Presenting Precision Glycomacromolecules on Gold Nanoparticles for Increased Lectin Binding




Abstract
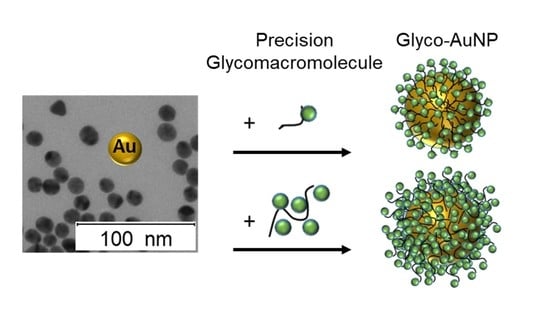
:
1. Introduction
2. Materials and Methods
2.1. Nuclear Magnetic Resonance Spectroscopy (NMR)
2.2. Mass Spectrometry (ESI, MALDI-TOF-MS, HR-MS)
2.3. Reversed Phase High Pressure Liquid Chromatography—Mass Spectrometry (RP-HPLC-MS)
2.4. Preparative Reversed Phase High Pressure Liquid Chromatography (Prep-RP-HPLC)
2.5. UV-Vis Spectroscopy
2.6. Microtiter Plate Spectrophotometer
2.7. Freeze Dryer
2.8. Dynamic Light Scattering
2.9. Transmission Electron Microscopy
2.10. Phenol-Sulphuric Acid Method
2.11. TBO Titration
2.12. Stability of Glyco-AuNPs in Dependence of Sodium Chloride Concentration
2.13. Stability of Glyco-AuNPs in Lectin Binding Buffer (LBB)
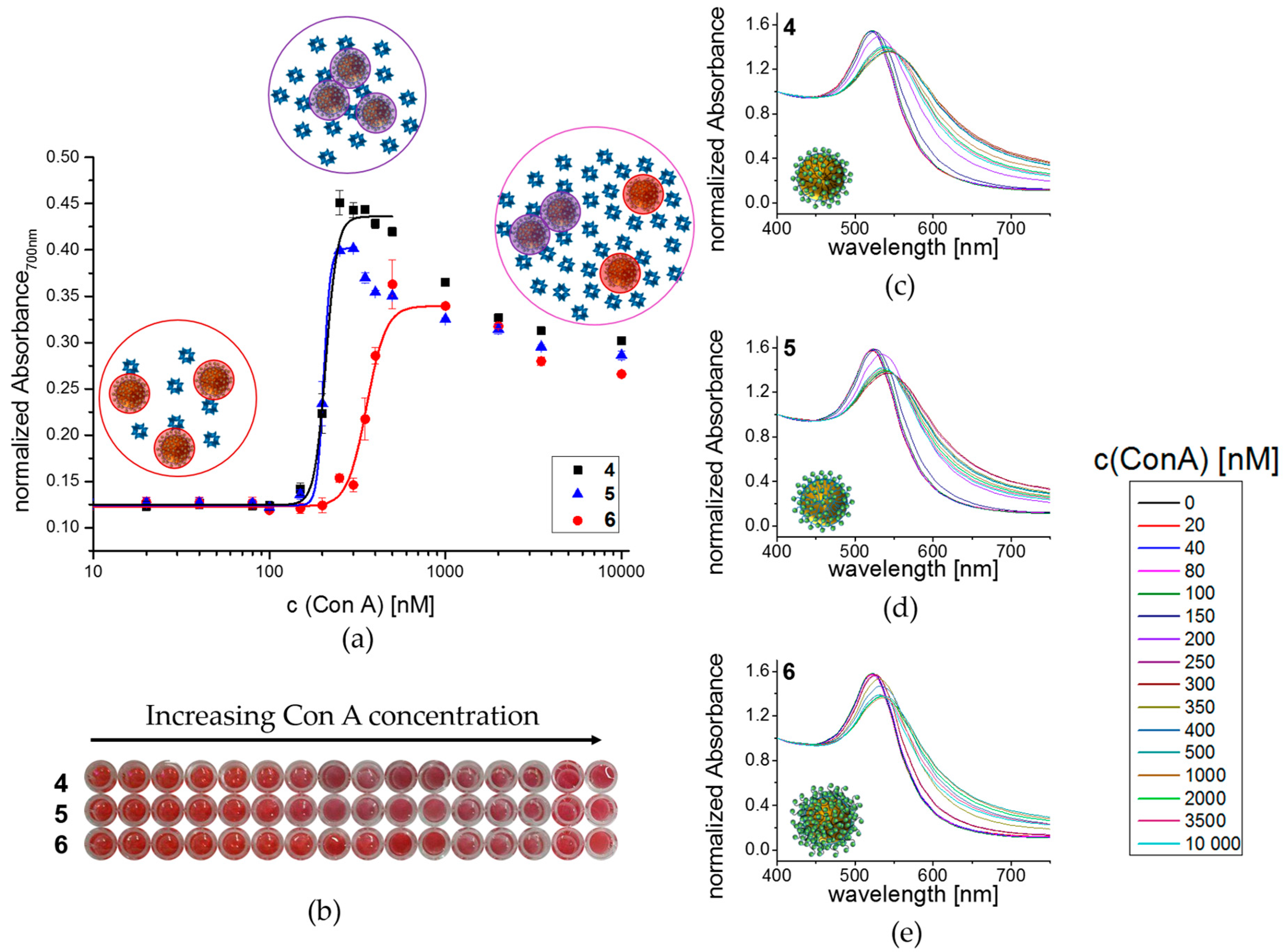
2.14. Plate-Based Colorimetric Aggregation Study with Con A
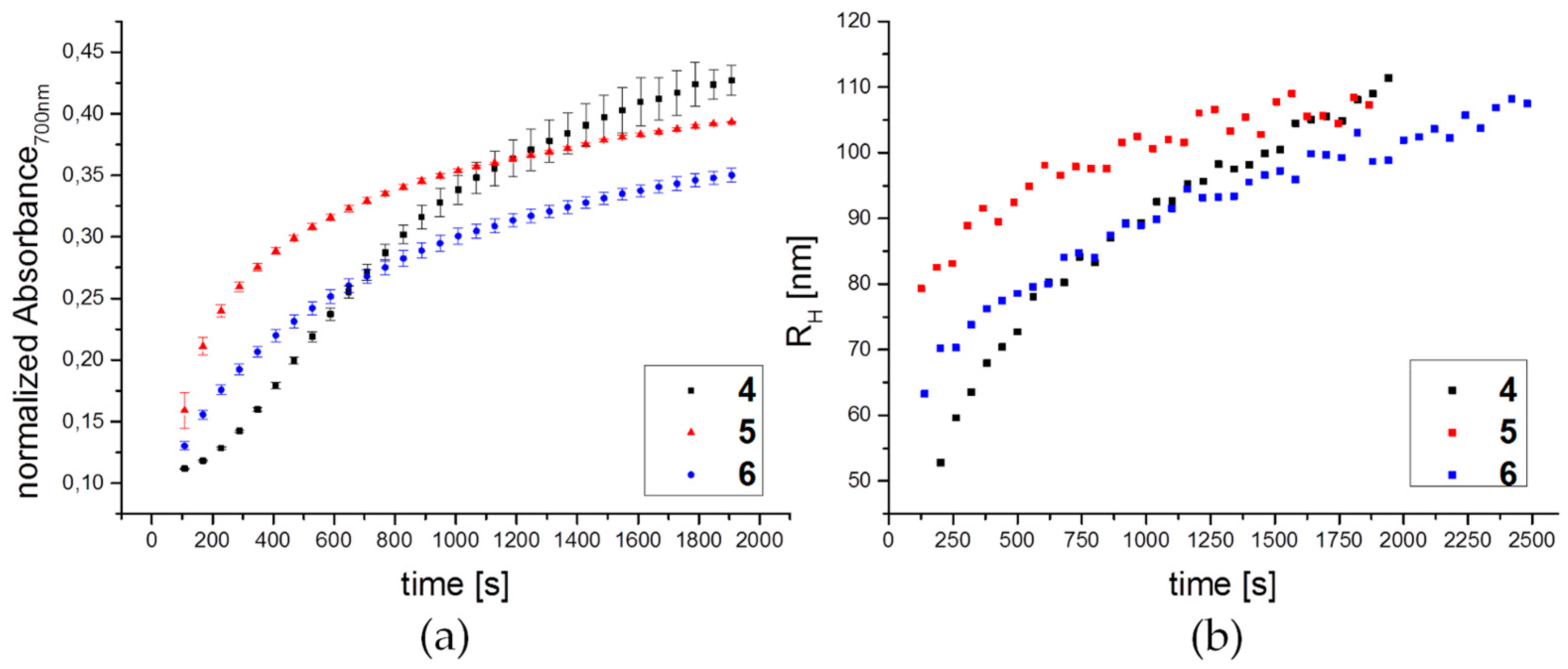
2.15. Kinetic Study of Aggregation Behavior with Con A by DLS and UV-Vis Spectroscopy
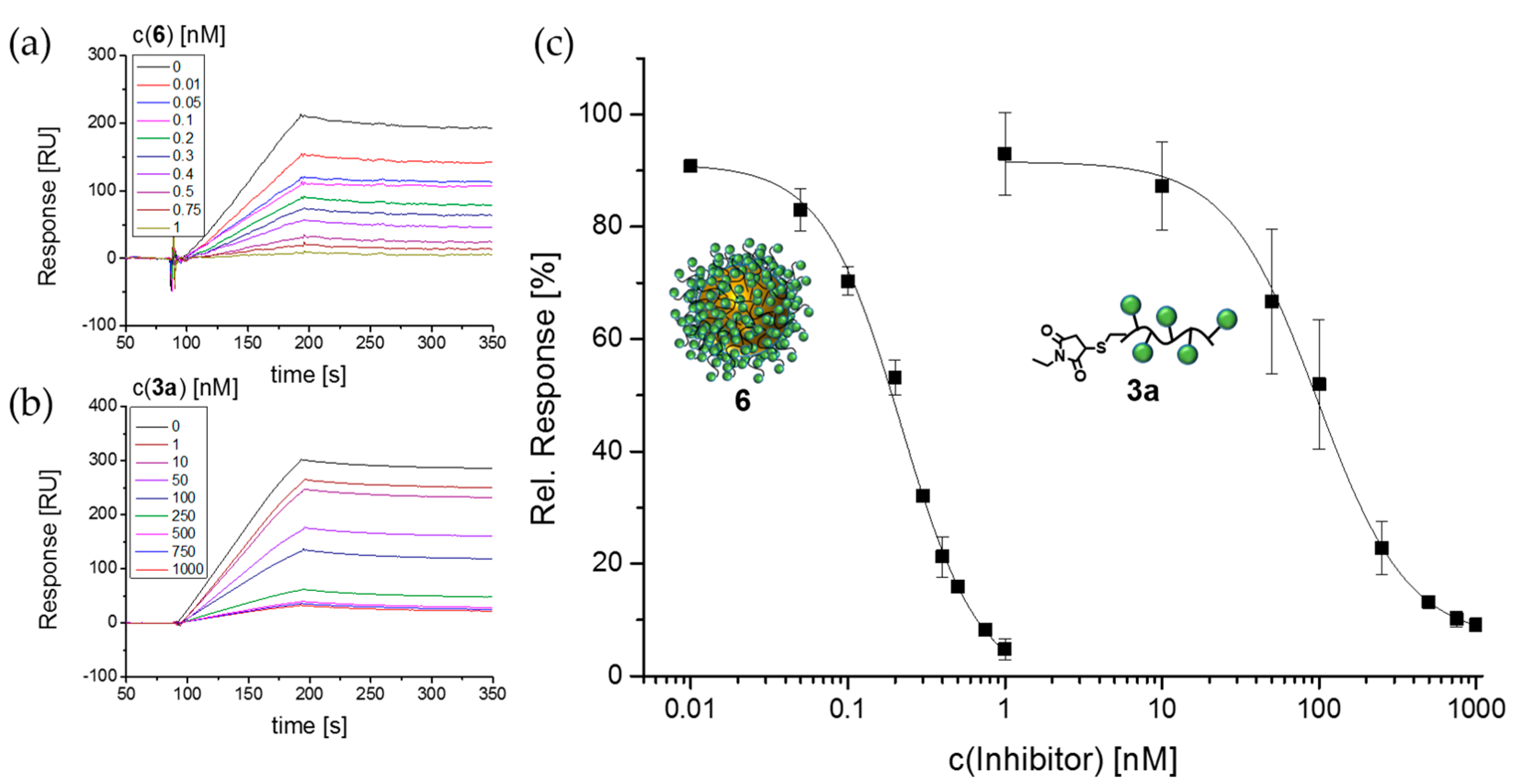
2.16. Surface Plasmon Resonance (SPR)
2.17. Syntheses
2.17.1. Syntheses of Building Blocks:
1-(Fluorenyl)-3,11-dioxo-7-(pent-4-ynoyl)-2-oxa-4,7,10-triazatetradecan-14-oic acid, TDS
1-(9H-Fluoren-9-yl)-3,14-dioxo-2,7,10-trioxa-4,13-diazaheptadecan-17-oic acid, EDS
Synthesis of ODS
N1-Trityloctan-1,8-diamine
(9H-Fluoren-9-yl)methyl (8-(tritylamino)octyl)carbamate
8-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)octan-1-aminium trifluoracetate
4-((8-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)octyl)amino)-4-oxobutanoic acid, ODS
(2-Azidoethyl)-2,3,4,6-tetra-O-acetyl-α-d-mannopyranoside
2.17.2. Solid Phase Polymer Synthesis
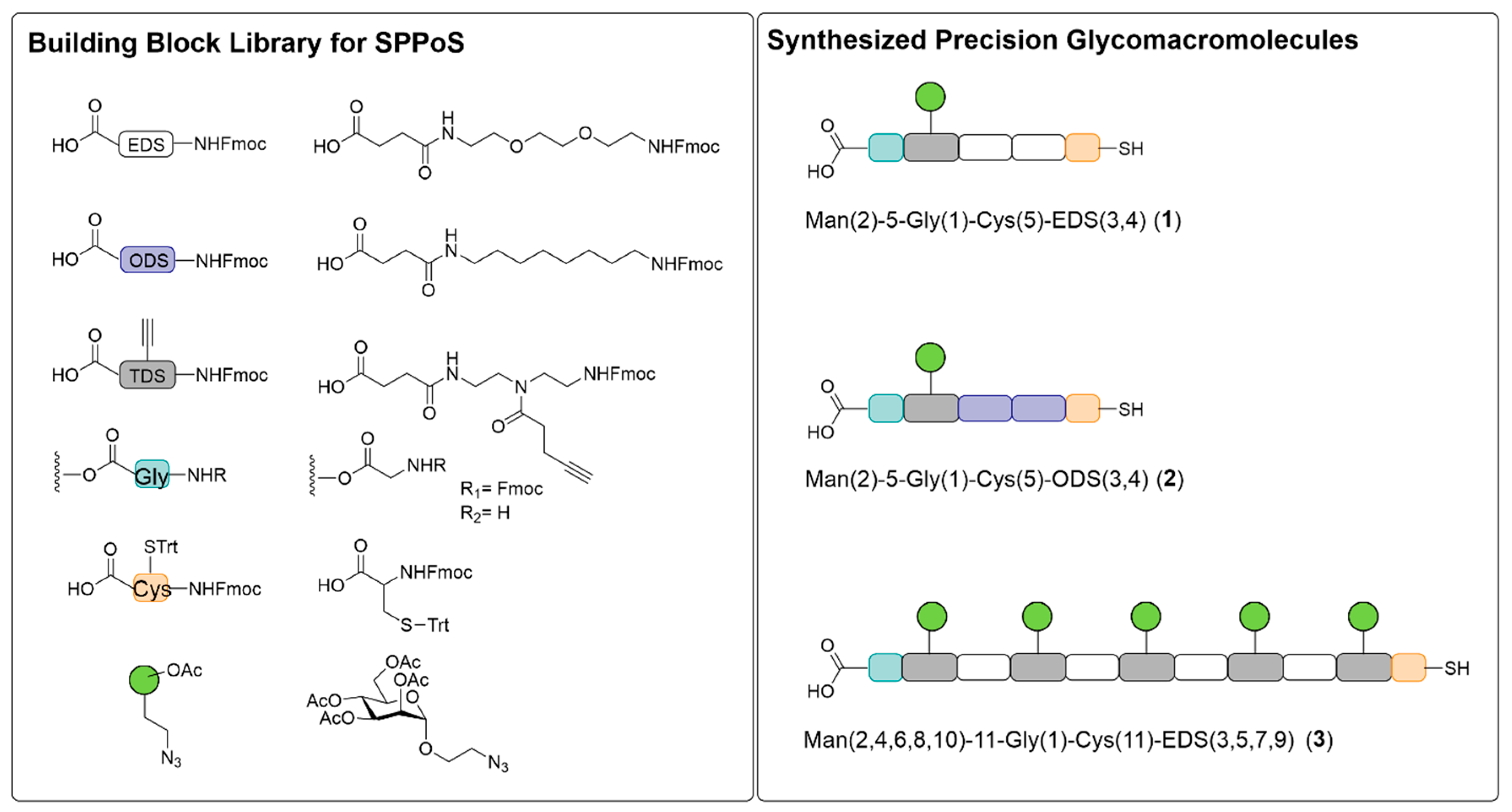
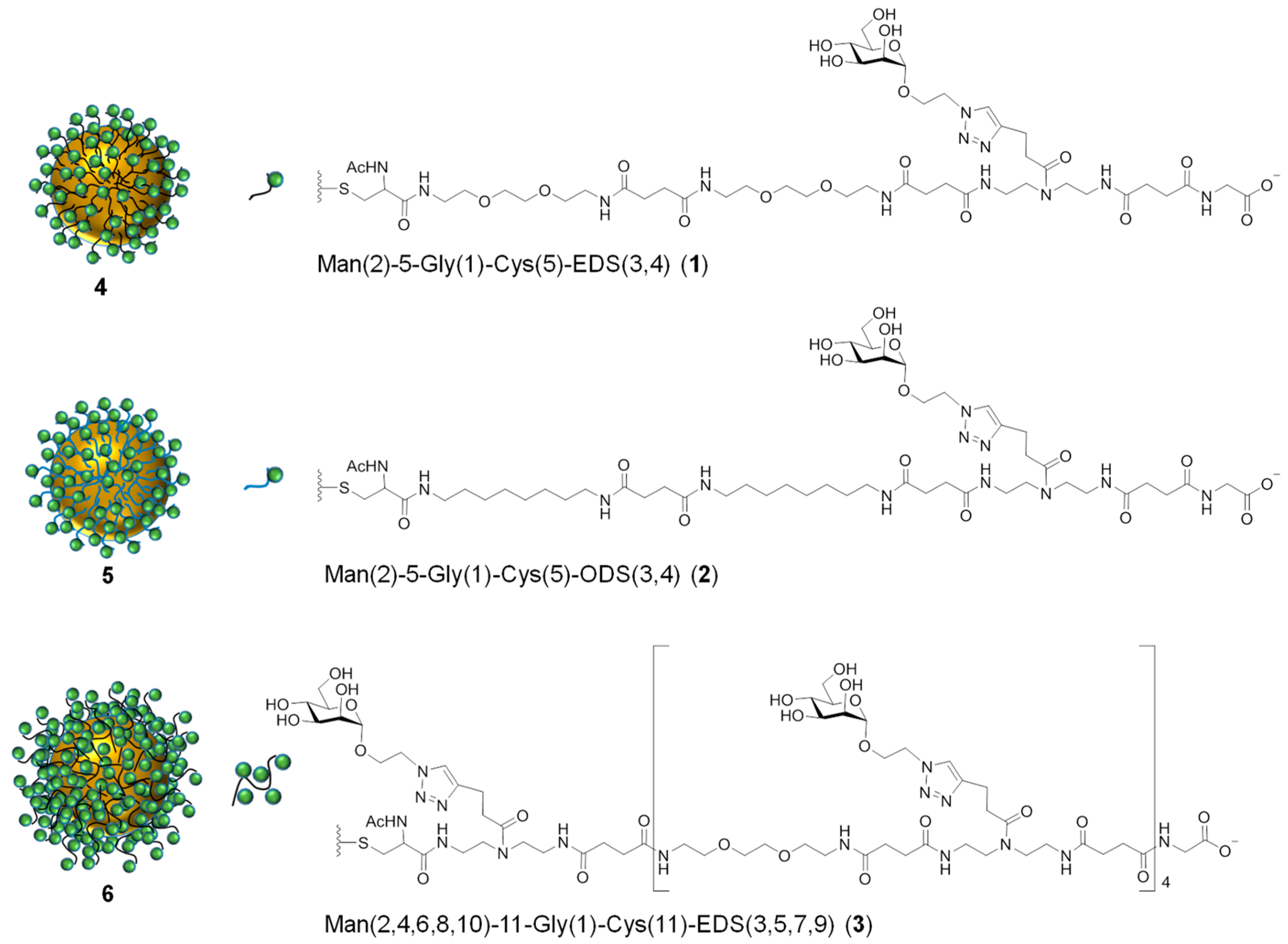
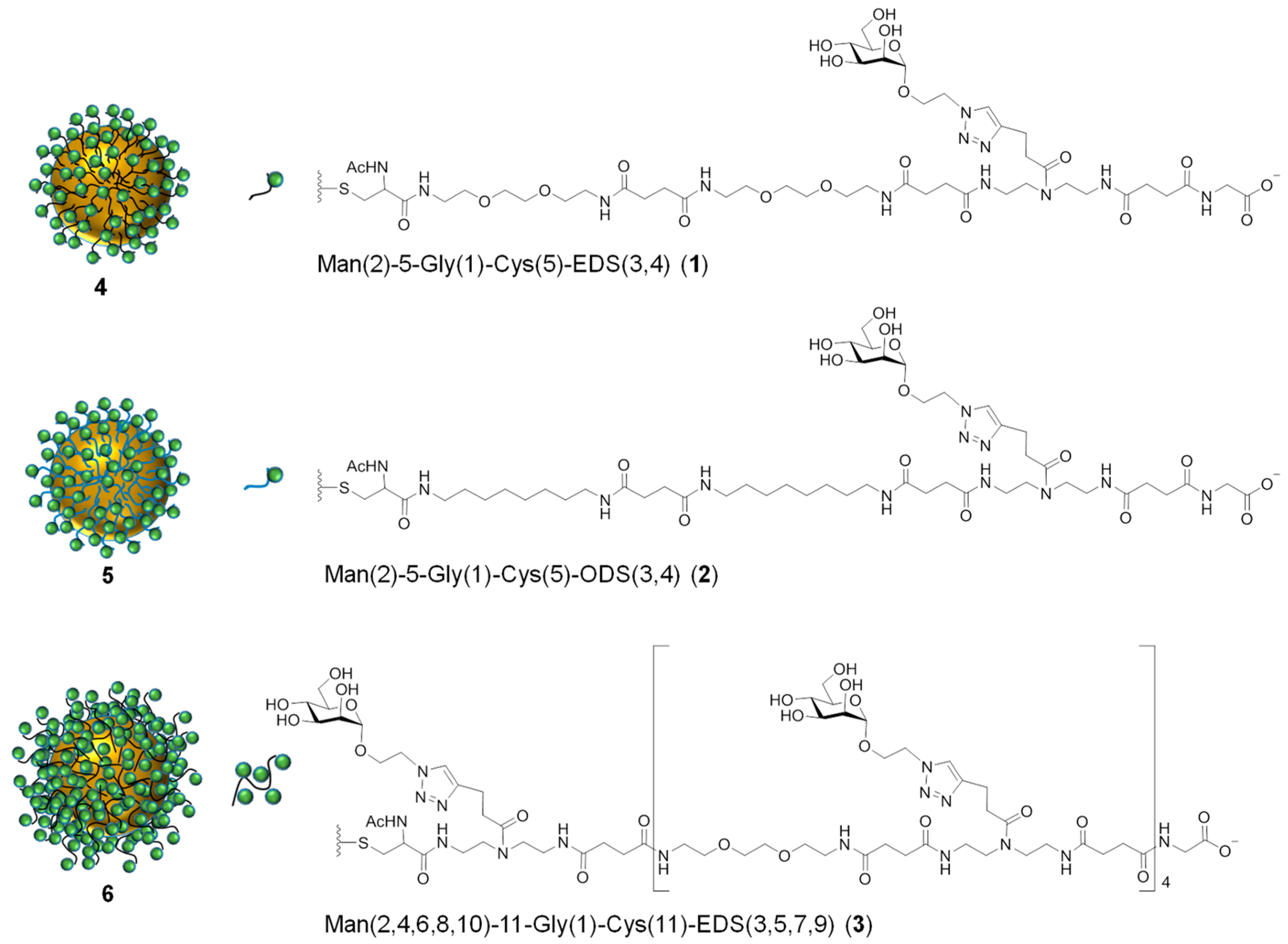
Man(2)-5-Gly(1)-EDS(3,4)-Cys(5), 1
Man(2)-5-Gly(1)-ODS(3,4)-Cys(5), 2
Man(2,4,6,8,10)-11-Gly(1)-EDS(3,5,7,9)-Cys(11), 3
2.17.3. Capping of Glycomacromolecule-Thiol Functionalities with N-ethylmaleimide
Man(2)-5-Gly(1)-EDS(3,4)-Cys(5)-N-ethylmaleimide, 1a
Man(2,4,6,8,10)-11-Gly(1)-EDS(3,5,7,9)-Cys(11)-N-ethylmaleimide, 3a
2.17.4. Synthesis of Citrate Stabilized AuNPs
2.17.5. Functionalization of AuNPs by Ligand Exchange
3. Results and Discussion
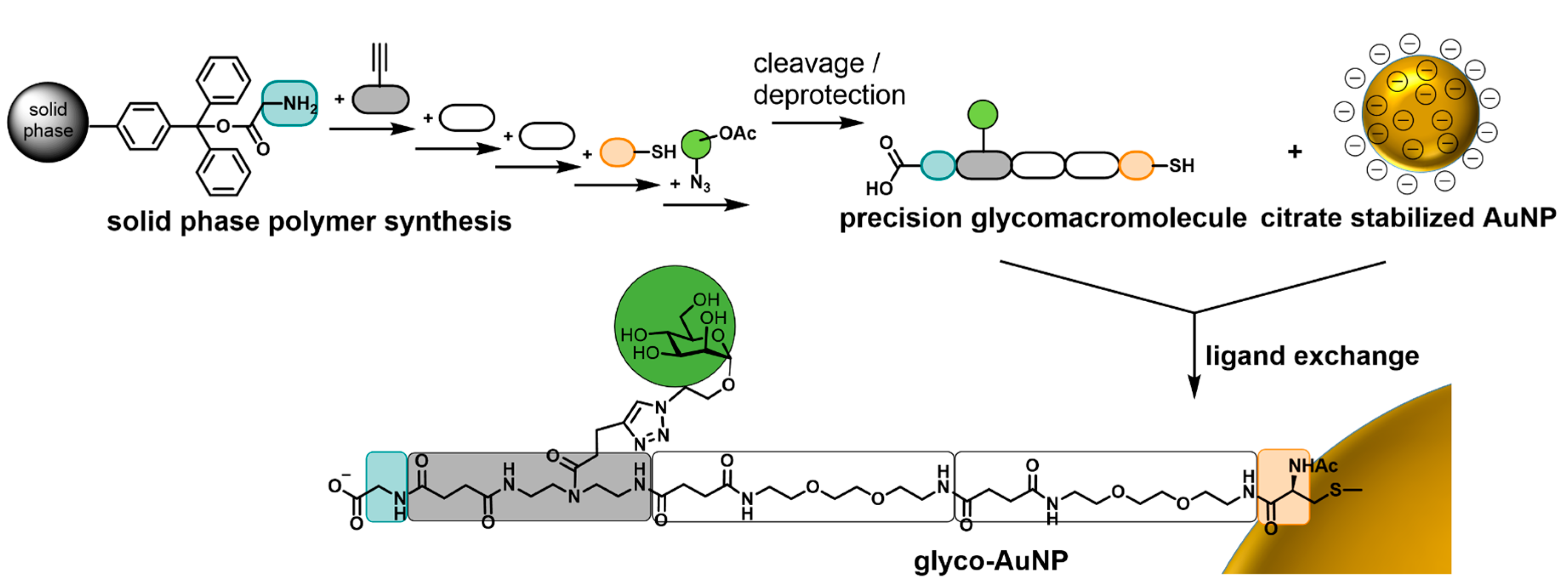
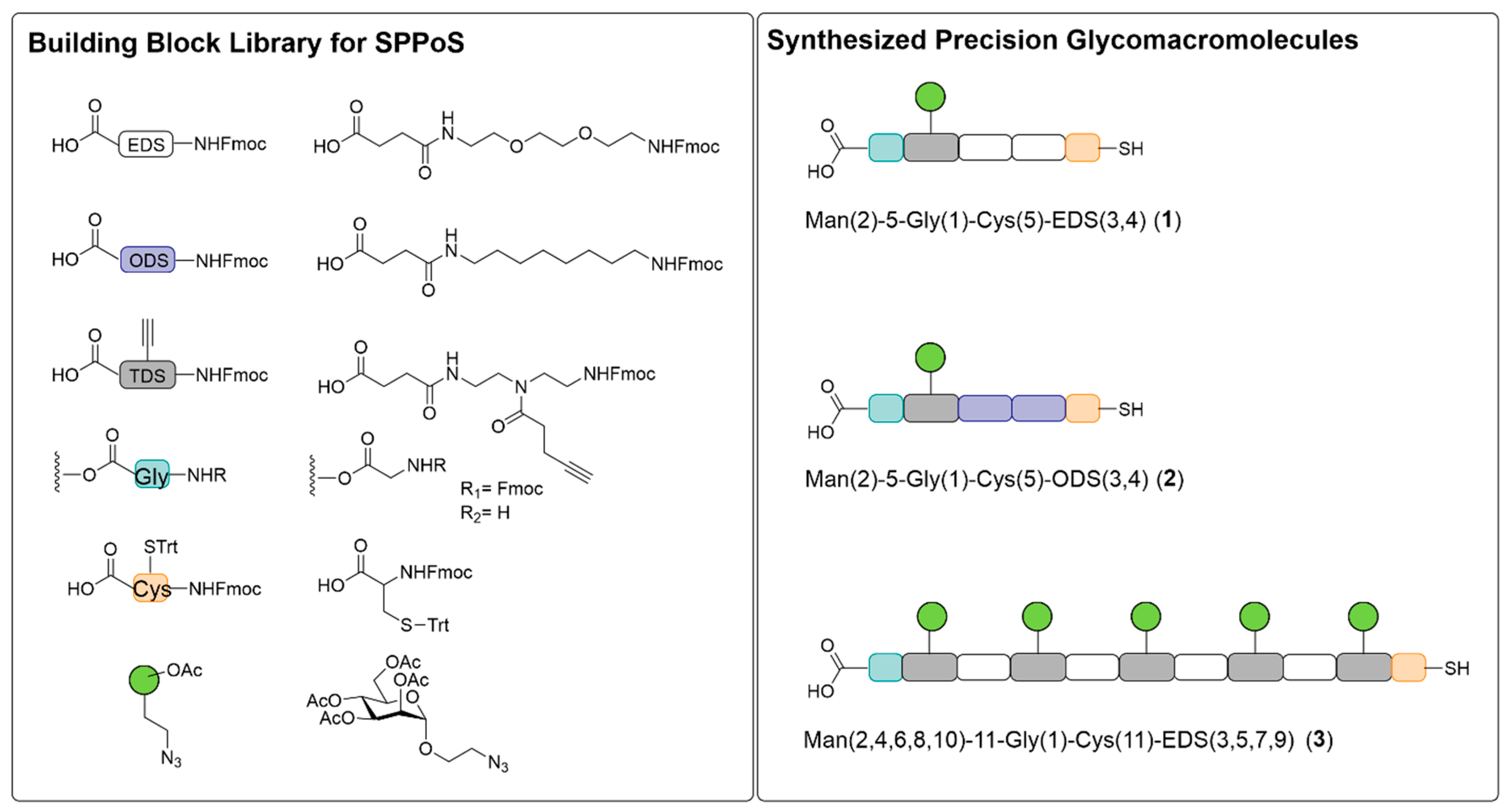
3.1. Synthesis of Thiol Functionalized Precision Glycomacromolecules
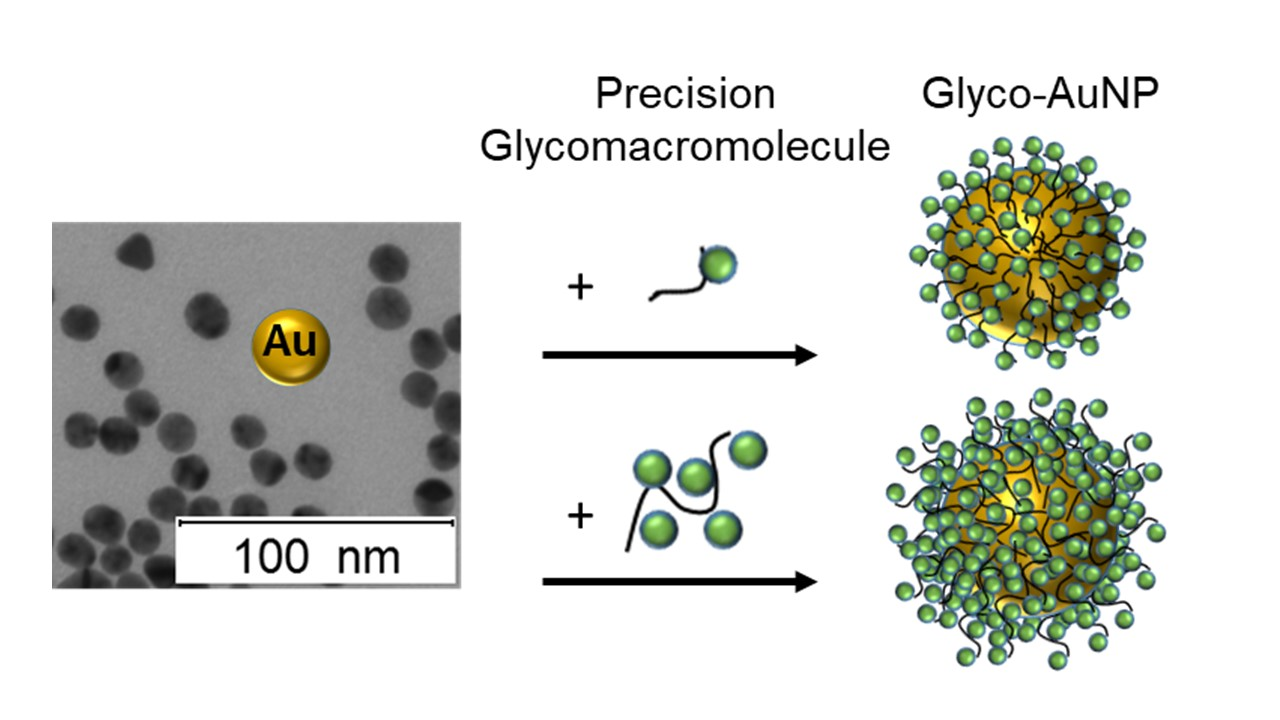
3.2. Preparation and Characterization of Glyco-AuNPs
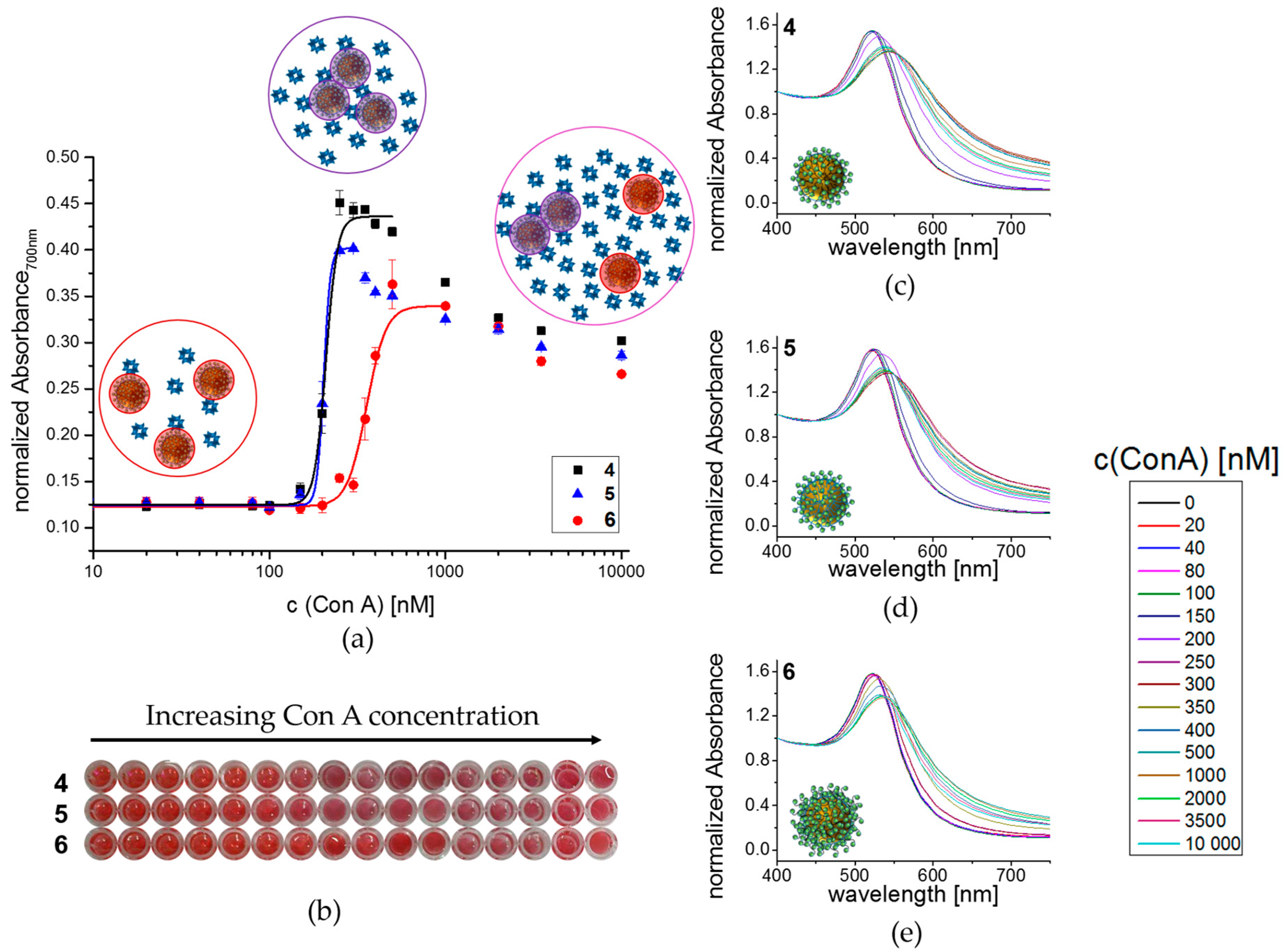
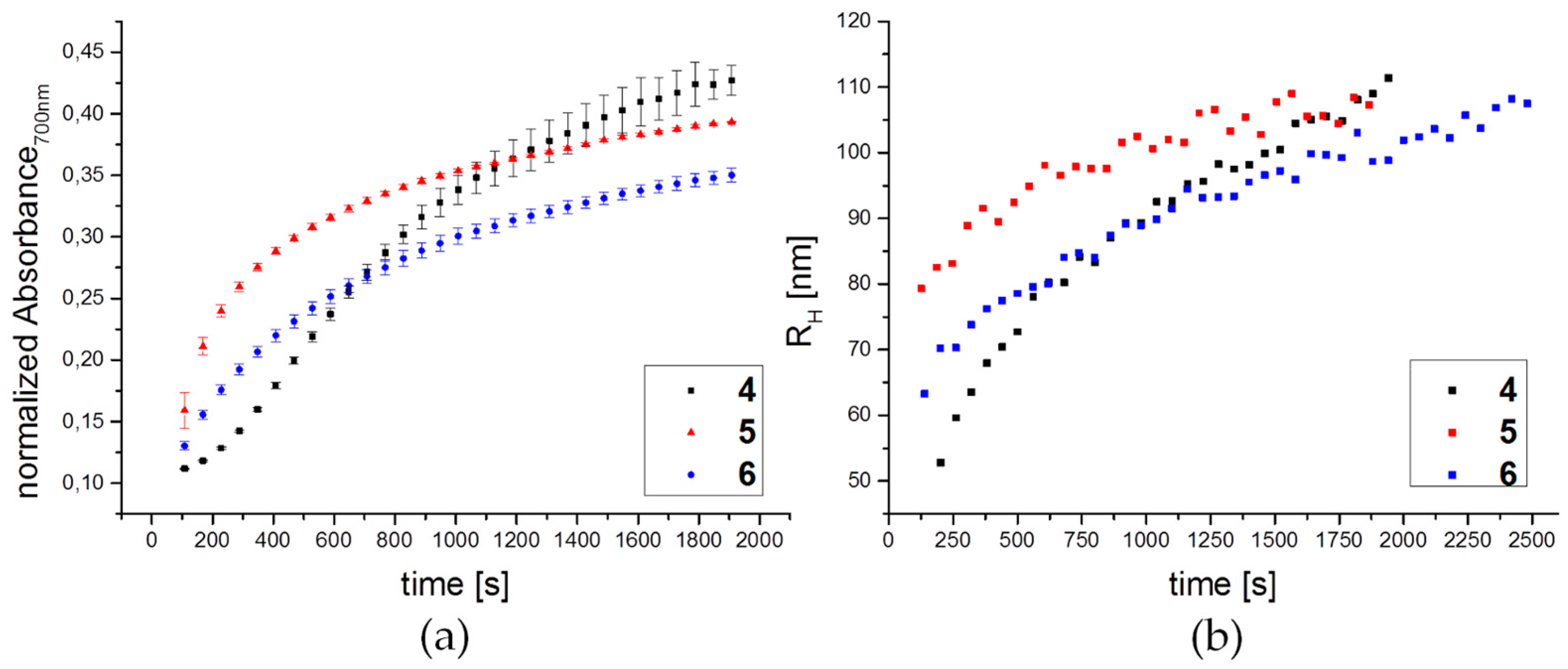
3.3. Lectin Binding Studies with Model Lectin Con A
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dwek, R.A. Glycobiology: Toward Understanding the Function of Sugars. Chem. Rev. 1996, 96, 683–720. [Google Scholar] [CrossRef] [PubMed]
- Imberty, A.; Varrot, A. Microbial recognition of human cell surface glycoconjugates. Curr. Opin. Struct. Biol. 2008, 18, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Fasting, C.; Schalley, C.A.; Weber, M.; Seitz, O.; Hecht, S.; Koksch, B.; Dernedde, J.; Graf, C.; Knapp, E.-W.; Haag, R. Multivalency as a Chemical Organization and Action Principle. Angew. Chem. Int. Ed. 2012, 51, 10472–10498. [Google Scholar] [CrossRef] [PubMed]
