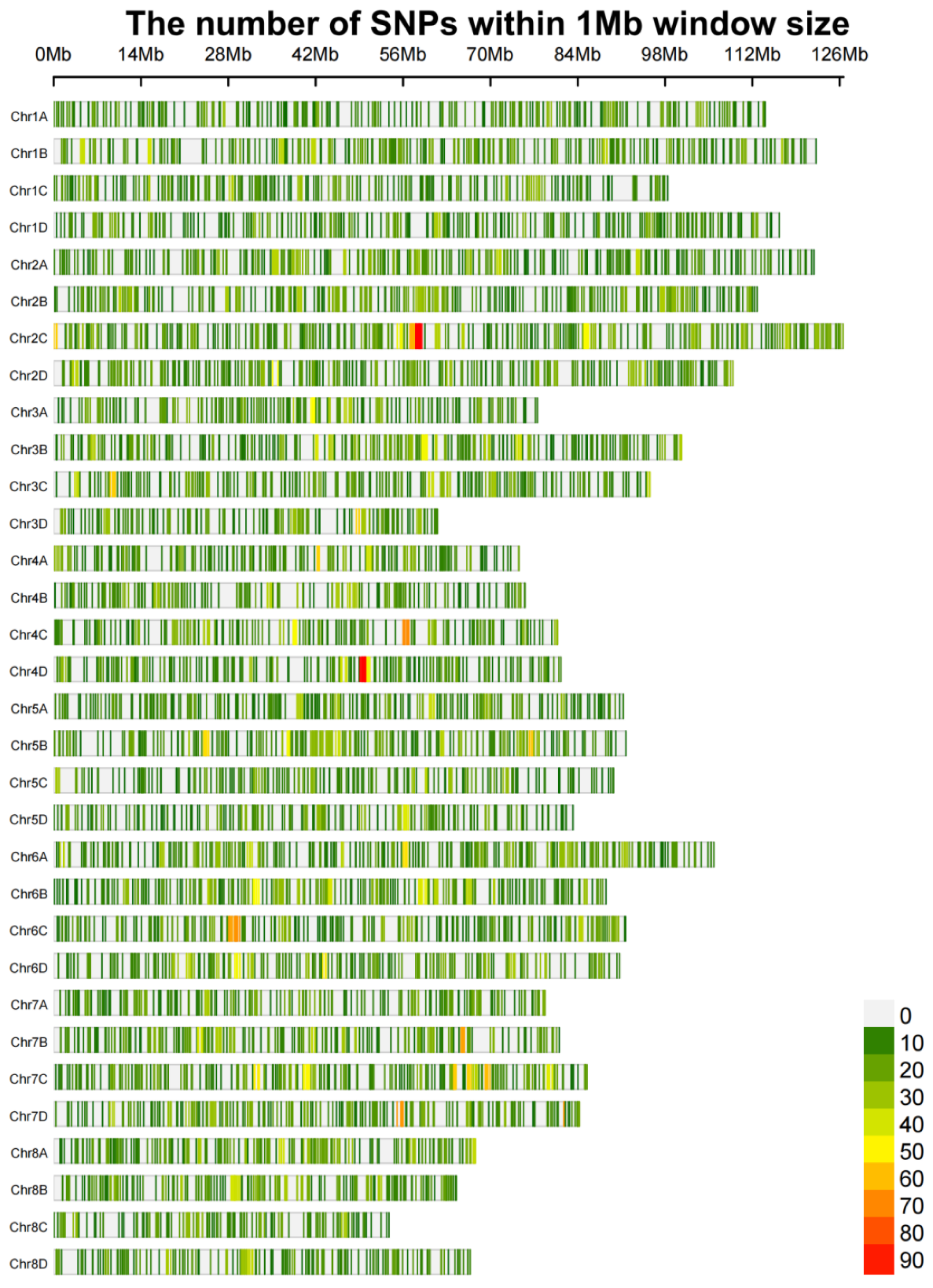
3.1. Phenotyping and Genotyping
3.2. Performance Comparison Between Genomic Selection Models
4. Discussion
4.1. Advancements in Genomic Selection Markers for Sugarcane
4.2. Model Comparisons and Prediction Accuracy in Genomic Selection
4.3. Enhancing Genomic Selection Through Weighted SNPs and Fixed Effects
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wu, Q.; Li, A.; Liu, J.; Zhao, Y.; Zhao, P.; Zhang, Y.; Que, Y. Sugarcane variety YZ05-51 with high yield and strong resistance: Breeding and cultivation perspectives. Trop. Plants 2024, 3. [Google Scholar] [CrossRef]
- Bao, Y.; Zhang, Q.; Huang, J.; Zhang, S.; Yao, W.; Yu, Z.; Deng, Z.; Yu, J.; Kong, W.; Yu, X.; et al. A chromosomal-scale genome assembly of modern cultivated hybrid sugarcane provides insights into origination and evolution. Nat. Commun. 2024, 15, 3041. [Google Scholar] [CrossRef]
- Zhao, Y.; Zan, F.; Deng, J.; Zhao, P.; Zhao, J.; Wu, C.; Liu, J.; Zhang, Y. Improvements in Sugarcane (Saccharum spp.) Varieties and Parent Traceability Analysis in Yunnan, China. Agronomy 2022, 12, 1211. [Google Scholar] [CrossRef]
- Wallace, J.G.; Rodgers-Melnick, E.; Buckler, E.S. On the Road to Breeding 4.0: Unraveling the Good, the Bad, and the Boring of Crop Quantitative Genomics. Annu. Rev. Genet. 2018, 52, 421–444. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Jiang, S.; Tian, Y.; Dong, X.; Xiao, J.; Lu, Y.; Liang, T.; Zhou, H.; Xu, D.; Zhang, H.; et al. Smart breeding driven by advances in sequencing technology. Mod. Agric. 2023, 1, 43–56. [Google Scholar] [CrossRef]
- Luo, H.; Xiong, F.; Qiu, L.; Liu, J.; Duan, W.; Gao, Y.; Qin, X.; Wu, J.; Li, Y.; Liu, J. Application Study of Molecular Markers Associated with Traits in Sugarcane Molecular Breeding. Crops 2022, 35–43. [Google Scholar] [CrossRef]
- Yadav, S.; Jackson, P.; Wei, X.; Ross, E.M.; Aitken, K.; Deomano, E.; Atkin, F.; Hayes, B.J.; Voss-Fels, K.P. Accelerating genetic gain in sugarcane breeding using genomic selection. Agronomy 2020, 10, 585. [Google Scholar] [CrossRef]
