
Analyses of Chloroplast Genome of Eutrema japonicum Provide New Insights into the Evolution of Eutrema Species



Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. DNA Extraction and Sequencing
2.3. Genome Assembly and Gene Annotation
2.4. Analysis of the Chloroplast Genome
2.5. Comparative Analysis of the Chloroplast Genome
2.5.1. Analysis of Non-Synonymous Mutation Rate (Ka), Synonymous Mutation Rates (Ks) and Ka/Ks, and Nucleotide Diversity (Pi) Value
2.5.2. Comparative Analysis of the Chloroplast Genome Structure
2.5.3. Genetic Distance Analysis
2.5.4. Phylogenetic Analysis
3. Results
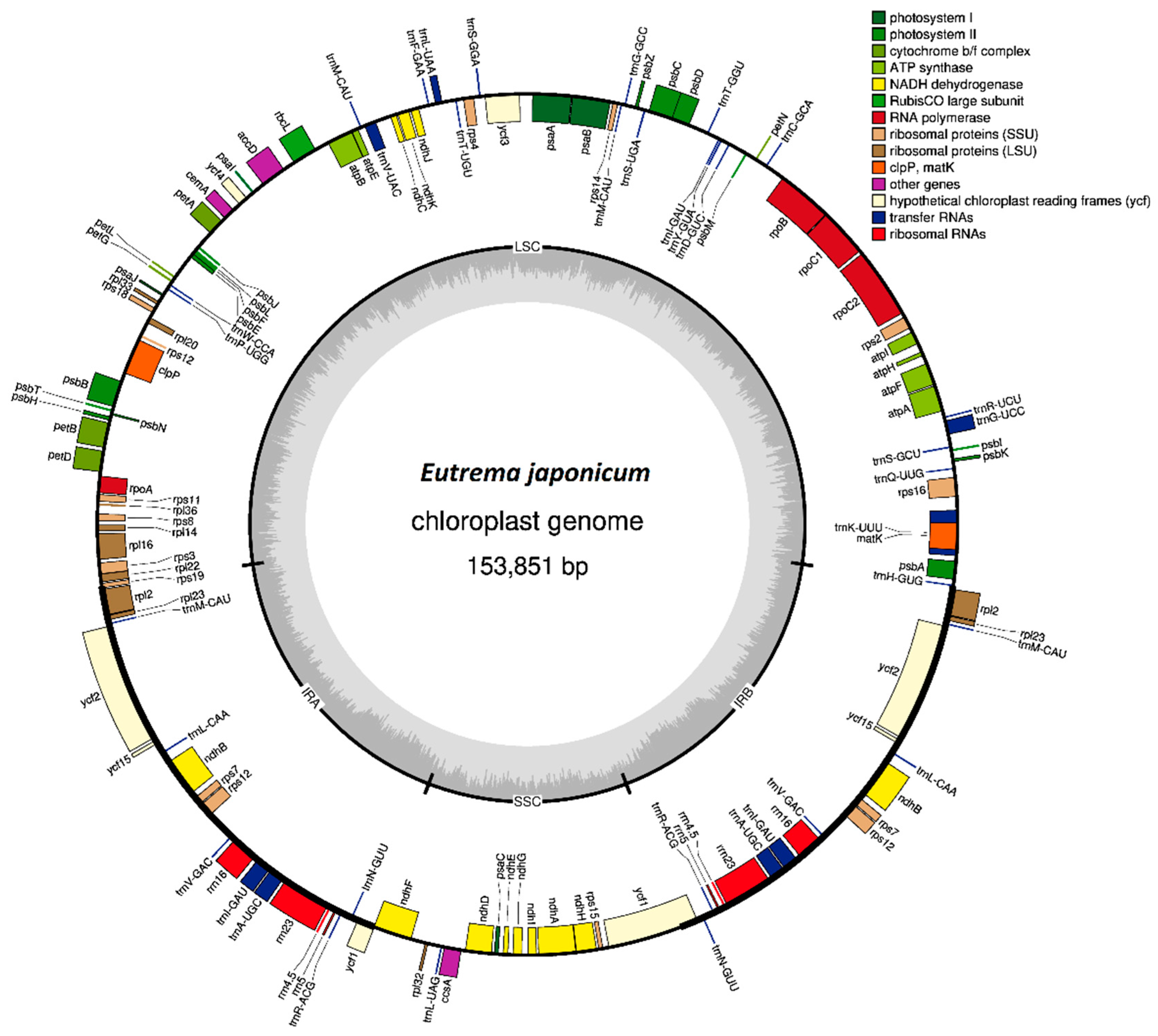
3.1. Annotation and Features of the Chloroplast Genome
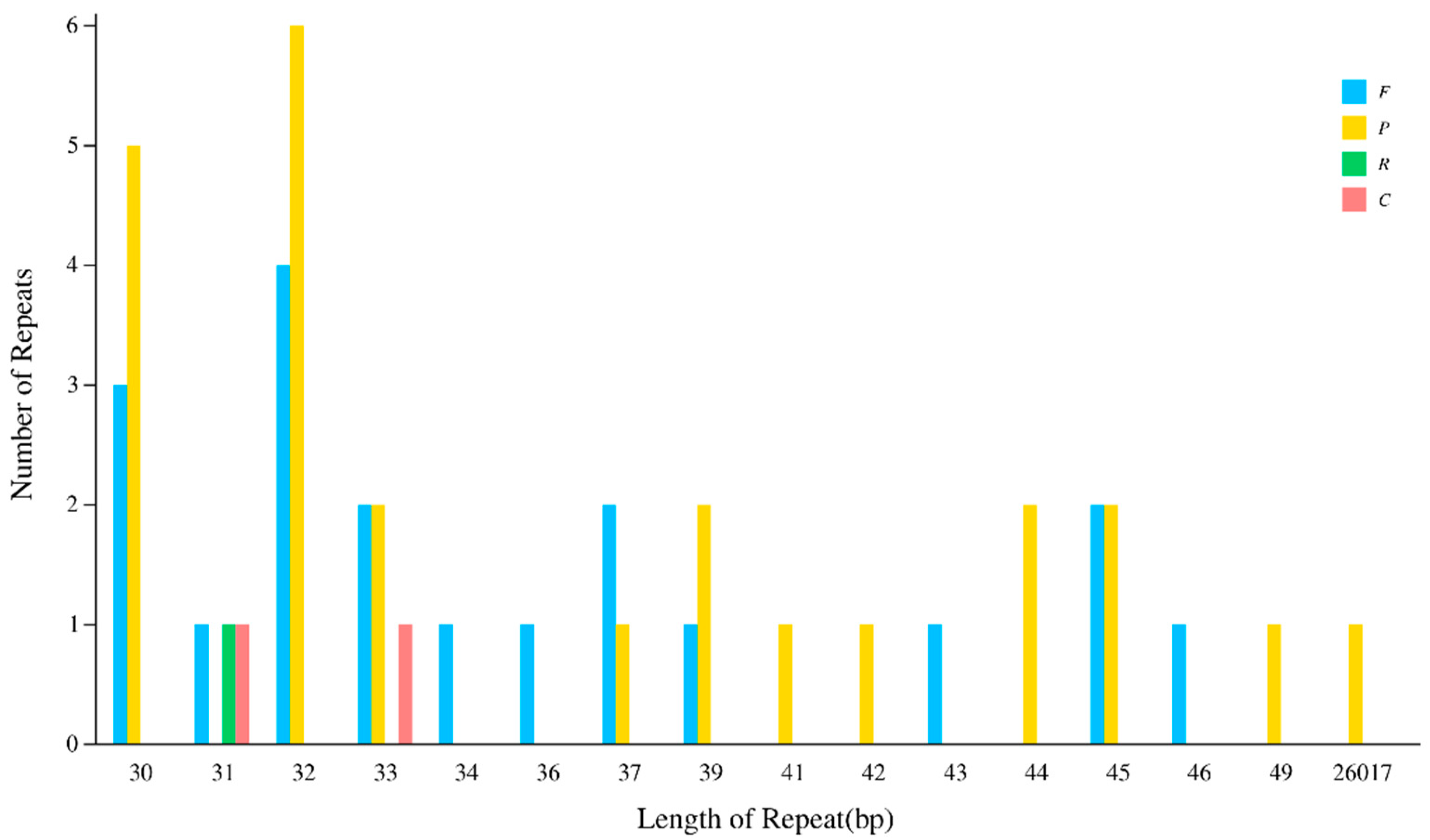
3.2. Analysis of Repeat Sequences
3.3. Codon Usage Analysis
3.4. Analysis of Synonymous and Non-Synonymous Substitution Rates
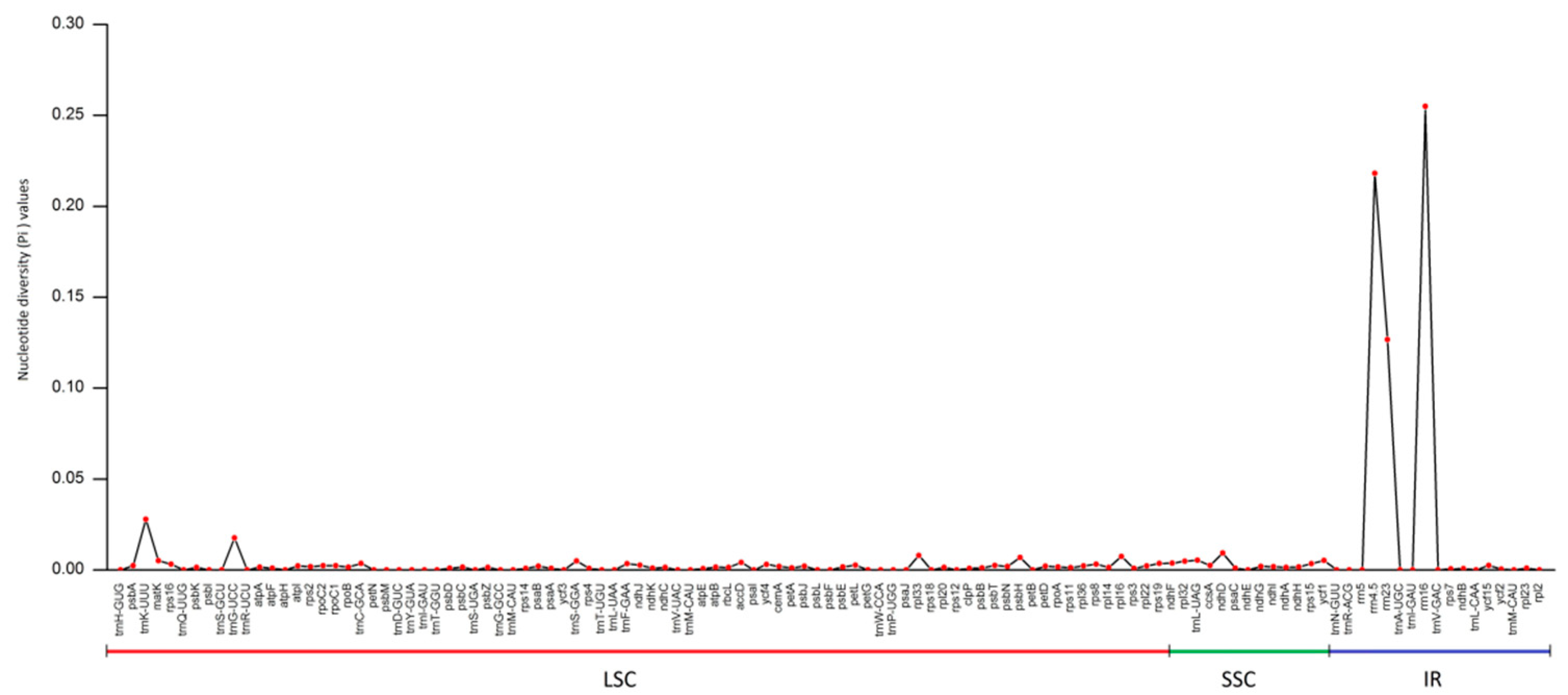
3.5. Nucleotide Diversity Analysis
3.6. Expansion and Contraction Analysis of the IR Regions
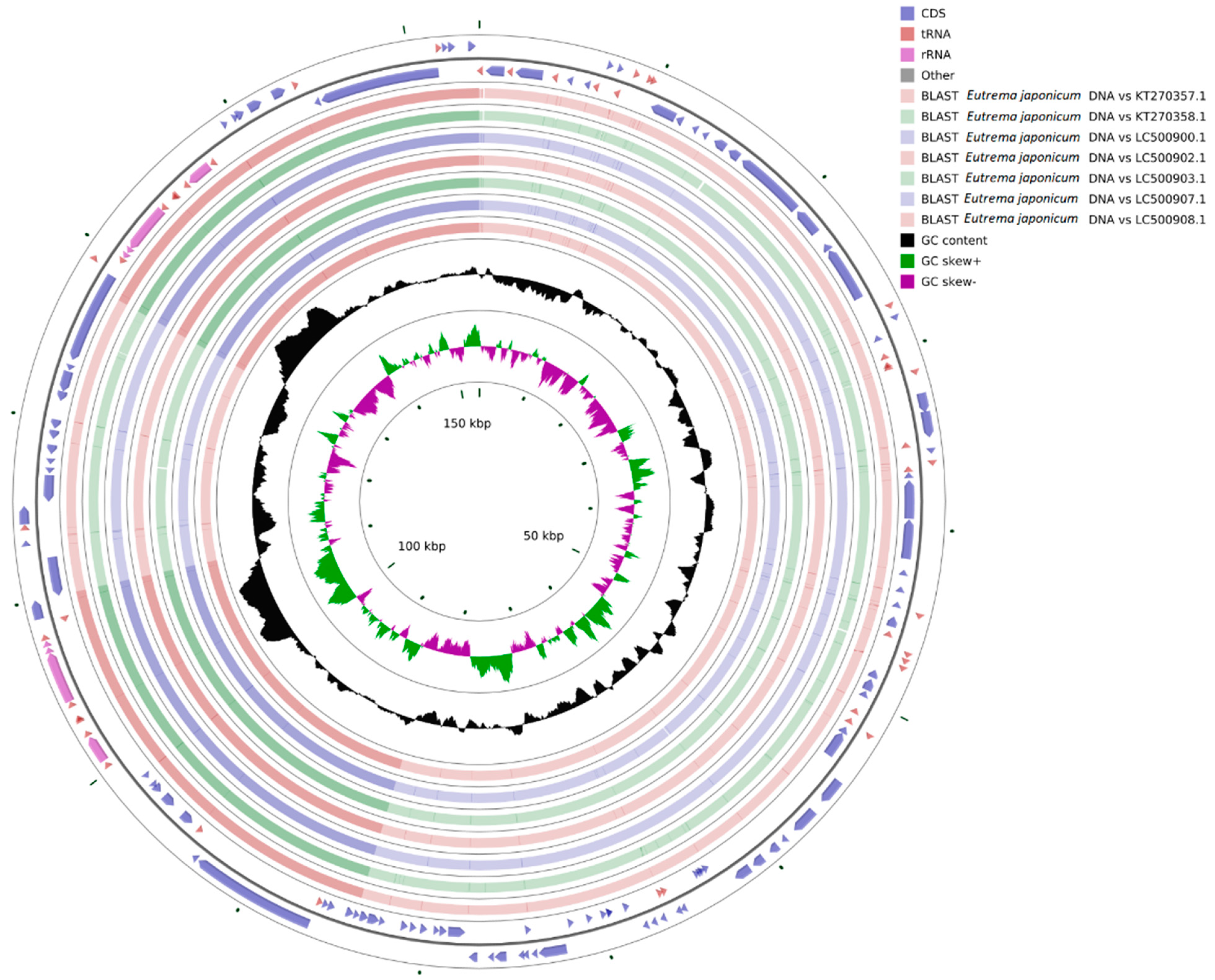
3.7. Genome Comparison and Collinearity Analysis
3.8. Genetic Divergence Analysis
3.9. Phylogenetic Analysis
4. Discussion
4.1. Evolution of the Chloroplast Genome
4.2. Comparison Analysis of the Chloroplast Genome
4.3. Phylogenetic Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hsuan, S.W.; Chyau, C.C.; Hung, H.Y.; Chen, J.H.; Chou, F.P. The induction of apoptosis and autophagy by Wasabia japonica extract in colon cancer. Eur. J. Nutr. 2016, 55, 491–503. [Google Scholar] [CrossRef] [PubMed]
- Murillo, G.; Mehta, R.G. Cruciferous Vegetables and Cancer Prevention. Nutr. Cancer 2001, 41, 17–18. [Google Scholar] [CrossRef]
- Mancuso, M. Wasaby (Wasabia japonica): A Natural Antimicrobial Agent for Food Preservation. J. Fish. Livest. Prod. 2017, 5, 1. [Google Scholar] [CrossRef]
- Nhung, N.H.; Yoshiaki, K.; Toshio, S.; Ryosuke, E. Effects of supporting materials in in vitro acclimatization stage on ex vitro growth of wasabi plants. Sci. Hortic. 2020, 261, 109042. [Google Scholar] [CrossRef]
- Li, J.L.; Lee, W.; Lee, W.J.; Auh, J.H.; Kim, S.; Yoon, J. Extraction of allyl isothiocyanate from wasabi (Wasabia japonica Matsum) using supercritical carbon dioxide. Food Sci. Biotechnol. 2010, 19, 405–410. [Google Scholar] [CrossRef]