- Kiessling, L.L.; Young, T.; Gruber, T.D.; Mortell, K.H. Multivalency in Protein–Carbohydrate Recognition. In Glycoscience, 2nd ed.; Fraser-Reid, B.O., Tatsuta, K., Thiem, J., Eds.; Springer: Berlin/Heidelberg, Germany, 2008; Volume 1, pp. 2483–2523. ISBN 978-3-540-30429-6. [Google Scholar]
- Lee, Y.C.; Lee, R.T. Carbohydrate-Protein Interactions: Basis of Glycobiology. Acc. Chem. Res. 1995, 28, 321–327. [Google Scholar] [CrossRef]
- Lundquist, J.J.; Toone, E.J. The Cluster Glycoside Effect. Chem. Rev. 2002, 102, 555–578. [Google Scholar] [CrossRef] [PubMed]
- Benaissa-Trouw, B.; Lefeber, D.J.; Kamerling, J.P.; Vliegenthart, J.F.G.; Kraaijeveld, K.; Snippe, H. Synthetic Polysaccharide Type 3-Related Di-, Tri-, and Tetrasaccharide–CRM197 Conjugates Induce Protection against Streptococcus pneumoniae Type 3 in Mice. Infect. Immun. 2001, 69, 4698–4701. [Google Scholar] [CrossRef] [PubMed]
- Rendle, P.M.; Seger, A.; Rodrigues, J.; Oldham, N.J.; Bott, R.R.; Jones, J.B.; Cowan, M.M.; Davis, B.G. Glycodendriproteins: A Synthetic Glycoprotein Mimic Enzyme with Branched Sugar-Display Potently Inhibits Bacterial Aggregation. J. Am. Chem. Soc. 2004, 126, 4750–4751. [Google Scholar] [CrossRef] [PubMed]
- Dam, T.K.; Gabius, H.-J.; André, S.; Kaltner, H.; Lensch, M.; Brewer, C.F. Galectins Bind to the Multivalent Glycoprotein Asialofetuin with Enhanced Affinities and a Gradient of Decreasing Binding Constants. Biochemistry 2005, 44, 12564–12571. [Google Scholar] [CrossRef] [PubMed]
- Doores, K.J.; Gamblin, D.P.; Davis, B.G. Exploring and Exploiting the Therapeutic Potential of Glycoconjugates. Chem. Eur. J. 2006, 12, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Herzner, H.; Reipen, T.; Schultz, M.; Kunz, H. Synthesis of Glycopeptides Containing Carbohydrate and Peptide Recognition Motifs. Chem. Rev. 2000, 100, 4495–4538. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.; Baek, M.-G. Glycodendrimers: Novel glycotope isosteres unmasking sugar coding. Case study with T-antigen markers from breast cancer MUC1 glycoprotein. Rev. Mol. Biotechnol. 2002, 90, 291–309. [Google Scholar] [CrossRef]
- Narain, R.; Armes, S.P. Synthesis and Aqueous Solution Properties of Novel Sugar Methacrylate-Based Homopolymers and Block Copolymers. Biomacromolecules 2003, 4, 1746–1758. [Google Scholar] [CrossRef] [PubMed]
- Bes, L.; Angot, S.; Limer, A.; Haddleton, D.M. Sugar-Coated Amphiphilic Block Copolymer Micelles from Living Radical Polymerization: Recognition by Immobilized Lectins. Macromolecules 2003, 36, 2493–2499. [Google Scholar] [CrossRef]
- Otsuka, I.; Blanchard, B.; Borsali, R.; Imberty, A.; Kakuchi, T. Enhancement of Plant and Bacterial Lectin Binding Affinities by Three-Dimensional Organized Cluster Glycosides Constructed on Helical Poly(phenylacetylene) Backbones. ChemBioChem 2010, 11, 2399–2408. [Google Scholar] [CrossRef] [PubMed]
- Kiessling, L.L.; Gestwicki, J.E.; Strong, L.E. Synthetic Multivalent Ligands as Probes of Signal Transduction. Angew. Chem. Int. Ed. 2006, 45, 2348–2368. [Google Scholar] [CrossRef] [PubMed]
- Nierengarten, J.-F.; Iehl, J.; Oerthel, V.; Holler, M.; Illescas, B.M.; Muñoz, A.; Martín, N.; Rojo, J.; Sánchez-Navarro, M.; Cecioni, S.; et al. Fullerene sugar balls. Chem. Commun. 2010, 46, 3860–3862. [Google Scholar] [CrossRef]
- Cecioni, S.; Oerthel, V.; Iehl, J.; Holler, M.; Goyard, D.; Praly, J.-P.; Imberty, A.; Nierengarten, J.-F.; Vidal, S. Synthesis of Dodecavalent Fullerene-Based Glycoclusters and Evaluation of Their Binding Properties towards a Bacterial Lectin. Chem. Eur. J. 2011, 17, 3252–3261. [Google Scholar] [CrossRef] [PubMed]
