
DNA Methylation Correlates with the Expression of Drought-Responsive Genes and Drought Resistance in Rice

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Growth Condition
2.2. δ13C Determination
2.3. Drought Stress Treatments
2.4. Photosynthesis Rate Measurements
2.5. Stomatal Aperture Determinations
2.6. RNA Extraction
2.7. Library Preparation and Illumina Hiseq 4000 Sequencing
2.8. Read Mapping and Differential Expression Analysis
2.9. GO and KEGG Analysis
2.10. qRT-PCR
2.11. Analysis of Genetic Diversity
2.12. DNA Methylome Analysis
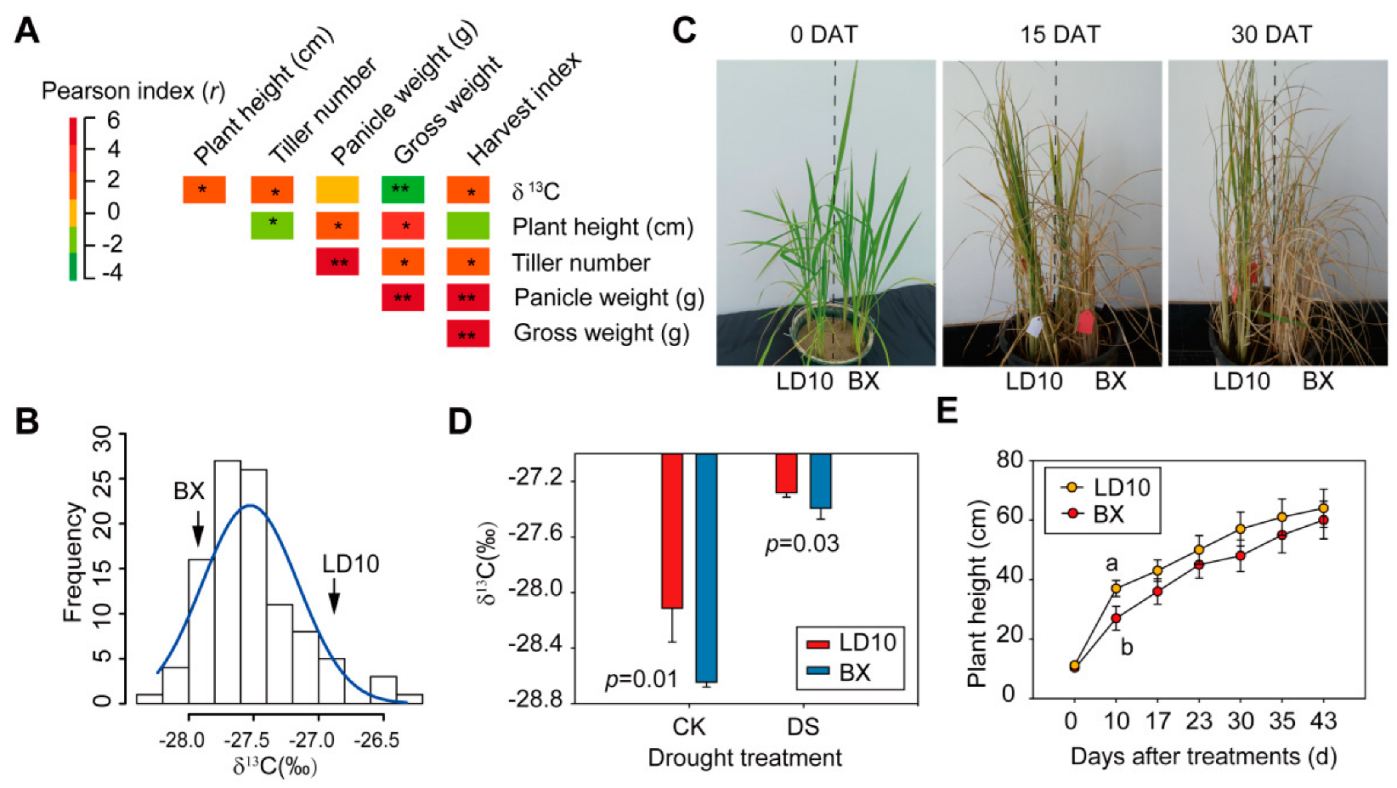
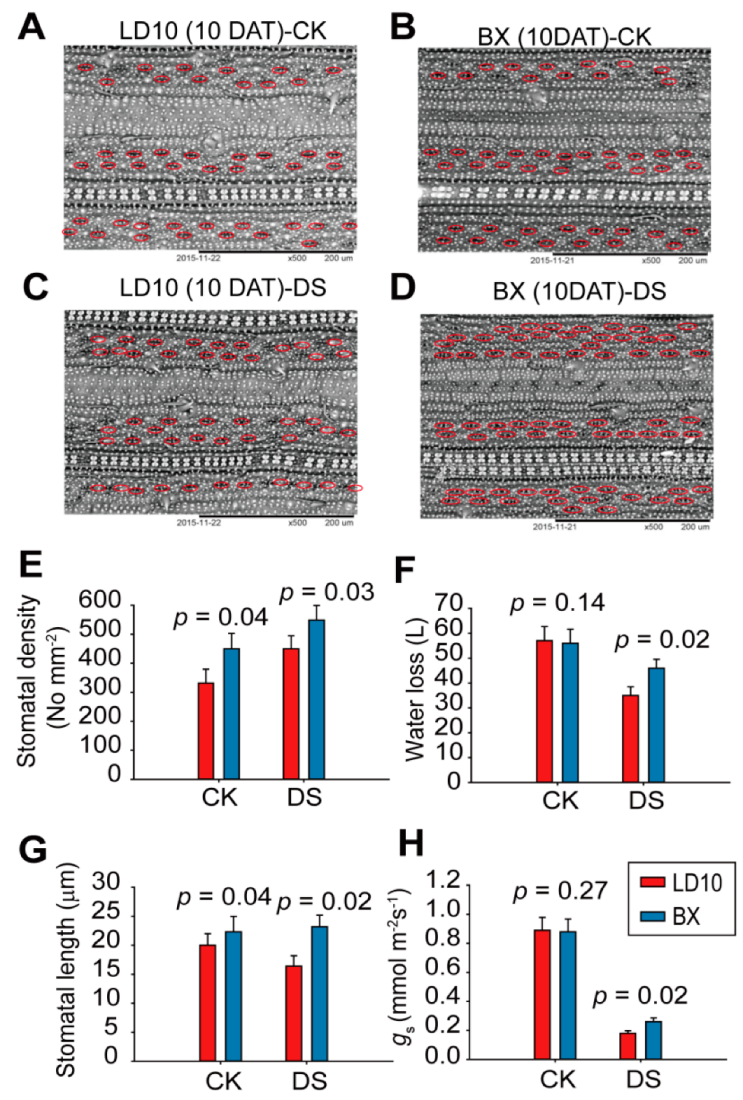
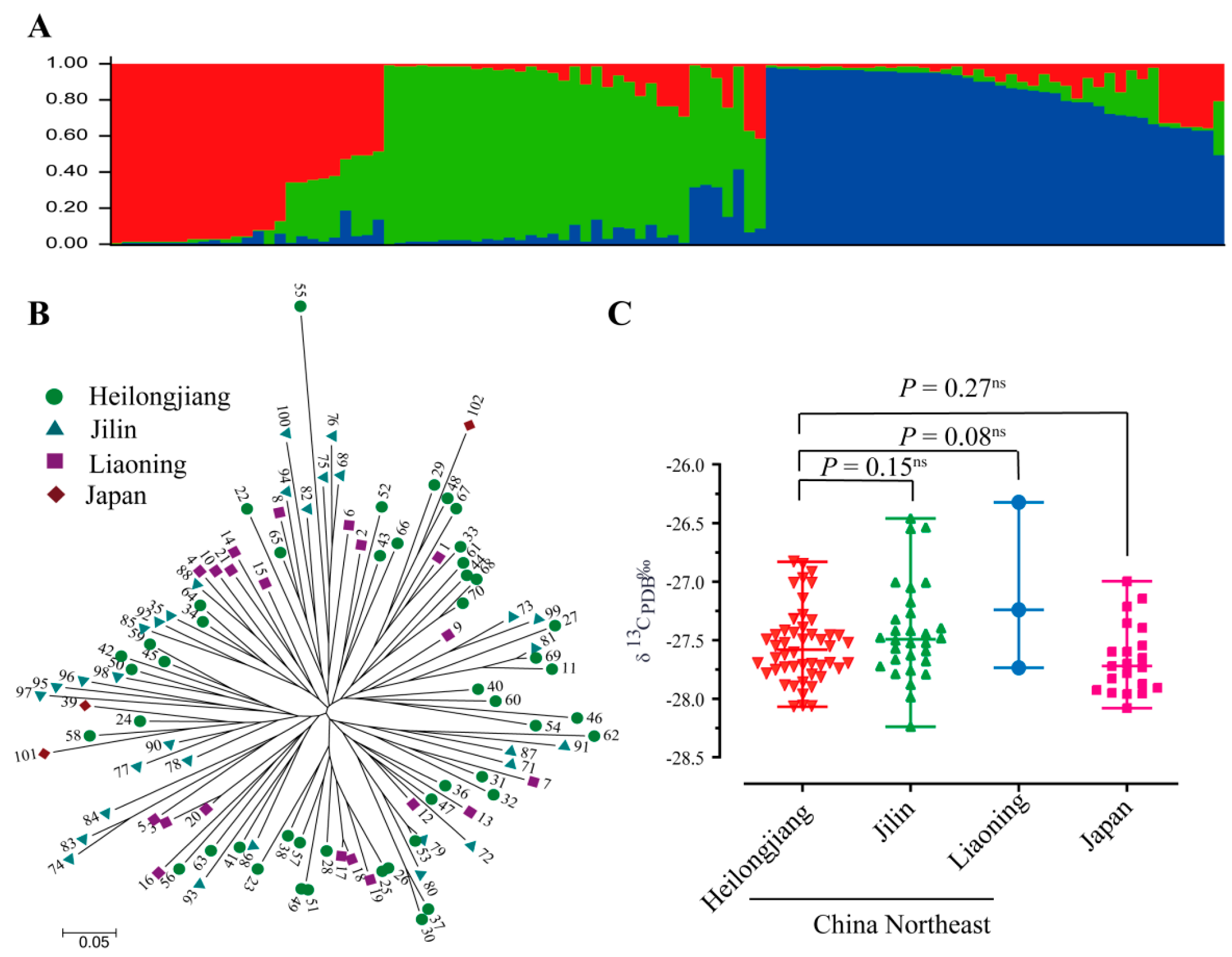
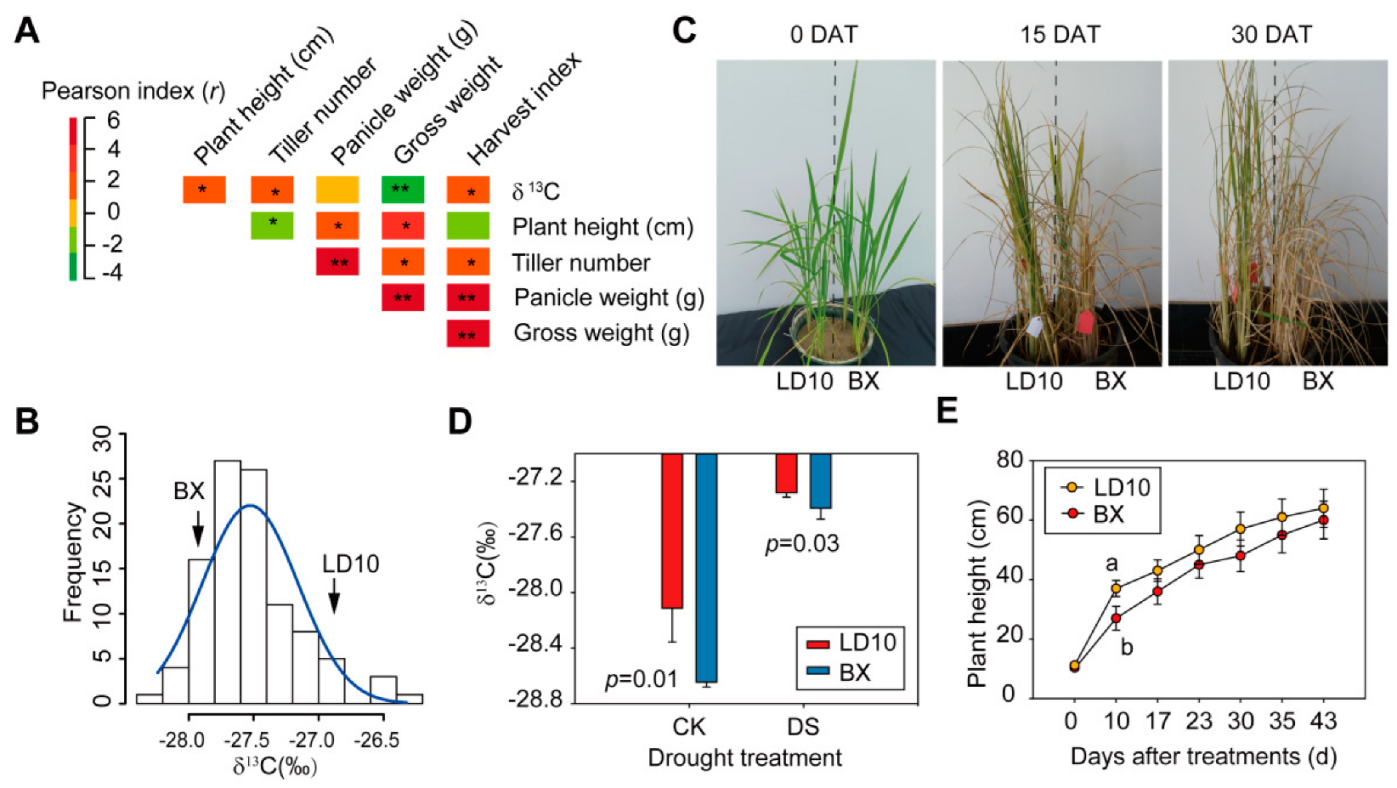
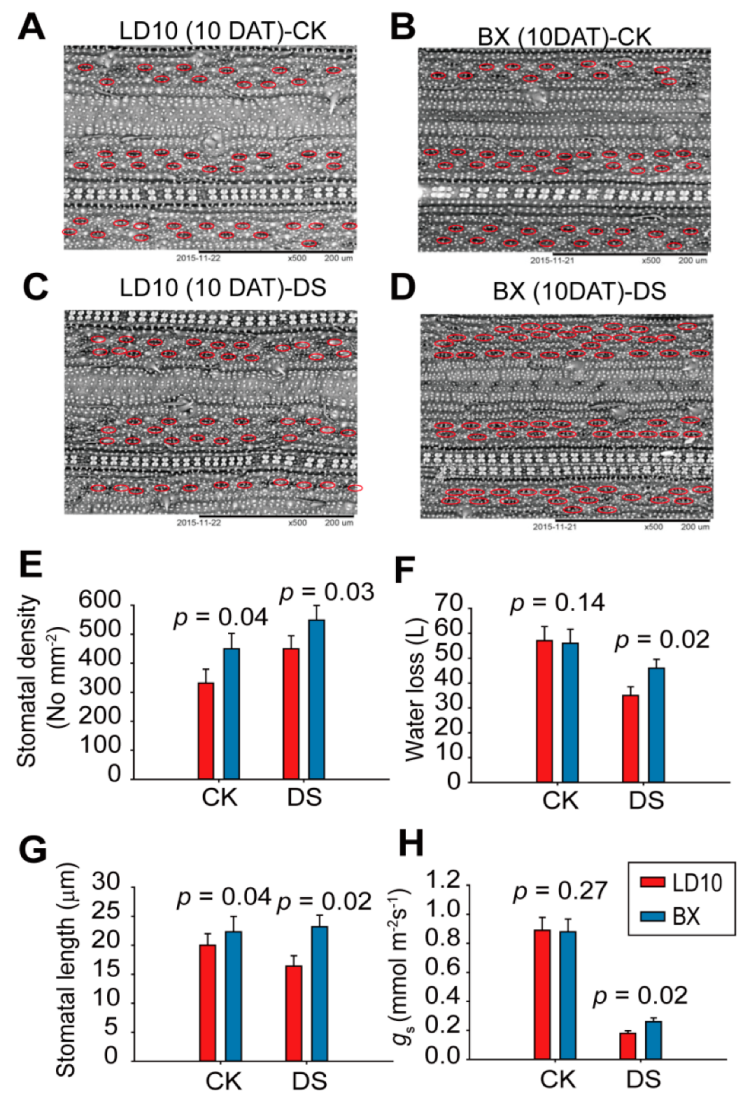
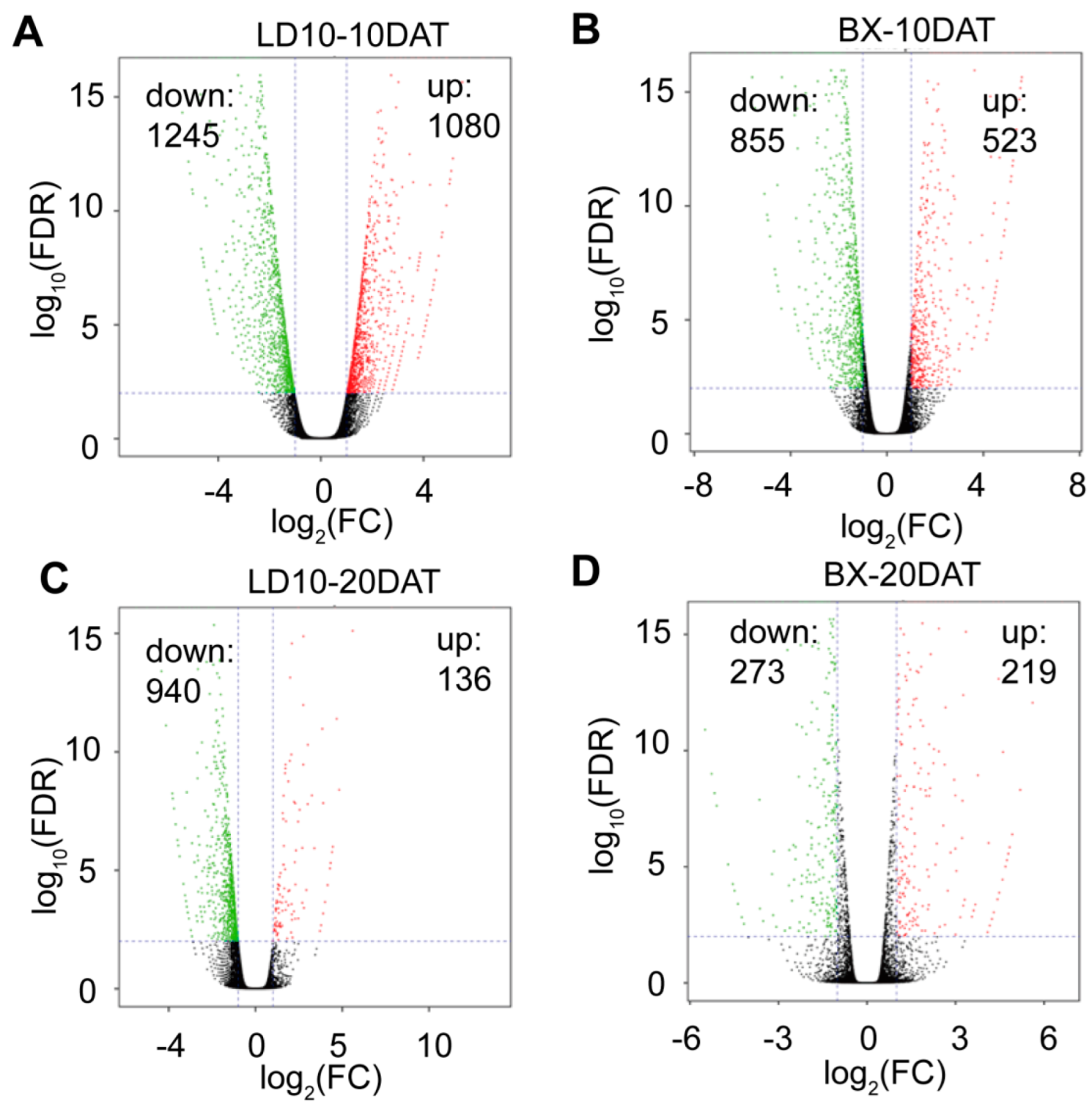
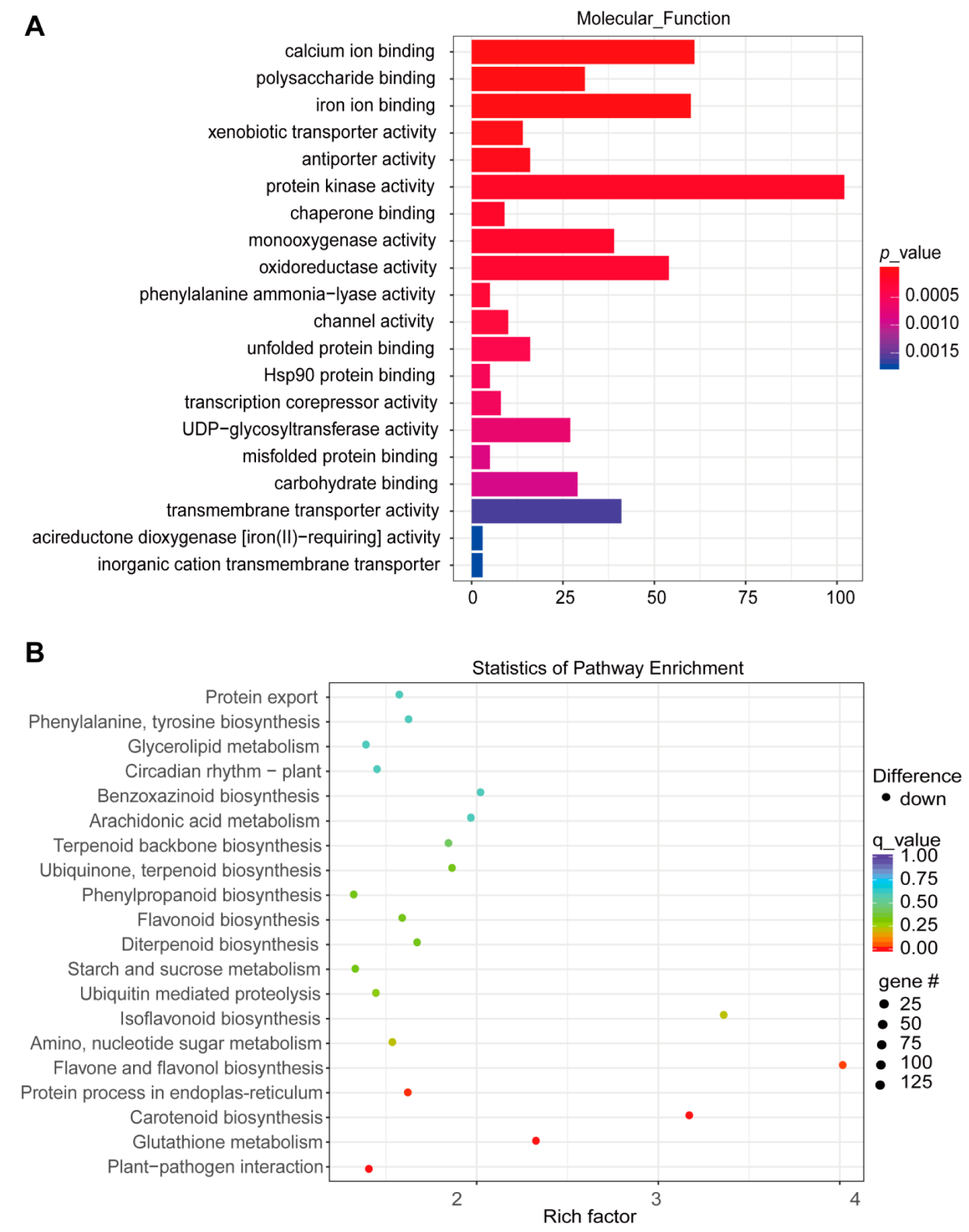
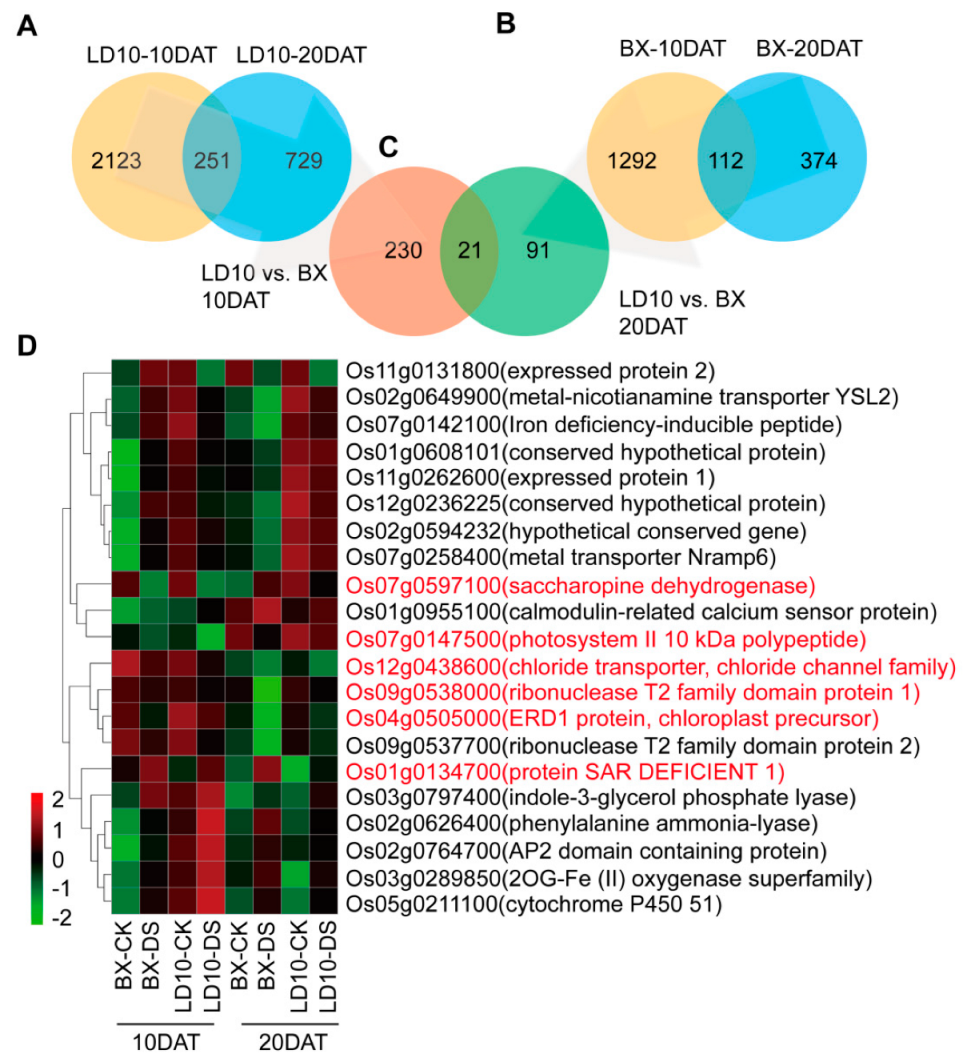