- Palmer, J. Germination and growth of wasabi (Wasabia japonica (Miq.) Matsumara). N. Z. J. Crop Hortic. Sci. 2012, 18, 2–164. [Google Scholar] [CrossRef][Green Version]
- Daniell, H.; Lin, C.S.; Yu, M.; Chang, W.J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 134. [Google Scholar] [CrossRef]
- Wen, F.; Wu, X.Z.; Jia, M.L.; Liu, X.S.; Liao, L. The complete chloroplast genome of Stauntonia chinensis and compared analysis revealed adaptive evolution of subfamily Lardizabaloideae species in China. BMC Genom. 2021, 22, 161. [Google Scholar] [CrossRef]
- Chen, N.; Sha, L.N.; Dong, Z.Z.; Tang, C.; Wang, Y.; Kang, H.Y.; Zhang, H.Q.; Yan, X.B.; Zhou, Y.H.; Fan, X. Complete structure and variation of the chloroplast genome of Agropyron cristatum (L.) Gaertn. Gene 2018, 640, 86–96. [Google Scholar] [CrossRef]
- Liang, C.L.; Wang, L.; Lei, J.; Duan, B.Z.; Ma, W.S.; Xiao, S.M.; Qi, H.J.; Wang, Z.; Liu, Y.Q.; Shen, X.F.; et al. A Comparative Analysis of the Chloroplast Genomes of Four Salvia Medicinal Plants. Engineering 2019, 5, 907–915. [Google Scholar] [CrossRef]
- Zuo, R.H.; Jiang, P.; Sun, C.B.; Chen, C.W.; Lou, X.J. Analysis of the chloroplast genome characteristics of Rhus chinensis by de novo sequencing. Chin. J. Biotechnol. 2020, 36, 772–781. [Google Scholar] [CrossRef]
- Deng, G.; Yang, M.; Zhao, K.Y.; Yang, Y.; Huang, X.; Cheng, X. The complete chloroplast genome of Cannabis sativa variety Yunma 7. Mitochondrial DNA B 2021, 6, 531–532. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.H.; Deng, G.Z.; Gao, J.Y.; Liu, H.M.; Zhou, X.Y.; Yue, J.Q.; Dong, M.C.; Yang, F.; Zhou, D.G.; Guo, L.N.; et al. Characterization of the complete chloroplast genome of ‘Yunning No.1′ lemon (Citrus limon). Mitochondrial DNA B 2021, 6, 425–427. [Google Scholar] [CrossRef]
- Rasheed, S.; Zaidi, S.; Azim, M.K. The chloroplast genome sequence of Momordica charantia L. (bitter gourd). Gene Rep. 2020, 21, 100963. [Google Scholar] [CrossRef]