- Compain, P.; Decroocq, C.; Iehl, J.; Holler, M.; Hazelard, D.; Barragán, T.M.; Mellet, C.O.; Nierengarten, J.-F. Glycosidase Inhibition with Fullerene Iminosugar Balls: A Dramatic Multivalent Effect. Angew. Chem. Int. Ed. 2010, 49, 5753–5756. [Google Scholar] [CrossRef] [PubMed]
- Baldini, L.; Casnati, A.; Sansone, F.; Ungaro, R. Calixarene-based multivalent ligands. Chem. Soc. Rev. 2007, 36, 254–266. [Google Scholar] [CrossRef] [PubMed]
- Cecioni, S.; Lalor, R.; Blanchard, B.; Praly, J.-P.; Imberty, A.; Matthews, S.E.; Vidal, S. Achieving High Affinity towards a Bacterial Lectin through Multivalent Topological Isomers of Calix[4]arene Glycoconjugates. Chem. Eur. J. 2009, 15, 13232–13240. [Google Scholar] [CrossRef] [PubMed]
- Dondoni, A.; Marra, A. Calixarene and Calixresorcarene Glycosides: Their Synthesis and Biological Applications. Chem. Rev. 2010, 110, 4949–4977. [Google Scholar] [CrossRef] [PubMed]
- Cecioni, S.; Faure, S.; Darbost, U.; Bonnamour, I.; Parrot-Lopez, H.; Roy, O.; Taillefumier, C.; Wimmerová, M.; Praly, J.-P.; Imberty, A.; et al. Selectivity among Two Lectins: Probing the Effect of Topology, Multivalency and Flexibility of “Clicked” Multivalent Glycoclusters. Chem. Eur. J. 2011, 17, 2146–2159. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.Y.; Tobias, G.; Al-jamal, K.T.; Ballesteros, B.; Ali-Boucetta, H.; Lozano-Perez, S.; Nellist, P.D.; Sim, R.B.; Finucane, C.; Mather, S.J.; et al. Filled and glycosylated carbon nanotubes for in vivo radioemitter localization and imaging. Nat. Mater. 2010, 9, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Robinson, A.; Fang, J.-M.; Chou, P.-T.; Liao, K.-W.; Chu, R.-M.; Lee, S.-J. Probing Lectin and Sperm with Carbohydrate-Modified Quantum Dots. ChemBioChem 2005, 6, 1899–1905. [Google Scholar] [CrossRef] [PubMed]
- De La Fuente, J.M.; Penadés, S. Glyco-quantum dots: A new luminescent system with multivalent carbohydrate display. Tetrahedron Asymmetry 2005, 16, 387–391. [Google Scholar] [CrossRef]
- Kikkeri, R.; Lepenies, B.; Adibekian, A.; Laurino, P.; Seeberger, P.H. In Vitro Imaging and in Vivo Liver Targeting with Carbohydrate Capped Quantum Dots. J. Am. Chem. Soc. 2009, 131, 2110–2112. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yu, M.; Yan, T.-T.; Zhao, Z.-H.; Sha, Y.-L.; Li, Z.-J. Characterization of multivalent lactose quantum dots and its application in carbohydrate–protein interactions study and cell imaging. Bioorg. Med. Chem. 2010, 18, 5234–5240. [Google Scholar] [CrossRef] [PubMed]
- Kikkeri, R.; Laurino, P.; Odedra, A.; Seeberger, P.H. Synthesis of Carbohydrate-Functionalized Quantum Dots in Microreactors. Angew. Chem. Int. Ed. 2010, 49, 2054–2057. [Google Scholar] [CrossRef] [PubMed]
- Saha, K.; Agasti, S.S.; Kim, C.; Li, X.; Rotello, V.M. Gold Nanoparticles in Chemical and Biological Sensing. Chem. Rev. 2012, 112, 2739–2779. [Google Scholar] [CrossRef] [PubMed]
- Katz, E.; Willner, I. Integrated Nanoparticle–Biomolecule Hybrid Systems: Synthesis, Properties, and Applications. Angew. Chem. Int. Ed. 2004, 43, 6042–6108. [Google Scholar] [CrossRef] [PubMed]
- Burda, C.; Chen, X.; Narayanan, R.; El-Sayed, M.A. Chemistry and Properties of Nanocrystals of Different Shapes. Chem. Rev. 2005, 105, 1025–1102. [Google Scholar] [CrossRef] [PubMed]