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Luo, L.J. Breeding for water-saving and drought-resistance rice (WDR) in China. J. Exp. Bot. 2010, 61, 3509–3517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; This, D.; Pausch, R.C.; Vonhof, W.M.; Coburn, J.R.; Comstock, J.P.; McCouch, S.R. Leaf-level water use efficiency determined by carbon isotope discrimination in rice seedlings: Genetic variation associated with population structure and QTL mapping. Theor. Appl. Genet. 2009, 118, 1065–1081. [Google Scholar] [CrossRef] [PubMed]
- Farquhar, G.; Richards, R. Isotopic Composition of Plant Carbon Correlates with Water-Use Efficiency of Wheat Genotypes. Funct. Plant Biol. 1984, 11, 539–552. [Google Scholar] [CrossRef]
- Qu, M.; Hamdani, S.; Li, W.; Wang, S.; Tang, J.; Chen, Z.; Song, Q.; Li, M.; Zhao, H.; Chang, T.; et al. Rapid stomatal response to fluctuating light: An under-explored mechanism to improve drought tolerance in rice. Funct. Plant Biol. 2016, 43, 727–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Zhao, X.; Zhai, L.; Shao, K.; Jiang, K.; Shen, C.; Chen, K.; Wang, S.; Wang, Y.; Xu, J. Genetic Bases of the Stomata-Related Traits Revealed by a Genome-Wide Association Analysis in Rice (Oryza sativa L.). Front. Genet. 2020, 11, 611. [Google Scholar] [CrossRef] [PubMed]
- Chater, C.; Gray, J.E. Stomatal Closure: The Old Guard Takes Up the SLAC. Curr. Biol. 2015, 25, R271–R273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.-K. Abiotic Stress Signaling and Responses in Plants. Cell 2016, 167, 313–324. [Google Scholar] [CrossRef] [Green Version]
- Qu, M.; Essemine, J.; Xu, J.; Ablat, G.; Perveen, S.; Wang, H.; Chen, K.; Zhao, Y.; Chen, G.; Chu, C.; et al. Alterations in stomatal response to fluctuating light increase biomass and yield of rice under drought conditions. Plant J. 2020, 104, 1334–1347. [Google Scholar] [CrossRef]
