Increased Surface Roughness in Polydimethylsiloxane Films by Physical and Chemical Methods



 ,
,
Abstract
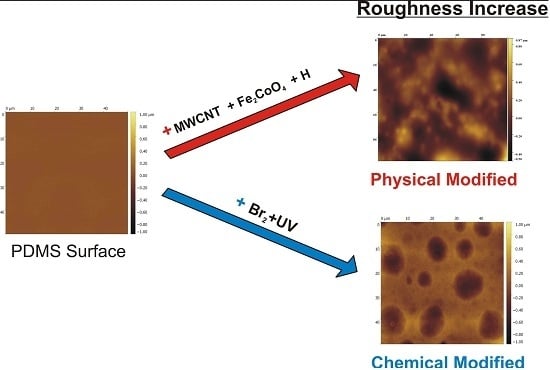
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Preparation of PDMS/Filler Composite Films
2.3. Exposition to Br2 Vapours and UV Reaction
2.4. Instrumentation
3. Results and Discussion
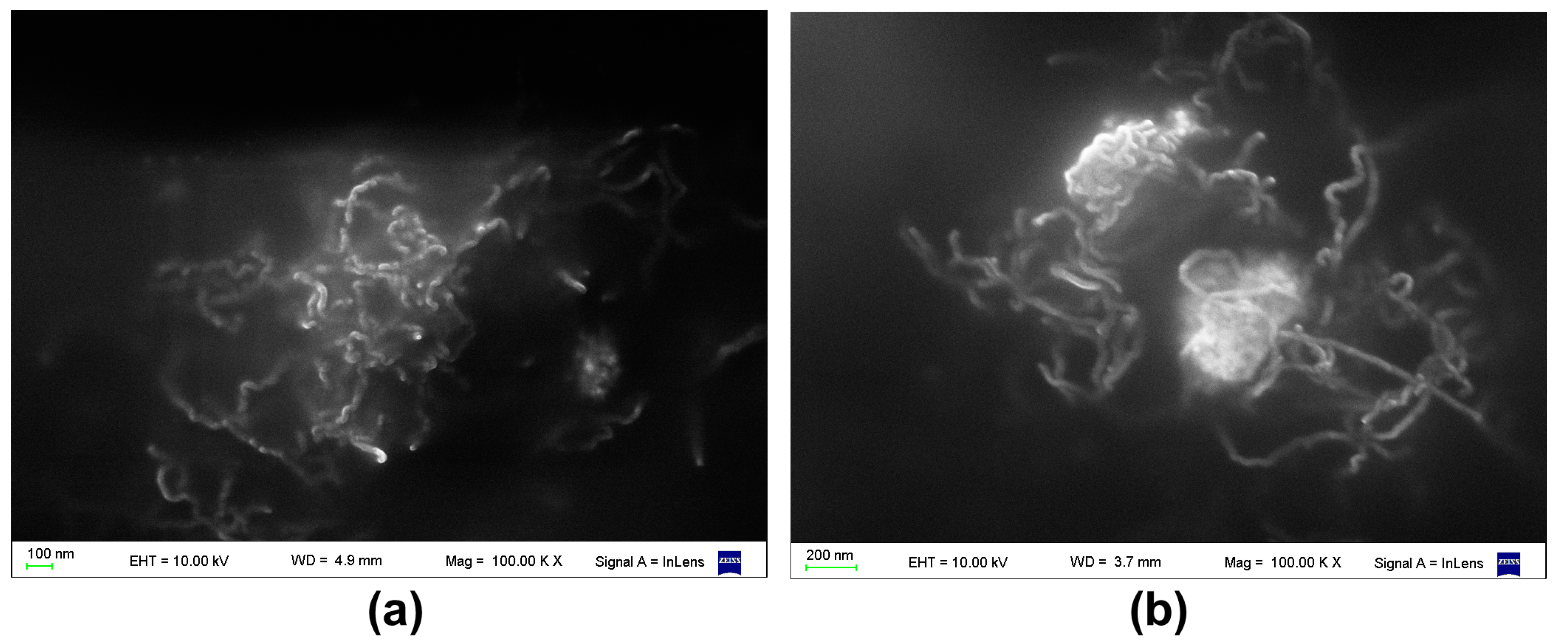
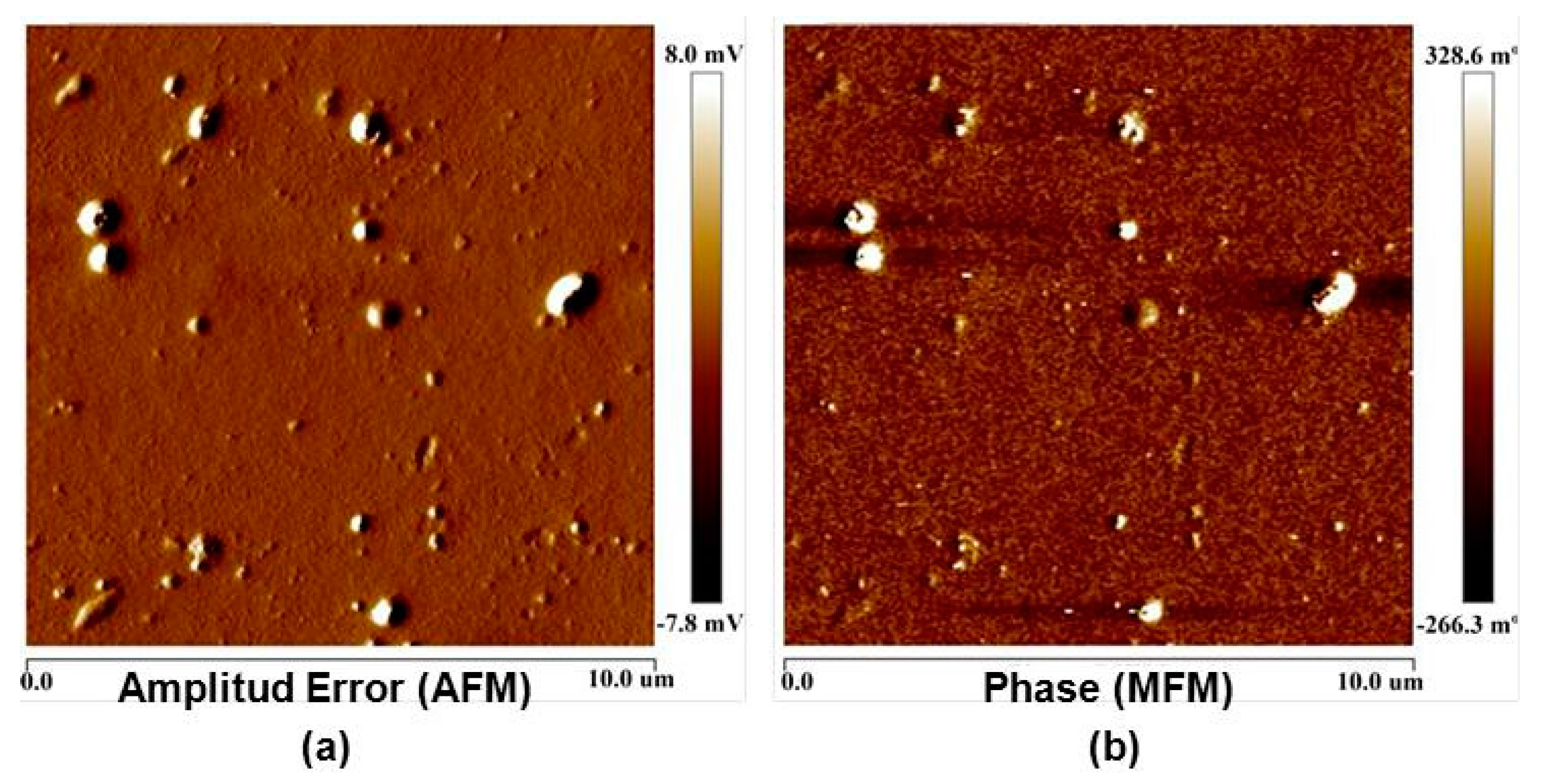
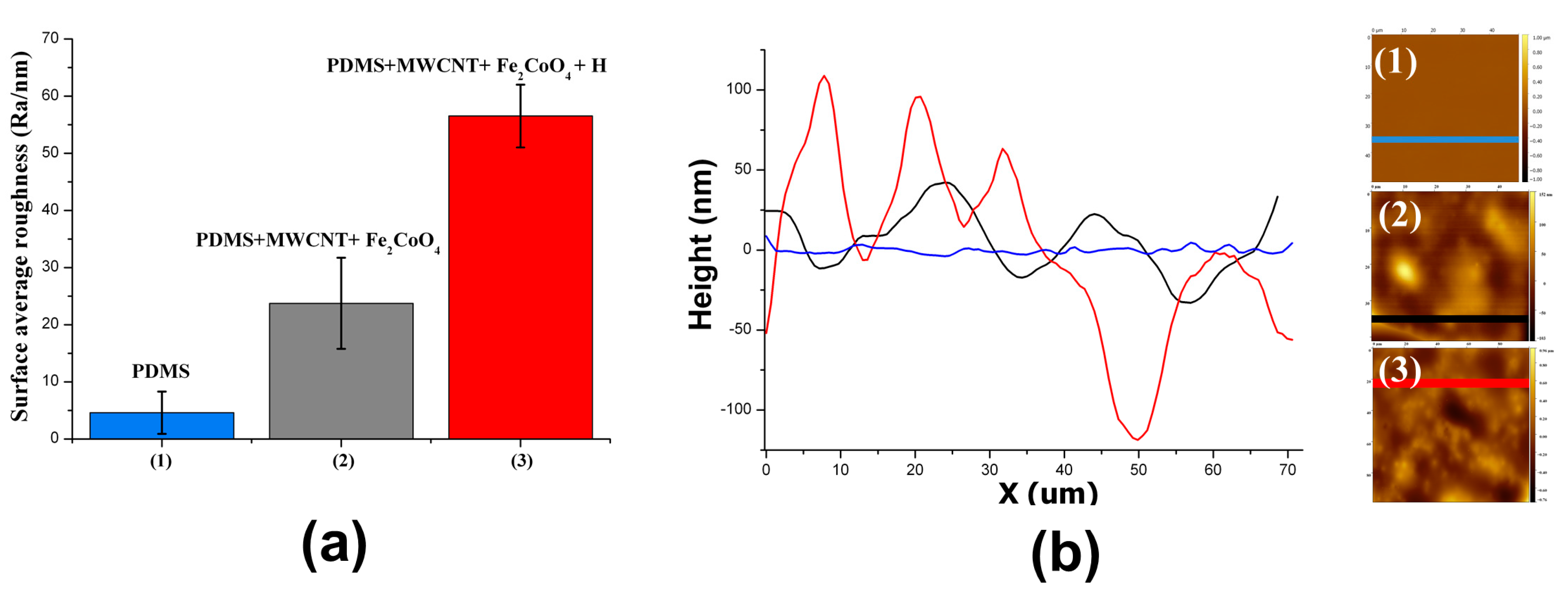
3.1. Surface Roughness in PDMS/Filler Composites
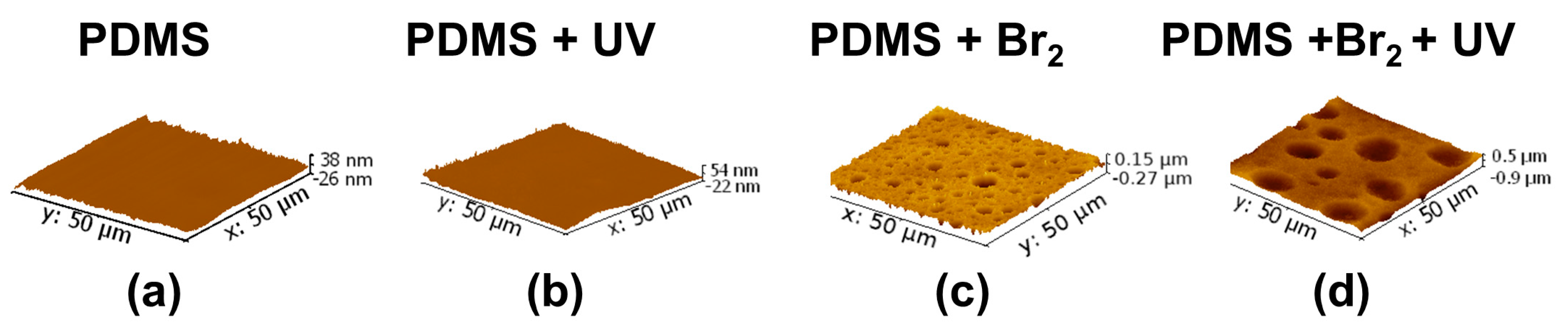
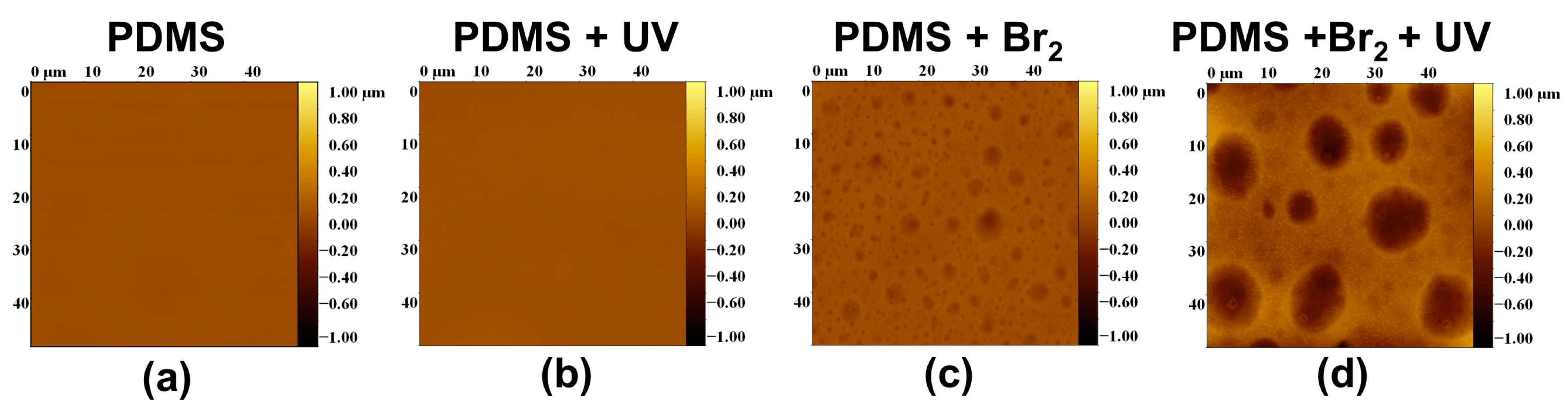
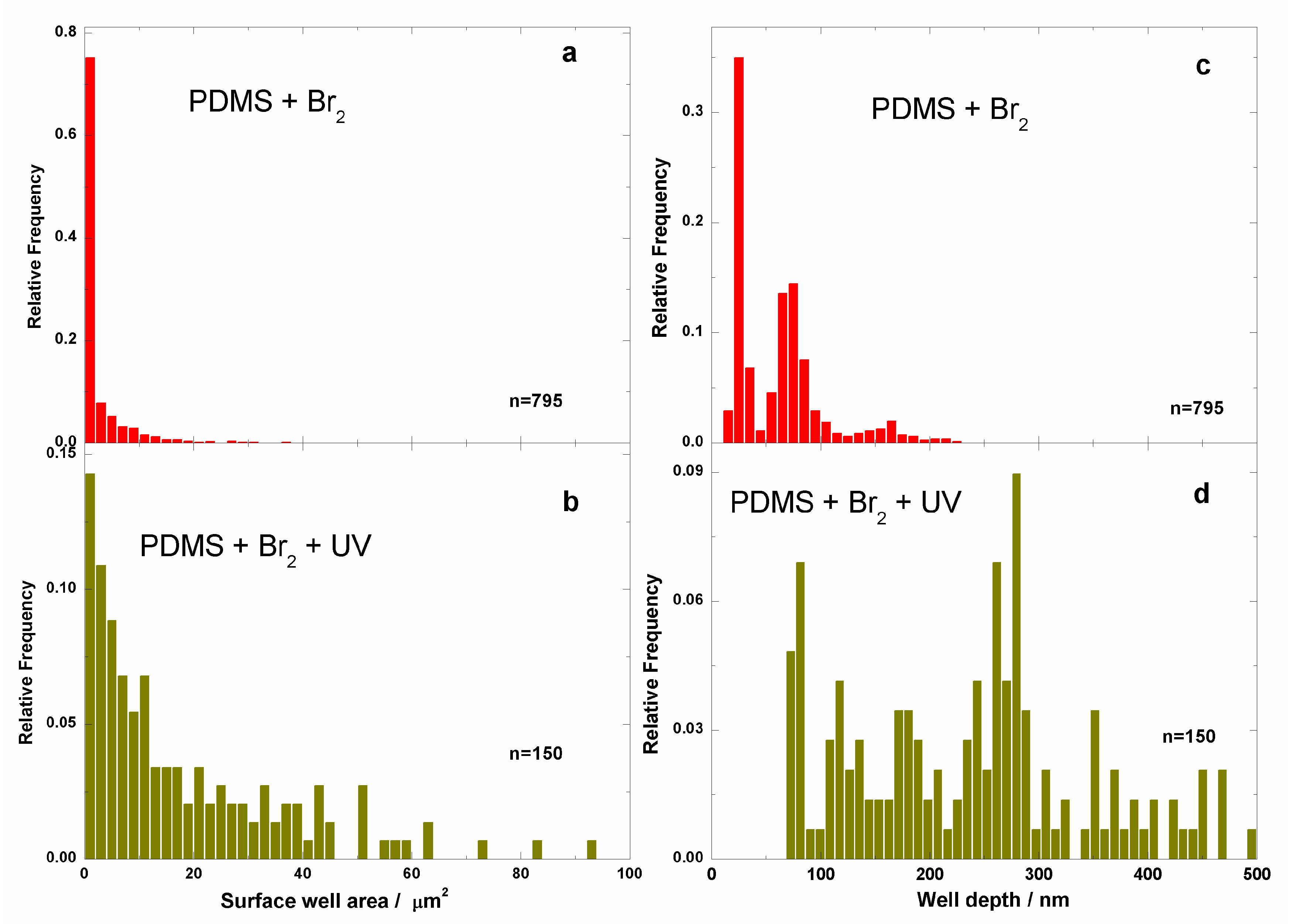
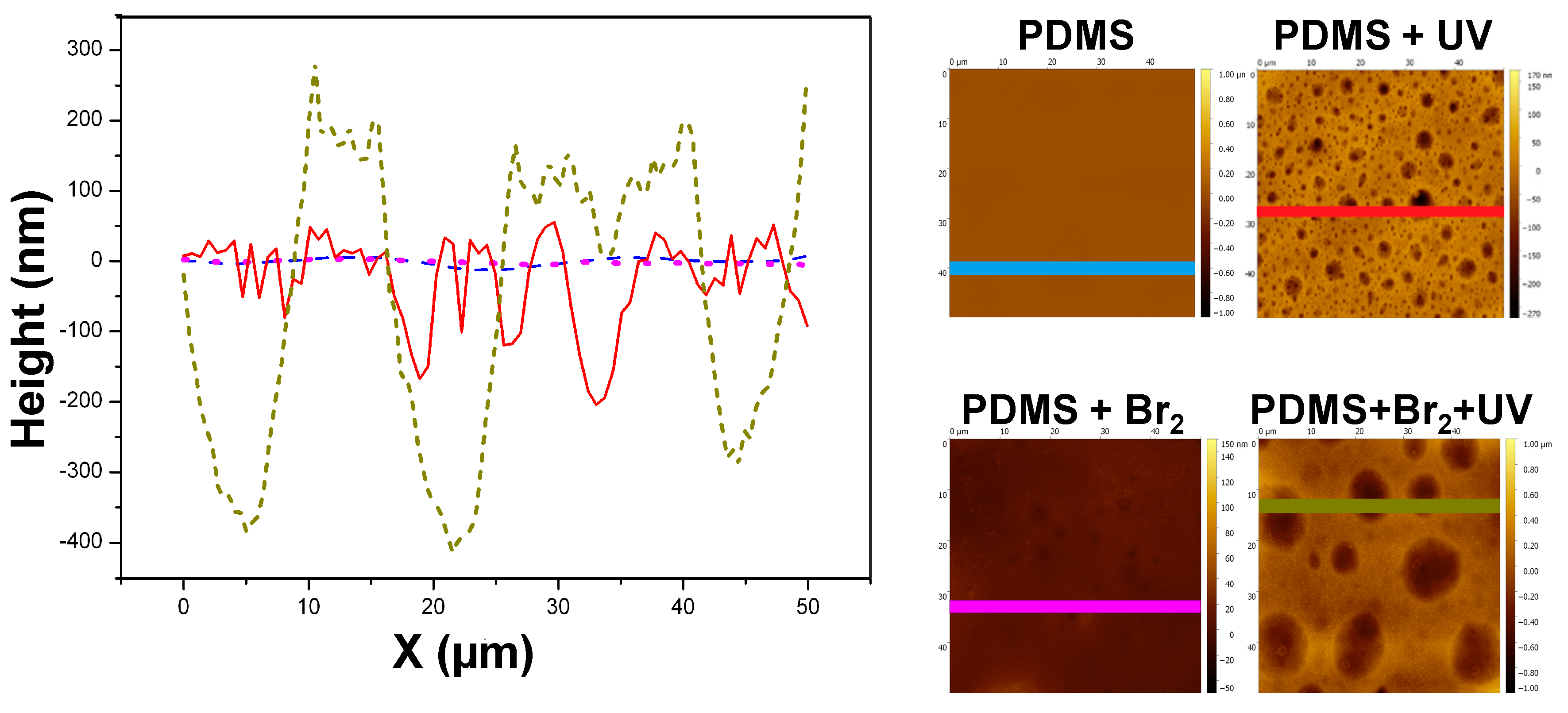
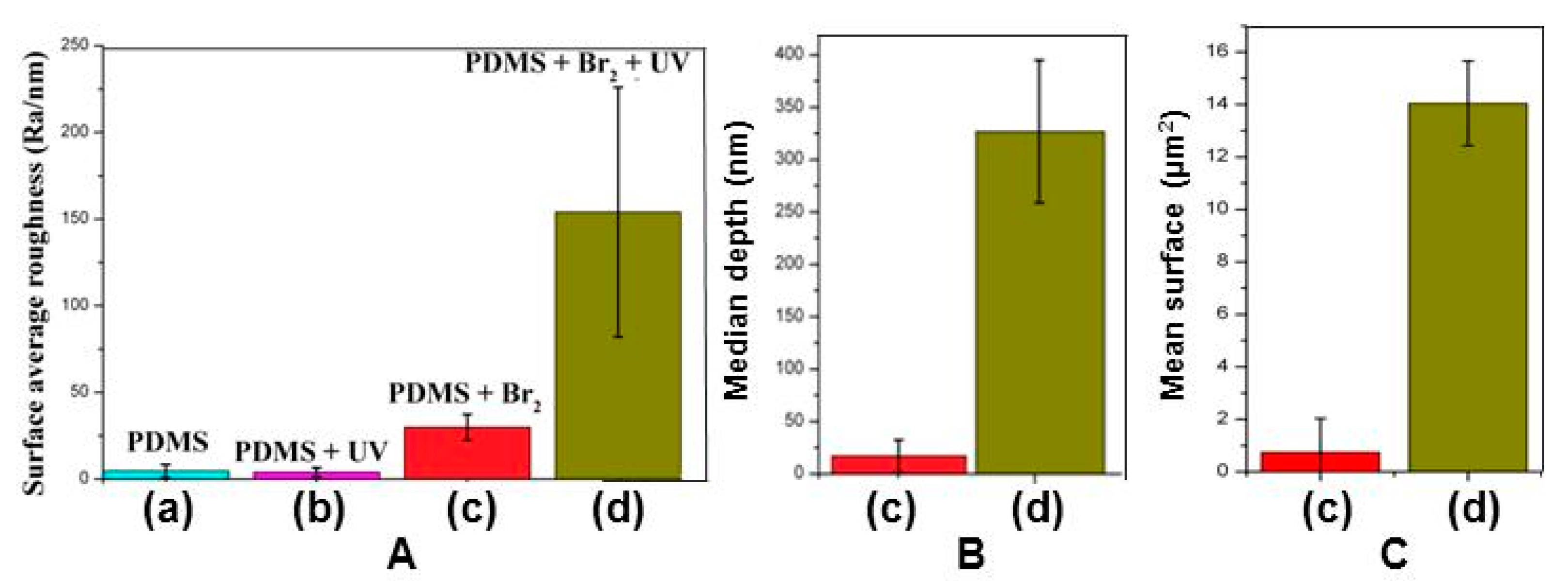
3.2. Films Exposed to Br2 (Liquid), Br2 (Gas) and UV

4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zhou, B.; Tian, J.; Wang, C.; Gao, Y.; Wen, W. A facile and cost-effective approach to engineer surface roughness for preparation of large-scale superhydrophobic substrate with high adhesive force. Appl. Surf. Sci. 2016, 389, 679–687. [Google Scholar] [CrossRef]
- Söz, C.K.; Yilgör, E.; Yilgör, I. Influence of the average surface roughness on the formation of superhydrophobic polymer surfaces through spin-coating with hydrophobic fumed silica. Polymer 2015, 62, 118–128. [Google Scholar]
- Wong, I.; Ho, C.M. Surface molecular property modifications for poly(dimethylsiloxane) (PDMS) based microfluidic devices. Microfluid. Nanofluid. 2009, 7, 291–306. [Google Scholar] [CrossRef] [PubMed]
