
Morphological, Molecular and Genomic Identification and Characterisation of Monilinia fructicola in Prunus persica from Portugal
 ,
,  ,
,  ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Isolation of Putative M. fructicola, Morphological Identification
2.3. DNA Extraction from Single Colony and Molecular Identification by PCR
2.4. Total DNA Extraction from Branches and Flower Extracts
2.5. Molecular Identification by Using Real-Time PCR
2.6. Pathogenicity Test
2.7. Genome Sequencing, Assembly, Annotation, and Functional Analysis
3. Results and Discussion
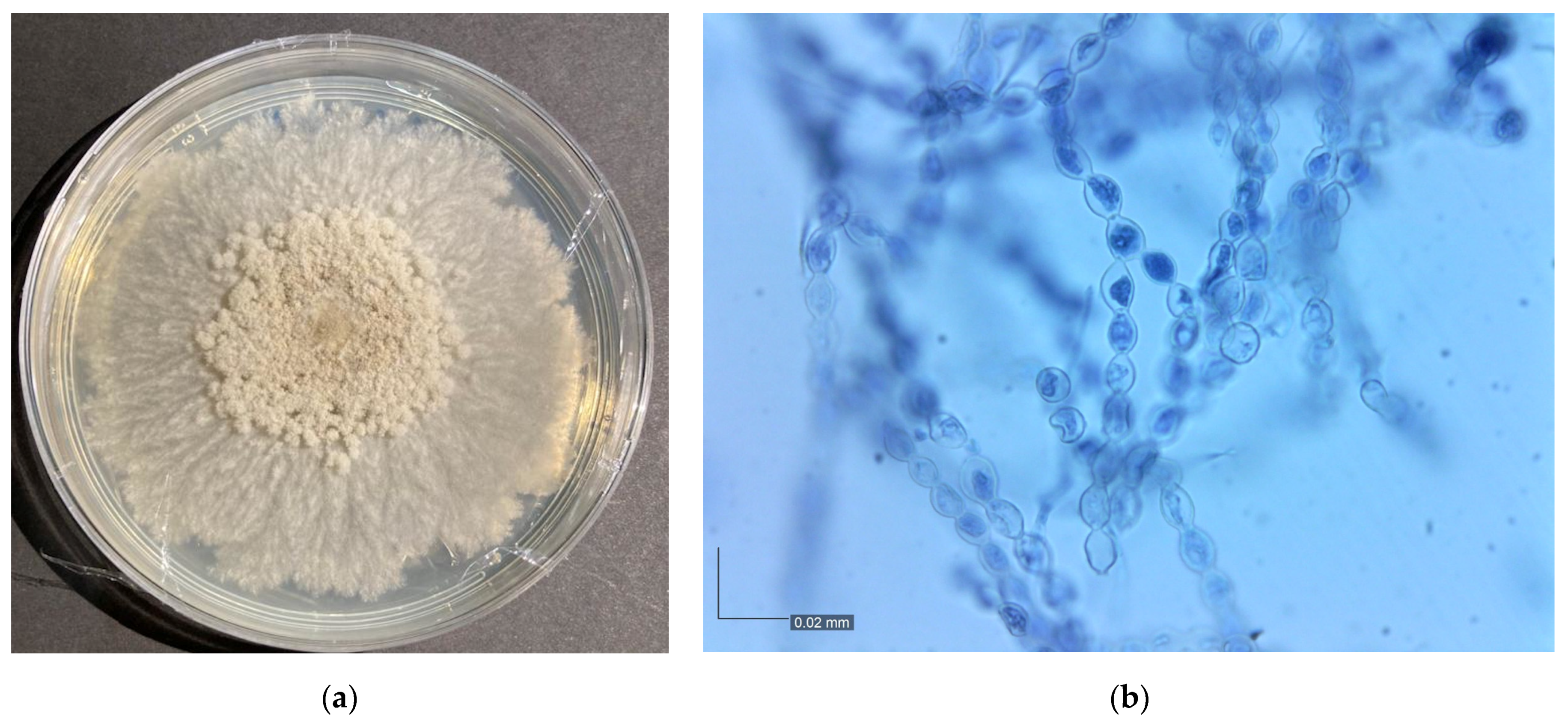
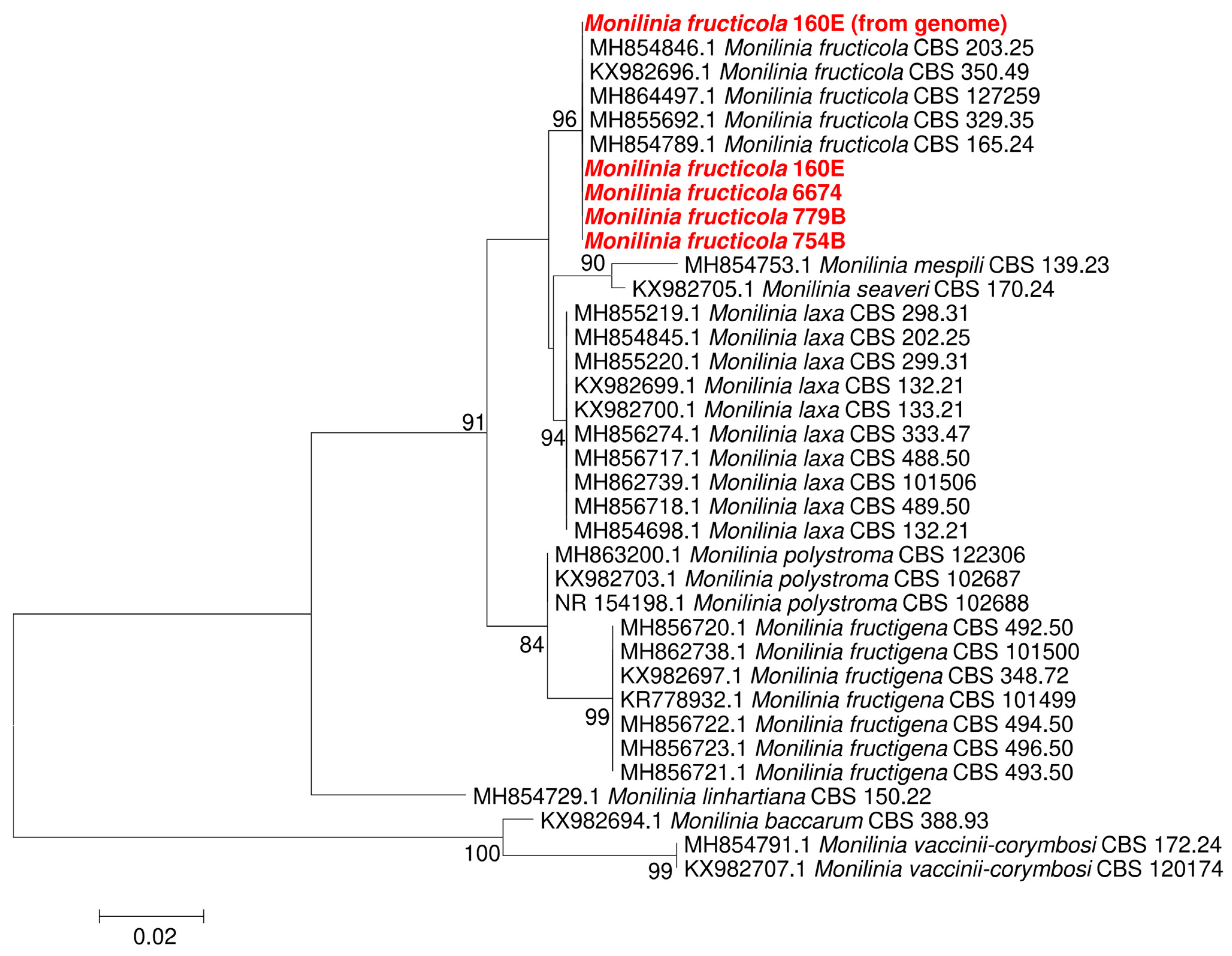
3.1. Monilinia Fructicola Isolation and Identification
3.2. Pathogenicity Tests