- Li, J.; Essemine, J.; Shang, C.; Zhang, H.; Zhu, X.; Yu, J.; Chen, G.; Qu, M.; Sun, D. Combined Proteomics and Metabolism Analysis Unravels Prominent Roles of Antioxidant System in the Prevention of Alfalfa (Medicago sativa L.) against Salt Stress. Int. J. Mol. Sci. 2020, 21, 909. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Zhang, F.; Wang, W.; Zhou, Y.; Fu, B.; Li, Z. Comparative transcriptome sequencing of tolerant rice introgression line and its parents in response to drought stress. BMC Genom. 2014, 15, 1026. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Lang, Z.; Zhu, J.-K. Dynamics and function of DNA methylation in plants. Nat. Rev. Mol. Cell Biol. 2018, 19, 489–506. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Dai, W.; Wu, C.; Song, X.; Qiang, S. Genetic diversity and origin of Japonica- and Indica-like rice biotypes of weedy rice in the Guangdong and Liaoning provinces of China. Genet. Resour. Crop Evol. 2012, 59, 399–410. [Google Scholar] [CrossRef]
- Qu, M.; Essemine, J.; Li, M.; Chang, S.; Chang, T.; Chen, G.Y.; Zhu, X.-G. Genome-wide association study unravels LRK1 as a dark respiration regulator in rice (Oryza sativa L.). Int. J. Mol. Sci. 2020, 21, 4930. [Google Scholar] [CrossRef] [PubMed]
- Qu, M.; Zheng, G.; Hamdani, S.; Essemine, J.; Song, Q.; Wang, H.; Chu, C.; Sirault, X.; Zhu, X.-G. Leaf Photosynthetic Parameters Related to Biomass Accumulation in a Global Rice Diversity Survey. Plant Physiol. 2017, 175, 248–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamdani, S.; Khan, N.; Perveen, S.; Qu, M.; Jiang, J.; Govindjee; Zhu, X.-G. Changes in the photosynthesis properties and photoprotection capacity in rice (Oryza sativa) grown under red, blue, or white light. Photosynth. Res. 2019, 139, 107–121. [Google Scholar] [CrossRef]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef]
- Wagner, G.P.; Kin, K.; Lynch, V.J. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci. 2012, 131, 281–285. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.-Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-S.; Su, W.