
Comparative Metagenomic and Metabolomic Profiling of Rhizospheres of Panax notoginseng Grown under Forest and Field Conditions

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Sites and Collection of Rhizosphere Samples
2.2. DNA Extraction, Library Preparation and Metagenomic Sequencing
2.3. Sequence Quality Control and Assembly
2.4. Gene Prediction and Functional Annotation
2.5. Statistical Analyses of Metagenomic Data
2.6. Metabolomic Analysis of Rhizosphere Samples
2.6.1. Sample Preparation
2.6.2. Sample Extraction
2.6.3. GC–MS/MS Analysis
2.6.4. Statistical Analysis of Metabolome Data
3. Results
3.1. Growth and Physical Characteristics of P. notoginseng
3.2. Metagenomic Sequencing, Assembly and Annotation
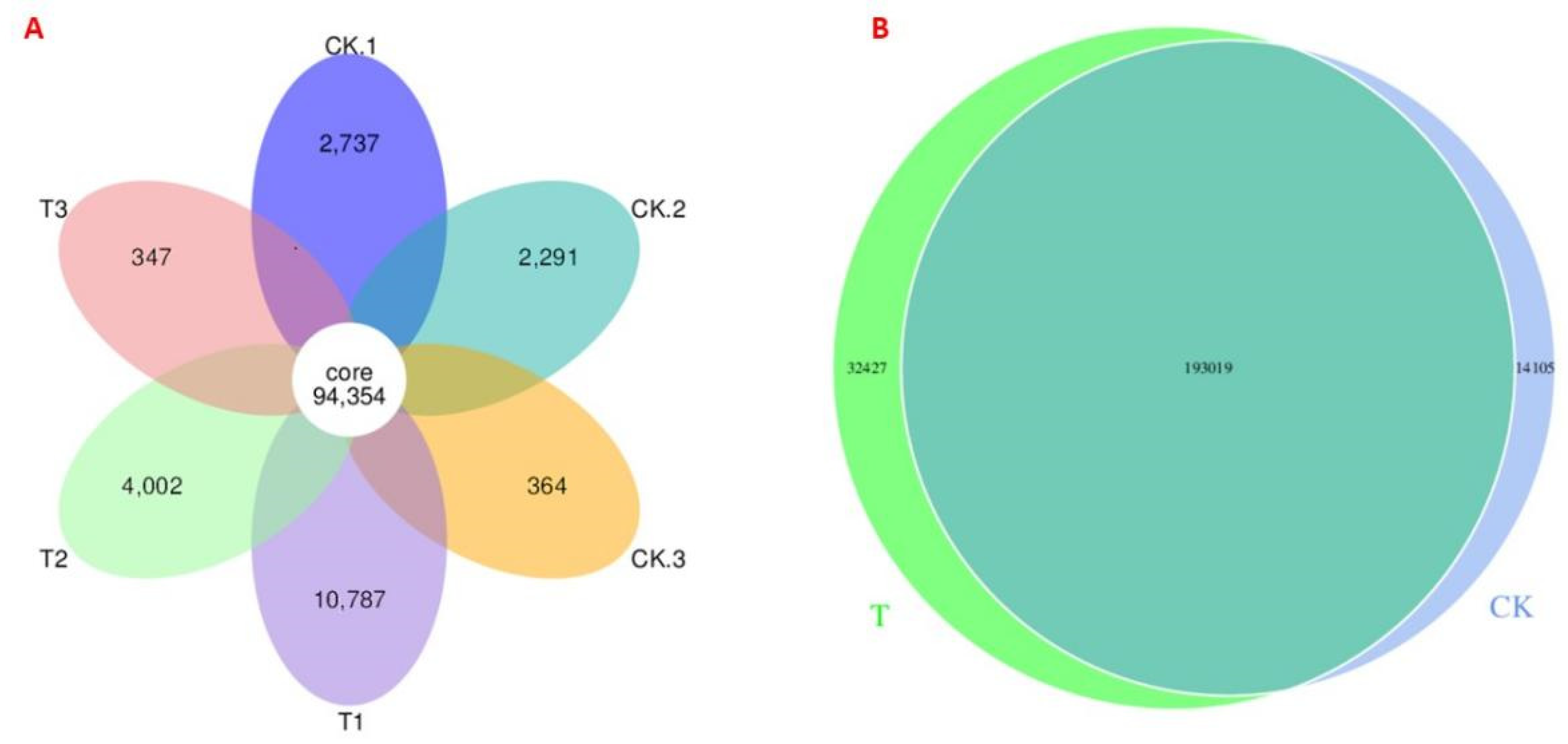
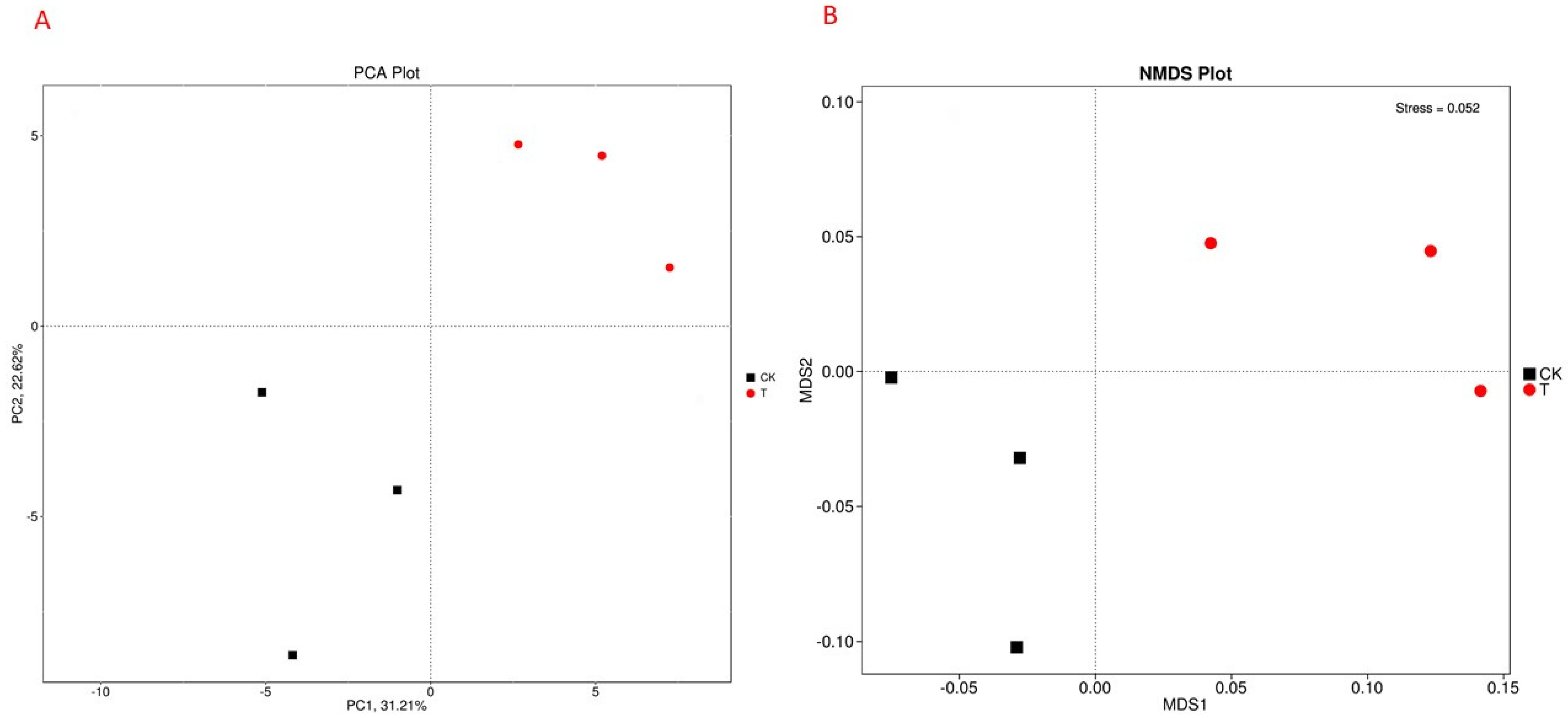
3.3. Pan/Core and Venn Diagram Analysis of Microbial Taxa
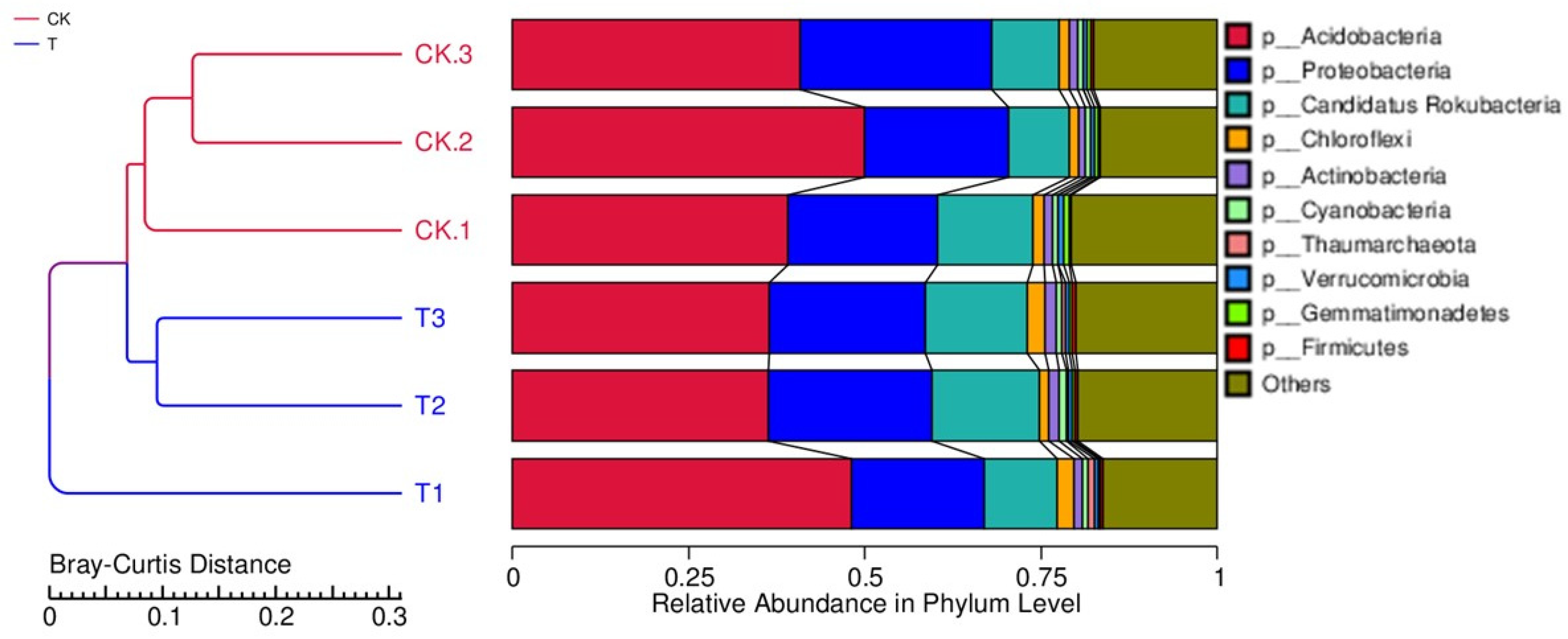
3.4. Phylogenetic Analysis of Rhizosphere Microbial Communities under Field and Forest Conditions
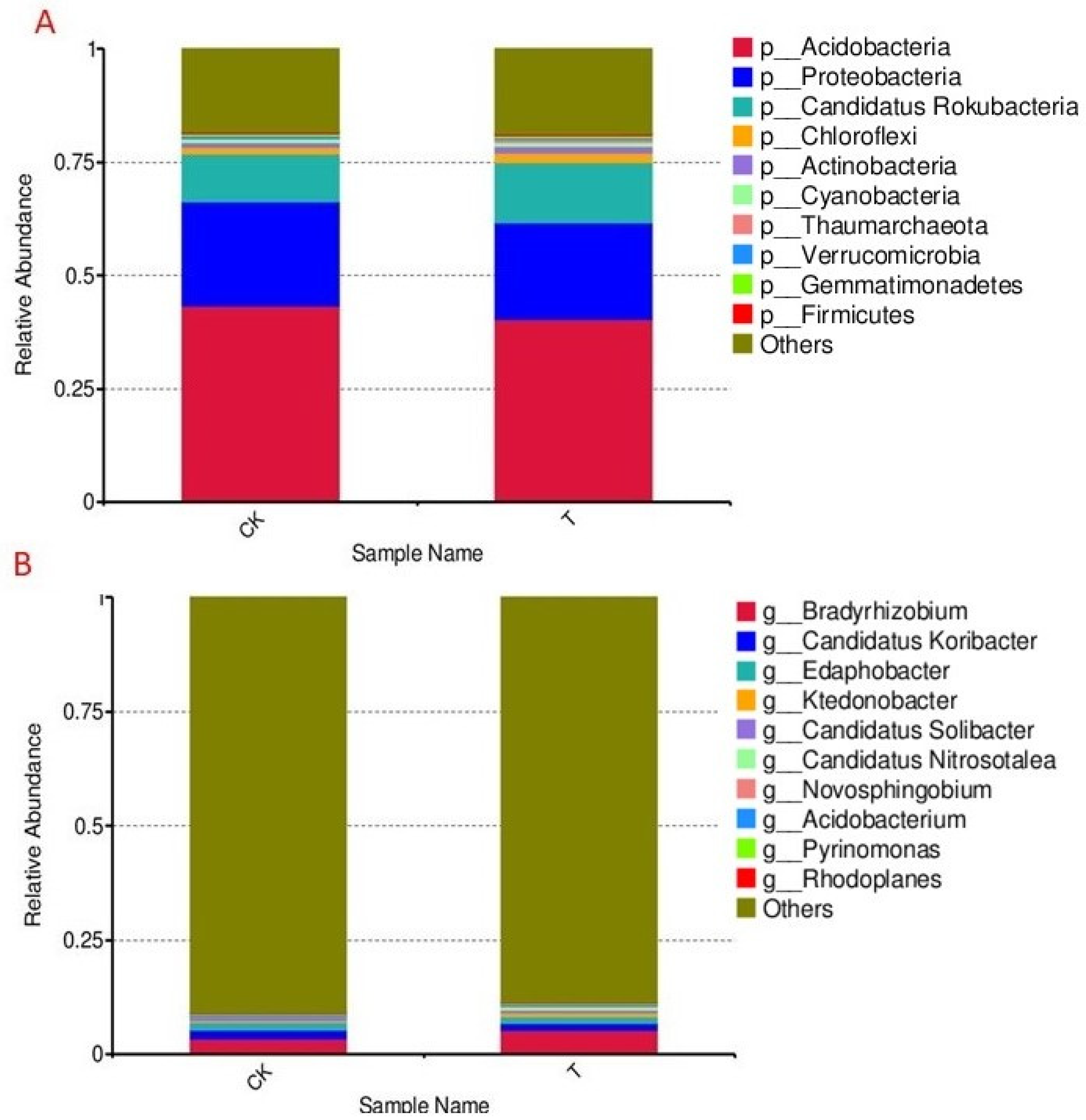
3.5. Relative Abundance of Microbial Communities
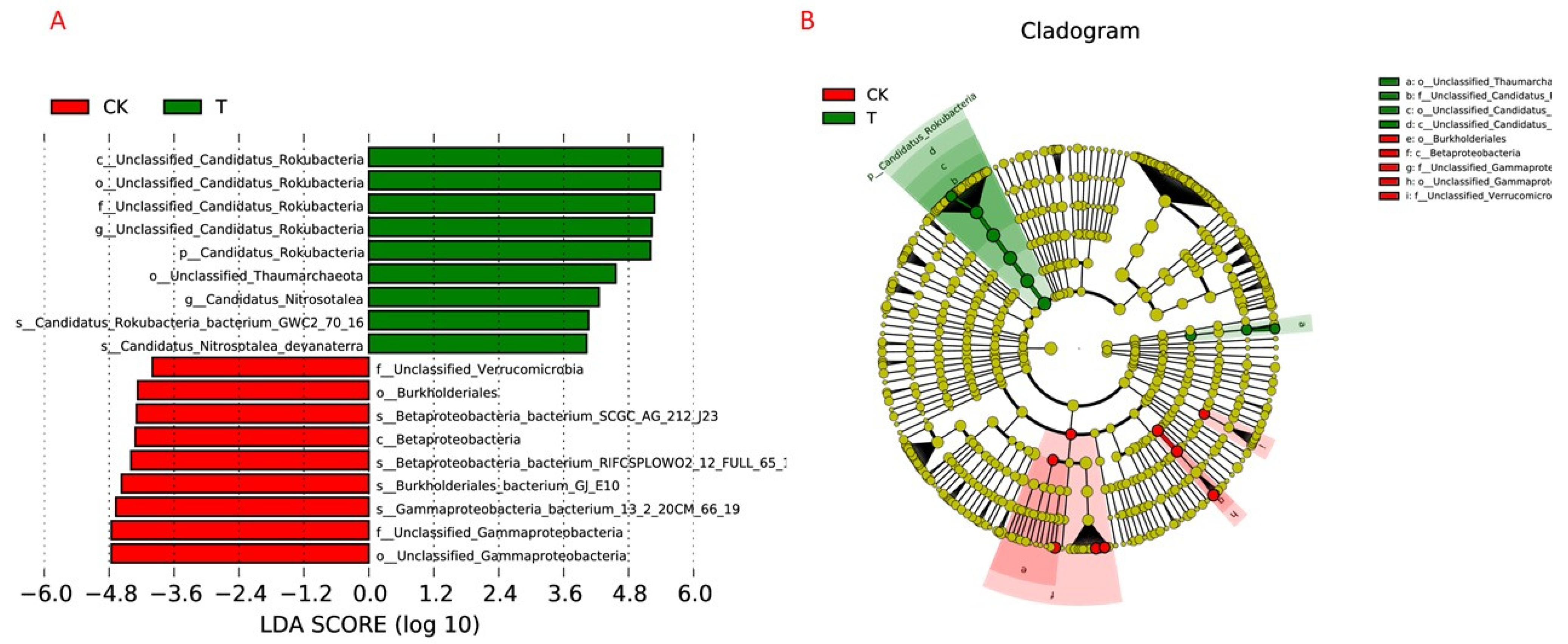
3.6. Biomarker Bacterial Taxa in the P. ginseng Rhizosphere Soil under Field and Forest Conditions
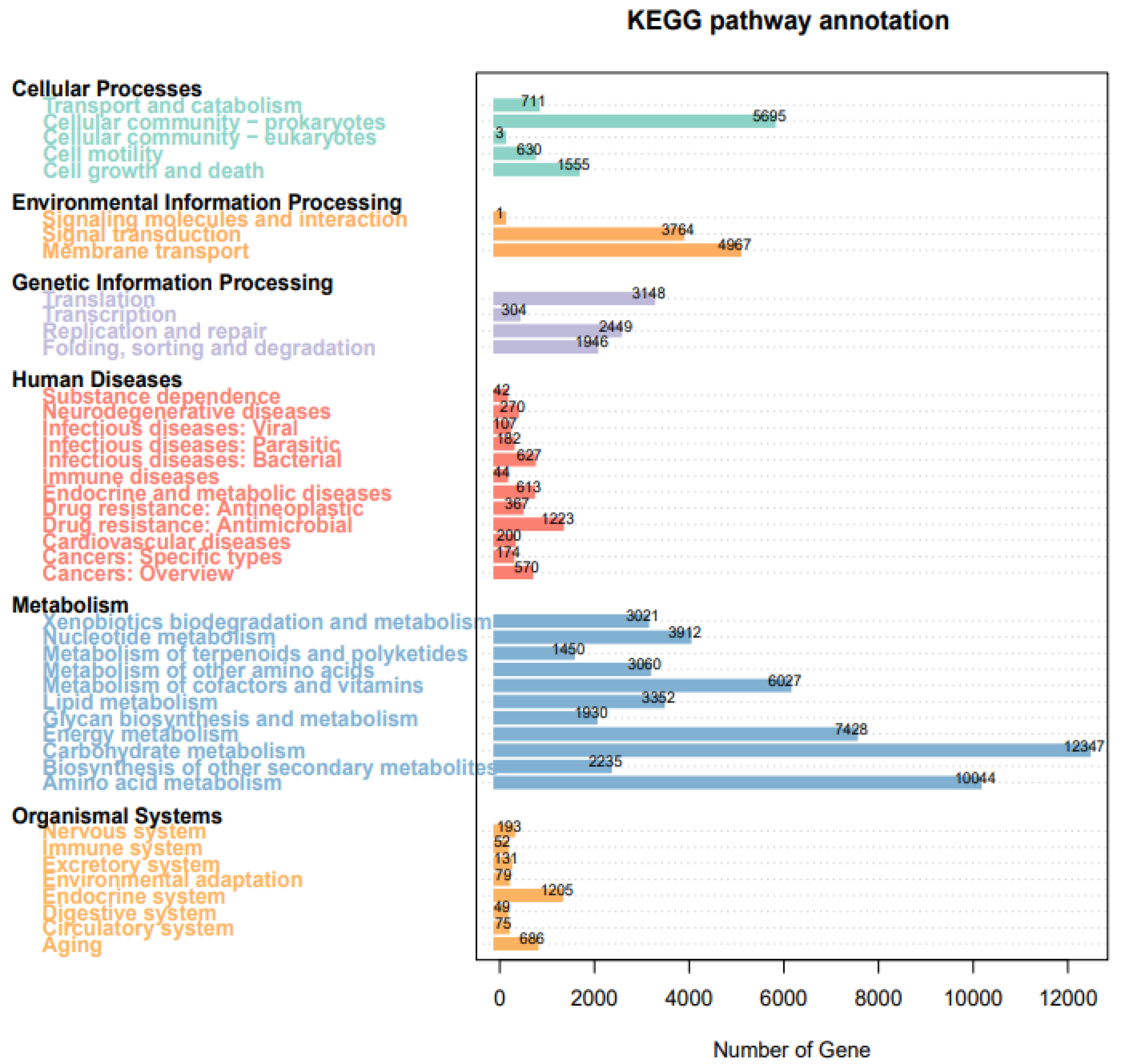
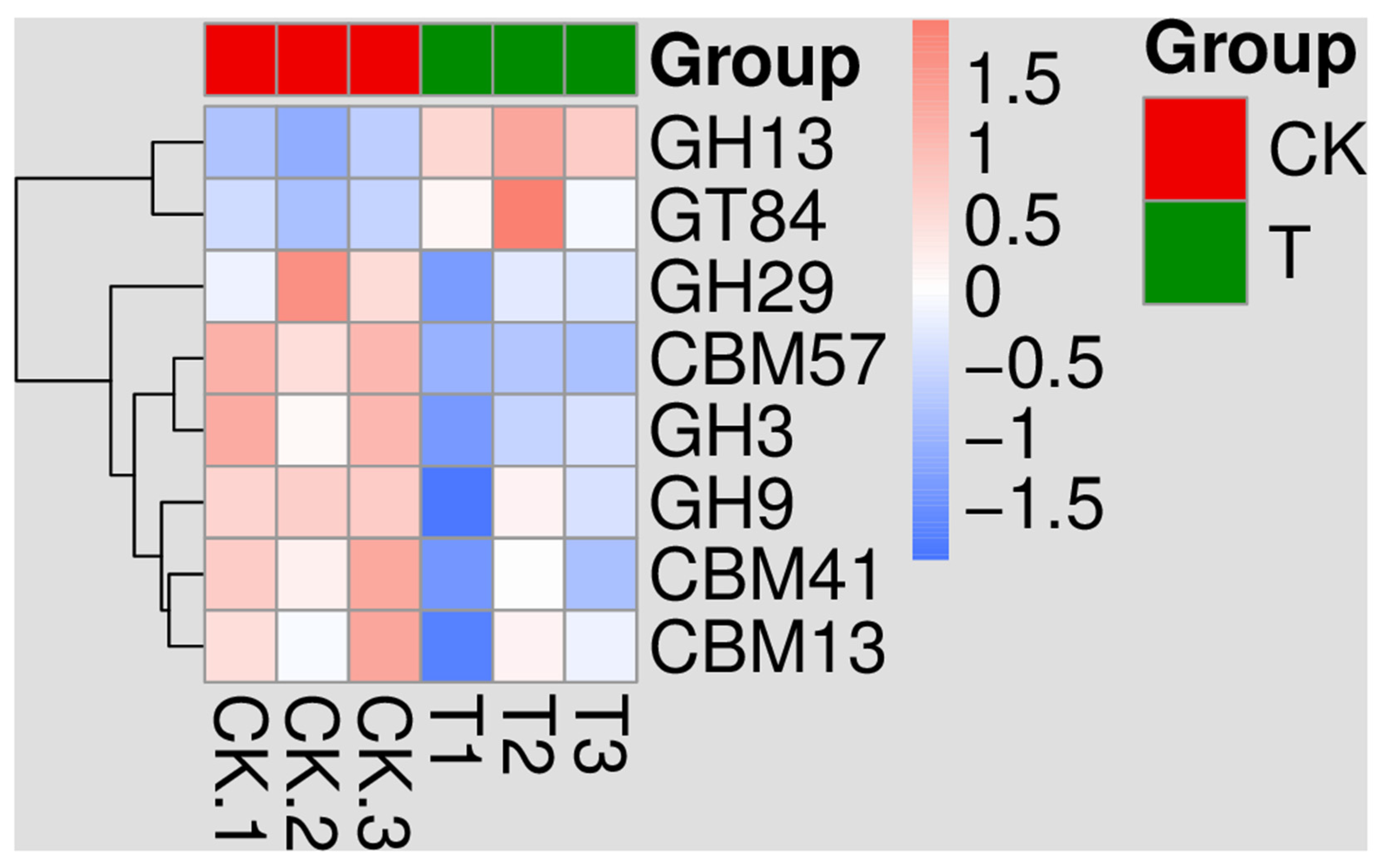
3.7. Kyoto Encyclopedia of Genes and Genomes (KEGG) Analysis
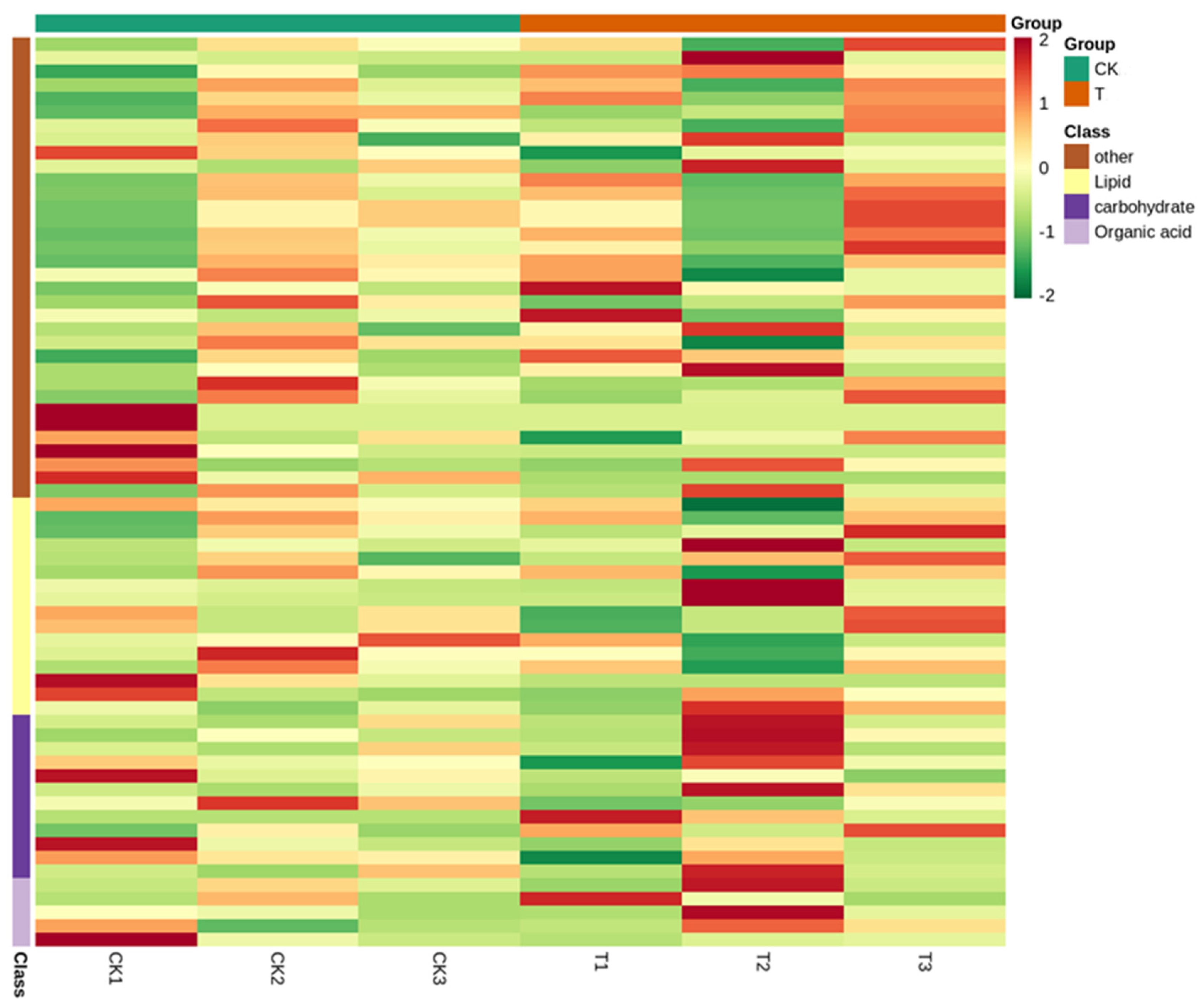
3.8. Differential Metabolite Analysis
4. Discussion
4.1. Growth and Physical Characteristics of P. notoginseng
4.2. Relative Abundance of Microbial Communities
4.3. Biomarker Bacterial Taxa in the P. ginseng Rhizosphere Soil under Field and Forest Conditions
4.4. Differential Metabolite Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sharma, S.K.; Pandit, M. A New Species of Panax L. (Araliaceae) from Sikkim Himalaya, India. Syst. Bot. 2009, 34, 434–438. [Google Scholar] [CrossRef]
- Zhang, H.-Z.; Liu, D.-H.; Zhang, D.-K.; Wang, Y.-H.; Cao, L.-J.; Xiao, X.-H.; Huang, L.-Q.; Wang, J.-B. Quality Assessment of Panax notoginseng from Different Regions through the Analysis of Marker Chemicals, Biological Potency and Ecological Factors. PLoS ONE 2016, 11, e0164384. [Google Scholar] [CrossRef]
