
SNPs, InDels, and Microsatellites within and Near to Rice NBS-LRR Resistance Gene Candidates



Abstract
:1. Introduction
2. Materials and Methods
2.1. MEME/MAST/NLR-Parser R-Gene Search in Five Indica and One Japonica Reference Genomes
2.2. HMMER Motif Search in Five Indica and One Japonica Reference Genomes
2.3. Read Processing and Variant Calling in 75 Resequenced Indica Genomes
2.4. Identification of Candidate Genes
2.5. Ortholog Identification
2.6. Comparison with Cloned Resistance Genes
2.7. Sequence Variation Analysis
2.8. Genomic Locations of SSR Primers
3. Results
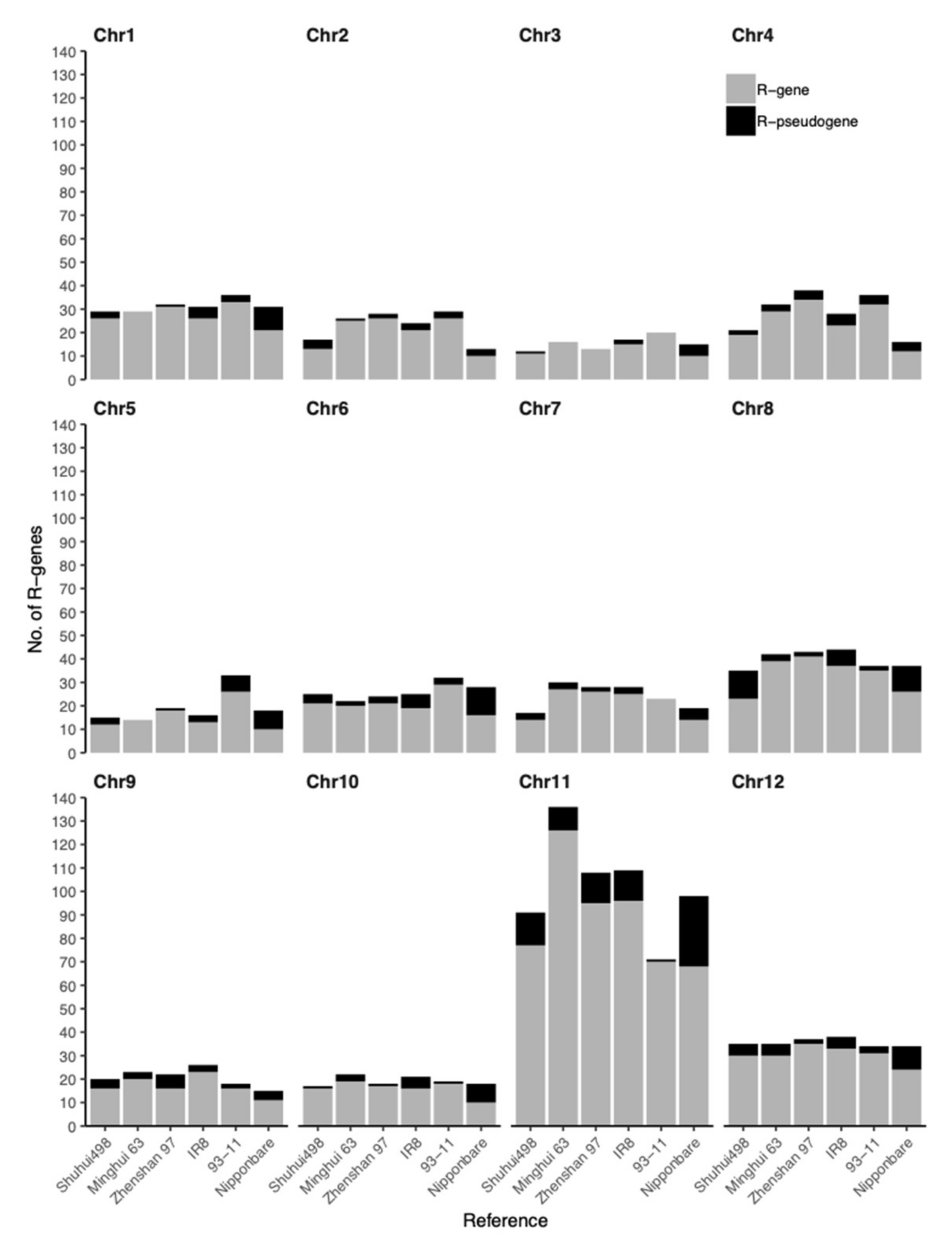
3.1. Identification of R-Genes, R-Pseudogenes, and Partial R-Genes
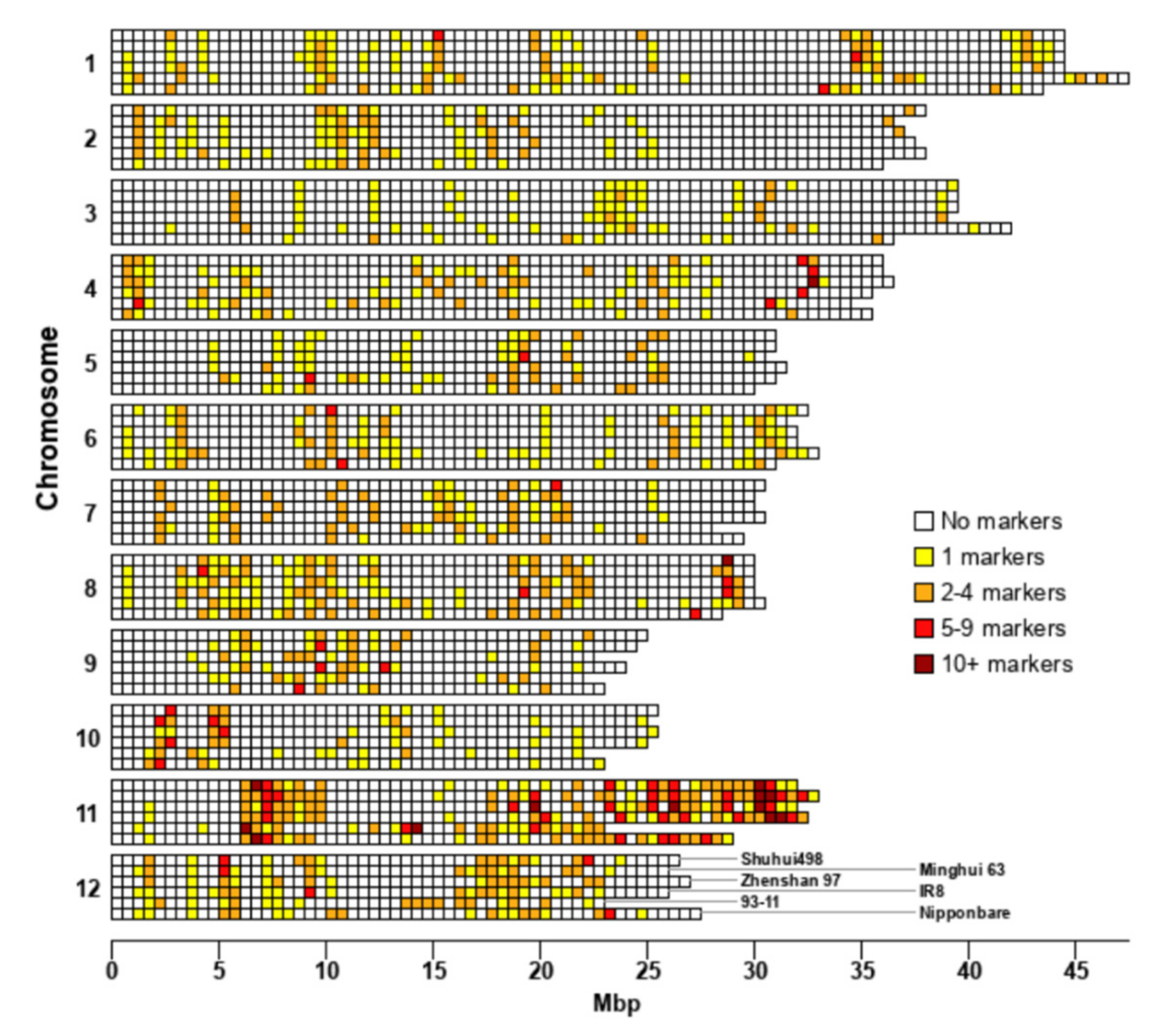
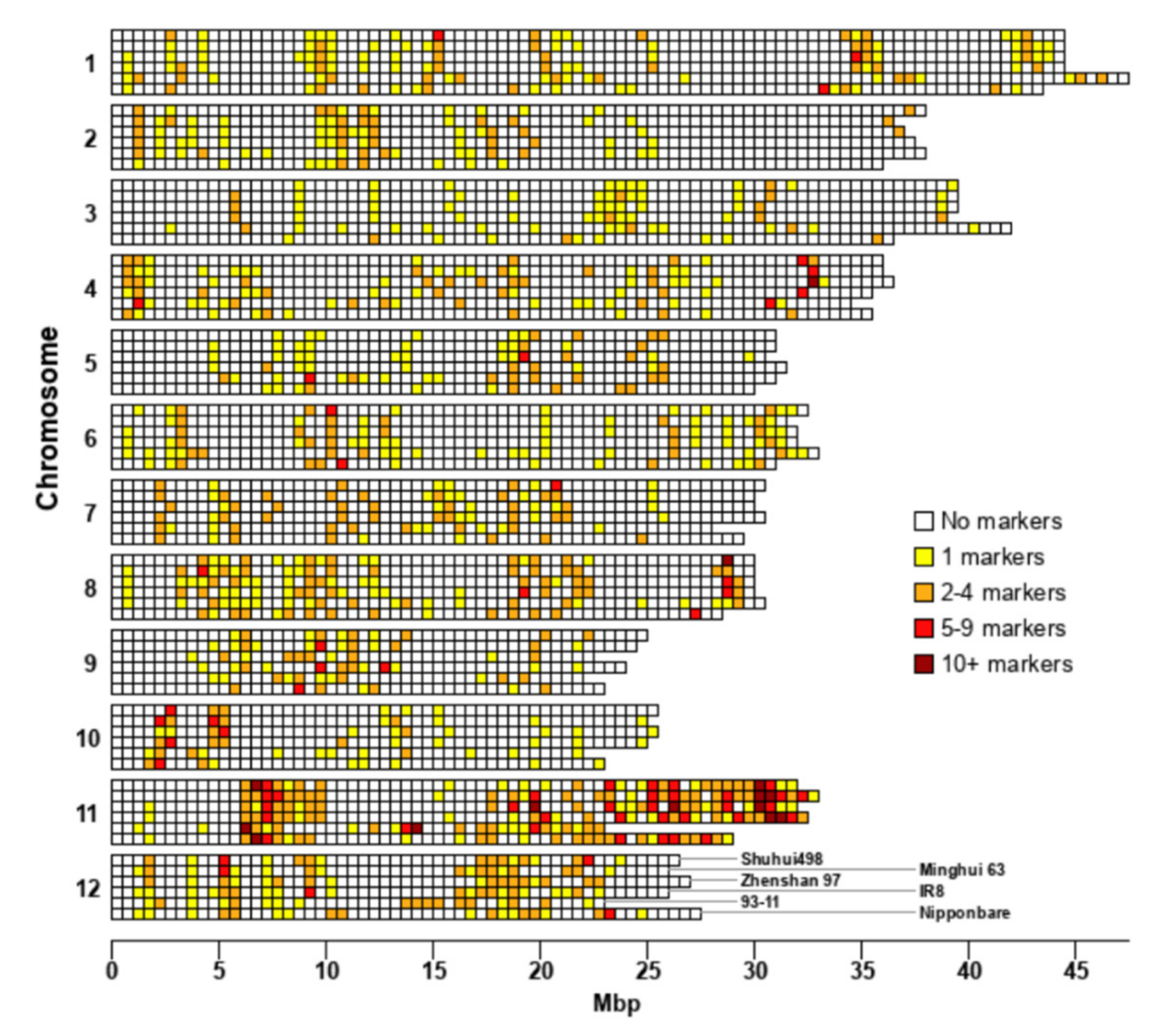
3.2. R-Gene Distribution
3.3. R-Gene Orthologs
3.4. Cloned R-Genes
3.5. Variation in Predicted R-Genes
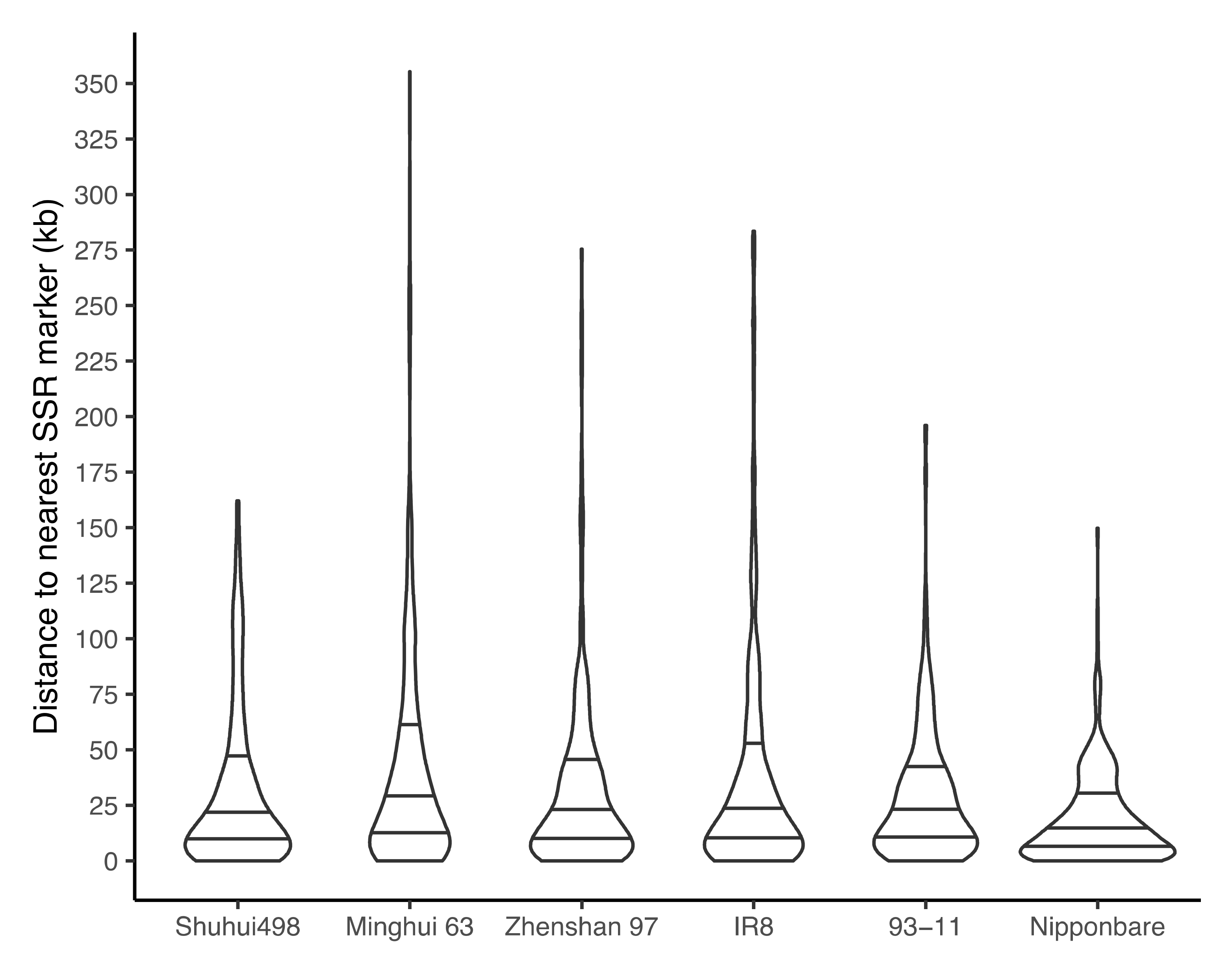
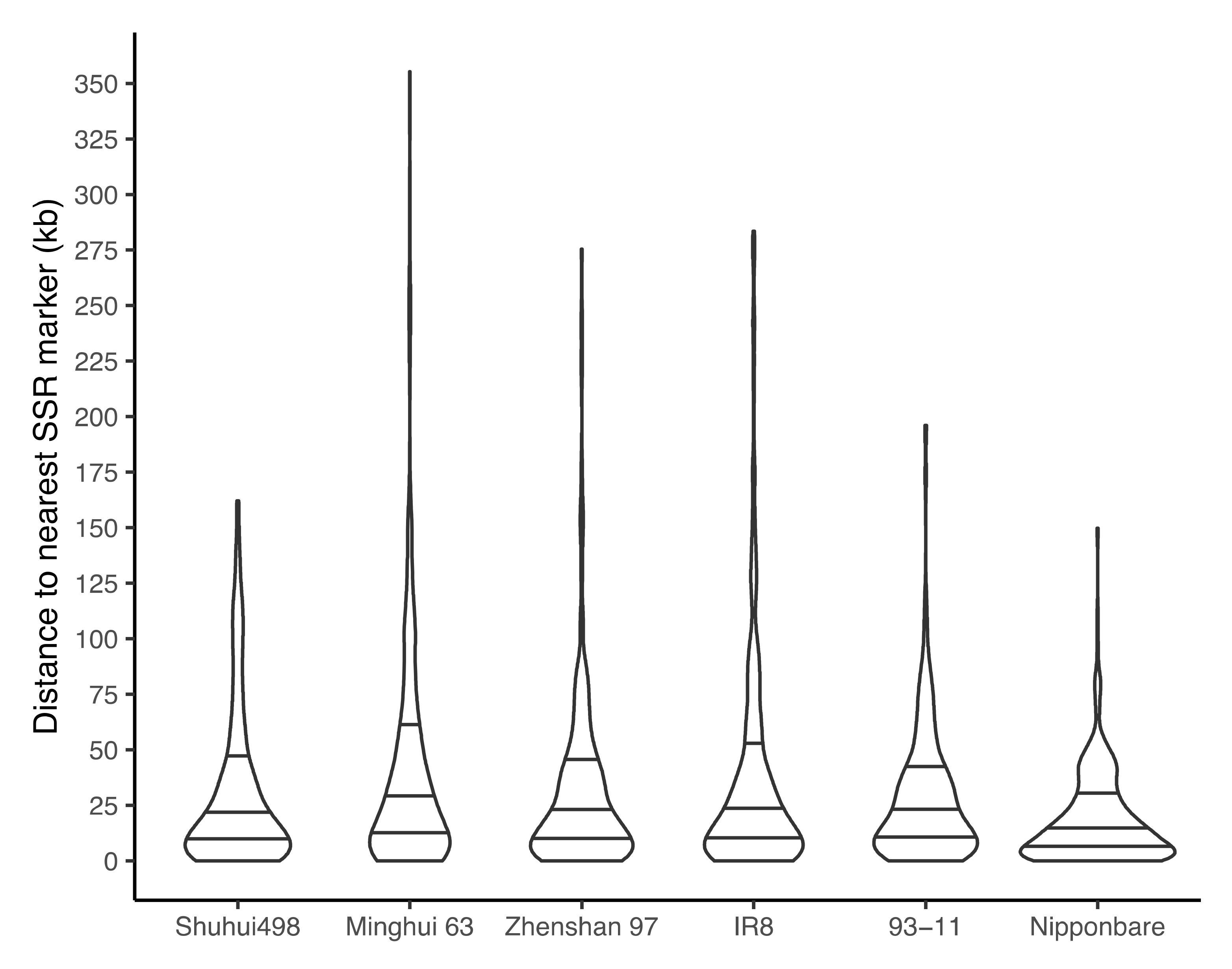
3.6. Proximity of SSR Markers to R-Genes
4. Discussion
4.1. Evaluation of In Silico Analysis Methods
4.2. R-Gene Variation between Rice Varieties
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Savary, S.; Willocquet, L.; Pethybridge, S.J.; Esker, P.; McRobers, N.; Nelson, A. The global burden of pathogens and pests on major food crops. Nat. Ecol. Evol. 2019, 3, 430–439. [Google Scholar] [CrossRef]
- Nelson, R.; Wiesner-Hanks, T.; Wisser, R.; Balint-Kurti, P. Navigating complexity to breed disease-resistant crops. Nat. Rev. Genet. 2018, 19, 21–33. [Google Scholar] [CrossRef]
- Yoshimura, S.; Yoshimura, A.; Iwata, N.; McCouch, S.R.; Lleva Abenes, M.; Baraoidan, M.R.; Mew, T.W.; Nelson, R.J. Tagging and combining bacterial blight resistance genes in rice using RAPD and RFLP markers. Mol. Breed. 1995, 1, 375–387. [Google Scholar] [CrossRef]
- Balachiranjeevi, C.H.; Bhaskar Naik, S.; Abhilash Kumar, V.; Harika, G.; Mahadev Swamy, H.K.; Hajira, S.; Dilip Kumar, T.; Anila, M.; Kale, R.R.; Yugender, A.; et al. Marker-assisted pyramiding of two major, broad-spectrum bacterial blight resistance genes, Xa21 and Xa33 into an elite maintainer line of rice, DRR17B. PLoS ONE 2018, 13, e0201271. [Google Scholar]
- Ballini, E.; Berruyer, R.; Morel, J.B.; Lebrun, M.H.; Nottéghem, J.L.; Tharreau, D. Modern elite rice varieties of the ‘Green Revolution’have retained a large introgression from wild rice around the Pi33 rice blast resistance locus. New Phytol. 2007, 175, 340–350. [Google Scholar] [CrossRef]
- Gottin, C.; Dievart, A.; Summo, M.; Droc, G.; Périn, C.; Ranwez, V.; Chantret, N. A New ‘Comprehensive’Annotation of Leucine-Rich Repeat-Containing Receptors in Rice. Plant J. 2021, 108, 492–508. [Google Scholar] [CrossRef]
