
Development of SSR Markers Based on Transcriptome Sequencing and Verification of Their Conservation across Species of Ornamental Pennisetum Rich. (Poaceae)

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material and DNA Extraction
2.2. Transcriptome Sequencing of P. setaceum cv. ‘Rubrum’
2.3. SSR Prediction, Selection, and Primer Design
2.4. Genetic Diversity Analysis
2.5. Unweighted Pair Group Method with Arithmetic Mean (UPGMA) Clustering Analysis and Principal Component Analysis (PCoA) of 38 Accessions
2.6. Construction of Fingerprints
3. Results
3.1. Illumina Sequencing and De Novo Transcriptome Assembly
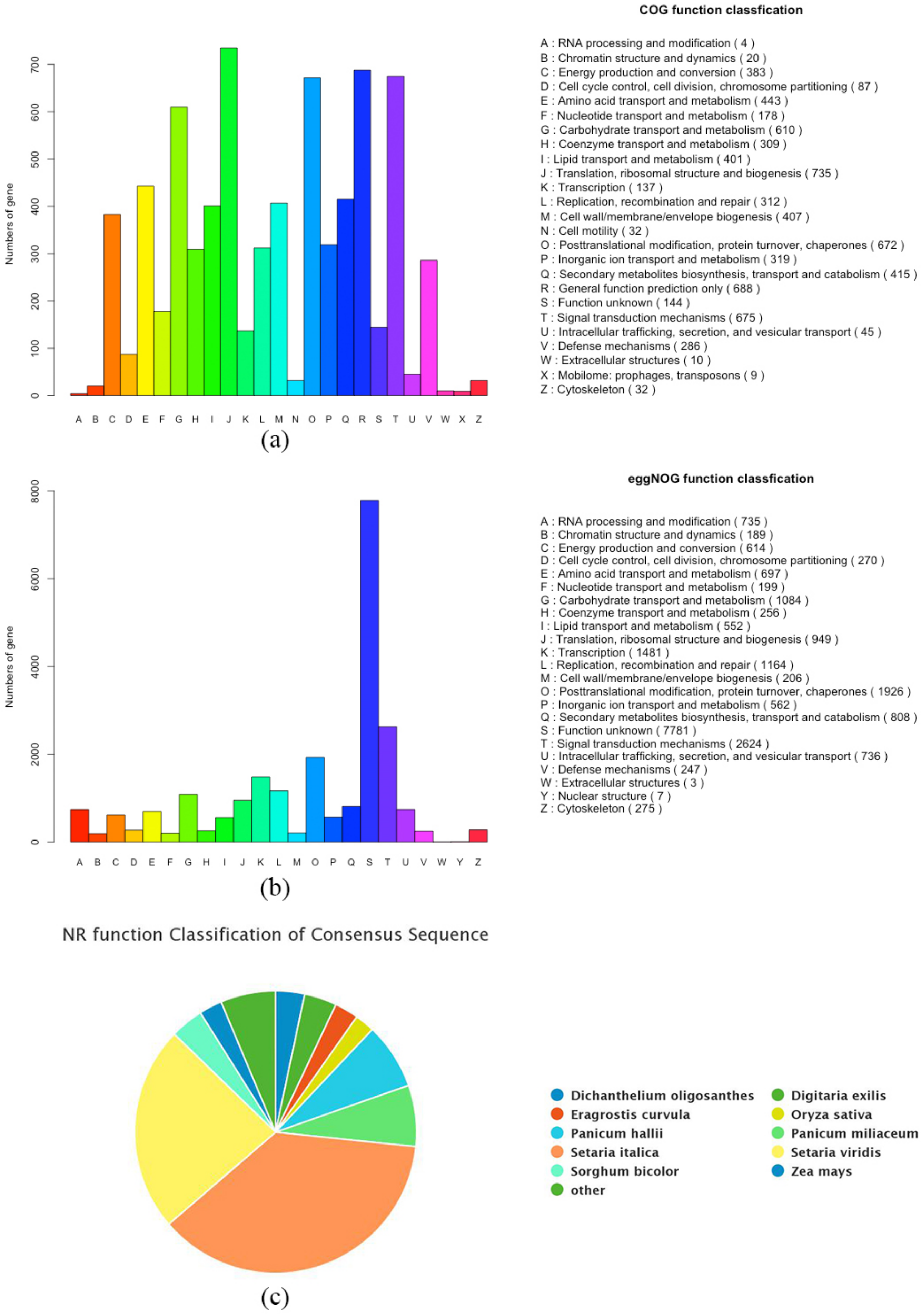
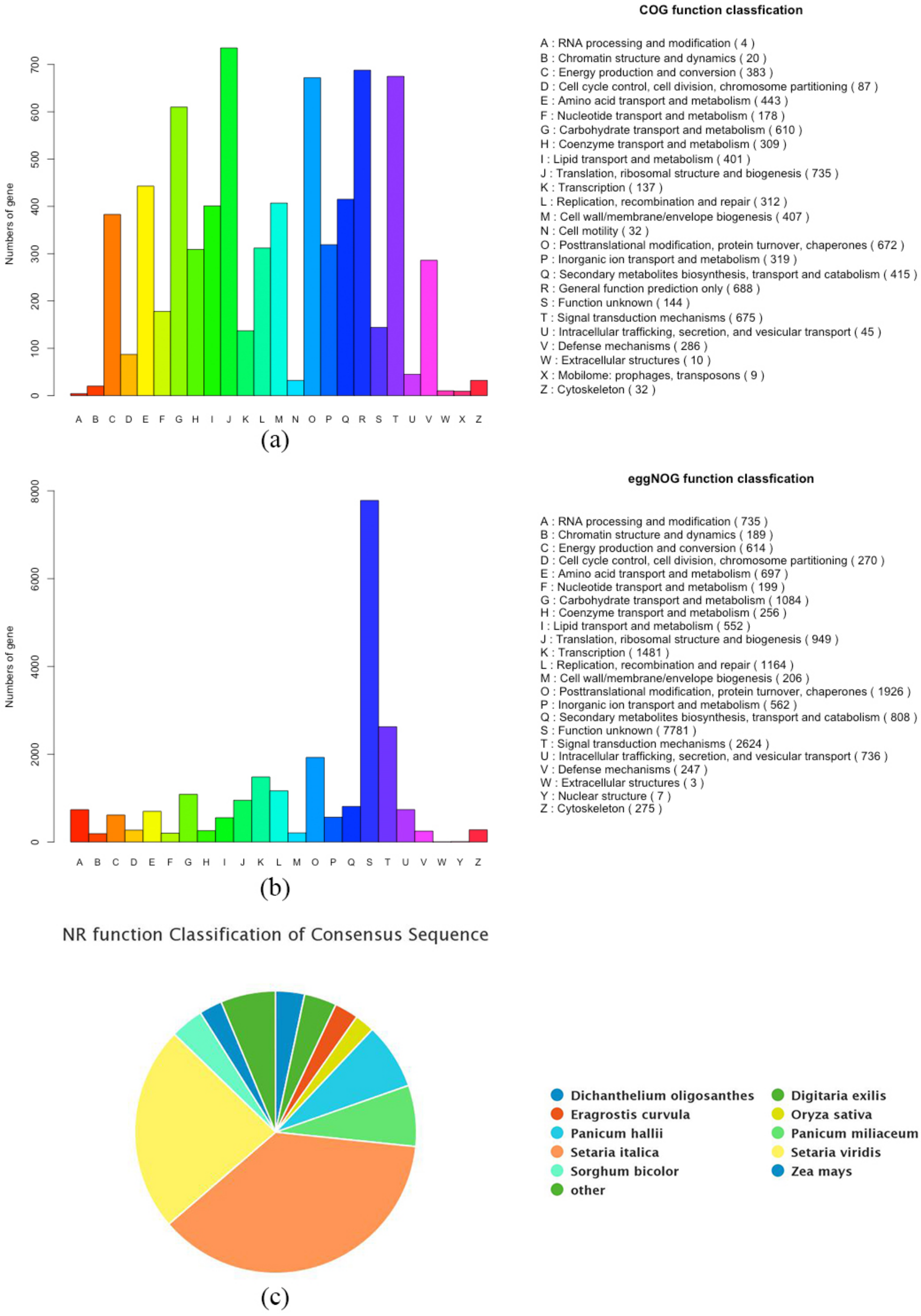
3.2. Functional Gene Annotation
3.3. Development of Novel SSRs and Analysis of Their Conservation
3.4. Genetic Diversity Statistics
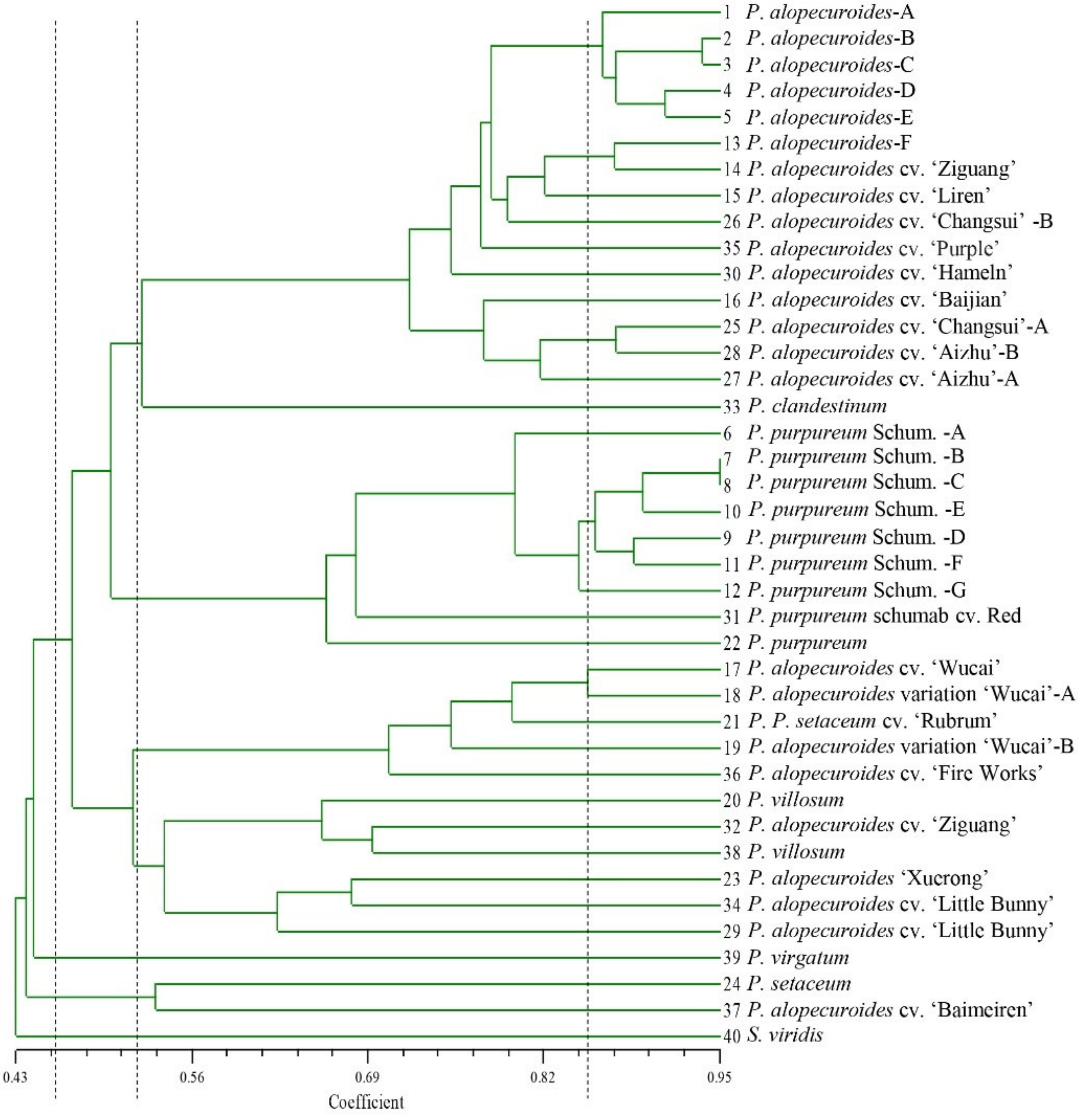
3.5. Cluster Analysis of 38 Pennisetum Accessions
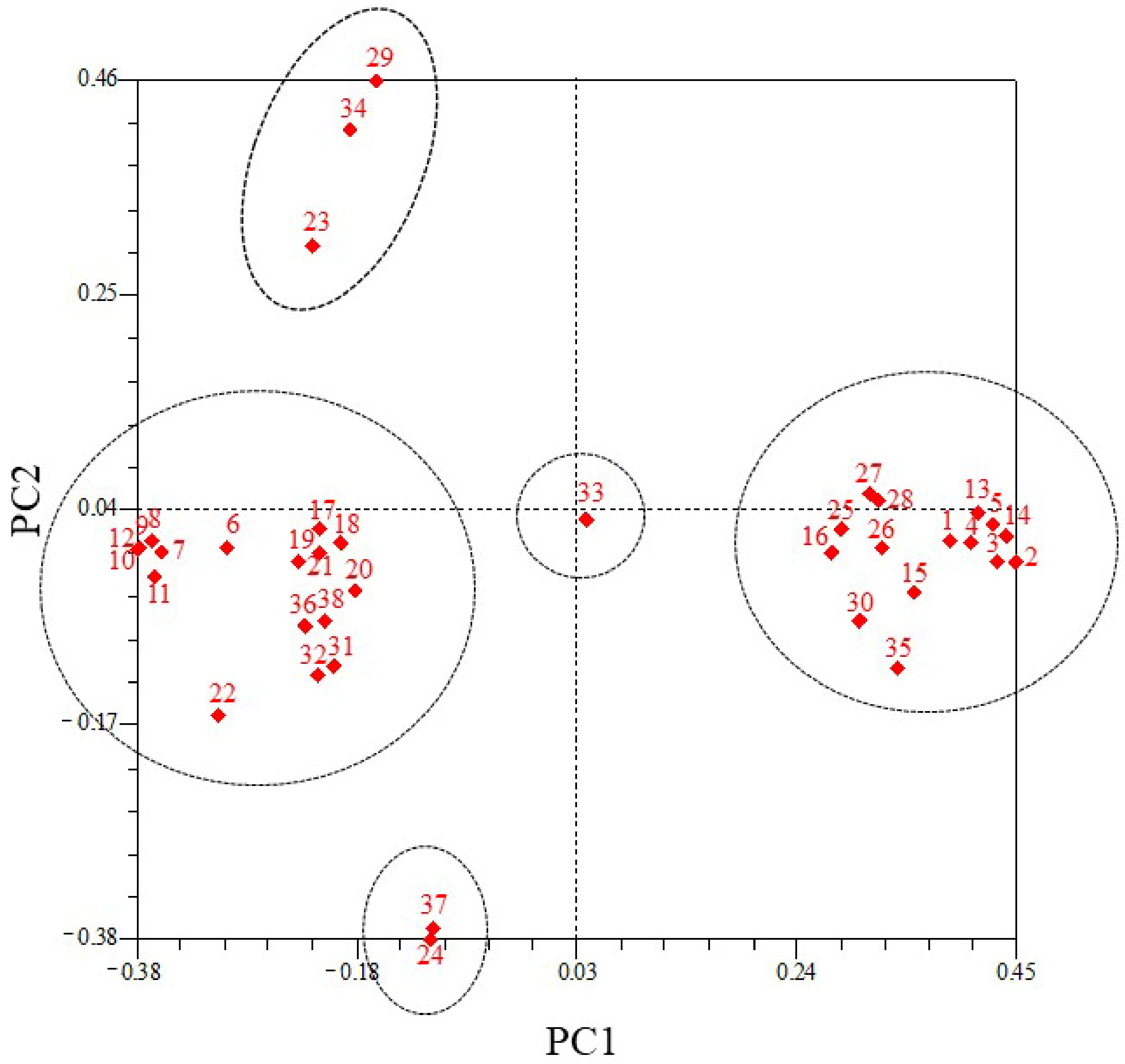
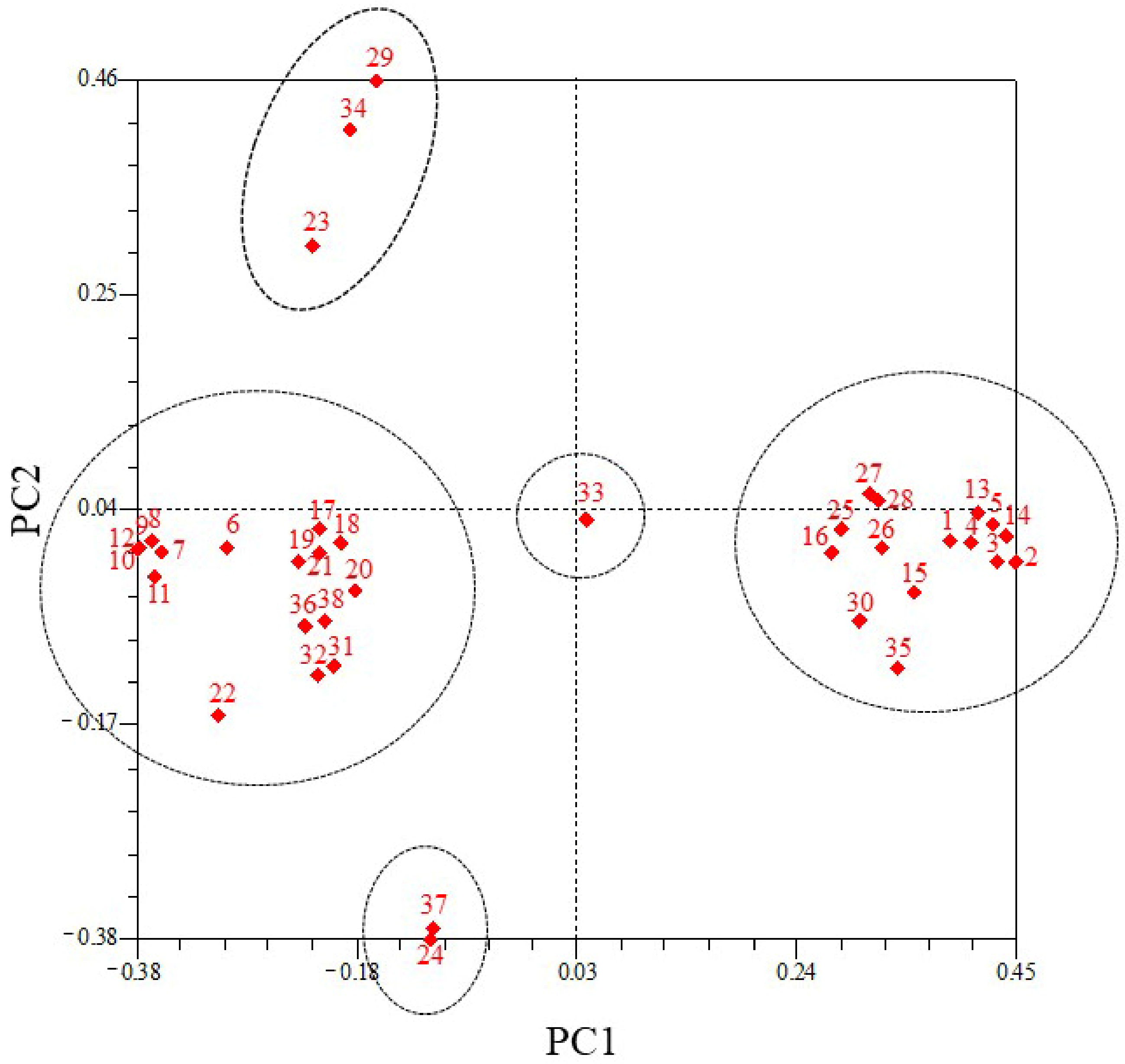
3.6. PCoA of 38 Pennisetum Accessions
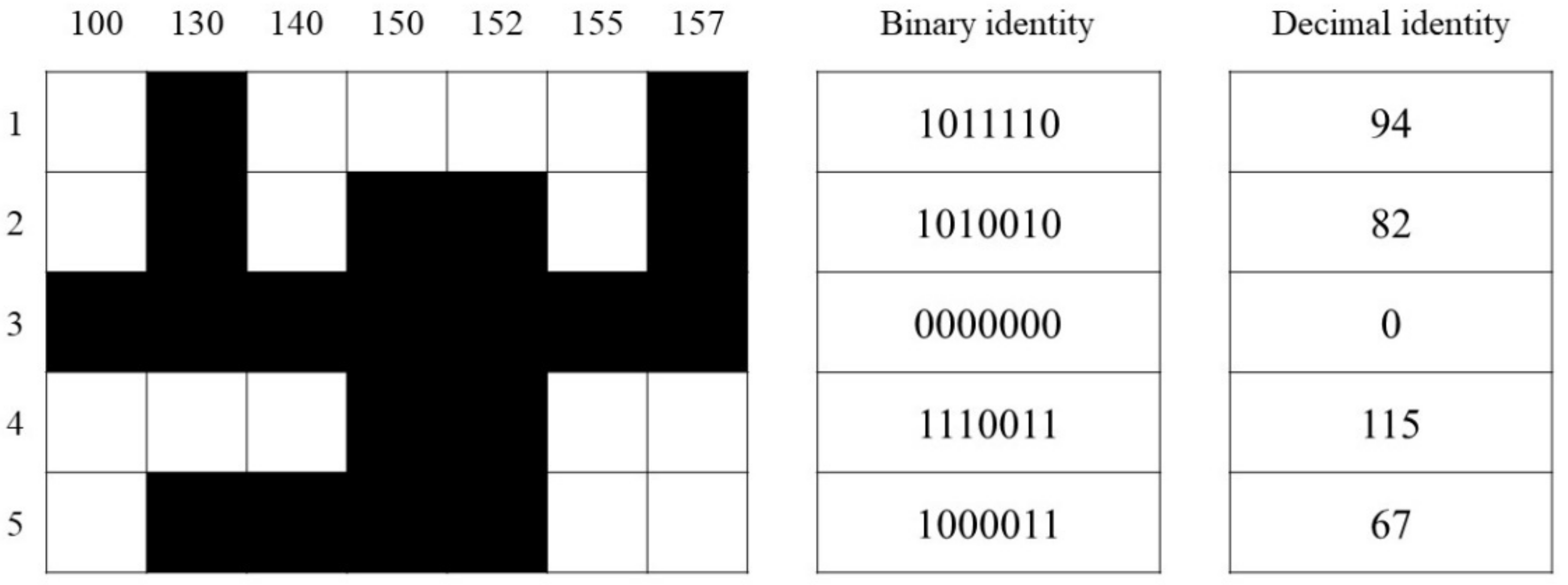
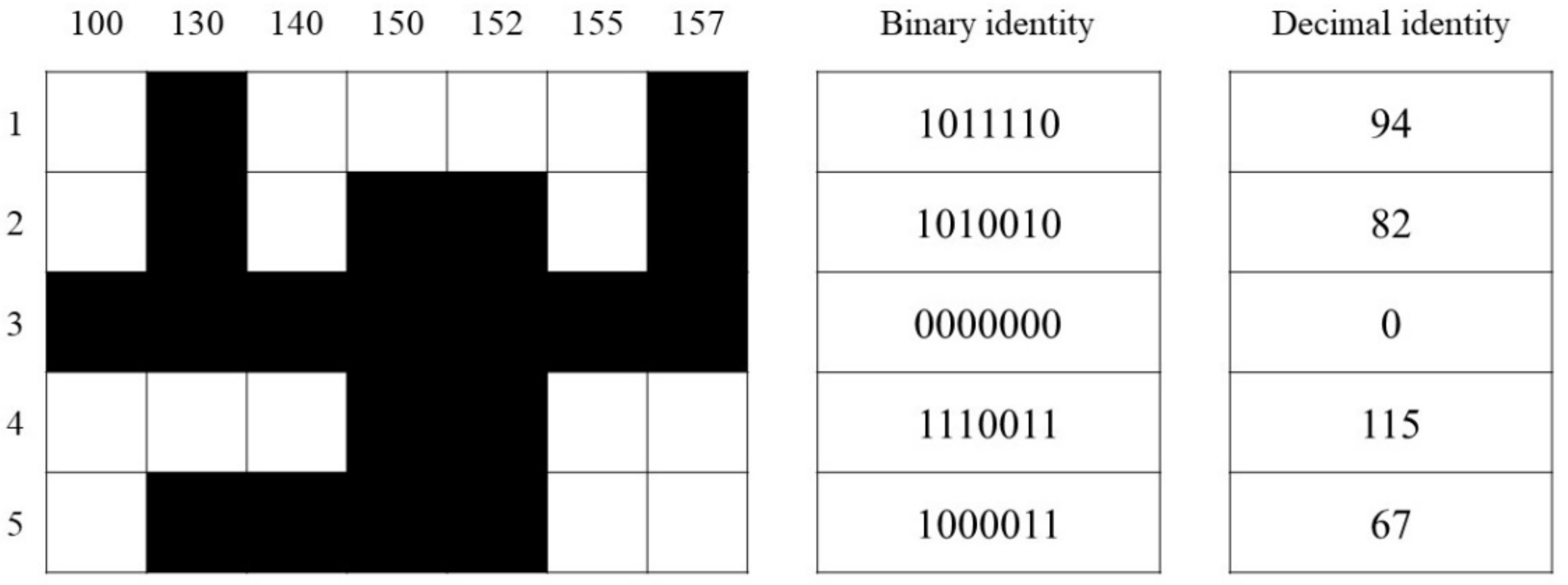
3.7. Fingerprint of Pennisetum Varieties with Similar Genetic Background
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Robert, T.; Khalfallah, N.; Martel, E.; Lamy, F.; Sarr, A. Pennisetum. In Wild Crop Relatives: Genomic and Breeding Resources; Springer: Berlin/Heidelberg, Germany, 2010; pp. 217–255. [Google Scholar]
- Tenorio, C.; Moya, R.; Tomazello, M.; Valaert, J. Quality of Pellets Made from Agricultural and Forestry Crops in Costa Rican Tropical Climates. Bioresources 2015, 10, 482–498. [Google Scholar] [CrossRef]
- Xu, J.; Liu, C.; Song, Y.; Li, M. Comparative Analysis of the Chloroplast Genome for Four Pennisetum Species: Molecular Structure and Phylogenetic Relationships. Front. Genet. 2021, 12, 687844. [Google Scholar] [CrossRef] [PubMed]
- Hayat, K.; Bundschuh, J.; Jan, F.; Menhas, S.; Zhou, P. Combating soil salinity with combining saline agriculture and phytomanagement with salt-accumulating plants. Crit. Rev. Env. Sci. Tec. 2019, 50, 1085–1115. [Google Scholar] [CrossRef]
- Gallego, L.J.; Escobar, A.; Penuela, M.; Pena, J.D.; Rios, L.A. King Grass: A promising material for the production of second-generation butanol. Fuel 2015, 143, 399–403. [Google Scholar] [CrossRef]