- Zhao, Q.; Wu, Y.-N.; Fan, Q.; Han, Q.-Q.; Paré, P.W.; Xu, R.; Wang, Y.-Q.; Wang, S.-M.; Zhang, J.-L. Improved Growth and Metabolite Accumulation in Codonopsis pilosula (Franch.) Nannf. by Inoculation of Bacillus amyloliquefaciens GB03. J. Agric. Food Chem. 2016, 64, 8103–8108. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Long, C.; Lam, E. Roles of Plant-Associated Microbiota in Traditional Herbal Medicine. Trends Plant Sci. 2018, 23, 559–562. [Google Scholar] [CrossRef]
- Dong, L.; Xu, J.; Zhang, L.; Cheng, R.; Wei, G.; Su, H.; Yang, J.; Qian, J.; Xu, R.; Chen, S. Rhizospheric microbial communities are driven by Panax ginseng at different growth stages and biocontrol bacteria alleviates replanting mortality. Acta Pharm. Sin. B 2018, 8, 272–282. [Google Scholar] [CrossRef]
- Köberl, M.; Schmidt, R.; Ramadan, E.M.; Bauer, R.; Berg, G. The microbiome of medicinal plants: Diversity and importance for plant growth, quality and health. Front. Microbiol. 2013, 4, 400. [Google Scholar] [CrossRef]
- Zhang, L. Study on discontinuous cultivating of Panax ginseng and its workable solution. J. Jilin Agric. Univ. 2008, 30, 481–485. [Google Scholar]
- Zhang, H.; Xue, Q. Research progress in control of continuous cropping obstacle in Ginseng. Acta Agric. Jiangxi 2010, 22, 68–71. [Google Scholar]
- Dong, L.; Xu, J.; Feng, G.; Li, X.; Chen, S. Soil bacterial and fungal community dynamics in relation to Panax notoginseng death rate in a continuous cropping system. Sci. Rep. 2016, 6, 31802. [Google Scholar] [CrossRef]
- Wu, L. A review on studies of the reason and control methods of succession cropping obstacle of Panax ginseng CA Mey. Spec. Wild Econ. Anim. Plant Res. 2008, 2, 68–72. [Google Scholar]
- Ying, Y. Influence of Panax ginseng continuous cropping on metabolic function of soil microbial communities. Chin. Herb. Med. 2012, 4, 329–334. [Google Scholar]
- Xun, W.; Liu, Y.; Li, W.; Ren, Y.; Xiong, W.; Xu, Z.; Zhang, N.; Miao, Y.; Shen, Q.; Zhang, R. Specialized metabolic functions of keystone taxa sustain soil microbiome stability. Microbiome 2021, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Randewig, D.; Marshall, J.D.; Näsholm, T.; Jämtgård, S. Combining microdialysis with metabolomics to characterize the in situ composition of dissolved organic compounds in boreal forest soil. Soil Biol. Biochem. 2019, 136, 107530. [Google Scholar] [CrossRef]
- Chen, W.; Kui, L.; Zhang, G.; Zhu, S.; Zhang, J.; Wang, X.; Yang, M.; Huang, H.; Liu, Y.; Wang, Y.; et al. Whole-Genome Sequencing and Analysis of the Chinese Herbal Plant Panax notoginseng. Mol. Plant 2017, 10, 899–902. [Google Scholar] [CrossRef] [PubMed]