- Jian, A.; Tang, X.; Feng, Q.; Duan, Q.; Ji, J.; Ma, Z.; Zhang, W.; Sang, S. A PDMS surface stress biosensor with optimized micro-membrane: Fabrication and application. Sens. Actuators B 2017, 242, 969–976. [Google Scholar] [CrossRef]
- Zhang, Y.; Karasu, F.; Rocco, C.; van der Ven, L.G.J.; van Benthem, R.A.T.M.; Allonas, X.; Croutxe-Barghorn, C.; Esteves, A.C.C.; de With, G. PDMS-based self-replenishing coatings. Polymer 2016, 107, 249–262. [Google Scholar] [CrossRef]
- Beigbeder, A.; Mincheva, R.; Pettitt, M.E.; Callow, M.E.; Callow, J.A.; Claes, M.; Dubois, P. Marine fouling release silicone/carbon nanotube nanocomposite coatings: On the importance of the nanotube dispersion state. J. Nanosci. Nanotechnol. 2010, 10, 2972–2978. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.G.; Chen, F.; Wu, C.Y.; Chang, M.H. Effect of surface roughness of chill wheel on ribbon formation in the planar flow casting process. J. Mater. Process. Technol. 2016, 229, 609–613. [Google Scholar] [CrossRef]
- Ammar, Y.; Swailes, D.; Bridgens, B.; Chen, J. Influence of surface roughness on the initial formation of biofilm. Surf. Coat. Technol. 2015, 284, 410–416. [Google Scholar] [CrossRef]
- Bullock, S.; Johnston, E.E.; Wilson, T.; Gatenholm, P.; Wynne, K.J. Surface science of a filled polydimethylsiloxane-based alkoxysilane-cured elastomer: RTV11. J. Colloid Int. Sci. 1999, 210, 18. [Google Scholar] [CrossRef] [PubMed]