- Zhang, Y.; Yuan, X.; Teng, W.; Chen, C.; Wu, J. Identification and Phylogenetic Classification of Pennisetum (Poaceae) Ornamental Grasses Based on SSR Locus Polymorphisms. Plant Mol. Biol. Rep. 2016, 34, 1181–1192. [Google Scholar] [CrossRef]
- Wu, J.; Teng, W.; Wang, Q. Basic Botanic Characters, Adaptabilities and Applying in Landscape Architecture of Pennisetum alopecuroides. Chin. Landsc. Archit. 2005, 21, 57–59. [Google Scholar]
- Song, X.; Zhong, Y.; Zhang, Q. Preliminary Studies on the Application of Ornamental Grassland in Landscape. Chin. Landsc. Archit. 2004, 3, 35–39. [Google Scholar]
- Yue, Y.; Fan, X.; Hu, Y.; Han, C.; Wu, J. In vitro induction and characterization of hexaploid Pennisetum × advena, an ornamental grass. Plant Cell Tiss. Org. 2020, 142, 221–228. [Google Scholar] [CrossRef]
- Owen, W.G.; Lopez, R.G. Geranium and Purple Fountain Grass Leaf Pigmentation Is Influenced by End-of-Production Supplemental Lighting with Red and Blue Light-emitting Diodes. HortScience 2017, 52, 236–244. [Google Scholar] [CrossRef] [Green Version]
- Hasanova, S.; Akparov, Z.; Mammadov, A.; Amirov, L.; Abbasov, M. Genetic diversity of chickpea genotypes as revealed by ISSR and RAPD markers. Genetika 2017, 49, 415–423. [Google Scholar] [CrossRef]
- Bortolini, F.; Agnol, M.D.; Schifino-Wittmann, M.T. Molecular Characterization of the USDA White Clover (Trifolium repens L.) Core Collection by RAPD Markers. Genet. Resour. Crop Evol. 2006, 53, 1081–1087. [Google Scholar] [CrossRef]
- Zaharieva, M.; Santoni, S.; David, J. Use of RFLP markers to study genetic diversity and to build a core-collection of the wild wheat relative Ae-geniculata Roth (=Ae ovata L.). Genet. Sel. Evol. 2001, 33, S269–S288. [Google Scholar] [CrossRef] [Green Version]
- Chavarriaga-Aguirre, P.; Maya, M.M.; Tohme, J.; Duque, M.C.; Iglesias, C.; Bonierbale, M.W.; Kresovich, S.; Kochert, G. Using microsatellites, isozymes and AFLPs to evaluate genetic diversity and redundancy in the cassava core collection and to assess the usefulness of DNA-based markers to maintain germplasm collections. Mol. Breed. 1999, 5, 263–273. [Google Scholar] [CrossRef]
- Wang, R.; Li, X.; Zhang, W.; Ou, J.; Fang, C.; Song, Q.; Zhou, H. SSR analysis and fingerprint construction to evaluate the genetic diversity of medicinal plum varieties. J. Plant Biochem. Biot. 2021, 31, 1–11. [Google Scholar] [CrossRef]