- Withers, E.; Hill, P.W.; Chadwick, D.R.; Jones, D.L. Use of untargeted metabolomics for assessing soil quality and microbial function. Soil Biol. Biochem. 2020, 143, 107758. [Google Scholar] [CrossRef]
- Xiao, C.; Yang, L.; Zhang, L.; Liu, C.; Han, M. Effects of cultivation ages and modes on microbial diversity in the rhizosphere soil of Panax ginseng. J. Ginseng Res. 2016, 40, 28–37. [Google Scholar] [CrossRef]
- Bian, X.; Zhao, Y.; Xiao, S.; Yang, H.; Han, Y.; Zhang, L. Metabolome and transcriptome analysis reveals the molecular profiles underlying the ginseng response to rusty root symptoms. BMC Plant Biol. 2021, 21, 215. [Google Scholar] [CrossRef]
- Wang, J.-H.; Lu, J.; Zhang, Y.-X.; Wu, J.; Luo, Y.; Liu, H. Metagenomic analysis of antibiotic resistance genes in coastal industrial mariculture systems. Bioresour. Technol. 2018, 253, 235–243. [Google Scholar] [CrossRef]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. GigaScience 2012, 1, 18. [Google Scholar] [CrossRef]
- Zhu, W.; Lomsadze, A.; Borodovsky, M. Ab initio gene identification in metagenomic sequences. Nucleic Acids Res. 2010, 38, e132. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Villar, E.; Farrant, G.K.; Follows, M.; Garczarek, L.; Speich, S.; Audic, S.; Bittner, L.; Blanke, B.; Brum, J.R.; Brunet, C.; et al. Environmental characteristics of Agulhas rings affect interocean plankton transport. Science 2015, 348, 1261447. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Huson, D.H.; Mitra, S.; Ruscheweyh, H.-J.; Weber, N.; Schuster, S.C. Integrative analysis of environmental sequences using MEGAN4. Genome Res. 2011, 21, 1552–1560. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, F.; Fåk, F.; Nookaew, I.; Tremaroli, V.; Fagerberg, B.; Petranovic, D.; Bäckhed, F.; Nielsen, J. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat. Commun. 2012, 3, 1245. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. Data, information, knowledge and principle: Back to metabolism in KEGG. Nucleic Acids Res. 2014, 42, D199–D205. [Google Scholar] [CrossRef] [PubMed]
- Powell, S. Eggnog v4. 0: Nested orthology inference across 3686 organisms. Nucleic Acids Res. 2014, 42, D231–D239. [Google Scholar] [CrossRef] [PubMed]
- White, J.R.; Nagarajan, N.; Pop, M. Statistical Methods for Detecting Differentially Abundant Features in Clinical Metagenomic Samples. PLoS Comput. Biol. 2009, 5, e1000352. [Google Scholar] [CrossRef]