- Yang, J.H.; Li, Z.P.; Lian, J.M.; Qi, G.N.; Shi, P.B.; He, J.W.; Hu, Z.Y.; Zhang, M.F. Brassicaceae transcriptomes reveal convergent evolution of super-accumulation of sinigrin. Commun. Biol. 2020, 3, 779. [Google Scholar] [CrossRef]
- Hashimoto, K.; Tashima, K.; Imai, T.; Matsumoto, K.; Horie, S. The rodent model of impaired gastric motility induced by allyl isothiocyanate, a pungent ingredient of wasabi, to evaluate therapeutic agents for functional dyspepsia. J. Pharm. Sci. 2021, 145, 122–129. [Google Scholar] [CrossRef]
- Haga, N.; Kobayashi, M.; Michiki, N.; Takano, T.; Baba, F.; Kobayashi, K.; Ohynagi, H.; Ohgane, J.; Yano, K.; Ynmane, K. Complete chloroplast genome sequence and phylogenetic analysis of wasabi (Eutrema japonicum) and its relatives. Sci. Rep. 2019, 9, 14377. [Google Scholar] [CrossRef]
- Aboul-Maaty, N.A.F.; Oraby, H.A.S. Extraction of high-quality genomic DNA from different plant orders applying a modified CTAB-based method. Bull. NRC 2019, 43, 25. [Google Scholar] [CrossRef]
- Li, M.Y.; Xie, F.J.; Li, J.; Sun, B.; Luo, Y.; Zhang, Y.; Chen, Q.; Wang, Y.; Zhang, F.; Zhang, Y.T.; et al. Tumorous Stem Development of Brassica Juncea: A Complex Regulatory Network of Stem Formation and Identification of Key Genes in Glucosinolate Biosynthesis. Plants 2020, 9, 1006. [Google Scholar] [CrossRef]
- Yao, J.J.; Zhao, F.Y.; Xu, Y.J.; Zhao, K.H.; Quan, H.; Su, Y.J.; Hao, P.Y.; Liu, J.; Yu, B.X.; Yao, M.; et al. Complete Chloroplast Genome Sequencing and Phylogenetic Analysis of Two Plants. BioMed Res. Int. 2020, 2020, 4374801. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Boetzer, M.; Pirovano, W. Toward almost closed genomes with GapFiller. Genome Biol. 2012, 13, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.; Drechsel, O.; Bock, R. OrganellarGenomeDRAW (OGDRAW): A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 2007, 52, 267–274. [Google Scholar] [CrossRef]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef]
- Wu, M.L.; Li, Q.; Hu, Z.G.; Li, X.W.; Chen, S.L. The Complete Amomum kravanh Chloroplast Genome Sequence and Phylogenetic Analysis of the Commelinids. Molecules 2017, 22, 1875. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed]