- Fang, W.P.; Meinhardt, L.W.; Tan, H.W.; Zhou, L.; Mischke, S.; Zhang, D. Varietal identification of tea (Camellia sinensis) using nanofluidic array of single nucleotide polymorphism (SNP) markers. Hortic. Res. 2014, 1, 8. [Google Scholar] [CrossRef] [Green Version]
- Kaur, S.; Panesar, P.S.; Bera, M.B.; Kaur, V. Simple Sequence Repeat Markers in Genetic Divergence and Marker-Assisted Selection of Rice Cultivars: A Review. Crit. Rev. Food Sci. 2015, 55, 41–49. [Google Scholar] [CrossRef]
- Szewc-Mcfadden, A.K.; Kresovich, S.; Bliek, S.M.; Mitchell, S.E.; Mcferson, J.R. Identification of polymorphic, conserved simple sequence repeats (SSRs) in cultivated Brassica species. Theor. Appl. Genet. 1996, 93, 534–538. [Google Scholar] [CrossRef]
- Saha, M.C.; Mian, M.; Eujayl, I.; Zwonitzer, J.C.; Wang, L.; May, G.D. Tall fescue EST-SSR markers with transferability across several grass species. Appl. Genet. 2004, 109, 783–791. [Google Scholar] [CrossRef]
- Torre, S.; Tattini, M.; Brunetti, C.; Fineschi, S.; Fini, A.; Ferrini, F.; Sebastiani, F. RNA-Seq Analysis of Quercus pubescens Leaves: De Novo Transcriptome Assembly, Annotation and Functional Markers Development. PLoS ONE 2014, 9, e112487. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Fan, X.; Tan, P.; Wu, J.; Teng, K. The development of SSR markers based on RNA-sequencing and its validation between and within Carex L. species. BMC Plant Biol. 2021, 21, 17. [Google Scholar] [CrossRef]
- Zhou, S.; Wang, C.; Yin, G.; Zhang, Y.; Huang, L. Phylogenetics and diversity analysis of Pennisetum species using Hemarthria EST-SSR marker. Grassl. Sci. 2018, 65, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Yan, H.; Ji, L.; Zhou, S.; Liu, T.; Zhang, X.; Huang, L. Genome survey sequencing of purple elephant grass (Pennisetum purpureum Schum ‘Zise’) and identification of its SSR markers. Mol. Breed. 2018, 38, 94. [Google Scholar] [CrossRef]
- Liu, L.; Teng, K.; Fan, X.; Han, C.; Zhang, H.; Wu, J.; Chang, Z. Combination analysis of single-molecule long-read and Illumina sequencing provides insights into the anthocyanin accumulation mechanism in an ornamental grass, Pennisetum setaceum cv. rubrum. Plant Mol. Biol. 2022, 109, 159–175. [Google Scholar] [CrossRef] [PubMed]
- Borba, T.; Brondani, R.; Rangel, P.; Brondani, C. Microsatellite marker-mediated analysis of the EMBRAPA Rice Core Collection genetic diversity. Genetica 2009, 137, 293–304. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Jianqi, L.I.; Songfeng, W.U.; Zhu, Y.; Chen, Y.; Fuchu, H.E. Integrated nr Database in Protein Annotation System and Its Localization. Comput. Eng. 2006, 32, 71–72. [Google Scholar]