- McHale, L.; Tan, X.; Koehl, P.; Michelmore, R.W. Plant NLR proteins: Adaptable guards. Genome Biol. 2006, 7, 212. [Google Scholar] [CrossRef] [Green Version]
- Bai, J.; Pennill, L.A.; Ning, J.; Lee, S.W.; Ramalingam, J.; Webb, C.A.; Zhao, B.; Sun, Q.; Nelson, J.C.; Leach, J.E.; et al. Diversity in nucleotide binding site-leucine-rich repeat genes in cereals. Genome Res. 2002, 12, 1871–1884. [Google Scholar] [CrossRef] [Green Version]
- Aarts, M.G.M.; Hekkert, B.L.; Holub, E.B.; Beynon, J.L.; Stiekema, W.J.; Pereira, A. Identification of r-gene homologous DNA fragments linked to disease resistance loci in Arabidopsis thaliana. Mol. Plant Microbe Interact. 1998, 11, 251–258. [Google Scholar] [CrossRef] [Green Version]
- Vossen, J.H.; Dezhsetan, S.; Esselink, D.; Arens, M.; Sanz, M.J.; Verweij, W.; Verzaux, E.; van der Linden, C.G. Novel applications of motif-directed profiling to identify disease resistance genes in plants. Plant Methods 2013, 9, 37. [Google Scholar] [CrossRef] [Green Version]
- Sallaud, C.; Lorieux, M.; Roumen, E.; Tharreau, D.; Berruyer, R.; Svestasrani, P.; Garsmeur, O.; Ghesquiere, A.; Notteghem, J.L. Identification of five new blast resistance genes in the highly blast-resistant rice variety IR64 using a QTL mapping strategy. Theor. Appl. Genet. 2003, 106, 794–803. [Google Scholar] [CrossRef]
- Wan, H.; Yuan, W.; Ye, Q.; Wang, R.; Ruan, M.; Li, Z.; Zhou, G.; Yao, Z.; Zhao, J.; Liu, S.; et al. Analysis of TIR- and non-TIR-NLR disease resistance gene analagous in pepper: Characterization, genetic variation, functional divergence and expression patterns. BMC Genom. 2012, 13, 502. [Google Scholar] [CrossRef] [Green Version]
- Sharma, R.; Rawat, V.; Suresh, C.G. Genome-wide identification and tissue-specific expression analysis of nucleotide binding site-leucine rich repeat gene family in Cicer arietinum (kabuli chickpea). Genom. Data 2017, 14, 24–31. [Google Scholar] [CrossRef]
- de Araújo, A.C.; Fonseca, F.C.D.A.; Cotta, M.G.; Alves, G.S.C.; Miller, R.N.G. Plant NLR receptor proteins and their potential in the development of durable genetic resistance to biotic stresses. Biotechnol. Res. Innov. 2019, 3, 80–94. [Google Scholar] [CrossRef]
- Mizuno, H.; Katagiri, S.; Kanamori, H.; Mukai, Y.; Sasaki, T.; Matsumoto, T.; Wu, J. Evolutionary dynamics and impacts of chromosome regions carrying R-gene clusters in rice. Sci. Rep. 2020, 10, 872. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Tao, Y.; Chen, X.; Zou, Y.; Lei, C.; Wang, J.; Li, X.; Zhao, X.; Zhang, M.; Lu, Z.; et al. Identification of a new rice blast resistance gene, Pid3, by genomewide comparison of paired nucleotide-binding site-leucine-rich repeat genes and their pseudogene alleles between the two sequenced rice genomes. Genetics 2009, 182, 1303–1311. [Google Scholar] [CrossRef] [Green Version]