- Shenhar, R.; Rotello, V.M. Nanoparticles: Scaffolds and Building Blocks. Acc. Chem. Res. 2003, 36, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Daniel, M.-C.; Astruc, D. Gold Nanoparticles: Assembly, Supramolecular Chemistry, Quantum-Size-Related Properties, and Applications toward Biology, Catalysis, and Nanotechnology. Chem. Rev. 2004, 104, 293–346. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Yoshida, K.; Shinkawa, K.; Kumagawa, D.; Seguchi, H. Thioglucose-stabilized gold nanoparticles as a novel platform for colorimetric bioassay based on nanoparticle aggregation. Colloids Surf. B Biointerfaces 2010, 81, 570–577. [Google Scholar] [CrossRef] [PubMed]
- Chuang, Y.-J.; Zhou, X.; Pan, Z.; Turchi, C. A convenient method for synthesis of glyconanoparticles for colorimetric measuring carbohydrate–protein interactions. Biochem. Biophys. Res. Commun. 2009, 389, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Otten, L.; Richards, S.-J.; Fullam, E.; Besra, G.S.; Gibson, M.I. Gold nanoparticle-linked analysis of carbohydrate–protein interactions, and polymeric inhibitors, using unlabelled proteins; easy measurements using a ‘simple’ digital camera. J. Mater. Chem. B 2013, 1, 2665–2672. [Google Scholar] [CrossRef]
- Schofield, C.L.; Mukhopadhyay, B.; Hardy, S.M.; McDonnell, M.B.; Field, R.A.; Russell, D.A. Colorimetric detection of Ricinus communis Agglutinin 120 using optimally presented carbohydrate-stabilised gold nanoparticles. Analyst 2008, 133, 626–634. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-C.; Yeh, Y.-C.; Yang, C.-Y.; Chen, G.-F.; Chen, Y.-C.; Wu, Y.-C.; Chen, C.-C. Quantitative analysis of multivalent interactions of carbohydrate-encapsulated gold nanoparticles with concanavalin A. Chem. Commun. 2003, 2920–2921. [Google Scholar] [CrossRef]
- Dykman, L.; Khlebtsov, N. Gold nanoparticles in biomedical applications: Recent advances and perspectives. Chem. Soc. Rev. 2012, 41, 2256–2282. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.-J.; Fullam, E.; Besra, G.S.; Gibson, M.I. Discrimination between bacterial phenotypes using glyco-nanoparticles and the impact of polymer coating on detection readouts. J. Mater. Chem. B 2014, 2, 1490–1498. [Google Scholar] [CrossRef]
- Richards, S.-J.; Gibson, M.I. Optimization of the polymer coating for glycosylated gold nanoparticle biosensors to ensure stability and rapid optical readouts. ACS Macro Lett. 2014, 3, 1004–1008. [Google Scholar] [CrossRef]
- Otten, L.; Vlachou, D.; Richards, S.-J.; Gibson, M.I. Glycan heterogeneity on gold nanoparticles increases lectin discrimination capacity in label-free multiplexed bioassays. Analyst 2016, 141, 4305–4312. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.-J.; Otten, L.; Gibson, M.I. Glycosylated gold nanoparticle libraries for label-free multiplexed lectin biosensing. J. Mater. Chem. B. 2016, 4, 3046–3053. [Google Scholar] [CrossRef] [PubMed]
- Tsutsumi, H.; Ohkusa, H.; Park, H.; Takahashi, T.; Yuasa, H.; Mihara, H. Gold nanoparticles conjugated with monosaccharide-modified peptide for lectin detection. Bioorg. Med. Chem. Lett. 2012, 22, 6825–6827. [Google Scholar] [CrossRef] [PubMed]
- Arnáiz, B.; Martínez-Ávila, O.; Falcon-Perez, J.M.; Penadés, S. Cellular Uptake of Gold Nanoparticles Bearing HIV gp120 Oligomannosides. Bioconj. Chem. 2012, 23, 814–825. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.L.; Goforth, A.M. pH-Dependent Synthesis and Stability of Aqueous, Elemental Bismuth Glyconanoparticle Colloids: Potentially Biocompatible X-ray Contrast Agents. Chem. Mater. 2012, 24, 1599–1605. [Google Scholar] [CrossRef]