- Rolf, A.; Amos, B.; Wu, C.H.; Barker, W.C.; Brigitte, B.; Serenella, F.; Elisabeth, G.; Huang, H.; Rodrigo, L.; Michele, M. UniProt: The Universal Protein knowledgebase. Nucleic Acids Res. 2004, 32, D115–D119. [Google Scholar]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.; Li, K.; Huo, W.; Lu, X. De Novo Transcriptome Sequencing and Analysis to Identify Genes Involved in the Biosynthesis of Flavonoids in Abrus mollis Leaves. Russ. J. Plant Phys. 2018, 65, 333–344. [Google Scholar] [CrossRef]
- Jaime, H.C.; Damian, S.; Kristoffer, F.; Helen, C.; Davide, H.; Walter, M.C.; Thomas, R.; Mende, D.R.; Shinichi, S.; Michael, K. eggNOG 4.5: A hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2016, 44, D286–D293. [Google Scholar]
- Minoru, K.; Susumu, G.; Shuichi, K.; Yasushi, O.; Masahiro, H. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277. [Google Scholar]
- Huson, D.H.; Buchfink, B. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar]
- Yan, X.Q.; Min, L.U.; Hua-Ming, A.N.; University, G. Analysis on SSR Information in Transcriptome and Development of Molecular Markers in Rosa roxburghii. Acta Hortic. Sin. 2015, 42, 341–349. [Google Scholar]
- Guo, J.; Zhu, J.; Xie, S. Development of SSR Molecular Markers Based on Transcriptome and Analysis of Genetic Relationship of Germplasm Resources in Avocado. Acta Hortic. Sin. 2020, 47, 1552–1564. [Google Scholar] [CrossRef]
- Rohlf, F.; Rohlf, F.; Rohlf, F.; Rohlf, F.; Rohlf, F.J. NTSYS-pc, Numerical Taxonomy and Multivariate Analysis System, Version 2.1; Applied Biostatistics Inc.: New York, NY, USA, 2000. [Google Scholar]
- Wu, J.; Qian, C.; Liu, Z. De novo transcriptomic analysis for lignin synthesis in Cenchrus purpureus using RNA-seq. Acta Prataculturae Sin. 2019, 28, 150–161. [Google Scholar]
- Shivhare, R.; Lakhwani, D.; Asif, M.H.; Chauhan, P.S.; Lata, C. De novo assembly and comparative transcriptome analysis of contrasting pearl millet (Pennisetum glaucum L.) genotypes under terminal drought stress using illumina sequencing. Nucleus 2020, 63, 341–352. [Google Scholar] [CrossRef]