- Monosi, B.; Wisser, R.J.; Pennill, L.; Hulbert, S.H. Full-genome analysis of resistance gene homologues in rice. Theor. Appl. Genet. 2004, 109, 1434–1447. [Google Scholar] [CrossRef]
- Zhou, T.; Wang, Y.; Chen, J.-Q.; Araki, Z.; Jing, K.; Jiang, J.; Shen, J.; Tian, D. Genome-wide identification of NBS genes in japonica rice reveals significant expansion of divergent non-TIR NLR genes. Mol. Genet. Genom. 2004, 271, 402–415. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Shahid, M.Q.; Li, R.; Li, W.; Liu, W.; Ghouri, F.; Liu, X. Genome-wide analysis of genetic variations and the detection of rich variants of NLR encoding genes in common wild rice lines. Plant Mol. Biol. Report. 2018, 36, 618–630. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Hu, S.; Wang, J.; Wong, G.K.; Li, S.; Liu, B.; Deng, Y.; Dai, L.; Zhou, Y.; Zhang, X.; et al. A draft sequence of the rice genome (Oryza sativa L. ssp. Indica). Science 2002, 296, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Steele, K.A.; Quinton-Tulloch, M.J.; Amgai, R.B.; Dhakal, R.; Khatiwada, S.P.; Vyas, D.; Heine, M.; Witcombe, J.R. Accelerating public sector rice breeding with high-density KASP markers derived from whole genome sequencing of indica rice. Mol. Breed. 2018, 38, 38. [Google Scholar] [CrossRef] [Green Version]
- The 3,000 rice genomes project. The 3,000 rice genomes project. GigaScience 2014, 3, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jupe, F.; Pritchard, L.; Etherington, G.J.; MacKenzie, K.; Cock, P.J.A.; Wright, F.; Sharma, S.K.; Bolser, D.; Bryan, G.J.; Jones, J.D.G.; et al. Identification and localisation of the NB-LRR gene family within the potato genome. BMC Genom. 2012, 13, 75. [Google Scholar] [CrossRef] [Green Version]
- Bailey, T.; Gribskov, M. Combining evidence using p-values: Application to sequence homology searches. Bioinformatics 1998, 14, 48–54. [Google Scholar] [CrossRef] [Green Version]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. Meme suite: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Yu, Y.; Ma, Y.; Gao, Q.; Cao, Y.; Chen, Z.; Ma, B.; Qi, M.; Li, Y.; Zhao, X.; et al. Sequencing and de novo assembly of a near complete indica rice genome. Nat. Commun. 2017, 8, 15324. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, L.; Xing, F.; Kudrna, D.A.; Yao, W.; Copetti, D.; Mu, T.; Li, W.; Song, J.; Xie, W.; et al. Extensive sequence divergence between the reference genomes of two elite indica rice varieties Zhenshan 97 and Minghui 63. Proc. Natl. Acad. Sci. USA 2016, 113, E5163–E5171. [Google Scholar] [CrossRef] [Green Version]
- Stein, J.C.; Yu, Y.; Copetti, D.; Zwickl, D.J.; Zhang, L.; Zhang, C.; Chougule, K.; Gao, D.; Iwata, A.; Goicoechea, J.L.; et al. Genomes of 13 domesticated and wild rice relatives highlight genetic conservation, turnover and innovation across the genus Oryza. Nat. Genet. 2018, 50, 285–296. [Google Scholar] [CrossRef] [PubMed]