- Swenson, T.L.; Jenkins, S.; Bowen, B.P.; Northen, T.R. Untargeted soil metabolomics methods for analysis of extractable organic matter. Soil Biol. Biochem. 2015, 80, 189–198. [Google Scholar] [CrossRef]
- Berendsen, R.L.; Pieterse, C.M.J.; Bakker, P.A. The rhizosphere microbiome and plant health. Trends Plant Sci. 2012, 17, 478–486. [Google Scholar] [CrossRef]
- Priyadharsini, P. Mycorrhizosphere: The Extended Rhizosphere and Its Significance, in Plant-Microbe Interaction: An Approach to Sustainable Agriculture; Springer: Berlin/Heidelberg, Germany, 2016; pp. 97–124. [Google Scholar]
- Haichar, F.E.Z.; Marol, C.; Berge, O.; Rangel-Castro, J.I.; Prosser, J.; Balesdent, J.; Heulin, T.; Achouak, W. Plant host habitat and root exudates shape soil bacterial community structure. ISME J. 2008, 2, 1221–1230. [Google Scholar] [CrossRef] [PubMed]
- Narula, N.; Kothe, E.; Behl, R.K. Role of root exudates in plant-microbe interactions. J. Appl. Bot. Food Qual. 2012, 82, 122–130. [Google Scholar]
- Saleem, M.; Law, A.D.; Sahib, M.R.; Pervaiz, Z.H.; Zhang, Q. Impact of root system architecture on rhizosphere and root microbiome. Rhizosphere 2018, 6, 47–51. [Google Scholar] [CrossRef]
- Pieterse, C.M.; Zamioudis, C.; Does, D.V.; Van Wees, S. Signalling Networks Involved in Induced Resistance. Induced Resistance for Plant Defence: A Sustainable Approach to Crop Protection, 2nd ed.; John Wiley and Sons: Hoboken, NJ, USA, 2014; pp. 58–80. [Google Scholar]
- Yong, L. Microbial community diversity analysis of Panax ginseng rhizosphere and non-rhizosphere soil using randomly amplified polymorphic DNA method. Open J. Genet. 2012, 2, 20387. [Google Scholar]
- Toal, M.E.; Yeomans, C.; Killham, K.; Meharg, A. A review of rhizosphere carbon flow modelling. Plant Soil 2000, 222, 263–281. [Google Scholar] [CrossRef]
- Whipps, J.M. Microbial interactions and biocontrol in the rhizosphere. J. Exp. Bot. 2001, 52, 487–511. [Google Scholar] [CrossRef]
- Krzyzanowska, D.; Potrykus, M.; Golanowska, M.; Polonis, K.; Gwizdek-Wisniewska, A.; Lojkowska, E.; Jafra, S. Rhizosphere bacteria as potential biocontrol agents against soft rot caused by various pectobacterium and Dickeya spp. strains. J. Plant Pathol. 2012, 94, 367–378. [Google Scholar]
- Selvakumar, G.; Saha, S.; Kundu, S. Inhibitory activity of pine needle tannin extracts on some agriculturally resourceful microbes. Indian J. Microbiol. 2007, 47, 267–270. [Google Scholar] [CrossRef] [PubMed]