-J.; Wang, L.-J.; Lei, J.; Chai, S.-S.; Liu, Q.-C. Molecular diversity and genetic structure of 380 sweetpotato accessions as revealed by SSR markers. J. Integr. Agric. 2015, 14, 633–641. [Google Scholar] [CrossRef]
- Shen, Y.; Zhang, J.; Liu, Y.; Liu, S.; Liu, Z.; Duan, Z.; Wang, Z.; Zhu, B.; Guo, Y.-L.; Tian, Z. DNA methylation footprints during soybean domestication and improvement. Genome Biol. 2018, 19, 128. [Google Scholar] [CrossRef] [PubMed]
- Leakey, A.D.; Ferguson, J.N.; Pignon, C.P.; Wu, A.; Jin, Z.; Hammer, G.L.; Lobell, D.B. Water Use Efficiency as a Constraint and Target for Improving the Resilience and Productivity of C3 and C4 Crops. Annu. Rev. Plant Biol. 2019, 70, 781–808. [Google Scholar] [CrossRef]
- Agrama, H.A.; Yan, W.; Lee, F.; Fjellstrom, R.; Chen, M.-H.; Jia, M.; McClung, A. Genetic Assessment of a Mini-Core Subset Developed from the USDA Rice Genebank. Crop Sci. 2009, 49, 1336–1346. [Google Scholar] [CrossRef]