- Beigbeder, A.; Linares, M.; Devalckenaere, M.; Degée, P.; Claes, M.; Beljonne, D.; Lazzaroni, R.; Dubois, P. CH-pi Interactions as the Driving Force for Silicone-Based Nanocomposites with Exceptional Properties. Adv. Mater. 2008, 20, 1003. [Google Scholar] [CrossRef]
- Beigbeder, A.; Degée, P.; Conlan, S.L.; Mutton, R.; Clare, A.S.; Pettitt, M.E.; Callow, M.E.; Callow, J.A.; Dubois, P. Preparation and characterisation of silicone-based coatings filled with carbon nanotubes and natural sepiolite and their application as marine fouling-release coatings. Biofouling 2008, 24, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Ochs, H.; Vogelsang, J.; Meyer, G. Enhanced surface roughness of organic coatings due to UV-degradation: An unknown source of EIS-artifacts. Prog. Org. Coat. 2003, 46, 182–190. [Google Scholar] [CrossRef]
- Hu, S.; Ren, X.; Bachman, M.; Sims, C.E.; Li, G.P.; Allbritton, N.L. Surface-Directed, Graft Polymerization within Microfluidic Channels. Anal. Chem. 2004, 76, 1865–1870. [Google Scholar] [CrossRef] [PubMed]
- Richey, T.; Iwata, H.; Oowaki, H.; Uchida, E.; Matsuda, S.; Ikada, Y. Surface modification of polyethylene balloon catheters for local drug delivery. Biomaterials 2000, 21, 1057–1065. [Google Scholar] [CrossRef]
- Efimenko, K.; Wallace, W.E.; Genzer, J. Surface modification of Sylgard-184 poly(dimethylsiloxane) networks by ultraviolet and ultraviolet/ozone treatment. J. Colloid Interface Sci. 2002, 254, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Hillborg, H.; Tomczak, N.; Olah, A.; Schönherr, H.; Vancso, G.J. Nanoscale hydrophobic recovery: A chemical force microscopy study of UV/ozone treated cross-linked poly(dimethylsiloxane). Langmuir 2004, 20, 785–794. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Gu, Z.; Gracias, D.H. Kinetics of ultraviolet and plasma surface modification of poly(dimethylsiloxane) probed by sum frequency vibrational spectroscopy. Langmuir 2006, 22, 1863–1868. [Google Scholar] [CrossRef] [PubMed]
- Juárez-Moreno, A.J.; Ávila-Ortega, A.; Oliva, A.I.; Avilés, F.; Cauich-Rodríguez, J.V. Effect of wettability and surface roughness on the adhesion properties of collagen on PDMS films treated by capacitively coupled oxygen plasma. Appl. Surf. Sci. 2015, 349, 763–773. [Google Scholar] [CrossRef]
- Kwon, O.M.; See, S.J.; Kim, S.S.; Hwang, H.Y. Effects of surface treatment with coupling agents of PVDF-HFP fibers on the improvement of the adhesion characteristics on PDMS. Appl. Surf. Sci. 2014, 321, 378–386. [Google Scholar] [CrossRef]
- Ferhan, A.R.; Zainol, N.; Kim, D.-H. A facile method towards rough morphology polymer brush for increased mobility of embedded nanoparticles. Polymer 2015, 75, 57–63. [Google Scholar] [CrossRef]
- Li, J.; Wang, M.; Shen, Y. Chemical modification on top of nanotopography to enhance surface properties of PDMS. Surf. Coat. Technol. 2012, 206, 2161–2167. [Google Scholar] [CrossRef]
- Jang, Y.; Seo, J.-W.; Seok, J.; Lee, J.-Y.; Kim, K. Roughening Conjugated Polymer Surface for Enhancing the Charge Collection Efficiency of Sequentially Deposited Polymer/Fullerene Photovoltaics. Polymers 2015, 7, 1497–1509. [Google Scholar] [CrossRef]