- International Rice Genome Sequencing Project. The map-based sequence of the rice genome. Nature 2000, 436, 793–800. [Google Scholar]
- Steuernagel, B.; Jupe, F.; Witek, K.; Jones, J.D.G.; Wulff, B.B.H. NLR-parser: Rapid annotation of plant NLR complements. Bioinformatics 2015, 31, 1665–1667. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. Blast+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawahara, Y.; de la Bastide, M.; Hamilton, J.P.; Kanamori, H.; McCombie, W.R.; Ouyang, S.; Schwartz, D.C.; Tanaka, T.; Wu, J.; Zhou, S.; et al. Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice 2013, 6, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakai, H.; Lee, S.S.; Tanaka, T.; Numa, H.; Kim, J.; Kawahara, Y.; Wakimoto, H.; Yang, C.C.; Iwamoto, M.; Abe, T.; et al. Rice annotation project database (RAP-DB): An integrative and interactive database for rice genomics. Plant Cell Physiol. 2013, 54, e6. [Google Scholar] [CrossRef]
- Zhao, W.; Wang, J.; He, X.; Huang, X.; Jiao, Y.; Dai, M.; Wei, S.; Fu, J.; Chen, Y.; Ren, X.; et al. BGI-RIS: An integrated information resource and comparative analysis workbench for rice genomics. Nucleic Acids Res. 2004, 32, D377–D382. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.B.; Pickett, B.D.; Ridge, P.G. JustOrthologs: A fast, accurate and user-friendly ortholog identification algorithm. Bioinformatics 2019, 35, 546–552. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scaleable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Meyers, B.C.; Kozik, A.; Griego, A.; Kuang, H.; Michelmore, R.W. Genome-wide analysis of NLR-encoding genes in Arabidopsis. Plant Cell 2003, 15, 809–834. [Google Scholar] [CrossRef] [Green Version]
- Kuang, H.; Woo, S.S.; Meyers, B.C.; Nevo, E.; Michelmore, R.W. Multiple genetic processes result in heterogenous rates of evolution within the major cluster disease resistance genes in lettuce. Plant Cell 2004, 16, 2870–2894. [Google Scholar] [CrossRef] [Green Version]
- Luo, S.; Zhang, Y.; Hu, Q.; Chen, J.; Li, K.; Lu, C.; Liu, H.; Wang, W.; Kuang, H. Dynamic nucleotide-binding site and leucine-rich repeat-encoding genes in the grass family. Plant Physiol. 2012, 159, 197–210. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Xia, R.; Kuang, H.; Meyers, B.C. The diversification of plant NLR defense genes directs the evolution of microRNAs that target them. Mol. Biol. Evol. 2016, 33, 2692–2705. [Google Scholar] [CrossRef] [Green Version]
- Choi, K.; Reinhard, C.; Serra, H.; Ziolkowski, P.A.; Underwood, C.J.; Zhao, X.; Hardcastle, T.J.; Yelina, N.E.; Griffin, C.; Jackson, M.; et al. Recombination rate heterogeneity within Arabidopsis disease resistance genes. PLoS Genet. 2016, 12, e1006179. [Google Scholar] [CrossRef] [PubMed]
