Binding of the GTPase Sar1 to a Lipid Membrane Monolayer: Insertion and Orientation Studied by Infrared Reflection–Absorption Spectroscopy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Sar1 and AH Peptide Adsorption to the Air–Water Interface
2.2.2. Lipid Monolayer Preparation and Sar1 Insertion
2.2.3. IRRAS Measurements
2.2.4. IRRAS Data Analysis
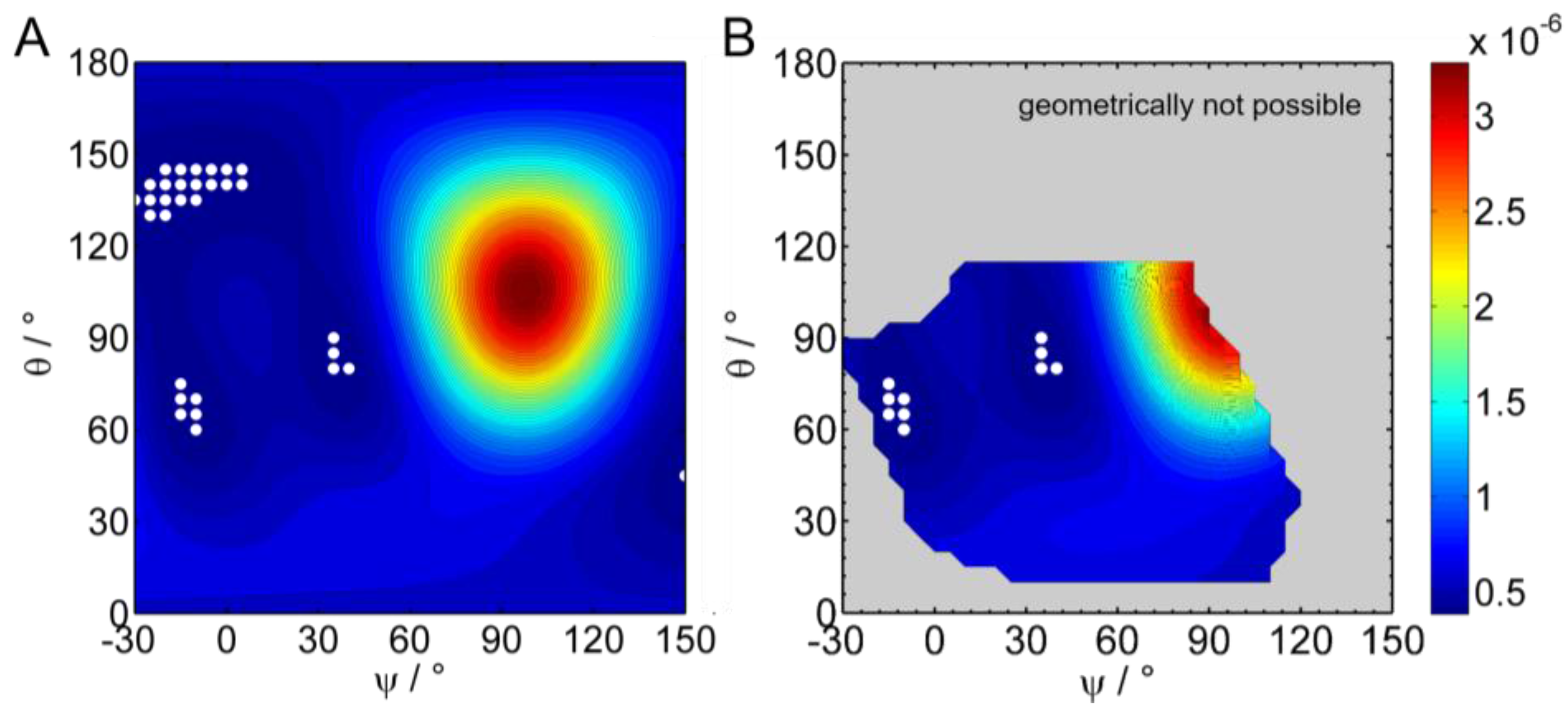
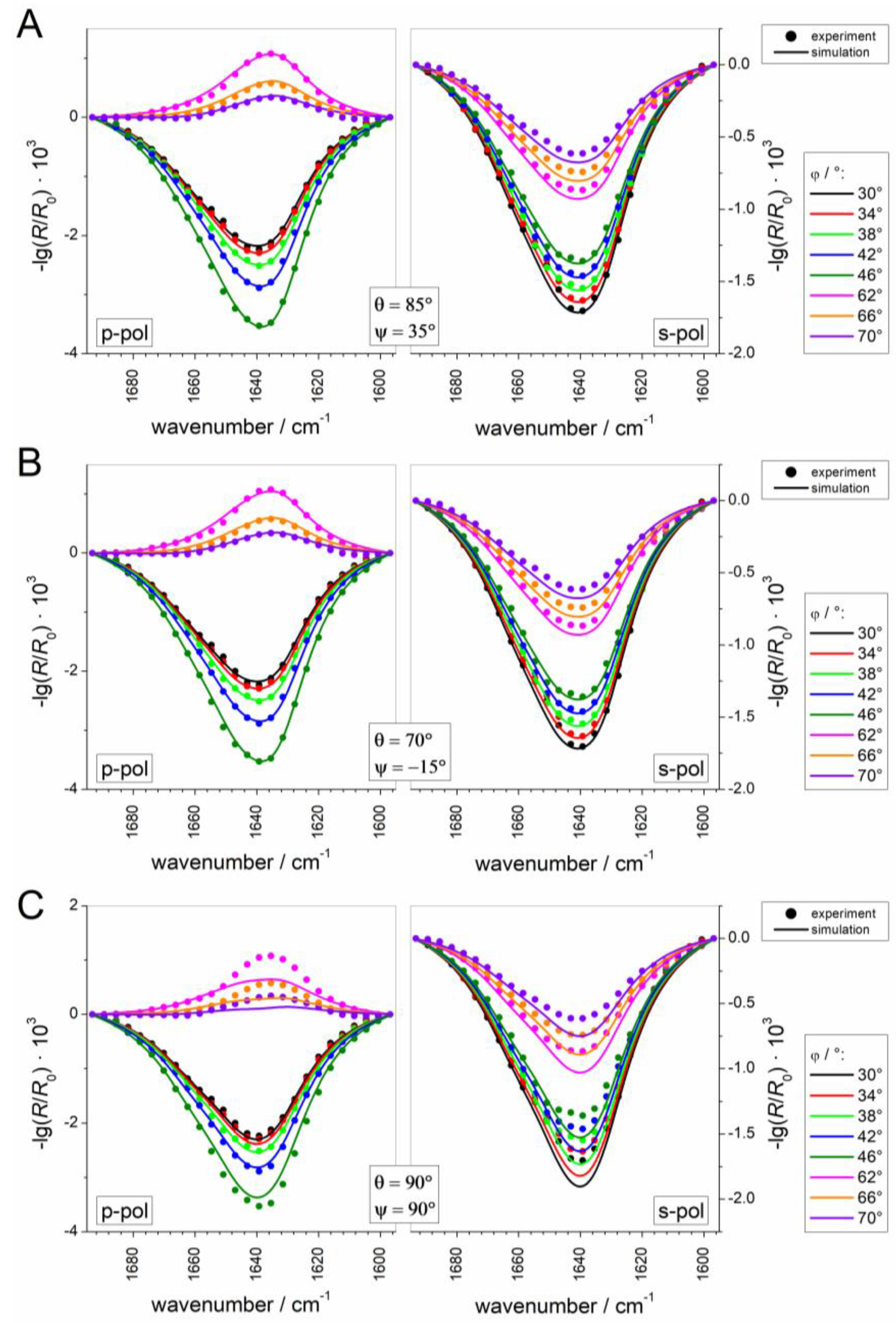
2.2.5. Simulation and Fitting of IRRA Spectra
3. Results and Discussion
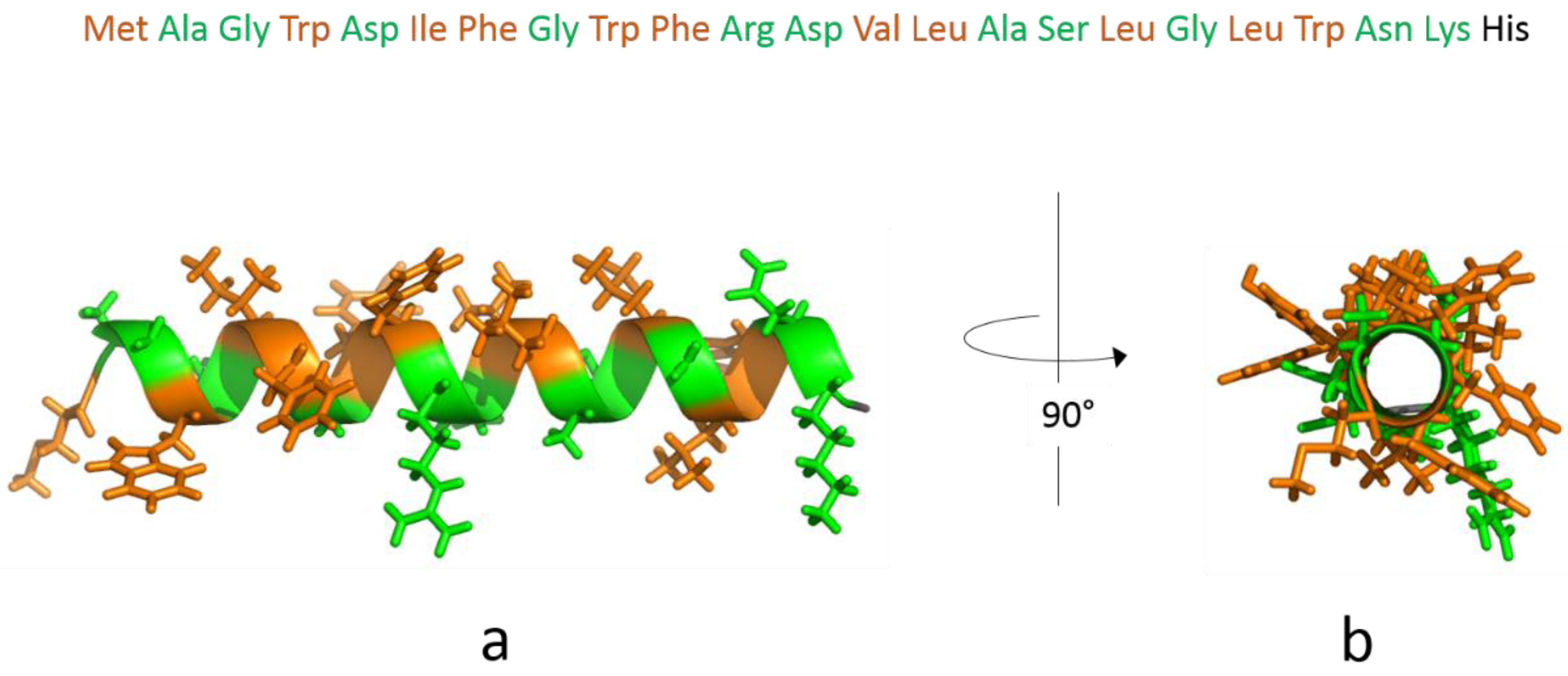
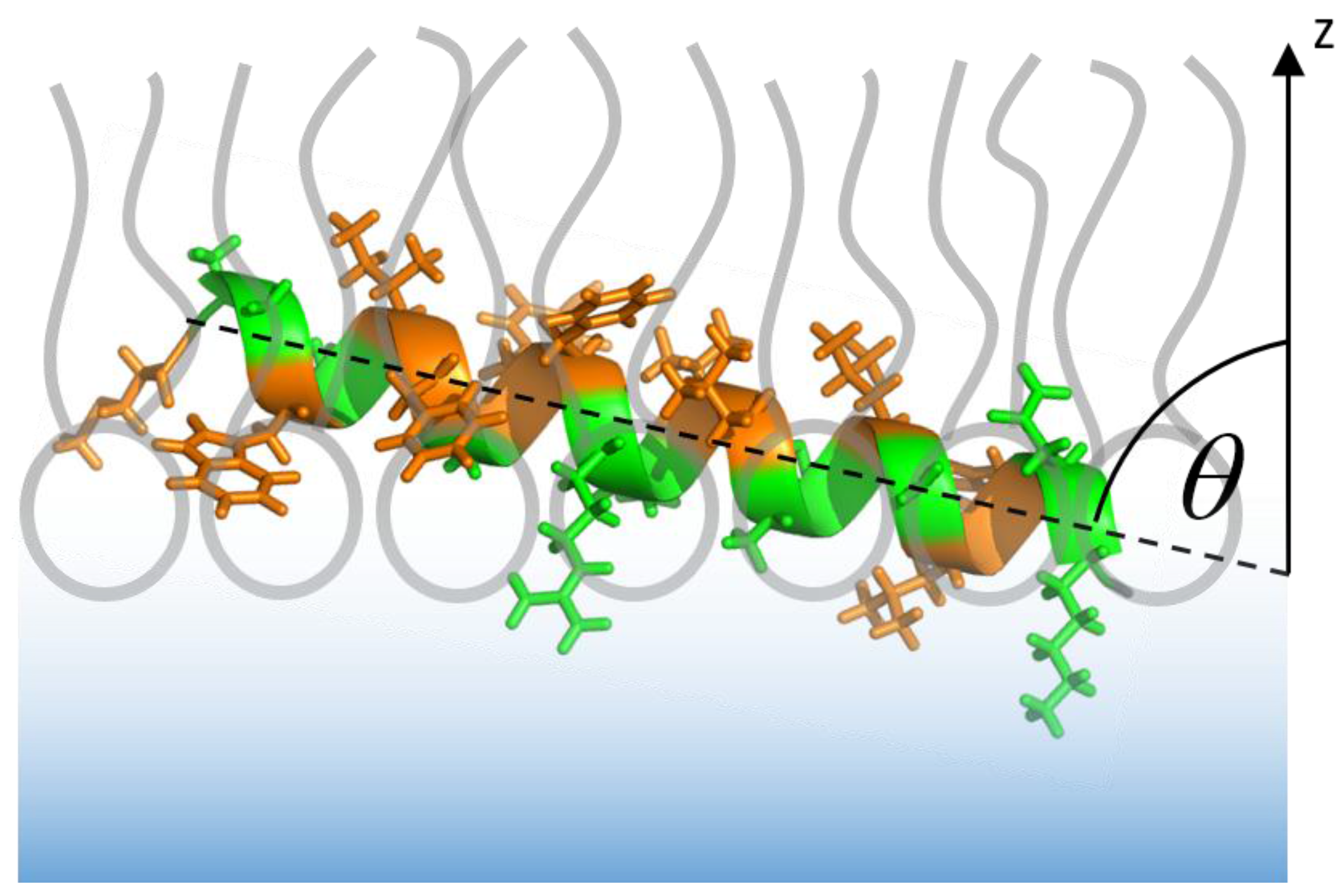
3.1. The Isolated Amphipathic Helix
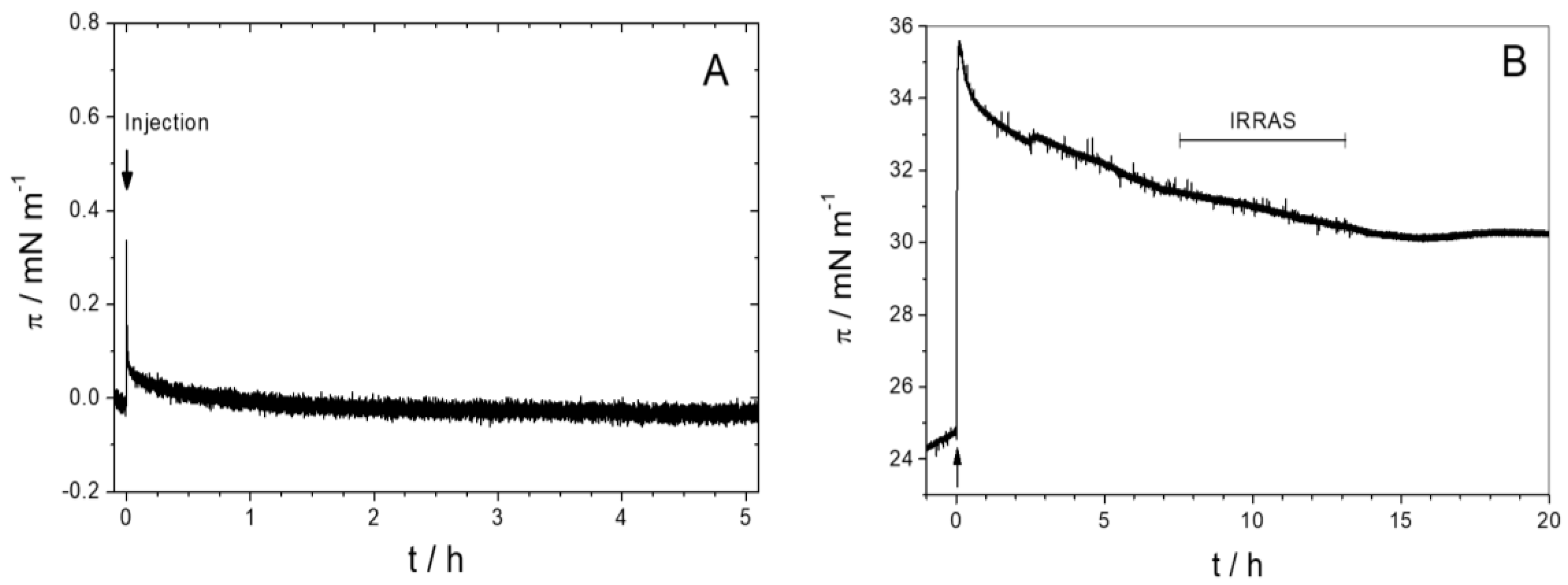
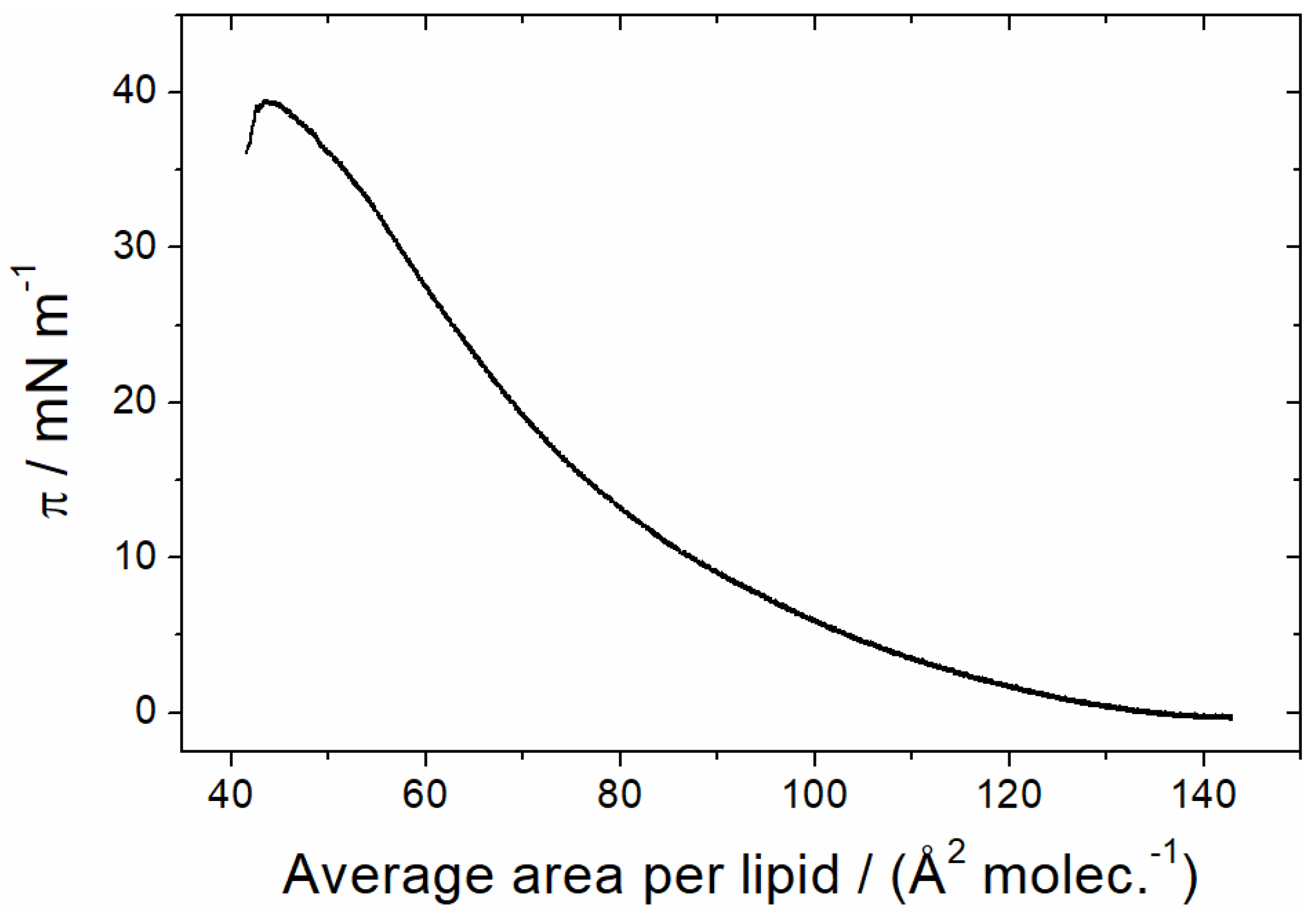
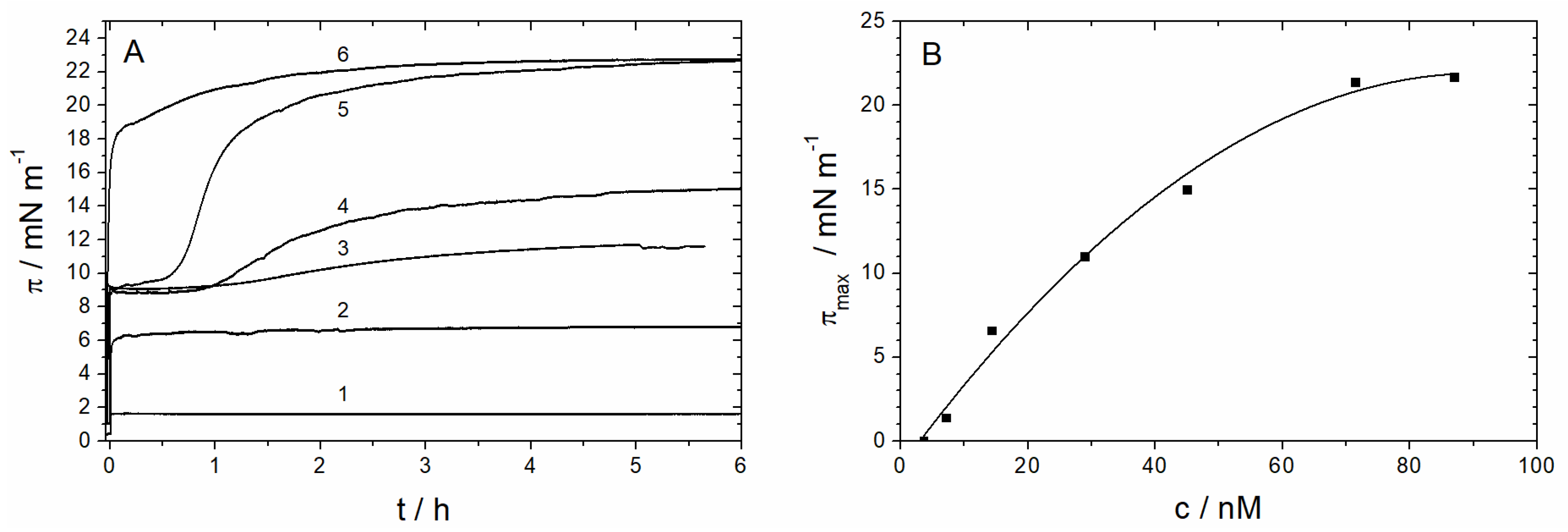
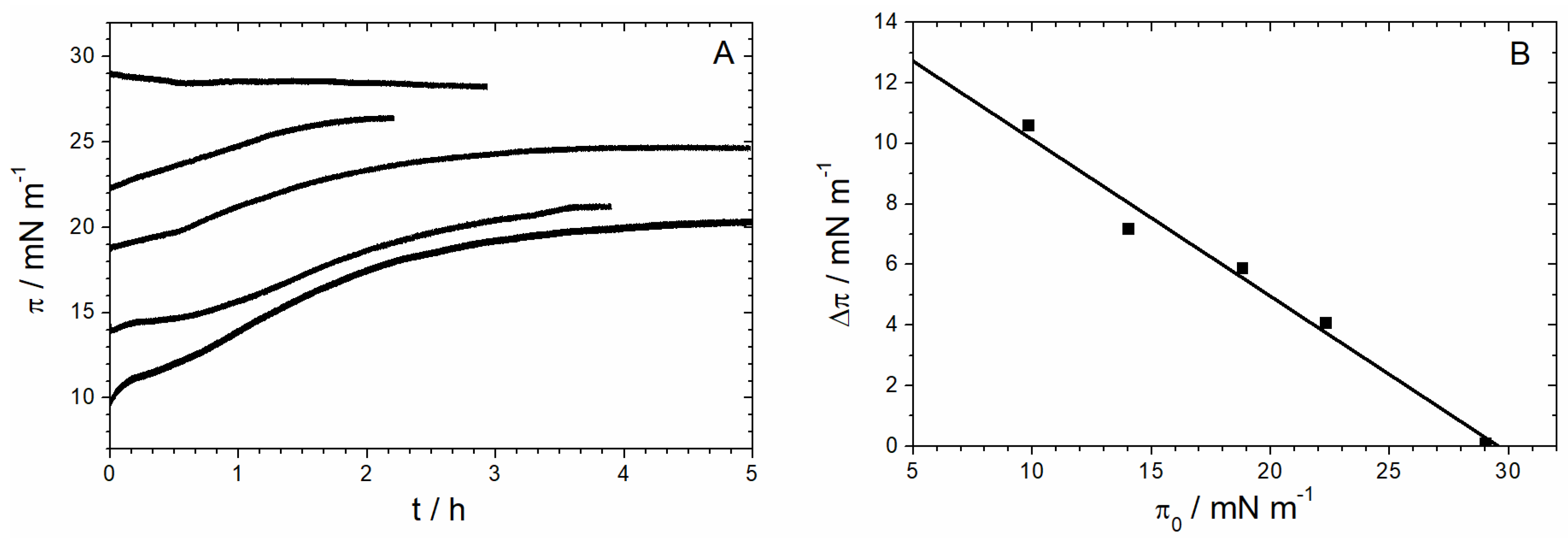
3.1.1. Monolayer Adsorption Experiments
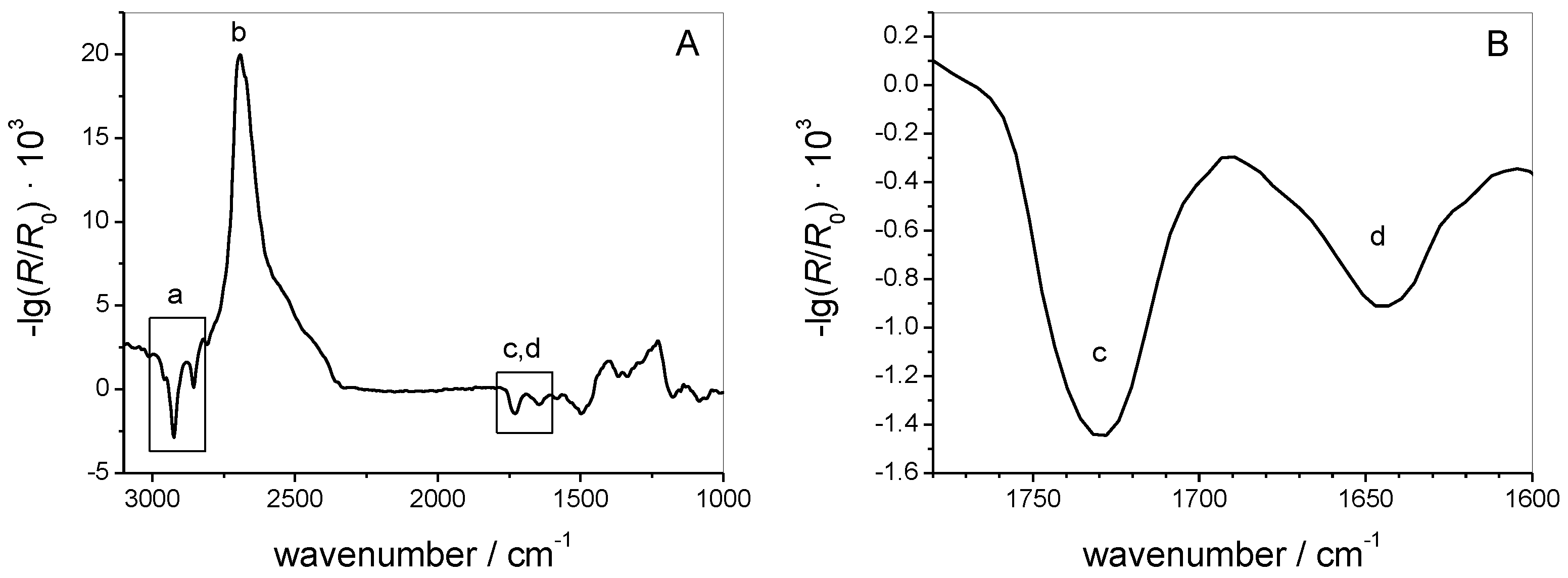
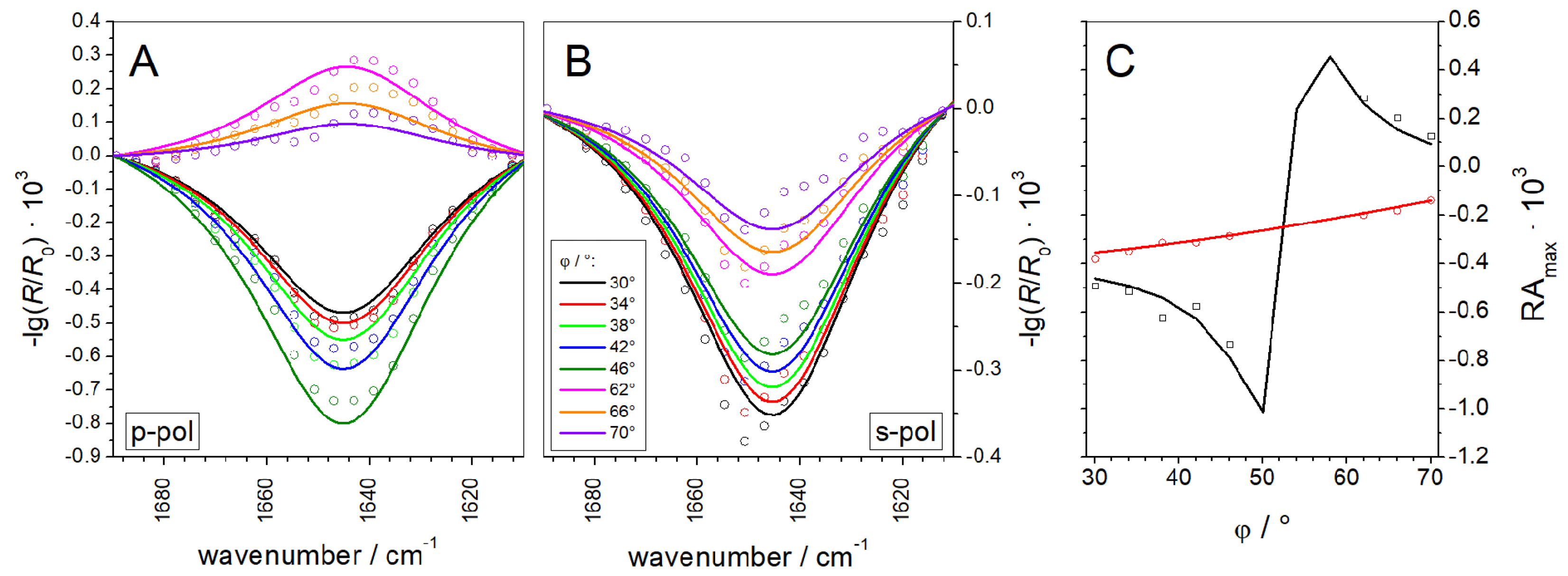
3.1.2. IRRAS
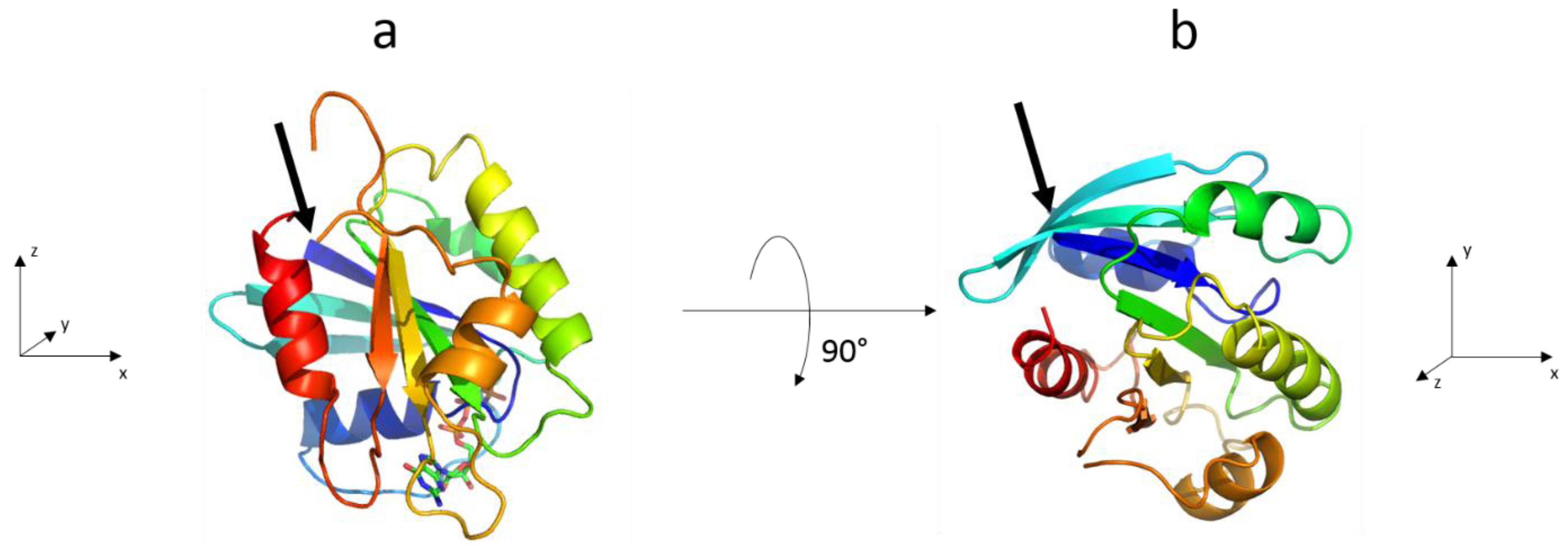
3.2. The Complete Sar1 Protein
3.2.1. Sar1 Adsorption at the Air–Water Interface
3.2.2. Sar1 Adsorption at the Lipid Monolayer
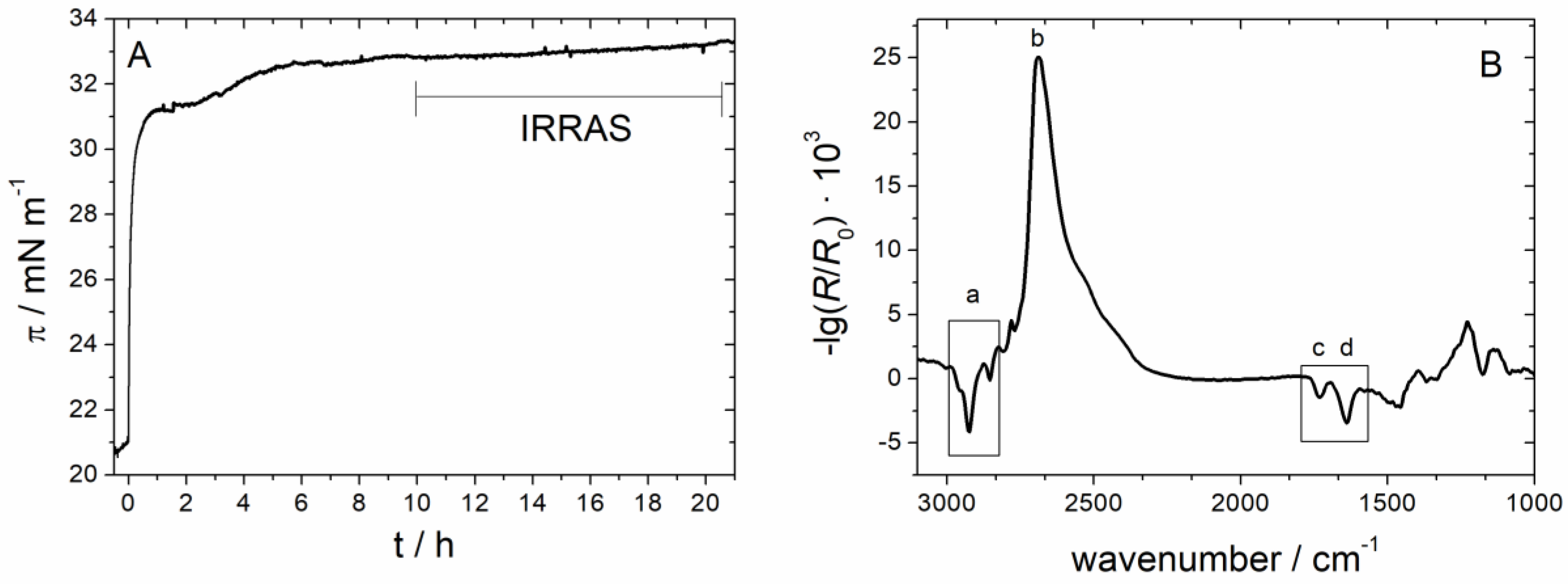
3.2.3. IRRAS
3.2.4. Time Dependency/Stability
3.2.5. Adsorption and Orientation of Sar1 in the Presence of GMP-PNP
3.2.6. Comparison to Sar1 Orientation after Sec23/24p and Sec13/31p Recruitment
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Matsuoka, K.; Orci, L.; Amherdt, M.; Bednarek, S.Y.; Hamamoto, S.; Schekman, R.; Yeung, T. COPII-coated vesicle formation reconstituted with purified coat proteins and chemically defined liposomes. Cell 1998, 93, 263–275. [Google Scholar] [CrossRef]
- Bi, X.; Corpina, R.A.; Goldberg, J. Structure of the Sec23/24-Sar1 pre-budding complex of the COPII vesicle coat. Nature 2002, 419, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Nakano, A.; Muramatsu, M. A Novel GTP-Binding Protein, Sar1p, Is Involved in Transport from the Endoplasmic-Reticulum to the Golgi-Apparatus. J. Cell Biol. 1989, 109, 2677–2691. [Google Scholar] [CrossRef] [PubMed]