- Ambika, D.; Harshraj, S.; Daisuke, T.; Liu, S.; Tetsuo, T.; Singh, Y.R. Transcriptomic analysis reveals the differentially expressed genes and pathways involved in drought tolerance in pearl millet [Pennisetum glaucum (L.) R. Br]. PLoS ONE 2018, 13, e195908. [Google Scholar]
- Wang, Z.; Yu, G.; Shi, B.; Wang, X.; Qiang, H.; Gao, H.; Zhou, F. Development and Characterization of Simple Sequence Repeat (SSR) Markers Based on RNA-Sequencing of Medicago sativa and in silico Mapping onto the M. truncatula Genome. PLoS ONE 2014, 9, e92029. [Google Scholar] [CrossRef]
- Zhao, N.; Yuan, X.; Miao, F. Development of SSR Molecular Markers Based on Transcriptome Data of Pinus kesiya var. langbianensis. Biotechnol. Bull. 2017, 33, 71–77. [Google Scholar]
- Li, Q.; Liu, T.; Zhang, S. Development and Application of Survey Sequencing and gSSR Molecular Marker of Pennisetum glaucum. Mol. Plant Breed. 2019, 17, 5375–5382. [Google Scholar]
- Gupta, P.K.; Rustgi, S.; Sharma, S.; Singh, R.; Kumar, N.; Balyan, H.S. Transferable EST-SSR markers for the study of polymorphism and genetic diversity in bread wheat. Mol. Genet. Genom. 2003, 270, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, Y.; Fan, S. Genetic Diversity Analysis of 73 Vitis amurensis and Its Hybrids Offsprings Based on SSR Molecular Markers. Biotechnol. Bull. 2021, 37, 189–197. [Google Scholar]
- Tan, L.Q.; Peng, M.; Xu, L.Y.; Wang, L.Y.; Chen, S.X.; Zou, Y.; Qi, G.N.; Cheng, H. Fingerprinting 128 Chinese clonal tea cultivars using SSR markers provides new insights into their pedigree relationships. Tree Genet. Genomes 2015, 11, 90. [Google Scholar] [CrossRef]
- Schüller, E.; Fernández, F.F.; Antanaviciute, L.; Anhalt-Brüderl, U.; Forneck, A. Autochthonous Austrian Varieties of Prunus avium L. Represent a Regional Gene Pool, Assessed Using SSR and AFLP Markers. Genes 2021, 12, 322. [Google Scholar] [CrossRef] [PubMed]
- Arango, J.; Lopez, A.; Marquez, E.; Echeverri, J. Development and validation of microsatellite markers for kikuyu grass using next generation sequencing technology. Crop Pasture Sci. 2022, 73, 415–424. [Google Scholar] [CrossRef]
- Zhang, Y.; Dai, S.; Yan, H.; Song, X.; Fang, D.D. Application of Genomic SSR Locus Polymorphisms on the Identification and Classification of Chrysanthemum Cultivars in China. PLoS ONE 2014, 9, e104856. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Sample Name | Sampling Location |
|---|---|---|
| 1 | P. alopecuroides—A | Beijing |
| 2 | P. alopecuroides—B | Beijing |