- Karasov, T.L.; Chae, E.; Herman, J.J.; Bergelson, J. Mechanisms to mitigate the trade-off between growth and defense. Plant Cell 2017, 29, 666–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonnessen, B.W.; Bossa-Castro, A.M.; Mauleon, R.; Alexandrov, N.; Leach, J.E. Shared cis-regulatory architecture identified across defense response genes is associated with broad-spectrum quantitative resistance in rice. Sci. Rep. 2019, 9, 1536. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Deng, Y.; Lin, H.; Kudrna, D.A.; Wing, R.A.; Li, L.; Zhang, Q.; Luo, M. Comparative BAC-based physical mapping of Oryza sativa ssp. indica var. 93-11 and evaluation of the two rice reference sequence assemblies. Plant J. 2013, 77, 795–805. [Google Scholar] [CrossRef]
- Wang, L.; Zhao, L.; Zhang, X.; Zhang, Q.; Jia, Y.; Wang, G.; Li, S.; Tian, D.; Li, W.H.; Yang, S. Large-scale identification and functional analysis of NLR genes in blast resistance in the Tetep rice genome sequence. Proc. Natl. Acad. Sci. USA 2019, 116, 18479–18487. [Google Scholar] [CrossRef] [Green Version]
- Fukuoka, S.; Saka, N.; Koga, H.; Ono, K.; Shimizu, T.; Ebana, K.; Hayashi, N.; Takahashi, A.; Hirochika, H.; Okuno, K.; et al. Loss of function of a proline-containing protein confers durable disease resistance in rice. Science 2009, 325, 998–1001. [Google Scholar] [CrossRef]
- Chen, X.; Shang, J.; Chen, D.; Lei, C.; Zou, Y.; Zhai, W.; Liu, G.; Xu, J.; Ling, Z.; Cao, G.; et al. A B-lectin receptor kinase gene conferring rice blast resistance. Plant J. 2006, 46, 794–804. [Google Scholar] [CrossRef]
- Vasudevan, K.; Gruissem, W.; Bhullar, N.K. Identification of novel alleles of the rice blast resistance gene Pi54. Sci. Rep. 2015, 5, 15678. [Google Scholar] [CrossRef] [Green Version]
- Zhai, C.; Lin, F.; Dong, Z.; He, X.; Yuan, B.; Zeng, X.; Wang, L.; Pan, Q. The isolation and characterization of Pik, a rice blast resistance gene which emerged after rice domestication. New Phytol. 2011, 189, 321–334. [Google Scholar] [CrossRef]
- Kim, S.-K.; Reinke, R.F. A novel resistance gene for bacterial blight in rice, Xa43(t) identified by GWAS, confirmed by QTL mapping using a bi-parental population. PLoS ONE 2019, 14, e0211775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marone, D.; Russo, M.A.; Laidò, G.; De Leonardis, A.M.; Mastrangelo, A.M. Plant nucelotide binding site- leucine-rich repeat (NLR) genes: Active guardians in host defense responses 2013. Int. J. Mol. Sci. 2013, 14, 7302–7326. [Google Scholar] [CrossRef] [Green Version]