- Aridor, M.; Balch, W.E. Principles of selective transport: Coat complexes hold the key. Trends Cell Biol. 1996, 6, 315–320. [Google Scholar] [CrossRef]
- Barlowe, C. COPII and selective export from the endoplasmic reticulum. Biochim. Biophys. Acta Mol. Cell Res. 1998, 1404, 67–76. [Google Scholar] [CrossRef]
- Springer, S.; Spang, A.; Schekman, R. A primer on vesicle budding. Cell 1999, 97, 145–148. [Google Scholar] [CrossRef]
- Bielli, A.; Haney, C.J.; Gabreski, G.; Watkins, S.C.; Bannykh, S.I.; Aridor, M. Regulation of Sar1 NH2 terminus by GTP binding and hydrolysis promotes membrane deformation to control COPII vesicle fission. J. Cell Biol. 2005, 171, 919–924. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.C.; Orci, L.; Hamamoto, S.; Futai, E.; Ravazzola, M.; Schekman, R. Sar1p N-terminal helix initiates membrane curvature and completes the fission of a COPII vesicle. Cell 2005, 122, 605–617. [Google Scholar] [CrossRef] [PubMed]
- Farsad, K.; Camilli, P.D. Mechanisms of membrane deformation. Curr. Opin. Cell Biol. 2003, 15, 372–381. [Google Scholar] [CrossRef]
- Sheetz, M.P.; Singer, S.J. Biological-Membranes as Bilayer Couples—Molecular Mechanism of Drug-Erythrocyte Interactions. Proc. Natl. Acad. Sci. USA 1974, 71, 4457–4461. [Google Scholar] [CrossRef] [PubMed]
- Zimmerberg, J.; Kozlov, M.M. How proteins produce cellular membrane curvature. Nat. Rev. Mol. Cell Biol. 2006, 7, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Campelo, F.; McMahon, H.T.; Kozlov, M.M. The hydrophobic insertion mechanism of membrane curvature generation by proteins. Biophys. J. 2008, 95, 2325–2339. [Google Scholar] [CrossRef] [PubMed]