- Grant, J.R.; Stothard, P. The CGView Server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 2008, 36, W181–W184. [Google Scholar] [CrossRef]
- Darling, A.C.E.; Mau, B.; Blattner, R.F.; Perna, T.N. Mauve: Multiple Alignment of Conserved Genomic Sequence With Rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvemems in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Cui, N.; Liao, B.S.; Liang, C.L.; Li, S.F.; Zhang, H.; Xu, J.; Li, X.W.; Chen, S.L. Complete chloroplast genome of Salvia plebeia: Organization, specific barcode and phylogenetic analysis. Chin. J. Nat. Med. 2020, 18, 563–572. [Google Scholar] [CrossRef]
- Jiao, Y.; Jia, H.M.; Li, X.W.; Chai, M.L.; Jia, H.J.; Chen, Z.; Wang, G.Y.; Chai, C.Y.; Weg, V.D.E.; Gao, Z.S. Development of simple sequence repeat (SSR) markers from a genome survey of Chinese bayberry (Myrica rubra). BMC Genom. 2012, 13, 201. [Google Scholar] [CrossRef]
- Singh, R.B.; Mahenderakar, M.D.; Jugran, A.K.; Singh, R.K.; Srivastava, R.K. Assessing genetic diversity and population structure of sugarcane cultivars, progenitor species and genera using microsatellite(SSR)markers. Gene 2020, 753, 144800. [Google Scholar] [CrossRef]
- Wang, L.; He, N.; Li, Y.; Fang, Y.M.; Zhang, F.L. Complete Chloroplast Genome Sequence of Chinese Lacquer Tree (Toxicodendron vernicifluum, Anacardiaceae) and Its Phylogenetic Significance. BioMed Res. Int. 2020, 2020, 9014873. [Google Scholar] [CrossRef]
- Gu, C.H.; Dong, B.; Xu, L.; Tenmbrock, L.R.; Zheng, S.Y.; Wu, Z.Q. The Complete Chloroplast Genome of Heimia myrtifolia and Comparative Analysis within Myrtales. Molecules 2018, 23, 846. [Google Scholar] [CrossRef]
- Jiang, W.L.; Tan, W.; Gao, H.; Yu, X.L.; Zhang, H.Y.; Bian, Y.H.; Wang, Y.; Tian, X.X. Transcriptome and complete chloroplast genome of Glycyrrhiza inflata and comparative analyses with the other two licorice species. Genomics 2020, 112, 4179–4188. [Google Scholar] [CrossRef]
- Sheng, J.J.; Yan, M.; Wang, J.; Zhao, L.L.; Zhou, F.S.; Hu, Z.L.; Jin, S.R.; Diao, Y. The complete chloroplast genome sequences of five Miscanthus species, and comparative analyses with other grass plastomes. Ind. Crop. Prod. 2021, 162, 113248. [Google Scholar] [CrossRef]