- Schatz, M.C.; Maron, L.G.; Stein, J.C.; Hernandez Wences, A.; Gurtowski, J.; Biggers, E.; Lee, H.; Kramer, M.; Antoniou, E.; Ghiban, E.; et al. Whole genome de novo assemblies of three divergent strains of rice, Oryza sativa, document novel gene space of aus and indica. Genome Biol. 2014, 15, 506. [Google Scholar] [PubMed]
- Cao, J.; Schneeberger, K.; Ossowksi, S.; Günther, T.; Bender, S.; Fitz, J.; Koenig, D.; Lanz, C.; Stegle, O.; Lippert, C.; et al. Whole-genome sequencing of multiple Arabidopsis thaliana populations. Nat. Genet. 2011, 43, 956–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, P.; Silverstein, K.A.T.; Ramaraj, T.; Guhlin, J.; Denny, R.; Liu, J.; Farmer, A.D.; Steele, K.P.; Stupar, R.M.; Miller, J.R.; et al. Exploring structural variation and gene family architecture with De Novo assemblies of 15 Medicago genomes. BMC Genom. 2017, 18, 261. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locus | Chromosome | Gene Allele | GenBank ID | Source Variety |
|---|---|---|---|---|
| Resistance Conferred against Rice Blast Pathogen (Magnaporthe grisea) | ||||
| Pi2 | 6 | Pi2 | ABC94599.1 | C101A51 (indica) |
| 6 | Pi9 | ABB88855.1 | 75-1-127 (indica) | |
| 6 | Pi50 | AKS24975.1 | NIL-e1 (japonica) | |
| 6 | Piz-t | ABC73398.1 | Toride 1 (japonica) | |
| Pi5-1 | 9 | Pi5-1 | ACJ54697.1 | RIL260 (japonica) |
| 9 | Pii | BAN59294.1 | Hitomebore (japonica) | |
| Pi5-2 | 9 | Pi5-2 | ACJ54698.1 | RIL260 (japonica) |
| Pi21 | 4 | Pi21 | Q7XL73.1 | (japonica) |
| Pi36 | 8 | Pi36 | ABI64281.1 | Kasalath (indica) |
| Pi37 | 1 | Pish | XP_015646397.1 | Nipponbare (japonica) |
| 1 | Pi37 | ABI94578.1 | St. No. 1 (japonica) | |
| Pi54 | 11 | Pi54 (partial) | AAY33493.1 | Tetep (indica) |
| 11 | Pi54 (partial) | CCD32366.1 | Basmati 386 (indica) | |
| Pi64 | 1 | Pi64 | BAS74649.1 | Nipponbare (japonica) |
| Pi-b | 2 | Pi-b | BAA85975.1 | BL-1 (japonica) |
| Pi-CO39 | 11 | Pi-CO39 | ARF20187.1 | Yixiang1B (indica) |
| Pid-2 | 6 | Pid-2 | ALU57428.1 | Nipponbare (japonica) |
| Pid3 | 6 | Pid3 | ACN62383.1 | Digu (indica) |
| 6 | Pid3-A4 | ACN62387.1 | A4 (O. rufipogon) | |
| 6 | Pi25 | AFM35701.1 | Zhongjian100 (indica) | |
| Pik-1 | 11 | Pik1-KA | BAL63005.1 | Kanto51 (japonica) |
| 11 | Pi1-5 | AEB00617.1 | C101LAC (indica) | |
| 11 | Pikm-1 | BAG72135.1 | Tsuyake (japonica) | |
| 11 | Pikp-1 | ADV58352.1 | (japonica) | |
| Pik-2 | 11 | Pik2-KA | BAL63006.1 | Kanto51 (japonica) |
| 11 | Pikp-2 | ADV58351.1 | (japonica) | |
| 11 | Pi1-6 | AEB00618.1 | C101LAC (indica) | |
| 11 | Pikm-2 | BAG72136.1 | Tsuyake (japonica) | |
| Pit | 12 | Pit | BAH20864.1 | Nipponbare (japonica) |
| Pi-ta | 12 | Pi-ta | AAO45178.1 | Tsuyake (japonica) |
| Pb1 | 11 | Pb1 | BAJ25849.1 | Yixiang1B (indica) |
| 11 | Pb1 | BAJ25848.1 | St. No. 1 (japonica) | |
| LABR_64-1 | 9 | LABR_64-1 | AIP86911.1 | 312007 (japonica) |
| 9 | LABR_64-1 | AIP86912.1 | 301279 (japonica) | |