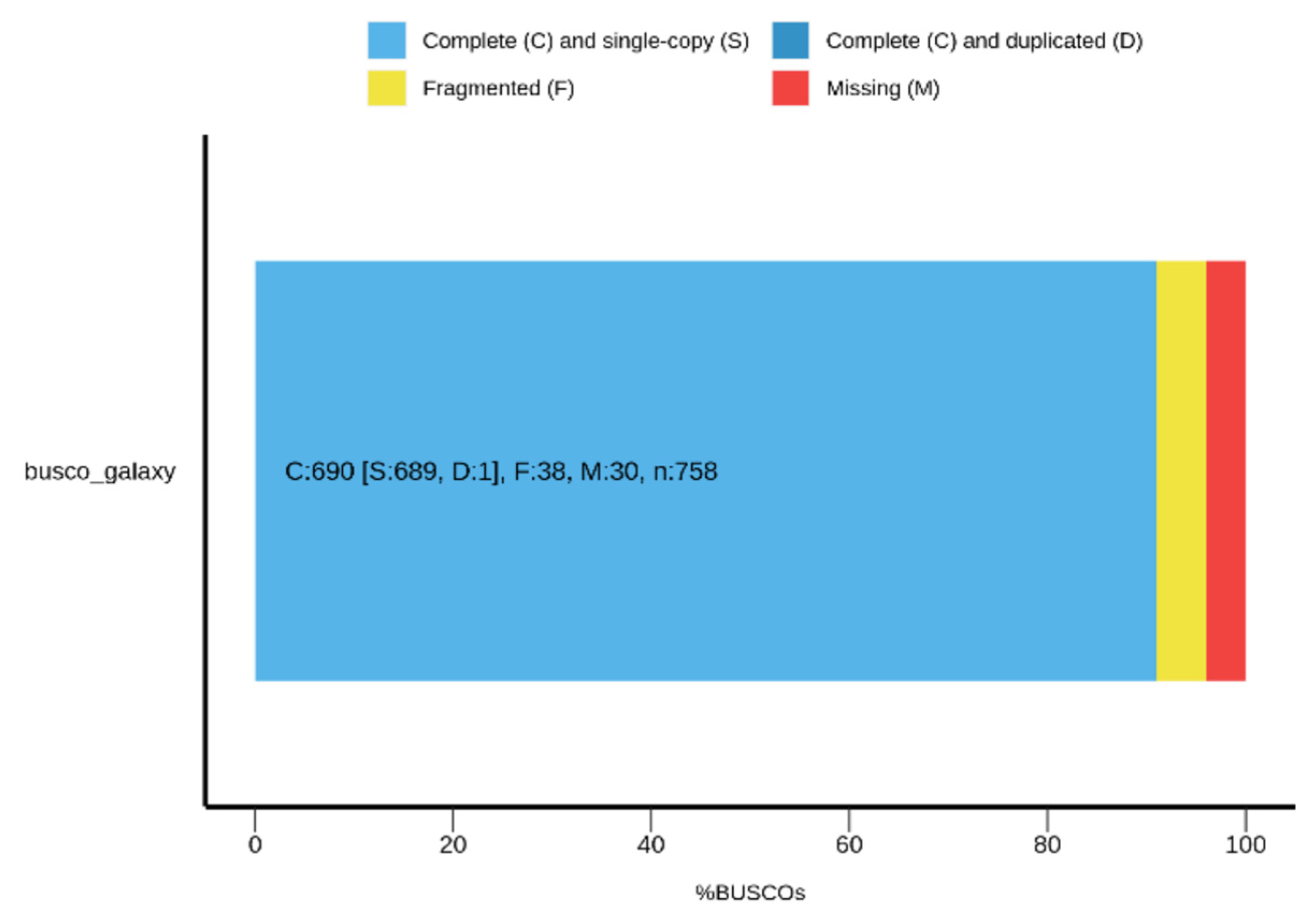
3.3. Genome Assembly
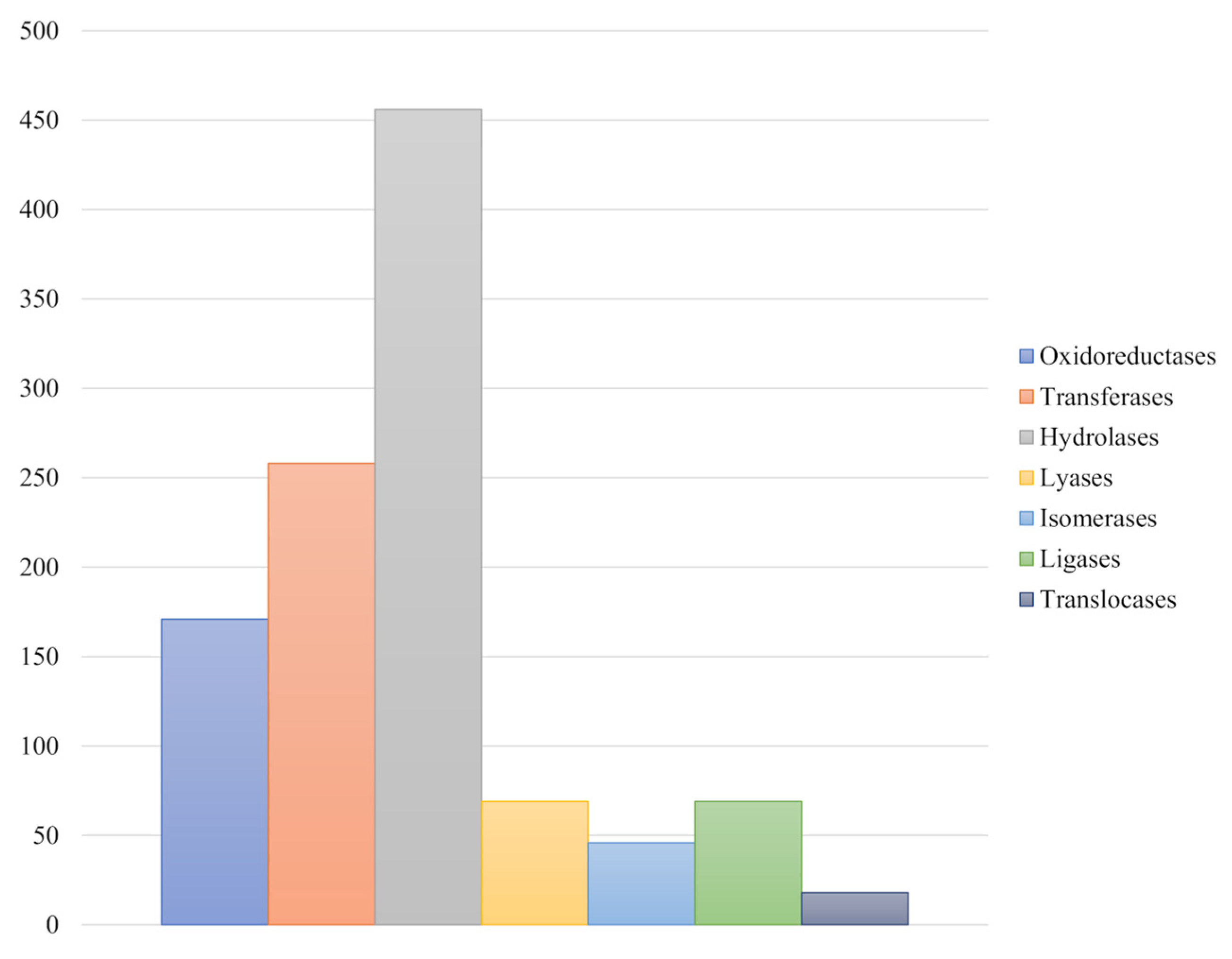
3.4. Genome Annotation and Functional Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Food and Agricultural Organization of the United Nations. 2020. Available online: https://www.fao.org/faostat/en/#data/QCL (accessed on 5 January 2023).
- Das, B. Prunus diversity- early and present development: A review. Int. J. Biodivers. Conserv. 2011, 3, 721–734. [Google Scholar] [CrossRef]
- Instituto Nacional de Estatistica. Estatisticas Agricolas 2020. Available online: https://www.ine.pt/xportal/xmain?xpid=INE&xpgid=ine_publicacoes&PUBLICACOESpub_boui=437147278&PUBLICACOEStema=55505&PUBLICACOESmodo=2 (accessed on 5 January 2023).