- Du, X.Y.; Zeng, T.; Feng, Q.; Hu, L.J.; Luo, X.; Weng, Q.B.; He, J.F.; Zhu, B. The complete chloroplast genome sequence of yellow mustard (Sinapis alba L.) and its phylogenetic relationship to other Brassicaceae species. Gene 2020, 731, 144340. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Feng, C.; Cai, M.M.; Chen, J.H.; Ding, P. Complete chloroplast genome sequence of Amomum villosum and comparative analysis with other Zingiberaceae plants. Chin. Herb. Med. 2020, 12, 375–383. [Google Scholar] [CrossRef]
- Jun, L.; Xie, D.F.; Guo, X.L.; Zheng, Z.Y.; He, X.J.; Zhou, S.D. Comparative Analysis of the Complete Plastid Genome of Five Bupleurum Species and New Insights into DNA Barcoding and Phylogenetic Relationship. Plants 2020, 9, 543. [Google Scholar] [CrossRef]
- Jeong, Y.M.; Chung, W.H.; Mum, J.H.; Kim, N.; Yu, H.J. De novo assembly and characterization of the complete chloroplast genome of radish (Raphanus sativus L.). Gene 2014, 551, 39–48. [Google Scholar] [CrossRef]
- Nie, X.J.; Lv, S.Z.; Zhang, Y.X.; Du, X.H.; Biradar, S.S.; Tan, X.F.; Wan, F.H.; Song, W.N. Complete chloroplast genome sequence of a major invasive species, crofton weed (Ageratina adenophora). PLoS ONE 2018, 7, e36869. [Google Scholar] [CrossRef]
- Yan, C.; Du, J.C.; Gao, L.; Li, Y.; Hou, X.L. The complete chloroplast genome sequence of watercress (Nasturtium officinale R. Br.): Genome organization, adaptive evolution and phylogenetic relationships in Cardamineae. Gene 2019, 699, 24–36. [Google Scholar] [CrossRef]
- Tan, W.; Gao, H.; Zhang, H.Y.; Yu, X.L.; Tian, X.X.; Jiang, W.L.; Zhou, K. The complete chloroplast genome of Chinese medicine (Psoralea corylifolia: Molecular structures, barcoding and phylogenetic analysis. Plant Gene 2020, 21, 100216. [Google Scholar] [CrossRef]
- Njuguna, A.W.; Li, Z.Z.; Saina, J.K.; Munywoki, J.M.; Gichira, A.W.; Gituru, R.W.; Wang, Q.F.; Chen, J.M. Comparative analyses of the complete chloroplast genomes of nymphoides and menyanthes species (menyanthaceae). Aquat. Bot. 2019, 156, 73–81. [Google Scholar] [CrossRef]