| LABR_64-2 | 9 | LABR_64-2 | AIP86925.1 | 312007 (japonica) |
| 9 | LABR_64-2 | AIP86926.1 | 301279 (japonica) | |
| SasRGA4 | 11 | SasRGA4 | BAK39922.1 | Sasanishiki, Aichi-asahi (japonica) |
| SasRGA5 | 11 | SasRGA | BAK39930.1 | Sasanishiki, Aichi-asahi (japonica) |
| Resistance Conferred against Bacterial Blight Pathogen (Xanthomonas oryzae pv. oryzae) | ||||
| Xa1 | 4 | Xa1 | BAA25068.1 | IR-BB1 (indica) |
| Xa3/Xa26 | 11 | Xa3/Xa26 | AYH53004.1 | Wase Aikoku 3 (japonica) |
| Xa4 | 11 | Xa4 | AQQ72929.1 | Nipponbare (japonica) |
| 11 | Xa4 | AQQ72925.1 | Minghui 63 (indica) | |
| Xa5 | 5 | Xa5 | AHC94895.1 | PB1 (indica) |
| Xa10 | 11 | Xa10 | AGE45112.1 | IRBB10A (indica) |
| Xa13 | 8 | Xa13 | ABD78944.1 | IR24 (indica) |
| Xa21 | 11 | Xa21 | AAC49123.1 | IRBB21 (indica) |
| Xa23 | 11 | Xa23 | AIX09985.1 | JG30 (indica) |
| Xa25 | 12 | Xa25 | AGS56391.1 | Zhenshan 97 (indica) |
| Xa27 | 6 | Xa27 | AFO69279.1 | Taichung Native 1 (indica) |
| Xa41 | 11 | Xa41(t) | B8BKP4.1 | 93-11 (indica) |
| Candidate Type | Motif Search | Gene Counts | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Search Sequence Type | NLR-Parser | HMMER NB-ARC | HMMER LRR | Shuhui498 | Minguhi 63 | Zhenshan 97 | IR8 | 93-11 | Nipponbare | |
| R-genes | Protein | Complete | + | + | 126 | 191 | 179 | 156 | 140 | 94 |
| Complete | + | - | 35 | 46 | 50 | 35 | 55 | 25 | ||
| Partial | + | + | 115 | 155 | 140 | 152 | 166 | 106 | ||
| - | + | + | 2 | 2 | 4 | 4 | 10 | 7 | ||
| R-genes totals | 278 | 394 | 373 | 347 | 371 | 232 | ||||
| R-pseudogenes | 6-frame translated gene DNA | Complete | + | + | 21 | 12 | 10 | 30 | 13 | 46 |
| Complete | + | - | 10 | 8 | 15 | 13 | 8 | 25 | ||
| Complete | - | + | 0 | 0 | 0 | 2 | 0 | 0 | ||
| Complete | - | - | 0 | 0 | 0 | 0 | 0 | 1 | ||
| Partial | + | + | 23 | 12 | 12 | 15 | 10 | 36 | ||
| - | + | + | 2 | 1 | 0 | 0 | 1 | 2 | ||
| R-pseudogenes totals | 56 | 33 | 37 | 60 | 32 | 110 | ||||
| Orthologs Identified | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Genome | Predicted Genes | Shuhui498 | Minghui 63 | Zhenshan 97 | IR8 | 93-11 | Nipponbare | ||
| Shuhui498 | R-genes | 278 | R-genes | - | 199 | 183 | 173 | 181 | 135 |
| R-pseudogenes | - | 9 | 8 | 16 | 14 | 41 | |||
| Partial R-genes | - | 9 | 13 | 10 | 28 | 17 | |||
| Non-R-genes | - | 7 | 5 | 2 | 3 | 9 | |||
| R-pseudogenes | 56 | R-genes | - | 26 | 26 | 16 | 24 | 12 | |
| R-pseudogenes | - | 10 | 7 | 14 | 5 | 20 | |||
| Partial R-genes | - | 3 | 1 | 2 | 3 | 3 | |||
| Non-R-genes | - | 2 | 1 | 3 | 2 | 3 | |||
| Minghui 63 | R-genes | 394 | R-genes | 198 | - | 257 | 248 | 244 | 125 |
| R-pseudogenes | 24 | - | 13 | 25 | 13 | 57 | |||
| Partial R-genes | 58 | - | 14 | 15 | 36 | 30 | |||
| Non-R-genes | 9 | - | 7 | 1 | 4 | 19 | |||