- Sisquella, M.; Viñas, I.; Picouet, P.; Torres, R.; Usall, J. Effect of host and Monilinia spp. variables on the efficacy of radio frequency treatment on peaches. Postharvest Biol. Technol. 2014, 87, 6–12. [Google Scholar] [CrossRef]
- Obi, V.I.; Barriuso, J.J.; Gogorcena, Y. Peach brown rot: Still in search of an ideal management option. J. Agric. Sci. 2018, 8, 125. [Google Scholar] [CrossRef]
- Martini, C.; Mari, M. Monilinia fructicola, Monilinia laxa (Monilinia Rot, Brown Rot). In Postharvest Decay: Control Strategies; Academic Press: Cambridge, MA, USA, 2014. [Google Scholar] [CrossRef]
- EPPO Standards. Diagnostics. PM 7/18 (3) Monilinia fructicola. EPPO Bull. 2020, 50, 5–18. [Google Scholar] [CrossRef]
- EPPO/CABI. Monilinia fructicola. In Quarantine Pests for Europe, 2nd ed.; Smith, I.M., McNamara, D.G., Scott, P.R., Holderness, M., Eds.; CAB International: Wallingford, UK, 1997; pp. 530–535. [Google Scholar]
- Lichou, J.; Mandrin, J.F.; Breniaux, D.; Mercier, V.; Giauque, P.; Desbrus, D.; Blanc, P.; Belluau, E. Une nouvelle moniliose: Monilia fructicola s’attaque aux arbres fruitiers à noyaux. Phytoma 2002, 547, 22–25. [Google Scholar]
- De Cal, A.; Melgarejo, P. Effects of long-wave UV light on Monilinia growth and identification of species. Plant Dis. 1999, 83, 62–65. [Google Scholar] [CrossRef] [PubMed]
- Duchoslavová, J.; Širučková, I.; Zapletalová, E.; Navrátil, M.; Šafářová, D. First Report of Brown Rot Caused by Monilinia fructicola on Various Stone and Pome Fruits in the Czech Republic. Plant Dis. 2007, 91, 907. [Google Scholar] [CrossRef]
- Munda, A.; Viršček Marn, M. First Report of Brown Rot Caused by Monilinia fructicola Affecting Peach Orchards in Slovenia. Plant Dis. 2010, 94, 1166. [Google Scholar] [CrossRef]
- Pellegrino, C.; Gullino, M.L.; Garibaldi, A.; Spadaro, D. First report of brown rot of stone fruit caused by Monilinia fructicola in Italy. Plant Dis. 2009, 93, 668. [Google Scholar] [CrossRef]
- Hrustić, J.; Mihajlović, M.; Tanović, B.; Delibašić, G.; Stanković, I.; Krstić, B.; Bulajić, A. First Report of Brown Rot Caused by Monilinia fructicola on Nectarine in Serbia. Plant Dis. 2013, 97, 147. [Google Scholar] [CrossRef]
- Ivić, D.; Fazinić, T.; Cole, J.; Novak, A. Monilinia species identified on peach and nectarine in Croatia, with the first record of Monilinia fructicola. EPPO Bull. 2014, 44, 70–72. [Google Scholar] [CrossRef]
- Bobev, S.B.; Angelov, L.T.; Van Poucke, K.; Maes, M. First Report of Brown Rot on Peach, Nectarine, Cherry, and Plum Fruits Caused by Monilinia fructicola in Bulgaria. Plant Dis. 2020, 104, 1561. [Google Scholar] [CrossRef]
- Ramos, N.; Soares, C. Ficha de Divulgação nº06/2013 (Moniliose). Direção Regional de Agricultura Do Algarve, 1-2. 2013. Available online: https://www.drapalgarve.gov.pt/images/pdf/Fitossanidade/avisos_agricolas/PRUN_FD_EAA_06Monilose.pdf (accessed on 5 May 2023).
- Eevers, N.; Gielen, M.; Sánchez-López, A.; Jaspers, S.; White, J.C.; Vangronsveld, J.; Weyens, N. Optimization of isolation and cultivation of bacterial endophytes through addition of plant extract to nutrient media. Microb. Biotechnol. 2015, 8, 707–715. [Google Scholar] [CrossRef]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J. Amplification and direct sequencing of fungal ribosomal rna genes for phylogenetics. In PCR Protocols; Academic Press: Cambridge, MA, USA, 1990. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Golosova, O.; Fursov, M. UGENE team Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Van Brouwershaven, I.R.; Bruil, M.L.; Van Leeuwen, G.C.M.; Kox, L.F.F. A real-time (TaqMan) PCR assay to differentiate Monilinia fructicola from other brown rot fungi of fruit crops. Plant Pathol. 2010, 59, 548–555. [Google Scholar] [CrossRef]
- De Coster, W.; D’Hert, S.; Schultz, D.T.; Cruts, M.; Van Broeckhoven, C. NanoPack: Visualizing and processing long-read sequencing data. Bioinformatics 2018, 34, 2666–2669. [Google Scholar] [CrossRef]
- Jalili, V.; Afgan, E.; Gu, Q.; Clements, D.; Blankenberg, D.; Goecks, J.; Taylor, J.; Nekrutenko, A. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2020 update. Nucleic Acids Res. 2020, 48, W395–W402. [Google Scholar] [CrossRef]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Kriventseva, E.V.; Kuznetsov, D.; Tegenfeldt, F.; Manni, M.; Dias, R.; Simão, F.A.; Zdobnov, E.M. OrthoDB v10: Sampling the diversity of animal, plant, fungal, protist, bacterial and viral genomes for evolutionary and functional annotations of orthologs. Nucleic Acids Res. 2019, 47, D807–D811. [Google Scholar] [CrossRef]
- Seemann, T. Barrnap 0.7: Rapid ribosomal RNA prediction. 2013. Available online: https://github.com/tseemann/barrnap (accessed on 3 March 2023).