- Brockman, H. Lipid monolayers: Why use half a membrane to characterize protein-membrane interactions? Curr. Opin. Struct. Biol. 1999, 9, 438–443. [Google Scholar] [CrossRef]
- Maget-Dana, R. The monolayer technique: A potent tool for studying the interfacial properties of antimicrobial and membrane-lytic peptides and their interactions with lipid membranes. Biochim. Biophys. Acta 1999, 1462, 109–140. [Google Scholar] [CrossRef]
- Calvez, P.; Bussières, S.; Demers, É.; Salesse, C. Parameters modulating the maximum insertion pressure of proteins and peptides in lipid monolayers. Biochimie 2009, 91, 718–733. [Google Scholar] [CrossRef] [PubMed]
- Boisselier, E.; Calvez, P.; Demers, E.; Cantin, L.; Salesse, C. Influence of the Physical State of Phospholipid Monolayers on Protein Binding. Langmuir 2012, 28, 9680–9688. [Google Scholar] [CrossRef] [PubMed]
- Dyck, M.; Kerth, A.; Blume, A.; Losche, M. Interaction of the neurotransmitter, neuropeptide Y, with phospholipid membranes: Infrared spectroscopic characterization at the air/water interface. J. Phys. Chem. B 2006, 110, 22152–22159. [Google Scholar] [CrossRef] [PubMed]
- Arouri, A.; Kerth, A.; Dathe, M.; Blume, A. The Binding of an Amphipathic Peptide to Lipid Monolayers at the Air/Water Interface Is Modulated by the Lipid Headgroup Structure. Langmuir 2011, 27, 2811–2818. [Google Scholar] [CrossRef] [PubMed]
- Cornut, I.; Desbat, B.; Turiet, J.M.; Dufourcq, J. In situ study by polarization modulated Fourier transform infrared spectroscopy of the structure and orientation of lipids and amphipathic peptides at the air-water interface. Biophys. J. 1996, 70, 305–312. [Google Scholar] [CrossRef]
- Meister, A.; Nicolini, C.; Waldmann, H.; Kuhlmann, J.; Kerth, A.; Winter, R.; Blume, A. Insertion of lipidated Ras proteins into lipid monolayers studied by infrared reflection absorption spectroscopy (IRRAS). Biophys. J. 2006, 91, 1388–1401. [Google Scholar] [CrossRef] [PubMed]