| 3 | P. alopecuroides—C | Beijing |
| 4 | P. alopecuroides—D | Beijing |
| 5 | P. alopecuroides—E | Beijing |
| 6 | P. purpureum Schum. —A | Beijing |
| 7 | P. purpureum Schum. —B | Beijing |
| 8 | P. purpureum Schum. —C | Beijing |
| 9 | P. purpureum Schum. —D | Beijing |
| 10 | P. purpureum Schum. —E | Beijing |
| 11 | P. purpureum Schum. —F | Beijing |
| 12 | P. purpureum Schum. —G | Beijing |
| 13 | P. alopecuroides—F | Anhui |
| 14 | P. alopecuroides cv. ‘Ziguang’ | Beijing |
| 15 | P. alopecuroides cv. ‘Liren’ | Beijing |
| 16 | P. alopecuroides cv. ‘Baijian’ | Beijing |
| 17 | P. alopecuroides cv. ‘Wucai’ | Beijing |
| 18 | P. alopecuroides variation ‘Wucai’ —A | Beijing |
| 19 | P. alopecuroides variation ‘Wucai’ —B | Beijing |
| 20 | P. villosum | Beijing |
| 21 | P. setaceum cv. ‘Rubrum’ | Beijing |
| 22 | P. purpureum | Beijing |
| 23 | P. orientale cv. ‘Xuerong’ | Beijing |
| 24 | P. setaceum | Beijing |
| 25 | P. alopecuroides cv. ‘Changsui’ —A | Beijing |
| 26 | P. alopecuroides cv. ‘Changsui’ —B | Beijing |
| 27 | P. alopecuroides cv. ‘Aizhu’ —A | Beijing |
| 28 | P. alopecuroides cv. ‘Aizhu’ —B | Beijing |
| 29 | P. alopecuroides cv. ‘Little Bunny’ | Beijing |
| 30 | P. alopecuroides cv. ‘Hameln’ | Beijing |
| 31 | P. purpureum schumab cv. Red | Kunming |
| 32 | P. alopecuroides cv. ‘Ziguang’ | Kunming |
| 33 | P. clandestinum | Kunming |
| 34 | P. alopecuroides cv. ‘Little Bunny’ | Kunming |
| 35 | P. alopecuroides cv. ‘Purple’ | Kunming |
| 36 | P. alopecuroides cv. ‘Fire Works’ | Kunming |
| 37 | P. alopecuroides cv. ‘Baimeiren’ | Kunming |
| 38 | P. villosum | Kunming |
| 39 | P. virgatum | Beijing |
| 40 | S. viridis | Beijing |
| Database | Annotated Number | 300 ≤ Length < 1000 | Length ≥ 1000 |
|---|---|---|---|
| COG Annotation | 6606 | 1553 | 5053 |
| GO Annotation | 22,845 | 8859 | 13,986 |
| KEGG Annotation | 17,384 | 5855 | 11,529 |
| KOG Annotation | 13,498 | 4152 | 9346 |
| Pfam Annotation | 18,502 | 5282 | 13,220 |
| Swissprot Annotation | 16,727 | 5347 | 11,380 |
| eggNOG Annotation | 23,365 | 8827 | 14,538 |
| Nr Annotation | 30,020 | 12,302 | 17,718 |
| All Annotated | 30,930 | 12,999 | 17,931 |
| Type | Number | Proportion |
|---|---|---|
| Mono nucleotide | 2011 | 39.26% |
| Di nucleotide | 753 | 14.70% |
| Tri nucleotide | 2148 | 41.94% |
| Tetra nucleotide | 42 | 0.82% |
| Penta nucleotide | 11 | 0.21% |
| Hexa nucleotide | 2 | 0.04% |