- Cursi, D.E.; Castillo, R.O.; Tarumoto, Y.; Umeda, M.; Tippayawat, A.; Ponragdee, W.; Racedo, J.; Perera, M.F.; Hoffmann, H.P.; Carneiro, M.S. Origin, genetic diversity, conservation, and traditional and molecular breeding approaches in sugarcane. Cash Crops Genet. Divers. Eros. Conserv. Util. 2022, 83–116. [Google Scholar] [CrossRef]
- Batista, L.G.; Mello, V.H.; Souza, A.P.; Margarido, G.R. Genomic prediction with allele dosage information in highly polyploid species. Theor. Appl. Genet. 2022, 135, 723–739. [Google Scholar] [CrossRef]
- Zhang, J.; Nagai, C.; Yu, Q.; Pan, Y.; Ayala-Silva, T.; Schnell, R.J.; Comstock, J.C.; Arumuganathan, A.K.; Ming, R. Genome size variation in three Saccharum species. Euphytica 2012, 185, 511–519. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, X.; Tang, H.; Zhang, Q.; Hua, X.; Ma, X.; Zhu, F.; Jones, T.; Zhu, X.; Bowers, J.; et al. Allele-defined genome of the autopolyploid sugarcane Saccharum spontaneum L. Nat. Genet. 2018, 50, 1565–1573. [Google Scholar] [CrossRef] [PubMed]
- Gouy, M.; Rousselle, Y.; Bastianelli, D.; Lecomte, P.; Bonnal, L.; Roques, D.; Efile, J.; Rocher, S.; Daugrois, J.; Toubi, L. Experimental assessment of the accuracy of genomic selection in sugarcane. Theor. Appl. Genet. 2013, 126, 2575–2586. [Google Scholar] [CrossRef] [PubMed]
- Hayes, B.J.; Wei, X.; Joyce, P.; Atkin, F.; Deomano, E.; Yue, J.; Nguyen, L.; Ross, E.M.; Cavallaro, T.; Aitken, K.S. Accuracy of genomic prediction of complex traits in sugarcane. Theor. Appl. Genet. 2021, 134, 1455–1462. [Google Scholar] [CrossRef] [PubMed]
- Deomano, E.; Jackson, P.; Wei, X.; Aitken, K.; Kota, R.; Pérez-Rodríguez, P. Genomic prediction of sugar content and cane yield in sugar cane clones in different stages of selection in a breeding program, with and without pedigree information. Mol. Breed. 2020, 40, 38. [Google Scholar] [CrossRef]
- Islam, M.S.; Mccord, P.H.; Olatoye, M.O.; Qin, L.; Sood, S.; Lipka, A.E.; Todd, J.R. Experimental evaluation of genomic selection prediction for rust resistance in sugarcane. Plant Genome 2021, 14, e20148. [Google Scholar] [CrossRef]
- Murray, M.G.; Thompson, W.F. Rapid isolation of high molecular weight plant DNA. Nucleic. Acids. Res. 1980, 8, 4321–4326. [Google Scholar] [CrossRef] [PubMed]