- Shi, Y.; Chang, Y.-L.; Wu, H.-T.; Shalmani, A.; Liu, W.-T.; Li, W.-Q.; Xu, J.-W.; Chen, K.-M. OsRbohB-mediated ROS production plays a crucial role in drought stress tolerance of rice. Plant Cell Rep. 2020, 39, 1767–1784. [Google Scholar] [CrossRef]
- Cao, M.; Liu, X.; Zhang, Y.; Xue, X.; Zhou, X.E.; Melcher, K.; Gao, P.; Wang, F.; Zeng, L.; Zhao, Y.; et al. An ABA-mimicking ligand that reduces water loss and promotes drought resistance in plants. Cell Res. 2013, 23, 1043–1054. [Google Scholar] [CrossRef] [Green Version]
- De Angeli, A.; Zhang, J.; Meyer, S.; Martinoia, E. AtALMT9 is a malate-activated vacuolar chloride channel required for stomatal opening in Arabidopsis. Nat. Commun. 2013, 4, 1804. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, G.; Cao, L.; Zhou, J.; Li, Z.; Lai, Y.; Liu, K.; Luo, Y.; Bai, L.; Wang, X.; Wang, T.; et al. DNA Methylation Correlates with the Expression of Drought-Responsive Genes and Drought Resistance in Rice. Agronomy 2022, 12, 1445. https://doi.org/10.3390/agronomy12061445
Ding G, Cao L, Zhou J, Li Z, Lai Y, Liu K, Luo Y, Bai L, Wang X, Wang T, et al. DNA Methylation Correlates with the Expression of Drought-Responsive Genes and Drought Resistance in Rice. Agronomy. 2022; 12(6):1445. https://doi.org/10.3390/agronomy12061445
Chicago/Turabian StyleDing, Guohua, Liangzi Cao, Jinsong Zhou, Zhugang Li, Yongcai Lai, Kai Liu, Yu Luo, Liangming Bai, Xueyang Wang, Tongtong Wang, and et al. 2022. "DNA Methylation Correlates with the Expression of Drought-Responsive Genes and Drought Resistance in Rice" Agronomy 12, no. 6: 1445. https://doi.org/10.3390/agronomy12061445
APA StyleDing, G., Cao, L., Zhou, J., Li, Z., Lai, Y., Liu, K., Luo, Y., Bai, L., Wang, X., Wang, T., Wang, R., Yang, G., & Sun, S. (2022). DNA Methylation Correlates with the Expression of Drought-Responsive Genes and Drought Resistance in Rice. Agronomy, 12(6), 1445. https://doi.org/10.3390/agronomy12061445