- Vendan, R.T.; Lee, S.H.; Yu, Y.J.; Rhee, Y.H. Analysis of Bacterial Community in the Ginseng Soil Using Denaturing Gradient Gel Electrophoresis (DGGE). Indian J. Microbiol. 2011, 52, 286–288. [Google Scholar] [CrossRef]
- Marilley, L.; Aragno, M. Phylogenetic diversity of bacterial communities differing in degree of proximity of Lolium perenne and Trifolium repens roots. Appl. Soil Ecol. 1999, 13, 127–136. [Google Scholar] [CrossRef]
- Berggren, I.; Van Vuurde, J.; Mãrtensson, A.M.; Alström, S. Rhizoplane colonisation of peas by Rhizobium leguminosarum bv. viceae and a deleterious Pseudomonas putida. FEMS Microbiol. Ecol. 2005, 52, 71–78. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Smit, E.; Leeflang, P.; Gommans, S.; Broek, J.V.D.; van Mil, S.; Wernars, K. Diversity and Seasonal Fluctuations of the Dominant Members of the Bacterial Soil Community in a Wheat Field as Determined by Cultivation and Molecular Methods. Appl. Environ. Microbiol. 2001, 67, 2284–2291. [Google Scholar] [CrossRef]
- Castro, H.F.; Classen, A.; Austin, E.E.; Norby, R.; Schadt, C.W. Soil Microbial Community Responses to Multiple Experimental Climate Change Drivers. Appl. Environ. Microbiol. 2010, 76, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Hedrich, S.; Schlömann, M.; Johnson, D.B. The iron-oxidizing proteobacteria. Microbiology 2011, 157, 1551–1564. [Google Scholar] [CrossRef] [PubMed]
- Rosenzweig, N.; Tiedje, J.M.; Quensen, J.F.; Meng, Q.; Hao, J.J. Microbial Communities Associated with Potato Common Scab-Suppressive Soil Determined by Pyrosequencing Analyses. Plant Dis. 2012, 96, 718–725. [Google Scholar] [CrossRef]
- She, S.; Niu, J.; Zhang, C.; Xiao, Y.; Chen, W.; Dai, L.; Liu, X.; Yin, H. Significant relationship between soil bacterial community structure and incidence of bacterial wilt disease under continuous cropping system. Arch. Microbiol. 2016, 199, 267–275. [Google Scholar] [CrossRef]
- Tian, Y.; Gao, L. Bacterial Diversity in the Rhizosphere of Cucumbers Grown in Soils Covering a Wide Range of Cucumber Cropping Histories and Environmental Conditions. Microb. Ecol. 2014, 68, 794–806. [Google Scholar] [CrossRef]
- Xiong, W.; Zhao, Q.; Zhao, J.; Xun, W.; Li, R.; Zhang, R.; Wu, H.; Shen, Q. Different Continuous Cropping Spans Significantly Affect Microbial Community Membership and Structure in a Vanilla-Grown Soil as Revealed by Deep Pyrosequencing. Microb. Ecol. 2014, 70, 209–218. [Google Scholar] [CrossRef]