- Zou, M.; Xia, Z. Hyper-seq: A novel, effective, and flexible marker-assisted selection and genotyping approach. Innov. Amst. 2022, 3, 100254. [Google Scholar] [CrossRef] [PubMed]
- Vasimuddin, M.; Misra, S.; Li, H.; Aluru, S. Efficient Architecture-Aware Acceleration of BWA-MEM for Multicore Systems. In Proceedings of the 2019 IEEE International Parallel and Distributed Processing Symposium (IPDPS), Rio de Janeiro, Brazil, 20–24 May 2019; pp. 314–324. [Google Scholar] [CrossRef]
- Depristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; Del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Chrigui, N.; Sari, D.; Sari, H.; Eker, T.; Cengiz, M.F.; Ikten, C.; Toker, C. Introgression of Resistance to Leafminer (Liriomyza cicerina Rondani) from Cicer reticulatum Ladiz. to C. arietinum L. and Relationships between Potential Biochemical Selection Criteria. Agronomy 2021, 11, 57. [Google Scholar] [CrossRef]
- Vanraden, P.M. Efficient Methods to Compute Genomic Predictions. J. Dairy. Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef]
- Li, H.; Li, X.; Zhang, P.; Feng, Y.; Mi, J.; Gao, S.; Sheng, L.; Ali, M.; Yang, Z.; Li, L.; et al. Smart Breeding Platform: A web-based tool for high-throughput population genetics, phenomics, and genomic selection. Mol. Plant. 2024, 17, 677–681. [Google Scholar] [CrossRef] [PubMed]
- Misztal, I.; Tsuruta, S.; Strabel, T.; Auvray, B.; Druet, T.; Lee, D. BLUPF90 and Related Programs. In Proceedings of the 7th World Congress on Genetics Applied to Livestock Production, Montpellier, France, 19–23 August 2002; p. 743. Available online: https://hdl.handle.net/2268/84980 (accessed on 18 October 2024).
- Irvine, J.E. Saccharum species as horticultural classes. Theor. Appl. Genet. 1999, 98, 186–194. [Google Scholar] [CrossRef]
- Palhares, A.C.; Rodrigues-Morais, T.B.; Van Sluys, M.A.; Domingues, D.S.; Maccheroni, W.J.; Jordao, H.J.; Souza, A.P.; Marconi, T.G.; Mollinari, M.; Gazaffi, R.; et al. A novel linkage map of sugarcane with evidence for clustering of retrotransposon-based markers. BMC Genet. 2012, 13, 51. [Google Scholar] [CrossRef] [PubMed]