- Niu, Y.T.; Jabbour, F.; Barrett, R.L.; Ye, J.F.; Zhang, Z.Z.; Lu, K.Q.; Lu, L.M.; Chen, Z.D. Combining complete chloroplast genome sequences with target loci data and morphology to resolve species limits in Triplostegia (Caprifoliaceae). Mol. Phylogenet. Evol. 2018, 129, 15–26. [Google Scholar] [CrossRef]
- Wang, W.C.; Chen, S.Y.; Zhang, X.Z. Whole-Genome Comparison Reveals Heterogeneous Divergence and Mutation Hotspots in Chloroplast Genome of Eucommia ulmoides Oliver. Int. J. Mol. Sci. 2018, 19, 1037. [Google Scholar] [CrossRef]
- Cui, Y.X.; Zhou, J.G.; Chen, X.L.; Xu, Z.C.; Wang, Y.; Sun, W.; Song, J.Y.; Yao, H. Complete chloroplast genome and comparative analysis of three Lycium (Solanaceae) species with medicinal and edible properties. Gene Rep. 2019, 17, 100464. [Google Scholar] [CrossRef]
- Zhang, T.T.; Xing, Y.P.; Xu, L.; Bao, G.H.; Zhan, Z.L.; Yang, Y.Y.; Wang, J.H.; Li, S.N.; Zhang, D.C.; Kang, T.G. Comparative analysis of the complete chloroplast genome sequences of six species of Pulsatilla Miller, Ranunculaceae. Chin. Med. 2019, 14, 104–802. [Google Scholar] [CrossRef]
- Liu, H.Y.; Yu, Y.; Deng, Y.Q.; Li, J.; Huang, Z.X.; Zhou, S.D. The Chloroplast Genome of Lilium henrici: Genome Structure and Comparative Analysis. Molecules 2018, 23, 1276. [Google Scholar] [CrossRef] [PubMed]
- Cheeseman, J.M. The evolution of halophytes, glycophytes and crops, and its implications for food security under saline conditions. New Phytol. 2015, 206, 557–570. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Gene Group | Gene Name |
|---|---|---|
| Photosynthesis | Subunits of photosystem I | psaA, psaB, psaC, psaI, psaJ |
| Subunits of photosystem II | psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ | |
| Subunits of NADH dehydrogenase | ndhA *, ndhB * (2), ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK | |
| Subunits of cytochrome b/f complex | petA, petB *, petD *, petG, petL, petN | |
| Subunits of ATP synthase | atpA, atpB, atpE, atpF *, atpH, atpI | |
| Large subunit of rubisco | rbcL | |
| Subunits photochlorophyllide reductase | ||
| Self-replication | Proteins of large ribosomal subunit | rpl14, rpl16 *, rpl2 * (2), rpl20, rpl22, rpl23(2), rpl32, rpl33, rpl36 |
| Proteins of small ribosomal subunit | rps11, rps12 * (2), rps14, rps15, rps16 *, rps18, rps19, rps2, rps3, rps4, rps7(2), rps8 | |