- Laslett, D.; Canback, B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Flynn, J.M.; Hubley, R.; Goubert, C.; Rosen, J.; Clark, A.G.; Feschotte, C.; Smit, A.F. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 2010, 117, 9451–9457. [Google Scholar] [CrossRef] [PubMed]
- Smit, A.; Hubley, R.; Green, P. RepeatMasker Open-4.0. (2013–2015). Available online: http://www.repeatmasker.org (accessed on 3 March 2023).
- Stanke, M.; Morgenstern, B. AUGUSTUS: A web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 2005, 33, W465–W467. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef]
- Götz, S.; García-Gómez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talón, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef]
- Cantalapiedra, C.P.; Hernández-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. EggNOG-mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed]
- Blum, M.; Chang, H.Y.; Chuguransky, S.; Grego, T.; Kandasaamy, S.; Mitchell, A.; Nuka, G.; Paysan-Lafosse, T.; Qureshi, M.; Raj, S.; et al. The InterPro protein families and domains database: 20 years on. Nucleic Acids Res. 2021, 49, D344–D354. [Google Scholar] [CrossRef]
- Aramaki, T.; Blanc-Mathieu, R.; Endo, H.; Ohkubo, K.; Kanehisa, M.; Goto, S.; Ogata, H. KofamKOALA: KEGG Ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics 2020, 36, 2251–2252. [Google Scholar] [CrossRef]
- Teufel, F.; Almagro Armenteros, J.J.; Johansen, A.R.; Gíslason, M.H.; Pihl, S.I.; Tsirigos, K.D.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 6.0 Predicts All Five Types of Signal Peptides Using Protein Language Models. Nat. Biotechnol. 2022, 40, 1023–1025. [Google Scholar] [CrossRef] [PubMed]
- Hallgren, J.; Tsirigos, K.D.; Pedersen, M.D.; Armenteros, J.J.A.; Marcatili, P.; Nielsen, H.; Krogh, A.; Winther, O. DeepTMHMM Predicts Alpha and Beta Transmembrane Proteins Using Deep Neural Networks. BioRxiv 2022. [Google Scholar] [CrossRef]
- Sperschneider, J.; Dodds, P.N. EffectorP 3.0: Prediction of Apoplastic and Cytoplasmic Effectors in Fungi and Oomycetes. MPMI 2022, 35, 146–156. [Google Scholar] [CrossRef]
- Urban, M.; Cuzick, A.; Seager, J.; Wood, V.; Rutherford, K.; Venkatesh, S.Y.; De Silva, N.; Martinez, M.C.; Pedro, H.; Yates, A.D.; et al. PHI-Base: The Pathogen–Host Interactions Database. Nucleic Acids Res. 2020, 48, D613–D620. [Google Scholar] [CrossRef]
- Zhang, H.; Yohe, T.; Huang, L.; Entwistle, S.; Wu, P.; Yang, Z.; Busk, P.K.; Xu, Y.; Yin, Y. DbCAN2: A Meta Server for Automated Carbohydrate-Active Enzyme Annotation. Nucleic Acids Res. 2018, 46, W95–W101. [Google Scholar] [CrossRef]
- Blin, K.; Shaw, S.; Kloosterman, A.M.; Charlop-Powers, Z.; van Wezel, G.P.; Medema, M.H.; Weber, T. AntiSMASH 6.0: Improving Cluster Detection and Comparison Capabilities. Nucleic Acids Res. 2021, 49, W29–W35. [Google Scholar] [CrossRef]
- Sagasta, M.E. Monilinia disease. EPPO Bull. 1997, 7, 105–116. [Google Scholar] [CrossRef]
- Van Leeuwen, G.C.M.; Van Kesteren, H.A. Delineation of the three brown rot fungi of fruit crops (Monilinia spp.) on the basis of quantitative characteristics. Can. J. Bot. 1998, 76, 2042–2050. [Google Scholar] [CrossRef]