- Scaffaro, R.; Maio, A.; Lopresti, F.; Botta, L. Nanocarbons in Electrospun Polymeric Nanomats for Tissue Engineering: A Review. Polymers 2017, 9, 76. [Google Scholar] [CrossRef]
- Khelifa, F.; Habibi, Y.; Leclere, P.; Dubois, P. Convection-assisted assembly of cellulose nanowhiskers embedded in an acrylic copolymer. Nanoscale 2013, 5, 1082–1090. [Google Scholar] [CrossRef] [PubMed]
- Khelifa, F.; Druart, M.E.; Habibi, Y.; Benard, F.; Leclere, P.; Olivier, M.; Dubois, P. Sol-gel incorporation of silica nanofillers for tuning the anti-corrosion protection of acrylate-based coatings. Prog. Org. Coat. 2013, 76, 900–911. [Google Scholar] [CrossRef]
- Chakrabarty, S.; Zhang, X.; Bharti, P.; Chujo, Y.; Miyake, J.; Wynne, K.J.; Yadavalli, V. Processing dependence of surface morphology in condensation cured PDMS nanocomposites. Polymer 2010, 51, 5756–5763. [Google Scholar] [CrossRef]
- Khelifa, F.K.; Druart, M.E.; Hhabibi, Y.; Rioboo, R.; Olivier, M.; de Coninck, J.; Dubois, P. Effect of photo-crosslinking on the performances of silica nanoparticle-filled epoxidized acrylic copolymers coating. J. Mater. Chem. A 2013, 1, 13334–13344. [Google Scholar] [CrossRef]
- Beigbeder, A.; Jeusette, M.; Mincheva, R.; Claes, M.; Brocorens, P.; Lazzaaroni, R. On the effect of carbon nanotubes on the wettability and surface morphology of hydrosilylation-curing silicone coatings. J. Nanostruct. Polym. Nanocomp. 2009, 5, 37–43. [Google Scholar]