- Liu, D.; Yang, Y.; An, S.; Wang, H.; Wang, Y. The Biogeographical Distribution of Soil Bacterial Communities in the Loess Plateau as Revealed by High-Throughput Sequencing. Front. Microbiol. 2018, 9, 2456. [Google Scholar] [CrossRef]
- Tong, A.-Z.; Liu, W.; Liu, Q.; Xia, G.-Q.; Zhu, J.-Y. Diversity and composition of the Panax ginseng rhizosphere microbiome in various cultivation modesand ages. BMC Microbiol. 2021, 21, 18. [Google Scholar] [CrossRef]
- Zhang, M.; Chai, L.; Huang, M.; Jia, W.; Guo, J.; Huang, Y. Deciphering the archaeal communities in tree rhizosphere of the Qinghai-Tibetan plateau. BMC Microbiol. 2020, 20, 235. [Google Scholar] [CrossRef]
- Bernard, L. Dynamics and identification of soil microbial populations actively assimilating carbon from 13C-labelled wheat residue as estimated by DNA-and RNA-SIP techniques. Environ. Microbiol. 2007, 9, 752–764. [Google Scholar] [CrossRef]
- Butterfield, C.N.; Li, Z.; Andeer, P.F.; Spaulding, S.; Thomas, B.C.; Singh, A.; Hettich, R.L.; Suttle, K.B.; Probst, A.J.; Tringe, S.G.; et al. Proteogenomic analyses indicate bacterial methylotrophy and archaeal heterotrophy are prevalent below the grass root zone. PeerJ 2016, 4, e2687. [Google Scholar] [CrossRef]
- Moissl-Eichinger, C.; Pausan, M.-R.; Taffner, J.; Berg, G.; Bang, C.; Schmitz, R.A. Archaea Are Interactive Components of Complex Microbiomes. Trends Microbiol. 2018, 26, 70–85. [Google Scholar] [CrossRef] [PubMed]
- Spang, A.; Saw, J.; Jørgensen, S.L.; Zaremba-Niedzwiedzka, K.; Martijn, J.; Lind, A.E.; Van Eijk, R.; Schleper, C.; Guy, L.; Ettema, T.J.G. Complex archaea that bridge the gap between prokaryotes and eukaryotes. Nat. Cell Biol. 2015, 521, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Harrison, K.A.; Bol, R.; Bardgett, R.D. Do plant species with different growth strategies vary in their ability to compete with soil microbes for chemical forms of nitrogen? Soil Biol. Biochem. 2008, 40, 228–237. [Google Scholar] [CrossRef]
- Chen, J.; Carrillo, Y.; Pendall, E.; Dijkstra, F.A.; Evans, R.D.; Morgan, J.A.; Williams, D.G. Soil Microbes Compete Strongly with Plants for Soil Inorganic and Amino Acid Nitrogen in a Semiarid Grassland Exposed to Elevated CO2 and Warming. Ecosystems 2015, 18, 867–880. [Google Scholar] [CrossRef]
- Vio, S.A. Paraburkholderia. In Beneficial Microbes in Agro-Ecology; Elsevier: Amsterdam, The Netherlands, 2020; pp. 271–311. [Google Scholar]
- Fradin, E.F.; Thomma, B.P. Physiology and molecular aspects of Verticillium wilt diseases caused by V. dahliae and V. albo-atrum. Mol. Plant Pathol. 2006, 7, 71–86. [Google Scholar] [CrossRef]