- Hermelink, A.; Kirsch, C.; Klinger, R.; Reiter, G.; Brezesinski, G. Adsorption of GST-PI3Kgamma at the air-buffer interface and at substrate and nonsubstrate phospholipid monolayers. Biophys. J. 2009, 96, 1016–1025. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Erbe, A.; Kerth, A.; Dathe, M.; Blume, A. Interactions of KLA Amphipathic Model Peptides with Lipid Monolayers. ChemBioChem 2009, 10, 2884–2892. [Google Scholar] [CrossRef] [PubMed]
- Hoernke, M.; Falenski, J.A.; Schwieger, C.; Koksch, B.; Brezesinski, G. Triggers for beta-Sheet Formation at the Hydrophobic-Hydrophilic Interface: High Concentration, In-Plane Orientational Order, and Metal Ion Complexation. Langmuir 2011, 27, 14218–14231. [Google Scholar] [CrossRef] [PubMed]
- Blume, A.; Kerth, A. Peptide and protein binding to lipid monolayers studied by FT-IRRA spectroscopy. Biochim. Biophys. Acta 2013, 1828, 2294–2305. [Google Scholar] [CrossRef] [PubMed]
- Mendelsohn, R.; Mao, G.R.; Flach, C.R. Infrared reflection-absorption spectroscopy: Principles and applications to lipid-protein interaction in Langmuir films. Biochim. Biophys. Acta Biomembr. 2010, 1798, 788–800. [Google Scholar] [CrossRef] [PubMed]
- Daum, S.; Kruger, D.; Meister, A.; Auerswald, J.; Prinz, S.; Briggs, J.A.G.; Bacia, K. Insights from reconstitution reactions of COPII vesicle formation using pure components and low mechanical perturbation. Biol. Chem. 2014, 395, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Schwieger, C.; Blaffert, J.; Li, Z.; Kressler, J.; Blume, A. Perfluorinated Moieties Increase the Interaction of Amphiphilic Block Copolymers with Lipid Monolayers. Langmuir 2016, 32, 8102–8115. [Google Scholar] [CrossRef] [PubMed]