- Healey, A.L.; Garsmeur, O.; Lovell, J.T.; Shengquiang, S.; Sreedasyam, A.; Jenkins, J.; Plott, C.B.; Piperidis, N.; Pompidor, N.; Llaca, V.; et al. The complex polyploid genome architecture of sugarcane. Nature 2024, 628, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Daugrois, J.H.; Grivet, L.; Roques, D.; Hoarau, J.Y.; Lombard, H.; Glaszmann, J.C.; D’Hont, A. A putative major gene for rust resistance linked with a RFLP marker in sugarcane cultivar ‘R570’. Theor. Appl. Genet. 1996, 92, 1059–1064. [Google Scholar] [CrossRef] [PubMed]
- Raboin, L.M.; Oliveira, K.M.; Lecunff, L.; Telismart, H.; Roques, D.; Butterfield, M.; Hoarau, J.Y.; D Hont, A. Genetic mapping in sugarcane, a high polyploid, using bi-parental progeny: Identification of a gene controlling stalk colour and a new rust resistance gene. Theor. Appl. Genet. 2006, 112, 1382–1391. [Google Scholar] [CrossRef] [PubMed]
- Costet, L.; Raboin, L.; Payet, M.; D Hont, A.; Nibouche, S. A major quantitative trait allele for resistance to the Sugarcane yellow leaf virus (Luteoviridae). Plant Breed. 2012, 131, 637–640. [Google Scholar] [CrossRef]
- Yadav, S.; Wei, X.; Joyce, P.; Atkin, F.; Deomano, E.; Sun, Y.; Nguyen, L.T.; Ross, E.M.; Cavallaro, T.; Aitken, K.S.; et al. Improved genomic prediction of clonal performance in sugarcane by exploiting non-additive genetic effects. Theor. Appl. Genet. 2021, 134, 2235–2252. [Google Scholar] [CrossRef]
- Bernardo, R. Genomewide Selection when Major Genes Are Known. Crop Sci. 2014, 54, 68–75. [Google Scholar] [CrossRef]
- Islam, M.S.; Fang, D.D.; Jenkins, J.N.; Guo, J.; Mccarty, J.C.; Jones, D.C. Evaluation of genomic selection methods for predicting fiber quality traits in Upland cotton. Mol. Genet. Genom. 2020, 295, 67–79. [Google Scholar] [CrossRef]
- Talaei Khoei, T.; Ould Slimane, H.; Kaabouch, N. Deep learning: Systematic review, models, challenges, and research directions. Neural Comput. Appl. 2023, 35, 23103–23124. [Google Scholar] [CrossRef]
- Meuwissen, T.H.E.; Hayes, B.J.; Goddard, M.E. Prediction of Total Genetic Value Using Genome-Wide Dense Marker Maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [CrossRef]