- Antonel, P.S.; Jorge, G.A.; Perez, O.; Butera, A.; Leyva, G.; Martín Negri, R. Magnetic and Elastic Properties of CoFe2O4-PDMS Magnetically Oriented Elastomer Nanocomposites. J. Appl. Phys. 2011, 110, 43920–43928. [Google Scholar] [CrossRef]
- Antonel, P.S.; Oliveira, C.L.P.; Jorge, G.A.; Perez, O.E.; Leyva, A.G.; Negri, R.M. Synthesis and Characterization of CoFe2O4 Magnetic Nanotubes, Nanorods and Nanowires. Formation of Magnetic Structured Elastomers by Magnetic-Field Induced Alignment of CoFe2O4 Nanorods. J. Nanopart. Res. 2015, 17, 294–307. [Google Scholar] [CrossRef]
- Mietta, J.L.; Ruiz, M.M.; Antonel, P.S.; Perez, O.E.; Butera, A.; Jorge, G.A.; Negri, R.M. Anisotropic Magnetoresistance and Piezoresistivity in Structured Fe3O4-Silver Particles in PDMS Elastomers at Room Temperature. Langmuir 2012, 28, 6985–6996. [Google Scholar] [CrossRef] [PubMed]
- Mietta, J.L.; Jorge, G.; Perez, O.E.; Maeder, T.; Negri, R.M. Superparamagnetic anisotropic elastomer connectors exhibiting reversible magneto-piezoresistivity. Sens. Actuators A 2013, 192, 34–41. [Google Scholar] [CrossRef]
- Mietta, J.L.; Jorge, G.; Negri, R.M. A flexible strain gauge exhibiting reversible piezoresistivity based on an anisotropic magnetorheological polymer. Smart Mater. Struct. 2014, 23, 85026–85038. [Google Scholar] [CrossRef]
- Mietta, J.L.; Tamborenea, P.I.; Negri, R.M. Anisotropic Magnetoresistivity in Structured Elastomer Composites: Modelling and Experiments. Soft Matter 2016, 12, 6430–6441. [Google Scholar] [CrossRef] [PubMed]