- Rabe, M.; Schwieger, C.; Zope, H.R.; Versluis, F.; Kros, A. Membrane Interactions of Fusogenic Coiled-Coil Peptides: Implications for Lipopeptide Mediated Vesicle Fusion. Langmuir 2014, 30, 7724–7735. [Google Scholar] [CrossRef] [PubMed]
- Flach, C.R.; Brauner, J.W.; Taylor, J.W.; Baldwin, R.C.; Mendelsohn, R. External Reflection FTIR of Peptide Monolayer Films at the Air/Water Interface: Experimental Design, Spectra-Structure Correlations, and Effects of Hydrogen-Deuterium Exchange. Biophys. J. 1994, 67, 402–410. [Google Scholar] [CrossRef]
- Kuzmin, V.L.; Mikhailov, A.V. Molecular theory of light reflection and applicability limits of the macroscopic approach. Opt. Spectrosc. (USSR) 1981, 51, 383–385. [Google Scholar]
- Kuzmin, V.L.; Romanov, V.P.; Michailov, A.V. Reflection of light at the boundary of liquid systems and structure of the surface layer: A review. Opt. Spectrosc. 1992, 73, 1–26. [Google Scholar]
- Bertie, J.E.; Ahmed, M.K.; Eysel, H.H. Infrared Intensities of Liquids. 5. Optical and Dielectric-Constants, Integrated-Intensities, and Dipole-Moment Derivatives of H2O and D2O at 22-Degrees-C. J. Phys. Chem. 1989, 93, 2210–2218. [Google Scholar] [CrossRef]
- Bertie, J.E.; Lan, Z.D. Infrared intensities of liquids. 20. The intensity of the OH stretching band of liquid water revisited, and the best current values of the optical constants of H2O(1) at 25 degrees C between 15,000 and 1 cm−1. Appl. Spectrosc. 1996, 50, 1047–1057. [Google Scholar] [CrossRef]
- Schwieger, C.; Chen, B.; Tschierske, C.; Kressler, J.; Blume, A. Organization of T-Shaped Facial Amphiphiles at the Air/Water Interface Studied by Infrared Reflection Absorption Spectroscopy. J. Phys. Chem. B 2012, 116, 12245–12256. [Google Scholar] [CrossRef] [PubMed]