| R-pseudogenes | 33 | R-genes | 9 | - | 16 | 11 | 14 | 7 | |
| R-pseudogenes | 10 | - | 10 | 8 | 1 | 7 | |||
| Partial R-genes | 3 | - | 0 | 1 | 2 | 4 | |||
| Non-R-genes | 2 | - | 0 | 3 | 1 | 4 | |||
| Zhenshan 97 | R-genes | 373 | R-genes | 184 | 253 | - | 243 | 249 | 144 |
| R-pseudogenes | 22 | 14 | - | 19 | 11 | 53 | |||
| Partial R-genes | 66 | 18 | - | 15 | 39 | 25 | |||
| Non-R-genes | 8 | 6 | - | 2 | 5 | 18 | |||
| R-pseudogenes | 37 | R-genes | 8 | 13 | - | 11 | 12 | 9 | |
| R-pseudogenes | 6 | 10 | - | 10 | 4 | 3 | |||
| Partial R-genes | 4 | 3 | - | 4 | 3 | 3 | |||
| Non-R-genes | 2 | 2 | - | 3 | 0 | 1 | |||
| IR8 | R-genes | 347 | R-genes | 170 | 246 | 238 | - | 233 | 130 |
| R-pseudogenes | 15 | 11 | 10 | - | 11 | 46 | |||
| Partial R-genes | 51 | 20 | 22 | - | 36 | 33 | |||
| Non-R-genes | 17 | 15 | 13 | - | 6 | 20 | |||
| R-pseudogenes | 60 | R-genes | 17 | 25 | 20 | - | 25 | 14 | |
| R-pseudogenes | 13 | 8 | 10 | - | 2 | 13 | |||
| Partial R-genes | 10 | 6 | 5 | - | 8 | 4 | |||
| Non-R-genes | 1 | 2 | 1 | - | 0 | 0 | |||
| 93-11 | R-genes | 371 | R-genes | 186 | 247 | 255 | 240 | - | 148 |
| R-pseudogenes | 20 | 15 | 13 | 25 | - | 58 | |||
| Partial R-genes | 61 | 16 | 19 | 12 | - | 28 | |||
| Non-R-genes | 8 | 4 | 3 | 1 | - | 13 | |||
| R-pseudogenes | 32 | R-genes | 11 | 13 | 12 | 11 | - | 10 | |
| R-pseudogenes | 5 | 1 | 4 | 2 | - | 7 | |||
| Partial R-genes | 3 | 5 | 4 | 1 | - | 3 | |||
| Non-R-genes | 0 | 0 | 1 | 0 | - | 1 | |||
| Nipponbare | R-genes | 232 | R-genes | 136 | 124 | 142 | 128 | 144 | - |
| R-pseudogenes | 11 | 8 | 8 | 14 | 10 | - | |||
| Partial R-genes | 20 | 18 | 15 | 10 | 25 | - | |||
| Non-R-genes | 4 | 5 | 5 | 0 | 1 | - | |||
| R-pseudogenes | 110 | R-genes | 45 | 60 | 60 | 49 | 60 | - | |
| R-pseudogenes | 20 | 8 | 4 | 14 | 6 | - | |||
| Partial R-genes | 8 | 4 | 5 | 3 | 8 | - | |||
| Non-R-genes | 4 | 1 | 1 | 2 | 1 | - | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quinton-Tulloch, M.J.; Steele, K.A. SNPs, InDels, and Microsatellites within and Near to Rice NBS-LRR Resistance Gene Candidates. Agronomy 2021, 11, 2297. https://doi.org/10.3390/agronomy11112297
Quinton-Tulloch MJ, Steele KA. SNPs, InDels, and Microsatellites within and Near to Rice NBS-LRR Resistance Gene Candidates. Agronomy. 2021; 11(11):2297. https://doi.org/10.3390/agronomy11112297
Chicago/Turabian StyleQuinton-Tulloch, Mark J., and Katherine A. Steele. 2021. "SNPs, InDels, and Microsatellites within and Near to Rice NBS-LRR Resistance Gene Candidates" Agronomy 11, no. 11: 2297. https://doi.org/10.3390/agronomy11112297
APA StyleQuinton-Tulloch, M. J., & Steele, K. A. (2021). SNPs, InDels, and Microsatellites within and Near to Rice NBS-LRR Resistance Gene Candidates. Agronomy, 11(11), 2297. https://doi.org/10.3390/agronomy11112297