- Coullerez, G.; Seeberger, P.H.; Textor, M. Merging Organic and Polymer Chemistries to Create Glycomaterials for Glycomics Applications. Macromol. Biosci. 2006, 6, 634–647. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ramström, O.; Yan, M. Glyconanomaterials: Synthesis, Characterization, and Ligand Presentation. Adv. Mater. 2010, 22, 1946–1953. [Google Scholar] [CrossRef] [PubMed]
- Gestwicki, J.E.; Cairo, C.W.; Strong, L.E.; Oetjen, K.A.; Kiessling, L.L. Influencing Receptor−Ligand Binding Mechanisms with Multivalent Ligand Architecture. J. Am. Chem. Soc. 2002, 124, 14922–14933. [Google Scholar] [CrossRef] [PubMed]
- Benito-Alifonso, D.; Tremel, S.; Hou, B.; Lockyear, H.; Mantell, J.; Fermin, D.J.; Verkade, P.; Berry, M.; Galan, M.C. Lactose as a “Trojan Horse” for Quantum Dot Cell Transport. Angew. Chem. Int. Ed. 2014, 53, 810–814. [Google Scholar] [CrossRef] [PubMed]
- Gerke, C.; Ebbesen, M.F.; Jansen, D.; Boden, S.; Freichel, T.; Hartmann, L. Sequence-Controlled Glycopolymers via Step-Growth Polymerization of Precision Glycomacromolecules for Lectin Receptor Clustering. Biomacromolecules 2017, 18, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Ponader, D.; Maffre, P.; Aretz, J.; Pussak, D.; Ninnemann, N.M.; Schmidt, S.; Seeberger, P.H.; Rademacher, C.; Nienhaus, G.U.; Hartmann, L. Carbohydrate-Lectin Recognition of Sequence-Defined Heteromultivalent Glycooligomers. J. Am. Chem. Soc. 2014, 136, 2008–2016. [Google Scholar] [CrossRef] [PubMed]
- Ponader, D.; Wojcik, F.; Beceren-Braun, F.; Dernedde, J.; Hartmann, L. Sequence-Defined Glycopolymer Segments Presenting Mannose: Synthesis and Lectin Binding Affinity. Biomacromolecules 2012, 13, 1845–1852. [Google Scholar] [CrossRef] [PubMed]
- Grünstein, D.; Maglinao, M.; Kikkeri, R.; Collot, M.; Barylyuk, K.; Lepenies, B.; Kamena, F.; Zenobi, R.; Seeberger, P.H. Hexameric Supramolecular Scaffold Orients Carbohydrates To Sense Bacteria. J. Am. Chem. Soc. 2011, 133, 13957–13966. [Google Scholar] [CrossRef] [PubMed]
- Sangabathuni, S.; Murthy, R.V.; Chaudhary, P.M.; Surve, M.; Banerjee, A.; Kikkeri, R. Glyco-gold nanoparticle shapes enhance carbohydrate-protein interactions in mammalian cells. Nanoscale 2016, 8, 12729–12735. [Google Scholar] [CrossRef] [PubMed]
- Toraskar, S.; Gade, M.; Sangabathuni, S.; Thulasiram, H.V.; Kikkeri, R. Exploring the influence of shapes and heterogeneity of glycol-gold nanoparticles on bacterial binding for preventing infections. ChemMedChem 2017, 12, 1116–1124. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.-D.; Adak, A.K.; Yu, C.-C.; Hsiao, W.-C.; Lin, H.-J.; Chen, M.-L.; Lin, C.-C. Fabrication of highly stable glycol-gold nanoparticles and development of a glyco-gold nanoparticle-based oriented immobilized antibody microarray for lectin (GOAL) assay. Chem. Eur. J. 2015, 21, 3956–3967. [Google Scholar] [CrossRef] [PubMed]
- Barrientos, A.G.; de la Fuente, J.M.; Rojas, T.C.; Fernández, A.; Penadés, S. Gold Glyconanoparticles: Synthetic Polyvalent Ligands Mimicking Glycocalyx-Like Surfaces as Tools for Glycobiological Studies. Chem. Eur. J. 2003, 9, 1909–1921. [Google Scholar] [CrossRef] [PubMed]
- Wojcik, F.; Lel, S.; O’Brien, A.G.; Seeberger, P.H.; Hartmann, L. Synthesis of homo- and heteromultivalent carbohydrate-functionalized oligo(amidoamines) using novel glyco-building blocks. Beilstein J. Org. Chem. 2013, 9, 2395–2403. [Google Scholar] [CrossRef] [PubMed]