- Flach, C.R.; Gericke, A.; Mendelsohn, R. Quantitative determination of molecular chain tilt angles in monolayer films at the air/water interface: Infrared reflection/absorption spectroscopy of behenic acid methyl ester. J. Phys. Chem. B 1997, 101, 58–65. [Google Scholar] [CrossRef]
- Barth, A.; Zscherp, C. What vibrations tell us about proteins. Q. Rev. Biophys. 2002, 35, 369–430. [Google Scholar] [CrossRef] [PubMed]
- Goormaghtigh, E.; Cabiaux, V.; Ruysschaert, J.-M. Determination of Soluble and Membrane Protein Structure by Fourier Transform Infrared Spectroscopy. III. Secondary Structures. In Sucellular Biochemistry, Volume 23: Physicochemical Methods in the Study of Biomembranes; Hilderson, H.J., Ralston, G.B., Eds.; Plenum Press: New York, NY, USA, 1994; pp. 405–450. [Google Scholar]
- Buffeteau, T.; Le Clavez, E.; Castano, S.; Desbat, B.; Blaudez, D.; Dufourcq, J. Anisotropic Optical Constants of a-Helix and b-Sheet Secondary Structure in the Infrared. J. Phys. Chem. B 2000, 104, 4537–4544. [Google Scholar] [CrossRef]
- Johnson, M.L.; Faunt, L.M. Parameter-Estimation by Least-Squares Methods. Method Enzymol. 1992, 210, 1–37. [Google Scholar]
- Huang, M.; Weissman, J.T.; Beraud-Dufour, S.; Luan, P.; Wang, C.; Chen, W.; Aridor, M.; Wilson, I.A.; Balch, W.E. Crystal structure of Sar1-GDP at 1.7 A resolution and the role of the NH2 terminus in ER export. J. Cell Biol. 2001, 155, 937–948. [Google Scholar] [CrossRef] [PubMed]
- Wimley, W.C.; Creamer, T.P.; White, S.H. Solvation Energies of Amino Acid Side Chains and Backbone in a Family of Host−Guest Pentapeptides. Biochemistry 1996, 35, 5109–5124. [Google Scholar] [CrossRef] [PubMed]
- White, S.H.; Wimley, W.C. Hydrophobic interactions of peptides with membrane interfaces. Biochim. Biophys. Acta 1998, 1376, 339–352. [Google Scholar] [CrossRef]
- Yau, W.M.; Wimley, W.C.; Gawrisch, K.; White, S.H. The preference of tryptophan for membrane interfaces. Biochemistry 1998, 37, 14713–14718. [Google Scholar] [CrossRef] [PubMed]