- Arruda, M.P.; Lipka, A.E.; Brown, P.J.; Krill, A.M.; Thurber, C.; Brown-Guedira, G.; Dong, Y.; Foresman, B.J.; Kolb, F.L. Comparing genomic selection and marker-assisted selection for Fusarium head blight resistance in wheat (Triticum aestivum L.). Mol. Breed 2016, 36, 84. [Google Scholar] [CrossRef]
- Li, D.; Xu, Z.; Gu, R.; Wang, P.; Lyle, D.; Xu, J.; Zhang, H.; Wang, G. Enhancing genomic selection by fitting large-effect SNPs as fixed effects and a genotype-by-environment effect using a maize BC1F3:4 population. PLoS ONE 2019, 14, e223898. [Google Scholar] [CrossRef] [PubMed]
- Merrick, L.F.; Burke, A.B.; Chen, X.; Carter, A.H. Breeding with Major and Minor Genes: Genomic Selection for Quantitative Disease Resistance. Front. Plant Sci. 2021, 12, 713667. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
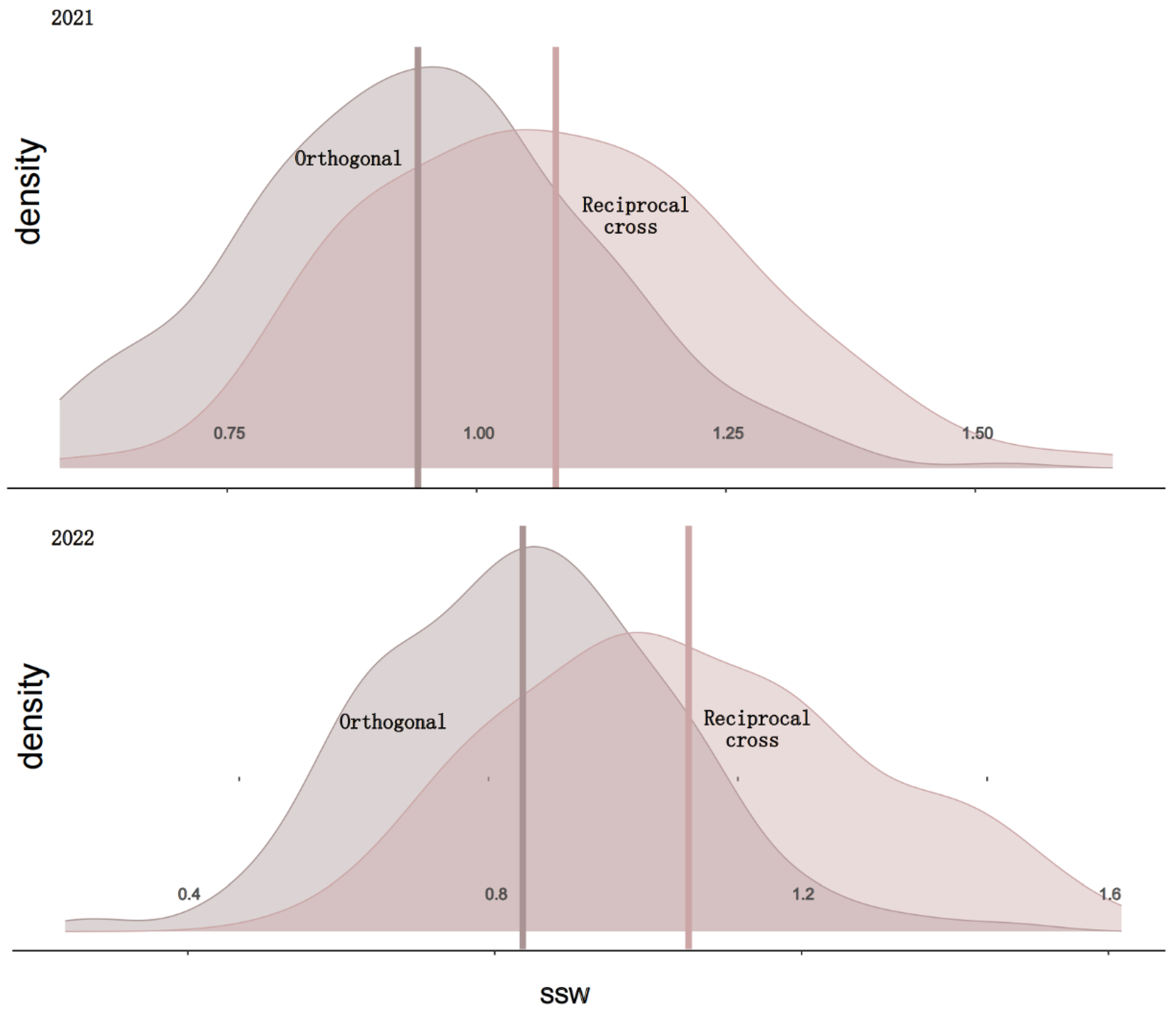
| Mean (kg) | Standard Deviation | Min | Max | CV (%) | H2 | |
|---|---|---|---|---|---|---|
| Orthogonal | 0.891 | 0.172 | 0.465 | 1.485 | 19.3% | 0.86 |
| Reciprocal cross | 1.067 | 0.189 | 0.683 | 1.592 | 17.7% | 0.88 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Z.; Xia, C.; Lu, Y.; Liu, Q.; Zou, M.; Zan, F.; Xia, Z. Optimizing Genomic Selection Methods to Improve Prediction Accuracy of Sugarcane Single-Stalk Weight. Agronomy 2024, 14, 2842. https://doi.org/10.3390/agronomy14122842
Wang Z, Xia C, Lu Y, Liu Q, Zou M, Zan F, Xia Z. Optimizing Genomic Selection Methods to Improve Prediction Accuracy of Sugarcane Single-Stalk Weight. Agronomy. 2024; 14(12):2842. https://doi.org/10.3390/agronomy14122842
Chicago/Turabian StyleWang, Zihao, Chengcai Xia, Yanjie Lu, Qi Liu, Meiling Zou, Fenggang Zan, and Zhiqiang Xia. 2024. "Optimizing Genomic Selection Methods to Improve Prediction Accuracy of Sugarcane Single-Stalk Weight" Agronomy 14, no. 12: 2842. https://doi.org/10.3390/agronomy14122842
APA StyleWang, Z., Xia, C., Lu, Y., Liu, Q., Zou, M., Zan, F., & Xia, Z. (2024). Optimizing Genomic Selection Methods to Improve Prediction Accuracy of Sugarcane Single-Stalk Weight. Agronomy, 14(12), 2842. https://doi.org/10.3390/agronomy14122842