- Igde, S.; Röblitz, S.; Müller, A.; Kolbe, K.; Boden, S.; Fessele, C.; Weber, M.; Lindhorst, T.K.; Hartmann, L. Linear Precision Glycomacromolecules with Varying Interligand Spacing and Linker Functionalities Binding to Concanavalin A and the Bacterial Lectin FimH. Macromol. Biosci. 2017. [Google Scholar] [CrossRef] [PubMed]
- Ponader, D.; Igde, S.; Wehle, M.; Märker, K.; Santer, M.; Bléger, D.; Hartmann, L. Photoswitchable precision glycooligomers and their lectin binding. Beilstein J. Org. Chem. 2014, 10, 1603–1612. [Google Scholar] [CrossRef] [PubMed]
- Merrifield, R.B. Solid Phase Peptide Synthesis. I. The Synthesis of a Tetrapeptide. J. Am. Chem. Soc. 1963, 85, 2149–2154. [Google Scholar] [CrossRef]
- Mosca, S.; Wojcik, F.; Hartmann, L. Precise Positioning of Chiral Building Blocks in Monodisperse, Sequence-Defined Polyamides. Macromol. Rapid Commun. 2011, 32, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Wojcik, F.; O´Brien, A.G.; Götze, S.; Seeberger, P.H.; Hartmann, L. Synthesis of Carbohydrate-Functionalised Sequence-Defined Oligo(amidoamine)s by Photochemical Thiol-Ene Coupling in a Continuous Flow Reactor. Chem. Eur. J. 2013, 19, 3090–3098. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, L. Polymers for Control Freaks: Sequence-Defined Poly(amidoamine)s and Their Biomedical Applications. Macromol. Chem. Phys. 2011, 212, 8–13. [Google Scholar] [CrossRef]
- Hartmann, L.; Börner, H.G. Precision Polymers: Monodisperse, Monomer-Sequence-Defined Segments to Target Future Demands of Polymers in Medicine. Adv. Mater. 2009, 21, 3425–3431. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, L.; Krause, E.; Antonietti, M.; Börner, H.G. Solid-Phase Supported Polymer Synthesis of Sequence-Defined, Multifunctional Poly(amidoamines). Biomacromolecules 2006, 7, 1239–1244. [Google Scholar] [CrossRef] [PubMed]
- Wojcik, F.; Mosca, S.; Hartmann, L. Solid-Phase Synthesis of Asymmetrically Branched Sequence-Defined Poly/Oligo(amidoamines). J. Org. Chem. 2012, 77, 4226–4234. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Bonilla, A.; Bordegé, V.; León, O.; Cuervo-Rodríguez, R.; Sánchez-Chaves, M.; Fernández-García, M. Influence of glycopolymers structure on the copolymerization reaction and on their binding behavior with lectins. Eur. Polym. J. 2012, 48, 963–973. [Google Scholar] [CrossRef]
- Ting, S.R.S.; Chen, G.; Stenzel, M.H. Synthesis of glycopolymers and their multivalent recognitions with lectins. Polym. Chem. 2010, 1, 1392–1412. [Google Scholar] [CrossRef]
- Patel, A.; Lindhorst, T.K. Multivalent glycomimetics: Synthesis of nonavalent mannoside clusters with variation of spacer properties. Carbohydr. Res. 2006, 341, 1657–1668. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.-L.; Fang, Y.; Wan, L.-S.; Huang, X.-J.; Xu, Z.-K. Glycopolymer Brushes for the Affinity Adsorption of RCA120: Effects of Thickness, Grafting Density, and Epitope Density. Langmuir 2012, 28, 13616–13623. [Google Scholar] [CrossRef] [PubMed]
- Cairo, C.W.; Gestwicki, J.E.; Kanai, M.; Kiessling, L.L. Control of Multivalent Interactions by Binding Epitope Density. J. Am. Chem. Soc. 2002, 124, 1615–1619. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.W.; Otten, L.; Richards, S.-J.; Lowery, R.; Philips, D.J.; Haddleton, D.M.; Gibson, M.I. Glycopolymers with secondary binding motifs mimic glycan branching and display bacterial lectin selectivity in addition to affinity. Chem. Sci. 2014, 5, 1611–1616. [Google Scholar] [CrossRef]