- Upchurch, R.; Chiu, C.-Y.; Everett, K.; Dyszynski, G.; Coleman, D.C.; Whitman, W.B. Differences in the composition and diversity of bacterial communities from agricultural and forest soils. Soil Biol. Biochem. 2008, 40, 1294–1305. [Google Scholar] [CrossRef]
- Priyadharsini, P.; Dhanasekaran, D. Diversity of soil Allelopathic Actinobacteria in Tiruchirappalli district, Tamilnadu, India. J. Saudi Soc. Agric. Sci. 2015, 14, 54–60. [Google Scholar] [CrossRef]
- Castañeda, L.E.; Barbosa, O. Metagenomic analysis exploring taxonomic and functional diversity of soil microbial communities in Chilean vineyards and surrounding native forests. PeerJ 2017, 5, e3098. [Google Scholar] [CrossRef]
- Fierer, N.; Leff, J.W.; Adams, B.J.; Nielsen, U.N.; Bates, S.T.; Lauber, C.L.; Owens, S.; Gilbert, J.A.; Wall, D.H.; Caporaso, J.G. Cross-biome metagenomic analyses of soil microbial communities and their functional attributes. Proc. Natl. Acad. Sci. USA 2012, 109, 21390–21395. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Rodriguez-R, L.M.; Johnston, E.; Wu, L.; Cheng, L.; Xue, K.; Tu, Q.; Deng, Y.; He, Z.; Shi, J.Z.; et al. Soil Microbial Community Responses to a Decade of Warming as Revealed by Comparative Metagenomics. Appl. Environ. Microbiol. 2013, 80, 1777–1786. [Google Scholar] [CrossRef]
- Thomas, T.; Gilbert, J.; Meyer, F. Metagenomics—A guide from sampling to data analysis. Microb. Inform. Exp. 2012, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Stam, M.R. Dividing the large glycoside hydrolase family 13 into subfamilies: Towards improved functional annotations of α-amylase-related proteins. Protein Eng. Des. Sel. 2006, 19, 555–562. [Google Scholar] [CrossRef]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy): An expert resource for Glycogenomics. Nucleic Acids Res. 2008, 37, D233–D238. [Google Scholar] [CrossRef] [PubMed]
- Arantes, V.; Saddler, J.N. Access to cellulose limits the efficiency of enzymatic hydrolysis: The role of amorphogenesis. Biotechnol. Biofuels 2010, 3, 1–11. [Google Scholar] [CrossRef]
- Reyes-Ortiz, V.; Heins, R.A.; Cheng, G.; Kim, E.Y.; Vernon, B.C.; Elandt, R.B.; Adams, P.D.; Sale, K.L.; Hadi, M.Z.; Simmons, B.A.; et al. Addition of a carbohydrate-binding module enhances cellulase penetration into cellulose substrates. Biotechnol. Biofuels 2013, 6, 93. [Google Scholar] [CrossRef]