| Subunits of RNA polymerase | rpoA, rpoB, rpoC1 *, rpoC2 | |
| Ribosomal RNAs | rrn16(2), rrn23(2), rrn4.5(2), rrn5(2) | |
| Transfer RNAs | trnA-UGC * (2), trnC-GCA, trnD-GUC, trnF-GAA, trnG-GCC, trnG-UCC *, trnH-GUG, trnI-GAU, trnI-GAU * (3), trnK-UUU *, trnL-CAA(2), trnL-UAA *, trnL-UAG, trnM-CAU(4), trnN-GUU(2), trnP-UGG, trnQ-UUG, trnR-ACG(2), trnR-UCU, trnS-GCU, trnS-GGA, trnS-UGA, trnT-GGU, trnT-UGU, trnV-GAC(2), trnV-UAC *, trnW-CCA, trnY-GUA | |
| Other genes | Maturase | matK |
| Protease | clpP ** | |
| Envelope membrane protein | cemA | |
| Acetyl-CoA carboxylase | accD | |
| C-type cytochrome synthesis gene | ccsA | |
| Translation initiation factor | - | |
| other | - | |
| Genes of unknown function | Conserved hypothetical chloroplast ORF | ycf1(2), ycf15(2), ycf2(2), ycf3 **, ycf4 |
| Amino Acid | Codon | Number | RSCU | Amino Acid | Condon | Number | RSCU |
|---|---|---|---|---|---|---|---|
| Ala | GCA | 388 | 1.128 | Pro | CCA | 305 | 1.1348 |
| GCC | 205 | 0.596 | CCC | 198 | 0.7368 | ||
| GCG | 156 | 0.4536 | CCG | 147 | 0.5468 | ||
| GCU | 627 | 1.8228 | CCU | 425 | 1.5812 | ||
| Cys | UGC | 85 | 0.5246 | Gln | CAA | 748 | 1.5682 |
| UGU | 239 | 1.4754 | CAG | 206 | 0.4318 | ||
| Asp | GAC | 189 | 0.361 | Arg | AGA | 476 | 1.8078 |
| GAU | 858 | 1.639 | AGG | 165 | 0.6264 | ||
| Glu | GAA | 1054 | 1.5122 | CGA | 366 | 1.3896 | |
| GAG | 340 | 0.4878 | CGC | 110 | 0.4176 | ||
| Phe | UUC | 528 | 0.6588 | CGG | 122 | 0.4632 | |
| UUU | 1075 | 1.3412 | CGU | 341 | 1.2948 | ||
| Gly | GGA | 738 | 1.6696 | Ser | AGC | 124 | 0.3654 |
| GGC | 163 | 0.3688 | AGU | 409 | 1.2048 | ||
| GGG | 282 | 0.638 | UCA | 411 | 1.2108 | ||
| GGU | 585 | 1.3236 | UCC | 308 | 0.9072 | ||
| His | CAC | 150 | 0.4816 | UCG | 202 | 0.5952 | |
| CAU | 473 | 1.5184 | UCU | 583 | 1.7172 | ||
| Ile | AUA | 738 | 0.9627 | Thr | ACA | 418 | 1.2268 |
| AUC | 424 | 0.5529 | ACC | 247 | 0.7248 | ||
| AUU | 1138 | 1.4844 | ACG | 146 | 0.4284 | ||
| Lys | AAA | 1160 | 1.5354 | Val | ACU | 552 | 1.62 |
| AAG | 351 | 0.4646 | GUA | 505 | 1.4296 | ||
| Leu | CUA | 395 | 0.8346 | GUC | 184 | 0.5208 | |