- Liu, X.; Atwater, M.; Wang, J.; Huo, Q. Extinction coefficient of gold nanoparticles with different sizes and different capping ligands. Colloids Surf. B Biointerfaces 2007, 58, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Dubois, M.; Gilles, K.A.; Hamilton, J.K.; Rebers, P.A.; Smith, F. Colorimetric Method for Determination of Sugars and Related Substances. Anal. Chem. 1956, 28, 350–356. [Google Scholar] [CrossRef]
- Gerchakov, S.M.; Hatcher, P.G. Improved technique for analysis of carbohydrates in sediments. Limnol. Oceanogr. 1972, 17, 938–943. [Google Scholar] [CrossRef]
- Valtola, L.; Rahikkala, A.; Raula, J.; Kauppinen, E.I.; Tenhu, H.; Hietala, S. Synthesis and lectin recognition of glycosylated amphiphilic nanoparticles. Eur. Polym. J. 2014, 59, 282–289. [Google Scholar] [CrossRef]
- Schmidt, S.; Wang, H.; Pussak, D.; Mosca, S.; Hartmann, L. Probing multivalency in ligand-receptor-mediated adhesion of soft, biomimetic interfaces. Beilstein J. Org. Chem. 2015, 11, 720–729. [Google Scholar] [CrossRef] [PubMed]
- Ebbesen, M.F.; Gerke, C.; Hartwig, P.; Hartmann, L. Biodegradable poly(amidoamine)s with uniform degradation fragments via sequence-controlled macromonomers. Polym. Chem. 2016, 7, 7086–7093. [Google Scholar] [CrossRef]
- Hayes, W.; Osborn, H.M.; Osborne, S.D.; Rastall, R.A.; Romagnoli, B. One-pot synthesis of multivalent arrays of mannose mono- and disaccharides. Tetrahedron 2003, 59, 7983–7996. [Google Scholar] [CrossRef]
- Turkevich, J.; Stevenson, P.C.; Hillier, J. A study of the nucleation and growth processes in the synthesis of colloidal gold. Discuss. Faraday Soc. 1951, 11, 55–75. [Google Scholar] [CrossRef]
- Trivedi, M.V.; Laurence, J.S.; Siahaan, T.J. The role of thiols and disulfides on stability. Curr. Protein Pept. Sci. 2009, 10, 614–625. [Google Scholar] [CrossRef] [PubMed]
- Compostella, F.; Pitirollo, O.; Silvestri, A.; Polito, L. Glyco-gold nanoparticles: Synthesis and applications. Beilstein J. Org. Chem. 2017, 13, 1008–1021. [Google Scholar] [CrossRef] [PubMed]
- Rahme, K.; Chen, L.; Hobbs, R.G.; Morris, M.A.; O’Driscoll, C.; Holmes, J.D. Correction: PEGylated gold nanoparticles: polymer quantification as a function of PEG lengths and nanoparticle dimensions. RSC Adv. 2017, 7, 8798–8799. [Google Scholar] [CrossRef]
- Kennedy, D.C.; Orts-Gil, G.; Lai, C.-H.; Müller, L.; Haase, A.; Luch, A.; Seeberger, P.H. Carbohydrate functionalization of silver nanoparticles modulates cytotoxicity and cellular uptake. J. Nanobiotechnol. 2014, 12, 59. [Google Scholar] [CrossRef]
- Ambrosi, M.; Cameron, N.R.; Davis, B.G. Lectins: Tools for the molecular understanding of the glycocode. Org. Biomol. Chem. 2005, 3, 1593–1608. [Google Scholar] [CrossRef] [PubMed]
- Brewer, C.F.; Brown, R.D.I.; Koenig, S.H. Metal Ion Binding and Conformational Transitions in Concanavalin A: A Structure-Function Study. J. Biomol. Struct. Dyn. 1983, 1, 961–997. [Google Scholar] [CrossRef] [PubMed]
- Loris, R.; Hamelryck, T.; Bouckaert, J.; Wyns, L. Legume lectin structure. Biochim. Biophys. Acta 1998, 1383, 9–36. [Google Scholar] [CrossRef]