| c 1 | 153 | 2.99% |
| c* 2 | 2 | 0.04% |
| Total number of identified SSRs | 5122 |
| Primer ID | Polymorphism | Forward Primer Sequence (5–3) | Reverse Primer Sequence (5–3) |
|---|---|---|---|
| PaSSR—1 | Yes | TATACTTGGTTGCCACGGGT | TTCATGGTGATGCGTCATTT |
| PaSSR—2 | Yes | AACCCCTAGCAGTCTCTCCC | GCGGTACTCGTACTGCTTGA |
| PaSSR—3 | No | TCCATGGAGTACCCGAAGAG | ACATCAACCACTGCAACCAA |
| PaSSR—4 | No | AAAATTAGGTCCGCTTGCCT | GACCGATTCCAATTCCGTTA |
| PaSSR—5 | No | CCCCTTTTTCTCTCACTCCC | CCACCAATTTGCCTTTCAGT |
| PaSSR—6 | No | AAAGAAAGAAAAGAAAACGCACA | CCTAGCTTGTCTGCCTCCTG |
| PaSSR—7 | Yes | GCGAGGAGATTCAGAGATCG | GGACGAACAAAGAGACCGAG |
| PaSSR—8 | Yes | TATGGGTTGCTCCTCGAATC | ATTGAACAGCTTCTGCGGAT |
| PaSSR—9 | Yes | TGGATGGAGGACAGTGATGA | ACGACCAGGAAAGCCTTACA |
| PaSSR—10 | Yes | TGTTCCGATATGCCTGTTTT | CTGCAACATTCTGCATGGAC |
| PaSSR—11 | Yes | AGCTAGGCACAAAGAAGGCA | CTAGCTTCATGATGCACGGA |
| PaSSR—12 | Yes | CTTTACCCAAACAGCCCCTC | TCTGGATTAACCACTTCGGC |
| PaSSR—13 | Yes | TGGTCAGTTGTCGACTCAGG | ACGCACTTGTACTGTGGCTG |
| PaSSR—14 | Yes | GTCCACGAGAGAGGGAAGAG | GTAGCATATCCCGCCTGTGT |
| PaSSR—15 | No | GGCTCAATTTGGTGCATTCT | TATTAAACCAGGGTGGCTGC |
| PaSSR—16 | Yes | AGCAGCAACAACTGCAACAG | GCTACAGGGTTTGCCACATT |
| PaSSR—17 | Yes | GACCAGTCGCTCTCGACC | TAATCCACCTTCCAAGCCAG |
| PaSSR—18 | Yes | CTCAGAAGGGTGGGTACGAA | TGTGCCAATGCAGAGAAGTC |
| PaSSR—19 | No | TCAACCAGGCCAGATCATAA | ACGAGGCCTCTACGACAGAA |
| PaSSR—20 | No | ACCTCTGCGTGGTGAAGAAT | CTCCAGAAGTAGCAGCAGCA |
| PaSSR—21 | Yes | GCTCTCGCAGTACATCTCCC | GCCACTTGACCTTCTCCTTG |
| PaSSR—22 | No | TCGTGGTCAAACTGATAGCG | CTCCAGAAGTAGCAGCAGCA |
| PaSSR—23 | No | CAGCAAATGCAGCCTATCAA | CTGTTGGTCACTGGTCCCTT |
| PaSSR—24 | Yes | AAGGGACCAGTGACCAACAG | CCAGATTCACGAACTGACCA |
| PaSSR—25 | No | GACAAAACTACGGGGGTCAA | CGGTGGGGAAGAAAGAAAAT |
| PaSSR—26 | Yes | AGACGAGCGGAGAGGAAAC | TCCGCTCCTTGATCTTTCTG |
| PaSSR—27 | Yes | GCACCACCACCTCTCTTCTT | CGAGGAGGAAGATCTCGATG |
| PaSSR—28 | Yes | AACCTCTTCGCTTCTCTCCC | CAGCAGGCACAACTTCCAT |
| PaSSR—29 | Yes | TTCGATTGCTTGTATGCTGC | CCGCACGTAGTTGTGAGTGT |
| PaSSR—30 | Yes | TTCTTCTTCGCCGTACGAAT | GATCGAGATGGCGACAAAAT |
| PaSSR—31 | Yes | GTTCCCCTCTGTATCTGGGC | GCTGGGGAAGGAAGACCTC |
| PaSSR—32 | Yes | AGTACGGCTGCCTCGTCTAC | TAGTTGCGGTCGAGAAGGAT |
| PaSSR—33 | Yes | ATCAGGTCGGTGGTGAGAAC | CCCATCTGATGCTCCAACTT |
| PaSSR—34 | Yes | TGCAGAGAAACCAATTGCAG | CCGGTTCATAAGCTGGTGTT |