| CUC | 184 | 0.3888 | GUG | 206 | 0.5832 | ||
| CUG | 173 | 0.3654 | GUU | 518 | 1.4664 | ||
| CUU | 601 | 1.2702 | Trp | UGG | 458 | 1 | |
| UUA | 954 | 2.016 | Tyr | UAC | 181 | 0.3706 | |
| UUG | 532 | 1.1244 | UAU | 796 | 1.6294 | ||
| Met | AUG | 606 | 1.9868 | Ter * | UAA | 49 | 1.6896 |
| GUG | 4 | 0.0132 | UAG | 24 | 0.8277 | ||
| Asn | AAC | 304 | 0.4662 | UGA | 14 | 0.4827 | |
| AAU | 1000 | 1.5338 |
| Groups | Each Gene | All Genes | ||||
|---|---|---|---|---|---|---|
| Ka/Ks > 1 | Ka/Ks = 1 | Ka/Ks < 1 | Ka | Ks | Ka/Ks | |
| Eutrema japonicum vs. KT270357.1 | 0 | 0 | 10 | 0.024612019 | 0.07334597 | 0.34 |
| Eutrema japonicum vs. KT270358.1 | 2 | 0 | 28 | 0.235550101 | 0.63714708 | 0.37 |
| Eutrema japonicum vs. LC500900.1 | 0 | 0 | 2 | 0.874397058 | 1.931823137 | 0.45 |
| Eutrema japonicum vs. LC500902.1 | 1 | 0 | 5 | 0.886763704 | 1.96469381 | 0.45 |
| Eutrema japonicum vs. LC500903.1 | 0 | 0 | 3 | 0.877017651 | 1.95558473 | 0.45 |
| Eutrema japonicum vs. LC500907.1 | 0 | 0 | 11 | 0.895988246 | 2.02793639 | 0.44 |
| Eutrema japonicum vs. LC500908.1 | 0 | 0 | 6 | 0.882924966 | 1.96288091 | 0.45 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, M.; Zhang, R.; Li, J.; Zheng, K.; Xiao, J.; Zheng, Y. Analyses of Chloroplast Genome of Eutrema japonicum Provide New Insights into the Evolution of Eutrema Species. Agronomy 2021, 11, 2546. https://doi.org/10.3390/agronomy11122546
Li M, Zhang R, Li J, Zheng K, Xiao J, Zheng Y. Analyses of Chloroplast Genome of Eutrema japonicum Provide New Insights into the Evolution of Eutrema Species. Agronomy. 2021; 11(12):2546. https://doi.org/10.3390/agronomy11122546
Chicago/Turabian StyleLi, Mengyao, Ran Zhang, Jie Li, Kaimin Zheng, Jiachang Xiao, and Yangxia Zheng. 2021. "Analyses of Chloroplast Genome of Eutrema japonicum Provide New Insights into the Evolution of Eutrema Species" Agronomy 11, no. 12: 2546. https://doi.org/10.3390/agronomy11122546
APA StyleLi, M., Zhang, R., Li, J., Zheng, K., Xiao, J., & Zheng, Y. (2021). Analyses of Chloroplast Genome of Eutrema japonicum Provide New Insights into the Evolution of Eutrema Species. Agronomy, 11(12), 2546. https://doi.org/10.3390/agronomy11122546