- Mandal, D.K.; Kishore, N.; Brewer, C.F. Thermodynamics of Lectin-Carbohydrate Interactions. Titration Microcalorimetry Measurements of the Binding of N-Linked Carbohydrates and Ovalbumin to Concanavalin A. Biochemistry 1994, 33, 1149–1156. [Google Scholar] [CrossRef] [PubMed]
- Bryce, R.A.; Hillier, I.H.; Naismith, J.H. Carbohydrate-Protein Recognition: Molecular Dynamics Simulations and Free Energy Analysis of Oligosaccharide Binding to Concanavalin A. Biophys. J. 2001, 81, 1373–1388. [Google Scholar] [CrossRef]
- Dam, T.K.; Brewer, C.F. Thermodynamic Studies of Lectin−Carbohydrate Interactions by Isothermal Titration Calorimetry. Chem. Rev. 2002, 102, 387–429. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, M.; Marradi, M.; Imberty, A.; Penadés, S.; Pérez, S. Multivalent Gold Glycoclusters: High Affinity Molecular Recognition by Bacterial Lectin PA-IL. Chem. Eur. J. 2012, 18, 4264–4273. [Google Scholar] [CrossRef] [PubMed]
- Baier, M.; Giesler, M.; Hartmann, L. Split & Combine Approach towards Branched Precision Glycomacromolecules and their Lectin Binding Behavior. Chem. Eur. J. 2017. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AuNP | RH in Water [nm] | Degree of Glycomacromolecule Functionalization per NP (PSA) | Degree of Glycomacromolecule Functionalization per NP (TBO) | Average Degree of Carbohydrate Functionalization per NP (PSA) | Average Occupied Surface Area per Glycomacro-Molecule (PSA) [nm2] |
|---|---|---|---|---|---|
| Citrate stabilized | 8.5 ± 0.1 | - | - | - | - |
| 4 | 11.7 ± 0.1 | 312 ± 218 | 2210 ± 553 | ~312 | ~2.0 |
| 5 | 13.3 ± 0.3 | 231 ± 98 | 1430 ± 377 | ~231 | ~2.7 |
| 6 | 13.5 ± 0.1 | 242 ± 2 | 1620 ± 206 | ~1210 | ~2.5 |
| Glyco-Au-NP | KD,app [nM] | KD,app per Glycomacromolecular Ligand [µM] | KD,app per Carbohydrate Ligand [µM] |
|---|---|---|---|
| 4 | 209 ± 12 | 65 | 65 |
| 5 | 203 ± 4 | 47 | 47 |
| 6 | 357 ± 12 | 86 | 432 |
| Glyco-Au-NP | RH in LBB [nm] | RH at KD,app conc. of Con A [nm] | RH after αMeMan Addition [nm] | Normalized Abs700 nm at KD,app conc. of Con A |
|---|---|---|---|---|
| 4 | 10.4 ± 0.2 | 106 ± 1 | 14.7 ± 0.7 | 0.43 ± 0.01 |
| 5 | 10.6 ± 0.1 | 112 ± 11 | 11.2 ± 0.2 | 0.39 ± 0.001 |
| 6 | 12.6 ± 0.1 | 101 ± 1 | 13.3 ± 0.2 | 0.35 ± 0.005 |
| Glyco-Au-NP | IC50 [nM] | RIP | RIP per Glycomacromolecular Ligand | RIP per Carbohydrate Ligand |
|---|---|---|---|---|
| αMeMan | (5.17 ± 0.84) × 105 | 1 | 1 | 1 |
| 1a 1 | (0.7 ± 0.5) × 105 | 5 | 5 | 5 |
| 3a 1 | 155 ± 78 | 2096 | 2096 | 419 |
| 4 | 0.78 ± 0.09 | 6.6·105 | 2115 | 2115 |
| 5 | 0.36 ± 0.10 | 14.4·105 | 6234 | 6234 |
| 6 | 0.33 ± 0.09 | 15.7·105 | 6487 | 1297 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boden, S.; Wagner, K.G.; Karg, M.; Hartmann, L. Presenting Precision Glycomacromolecules on Gold Nanoparticles for Increased Lectin Binding. Polymers 2017, 9, 716. https://doi.org/10.3390/polym9120716
Boden S, Wagner KG, Karg M, Hartmann L. Presenting Precision Glycomacromolecules on Gold Nanoparticles for Increased Lectin Binding. Polymers. 2017; 9(12):716. https://doi.org/10.3390/polym9120716
Chicago/Turabian StyleBoden, Sophia, Kristina G. Wagner, Matthias Karg, and Laura Hartmann. 2017. "Presenting Precision Glycomacromolecules on Gold Nanoparticles for Increased Lectin Binding" Polymers 9, no. 12: 716. https://doi.org/10.3390/polym9120716
APA StyleBoden, S., Wagner, K. G., Karg, M., & Hartmann, L. (2017). Presenting Precision Glycomacromolecules on Gold Nanoparticles for Increased Lectin Binding. Polymers, 9(12), 716. https://doi.org/10.3390/polym9120716