- Kerth, A.; Erbe, A.; Dathe, M.; Blume, A. Infrared reflection absorption spectroscopy of amphipathic model peptides at the air/water interface. Biophys. J. 2004, 86, 3750–3758. [Google Scholar] [CrossRef] [PubMed]
- Drin, G.; Antonny, B. Amphipathic helices and membrane curvature. FEBS Lett. 2010, 584, 1840–1847. [Google Scholar] [CrossRef] [PubMed]
- Wimley, W.C.; White, S.H. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat. Struct. Biol. 1996, 3, 842–848. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Brauner, J.W.; Mao, G.; Crouch, E.; Seaton, B.; Head, J.; Smith, K.; Flach, C.R.; Mendelsohn, R. Interaction of Recombinant Surfactant Protein D with Lipopolysaccharide: Conformation and Orientation of Bound Protein by IRRAS and Simulations. Biochemistry 2008, 47, 8103–8113. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, W.-P.; Vogel, H. Polarization-Modulated FTIR Spectroscopy of Lipid/Gramicidin Monolayers at the Air/Water Interface. Biophys. J. 1999, 76, 1639–1647. [Google Scholar] [CrossRef]
- Tamm, L.K.; Tatulian, S.A. Infrared spectroscopy of proteins and peptides in lipid bilayers. Q. Rev. Biophys. 1997, 30, 365–429. [Google Scholar] [CrossRef] [PubMed]
- Cameron, D.G.; Casal, H.L.; Mantsch, H.H.; Boulanger, Y.; Smith, I.C.P. The thermotropic behaviour of dipalmitoyl phosphatidylcholine bilayers. Biophys. J. 1981, 34, 1–16. [Google Scholar] [CrossRef]
- Hunt, R.; Mitchell, M.L.; Dluhy, R.A. The interfacial structure of phospholipid monolayer films: An infrared reflectance study. J. Mol. Struct. 1989, 214, 93–109. [Google Scholar] [CrossRef]
- Krimm, S.; Bandekar, J. Vibrational spectroscopy and conformation of peptides, polypeptides, and proteins. Adv. Protein Chem. 1986, 38, 181–363. [Google Scholar] [PubMed]