- De Miccolis Angelini, R.M.; Romanazzi, G.; Pollastro, S.; Rotolo, C.; Faretra, F.; Landi, L. New High-Quality Draft Genome of the Brown Rot Fungal Pathogen Monilinia fructicola. Genome Biol. Evol. 2019, 11, 2850–2855. [Google Scholar] [CrossRef]
- Vilanova, L.; Valero-Jiménez, C.A.; van Kan, J.A.L. Deciphering the Monilinia fructicola Genome to Discover Effector Genes Possibly Involved in Virulence. Genes 2021, 12, 568. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, M.M.; Moreno, L.F.; Stielow, B.J.; Muszewska, A.; Hainaut, M.; Gonzaga, L.; Abouelleil, A.; Patané, J.S.L.; Priest, M.; Souza, R.; et al. Exploring the Genomic Diversity of Black Yeasts and Relatives (Chaetothyriales, Ascomycota). Stud. Mycol. 2017, 86, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Baldrian, P.; Valášková, V. Degradation of Cellulose by Basidiomycetous Fungi. FEMS Microbiol. Rev. 2008, 32, 501–521. [Google Scholar] [CrossRef] [PubMed]
- De Miccolis Angelini, R.M.; Landi, L.; Raguseo, C.; Pollastro, S.; Faretra, F.; Romanazzi, G. Tracking of Diversity and Evolution in the Brown Rot Fungi Monilinia Fructicola, Monilinia Fructigena, and Monilinia Laxa. Front. Microbiol. 2022, 13, 680. [Google Scholar] [CrossRef]
- De Wit, P.J.G.M. Apoplastic fungal effectors in historic perspective; a personal view. New Phytol. 2016, 212, 805–813. [Google Scholar] [CrossRef]
- Tanaka, S.; Kahmann, R. Cell wall-associated effectors of plant-colonizing fungi. Mycologia 2021, 113, 247–260. [Google Scholar] [CrossRef]
- Marcet-Houben, M.; Villarino, M.; Vilanova, L.; De Cal, A.; van Kan, J.A.L.; Usall, J.; Gabaldón, T.; Torres, R. Comparative Genomics Used to Predict Virulence Factors and Metabolic Genes among Monilinia Species. J. Fungi 2021, 7, 464. [Google Scholar] [CrossRef]
- Medema, M.H.; Kottmann, R.; Yilmaz, P.; Cummings, M.; Biggins, J.B.; Blin, K.; de Bruijn, I.; Chooi, Y.H.; Claesen, J.; Coates, R.C.; et al. Minimum Information about a Biosynthetic Gene cluster. Nat. Chem. Biol. 2015, 11, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Kautsar, S.A.; Blin, K.; Shaw, S.; Navarro-Muñoz, J.C.; Terlouw, B.R.; van der Hooft, J.J.J.; van Santen, J.A.; Tracanna, V.; Suarez Duran, H.G.; Pascal Andreu, V.; et al. MIBiG 2.0: A repository for biosynthetic gene clusters of known function. Nucleic Acids Res. 2020, 48, D454–D458. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region | Orchard | Sampled Tree | Plant Material | Morphology | PCR (ITS) | RT-PCR |
|---|---|---|---|---|---|---|
| North | A | 1 | flowers | x | ||
| branch | x | |||||
| 2 | branch | x | ||||
| 3 | x | |||||
| 4 | x | |||||
| 5 | x | |||||
| 6 | x | |||||
| 7 | x | |||||
| 8 | x | |||||
| 9 | x | |||||
| 10 | x | |||||
| 11 | x | |||||
| 12 | x | |||||
| B | 1 | flowers | x | |||
| branch | x | |||||
| 2 | branch | x | x | x | ||
| 3 | x | |||||
| 4 | x | |||||
| 5 | x | x | x | |||
| 6 | x | |||||
| 7 | x | |||||
| 8 | x | x | x | |||
| C | 1 | branch | x | |||
| 2 | x | |||||
| D | 1 | branch | x | |||
| E | 1 | branch | x | |||
| 2 | x | |||||
| F | 1 | branch | x | x | x | |
| G | 1 | branch | x | |||
| H | 1 | branch | x | |||
| 2 | x | |||||
| I | 1 | branch | x | |||
| 2 | x | |||||
| 3 | x | |||||
| J | 1 | branch | x | |||
| 2 | x | |||||
| 3 | x | |||||