- Landa, R.A.; Antonel, P.S.; Ruiz, M.M.; Perez, O.E.; Butera, A.; Jorge, G.; Oliveira, C.L.P.; Negri, R.M. Magnetic and Elastic Anisotropy in Magnetorheological Elastomers using Nickel-based Nanoparticles and Nanochains. J. Appl. Phys. 2013, 114, 213912. [Google Scholar] [CrossRef]
- Ruiz, M.M.; Marchi, M.C.; Perez, O.E.; Jorge, G.A.; Fascio, M.; D’Accorso, N.; Negri, R.M. Structured elastomeric submillimeter films displaying magneto and piezo resistivity. J. Polym. Sci. B 2015, 53, 574–586. [Google Scholar] [CrossRef]
- Saleh Medina, L.S.; Jorge, G.A.; Rubi, D.; D’Accorso, N.; Negri, R.M. SBR/BiFeO3 Elastomer Capacitor Films Prepared under Magnetic and Electric Fields Displaying Magnetoelectric Coupling. J. Phys. Chem. C 2015, 119, 23319–23328. [Google Scholar] [CrossRef]
- Ruiz, M.M.; Antonel, P.S.; Perez, O.E.; Negri, R.M.; Jorge, G. Structural and magnetic properties of Fe2-xCoSmxO4—Nanoparticles and Fe2-xCoSmxO4—PDMS magnetoelastomers as a function of Sm content. J. Magn. Magn. Mater. 2012, 327, 11–19. [Google Scholar] [CrossRef]
- Fischer, J.; Bingle, J. The Vapor Pressure of Bromine from 24 to 116°. J. Am. Chem. Soc. 1955, 77, 6511–6512. [Google Scholar] [CrossRef]
- Puntes, V.F.; Gorostiza, P.; Aruguete, D.M.; Bastus, N.G.; Alivisatos, A.P. Collective behaviour in two-dimensional cobalt nanoparticle assemblies observed by magnetic force microscopy. Nat. Mater. 2004, 3, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Bellamy, L.J. The infrared spectra of complex molecules. In Advances in Infrared Group Frequencies; Springer: Berlin, Germany, 1980; Volume 2, ISBN 978-94-011-6522-8. [Google Scholar]