- Chalak, A.; Villares, A.; Moreau, C.; Haon, M.; Grisel, S.; D’Orlando, A.; Herpoël-Gimbert, I.; Labourel, A.; Cathala, B.; Berrin, J.-G. Influence of the carbohydrate-binding module on the activity of a fungal AA9 lytic polysaccharide monooxygenase on cellulosic substrates. Biotechnol. Biofuels 2019, 12, 1–10. [Google Scholar] [CrossRef]
- Sidar, A.; Albuquerque, E.D.; Voshol, G.P.; Ram, A.F.J.; Vijgenboom, E.; Punt, P.J. Carbohydrate Binding Modules: Diversity of Domain Architecture in Amylases and Cellulases From Filamentous Microorganisms. Front. Bioeng. Biotechnol. 2020, 8, 871. [Google Scholar] [CrossRef] [PubMed]
- Chapin, F.S.; Walker, B.H.; Hobbs, R.J.; Hooper, D.U.; Lawton, J.H.; Sala, O.E.; Tilman, D. Biotic Control over the Functioning of Ecosystems. Science 1997, 277, 500–504. [Google Scholar] [CrossRef]
- Tan, Y.; Cui, Y.; Li, H.; Kuang, A.; Li, X.; Wei, Y.; Ji, X. Rhizospheric soil and root endogenous fungal diversity and composition in response to continuous Panax notoginseng cropping practices. Microbiol. Res. 2017, 194, 10–19. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Traits | Field | Forest | SEM | p Value |
|---|---|---|---|---|
| Plant height (cm) | 30.20 | 25.47 | 1.42 | 0.07 |
| Root length (cm) | 7.20 | 6.98 | 0.55 | 0.79 |
| Root weight (g) | 6.18 | 8.91 | 0.20 | 0.001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, R.; Gu, H.; He, S.; Xiong, B.; Huang, Y.; Horowitz, A.R.; He, X. Comparative Metagenomic and Metabolomic Profiling of Rhizospheres of Panax notoginseng Grown under Forest and Field Conditions. Agronomy 2021, 11, 2488. https://doi.org/10.3390/agronomy11122488
Shi R, Gu H, He S, Xiong B, Huang Y, Horowitz AR, He X. Comparative Metagenomic and Metabolomic Profiling of Rhizospheres of Panax notoginseng Grown under Forest and Field Conditions. Agronomy. 2021; 11(12):2488. https://doi.org/10.3390/agronomy11122488
Chicago/Turabian StyleShi, Rui, Haiyan Gu, Shu He, Binjie Xiong, Youguo Huang, Abraham Rami Horowitz, and Xiahong He. 2021. "Comparative Metagenomic and Metabolomic Profiling of Rhizospheres of Panax notoginseng Grown under Forest and Field Conditions" Agronomy 11, no. 12: 2488. https://doi.org/10.3390/agronomy11122488
APA StyleShi, R., Gu, H., He, S., Xiong, B., Huang, Y., Horowitz, A. R., & He, X. (2021). Comparative Metagenomic and Metabolomic Profiling of Rhizospheres of Panax notoginseng Grown under Forest and Field Conditions. Agronomy, 11(12), 2488. https://doi.org/10.3390/agronomy11122488