| 4 | x | |||||
| K | 1 | branch | x | |||
| South | M | 1 | flowers | x | ||
| branch | x | |||||
| 2 | branch | x | ||||
| 3 | branch | x | ||||
| N | 1 | flowers | x | |||
| branch | x | |||||
| 2 | branch | x |
| Region | Orchard | Sampled Tree | Plant Material | Morphology | PCR (ITS) | RT- PCR |
|---|---|---|---|---|---|---|
| North | A | 6 | branch | x | ||
| B | 1 | flowers | x | |||
| branch | x | |||||
| 2 | branch | x | x | x | ||
| 4 | branch | x | ||||
| 5 | branch | x | x | x | ||
| 7 | branch | x | ||||
| 8 | branch | x | x | x | ||
| E | 1 | branch | x | |||
| F | 2 | branch | x | x | x |
| #Seq_Name | Length | Cov. | Circ. | Repeat |
|---|---|---|---|---|
| contig_26 | 4,277,937 | 88 | N | N |
| contig_24 | 3,718,282 | 89 | N | N |
| contig_28 | 3,587,349 | 88 | N | N |
| contig_8 | 3,247,611 | 88 | N | N |
| contig_2 | 2,689,953 | 91 | N | N |
| contig_30 | 2,601,703 | 89 | N | N |
| contig_7 | 2,592,405 | 91 | N | N |
| contig_9 | 2,581,112 | 89 | N | N |
| contig_21 | 2,547,891 | 90 | N | N |
| contig_14 | 2,388,942 | 90 | N | N |
| contig_11 | 2,329,967 | 90 | N | N |
| contig_29 | 2,262,380 | 88 | N | N |
| contig_13 | 2,234,929 | 91 | N | N |
| contig_25 | 2,104,283 | 92 | N | N |
| contig_20 | 1,976,944 | 93 | N | N |
| contig_5 | 1,931,526 | 92 | N | N |
| contig_17 | 355,259 | 130 | N | N |
| contig_15 | 352,833 | 106 | N | N |
| contig_27 | 325,972 | 129 | N | N |
| contig_4 | 262,987 | 122 | N | N |
| contig_31 | 155,463 | 2004 | Y | Y |
| contig_35 | 15,627 | 1304 | Y | Y |
| Info | Value |
|---|---|
| Total scaffold length | 44,541,355 |
| Largest scaffold | 4,277,937 |
| Scaffold N50 | 2,592,405 |
| Scaffold L50 | 7 |
| Initial number of reads | 1,003,591 reads |
| mean read_length | 4742.1 |
| mean qual | 12.5 |
| Reads >Q8 | 100% |
| Reads >Q10 | 90.0% |
| #N’s per 100 kbp | 0 |
| #N’s | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baltazar, E.; Rodrigues, S.; Ares, A.; Camelo, A.; Brandão, I.; Espirito Santo, C.; Trovão, J.; Garcia, E.; Costa, J. Morphological, Molecular and Genomic Identification and Characterisation of Monilinia fructicola in Prunus persica from Portugal. Agronomy 2023, 13, 1493. https://doi.org/10.3390/agronomy13061493
Baltazar E, Rodrigues S, Ares A, Camelo A, Brandão I, Espirito Santo C, Trovão J, Garcia E, Costa J. Morphological, Molecular and Genomic Identification and Characterisation of Monilinia fructicola in Prunus persica from Portugal. Agronomy. 2023; 13(6):1493. https://doi.org/10.3390/agronomy13061493
Chicago/Turabian StyleBaltazar, Elsa, Sara Rodrigues, Aitana Ares, Alexandra Camelo, Inês Brandão, Christophe Espirito Santo, João Trovão, Eva Garcia, and Joana Costa. 2023. "Morphological, Molecular and Genomic Identification and Characterisation of Monilinia fructicola in Prunus persica from Portugal" Agronomy 13, no. 6: 1493. https://doi.org/10.3390/agronomy13061493
APA StyleBaltazar, E., Rodrigues, S., Ares, A., Camelo, A., Brandão, I., Espirito Santo, C., Trovão, J., Garcia, E., & Costa, J. (2023). Morphological, Molecular and Genomic Identification and Characterisation of Monilinia fructicola in Prunus persica from Portugal. Agronomy, 13(6), 1493. https://doi.org/10.3390/agronomy13061493