| PaSSR—35 | Yes | ATGCTCTATGCACTCCCACC | TGAACCCTGATTTGAGGTCC |
| PaSSR—36 | Yes | CCGCTGTAACTCTCAGCCAC | CACTCCTTCACTCAGCCTCC |
| PaSSR—37 | No | CGCACCTCGTTCGATTTTT | GAACAGGTGCACAGGAGGAC |
| PaSSR—38 | Yes | TTACCCTCCCAGATTGCTTG | CGTGAAAAGAATAGTCGTCCG |
| PaSSR—39 | Yes | CACCACCACCTCTCCTCTTC | GAGAAGCTCATGTCGACGG |
| PaSSR—40 | Yes | TTCCACATCTCCGCTTCTCT | CCTTGAACTTCTCCTCGTCG |
| PaSSR—41 | Yes | TCGGGAAGAAAGCTGAAAAA | CTCGCCTCCTCTCCTCTCTT |
| PaSSR—42 | Yes | GAGGCCTCTCCCTCTCTCTC | GACCAAACCCAAACCCAAC |
| PaSSR—43 | Yes | CTCCGCTCATCCTACCCTC | TGGGTTCTAGGGTTCTGTCG |
| PaSSR—44 | Yes | CCAAATTTTCCAAGCCAAAA | ACTGGTGGATCTGCGCCT |
| PaSSR—45 | Yes | GCTCTTCATCATAGCGGTGG | AGACCGAGGACGTAGAGCAG |
| PaSSR—46 | Yes | AAATGCCATGACAACTGCTG | CAAGAACGCAGACGACAAAA |
| PaSSR—47 | Yes | CGGATTTCCTACAGCGAGAG | ATACCGACAAAAACCCGACA |
| PaSSR—48 | Yes | GTGCGTCTCACACACCACA | CCAAGTGGGGATGAACAGAG |
| PaSSR—49 | No | TAGACTTCGGTCGACTCGCT | AACGAACACCTGGCGTAGAT |
| PaSSR—50 | Yes | CAGGGTGCAGTTAAGGGTTC | CCATCTGTGTTCATATGGCG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, Y.; Liu, L.; Yue, Y.; Fan, X.; Teng, W.; Zhang, H.; Gao, K.; Guan, J.; Chang, Z.; Teng, K. Development of SSR Markers Based on Transcriptome Sequencing and Verification of Their Conservation across Species of Ornamental Pennisetum Rich. (Poaceae). Agronomy 2022, 12, 1683. https://doi.org/10.3390/agronomy12071683
Guo Y, Liu L, Yue Y, Fan X, Teng W, Zhang H, Gao K, Guan J, Chang Z, Teng K. Development of SSR Markers Based on Transcriptome Sequencing and Verification of Their Conservation across Species of Ornamental Pennisetum Rich. (Poaceae). Agronomy. 2022; 12(7):1683. https://doi.org/10.3390/agronomy12071683
Chicago/Turabian StyleGuo, Yidi, Lingyun Liu, Yuesen Yue, Xifeng Fan, Wenjun Teng, Hui Zhang, Kang Gao, Jin Guan, Zhihui Chang, and Ke Teng. 2022. "Development of SSR Markers Based on Transcriptome Sequencing and Verification of Their Conservation across Species of Ornamental Pennisetum Rich. (Poaceae)" Agronomy 12, no. 7: 1683. https://doi.org/10.3390/agronomy12071683
APA StyleGuo, Y., Liu, L., Yue, Y., Fan, X., Teng, W., Zhang, H., Gao, K., Guan, J., Chang, Z., & Teng, K. (2022). Development of SSR Markers Based on Transcriptome Sequencing and Verification of Their Conservation across Species of Ornamental Pennisetum Rich. (Poaceae). Agronomy, 12(7), 1683. https://doi.org/10.3390/agronomy12071683