- Shahzad, M.I.; Giorcelli, M.; Shahzad, N.; Guastella, S.; Castellino, M.; Jagdale, P.; Tagliaferro, A. Study of carbon nanotubes based Polydimethylsiloxane composite films. J. Phys. Conf. Ser. 2013, 439, 012010. [Google Scholar] [CrossRef]











© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cabrera, J.N.; Ruiz, M.M.; Fascio, M.; D’Accorso, N.; Mincheva, R.; Dubois, P.; Lizarraga, L.; Negri, R.M. Increased Surface Roughness in Polydimethylsiloxane Films by Physical and Chemical Methods. Polymers 2017, 9, 331. https://doi.org/10.3390/polym9080331
Cabrera JN, Ruiz MM, Fascio M, D’Accorso N, Mincheva R, Dubois P, Lizarraga L, Negri RM. Increased Surface Roughness in Polydimethylsiloxane Films by Physical and Chemical Methods. Polymers. 2017; 9(8):331. https://doi.org/10.3390/polym9080331
Chicago/Turabian StyleCabrera, Jorge Nicolás, Mariano M. Ruiz, Mirta Fascio, Norma D’Accorso, Rosica Mincheva, Philippe Dubois, Leonardo Lizarraga, and R. Martín Negri. 2017. "Increased Surface Roughness in Polydimethylsiloxane Films by Physical and Chemical Methods" Polymers 9, no. 8: 331. https://doi.org/10.3390/polym9080331
APA StyleCabrera, J. N., Ruiz, M. M., Fascio, M., D’Accorso, N., Mincheva, R., Dubois, P., Lizarraga, L., & Negri, R. M. (2017). Increased Surface Roughness in Polydimethylsiloxane Films by Physical and Chemical Methods. Polymers, 9(8), 331. https://doi.org/10.3390/polym9080331