- Marsh, D.; Muller, M.; Schmitt, F.J. Orientation of the infrared transition moments for an alpha-helix. Biophys. J. 2000, 78, 2499–2510. [Google Scholar] [CrossRef]
- Wang, J.; Lee, S.-H.; Chen, Z. Quantifying the Ordering of Adsorbed Proteins In Situ. J. Phys. Chem. B 2008, 112, 2281–2290. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.T.; Le Clair, S.P.V.; Ye, S.; Chen, Z. Orientation Determination of Protein Helical Secondary Structures Using Linear and Nonlinear Vibrational Spectroscopy. J. Phys. Chem. B 2009, 113, 12169–12180. [Google Scholar] [CrossRef] [PubMed]
- Blume, A. A comparative study of the phase transitions of phospholipid bilayers and monolayers. Biochim. Biophys. Acta Biomembr. 1979, 557, 32–44. [Google Scholar] [CrossRef]
- Seelig, A. Local anesthetics and pressure: A comparison of dibucaine binding to lipid monolayers and bilayers. Biochim. Biophys. Acta 1987, 899, 196–204. [Google Scholar] [CrossRef]
- Hoernke, M.; Mohan, J.; Larsson, E.; Blomberg, J.; Kahra, D.; Westenhoff, S.; Schwieger, C.; Lundmark, R. EHD2 restrains dynamics of caveolae by an ATP-dependent, membrane-bound, open conformation. Proc. Natl. Acad. Sci. USA 2017, 114, E4360–E4369. [Google Scholar] [CrossRef] [PubMed]
- Mcmahon, C.; Studer, S.M.; Clendinen, C.; Dann, G.P.; Jeffrey, P.D.; Hughson, F.M. The Structure of Sec12 Implicates Potassium Ion Coordination in Sar1 Activation. J. Biol. Chem. 2012, 287, 43599–43606. [Google Scholar] [CrossRef] [PubMed]
- Zanetti, G.; Prinz, S.; Daum, S.; Meister, A.; Schekman, R.; Bacia, K.; Briggs, J.A. The structure of the COPII transport-vesicle coat assembled on membranes. Elife 2013, 2, e00951. [Google Scholar] [CrossRef] [PubMed]
- Schwieger, C.; Blume, A. Interaction of poly(l-arginine) with negatively charged bilayers studied by FT-IR spectroscopy. Eur. Biophys. J. Biophys. Lett. 2011, 40, 204. [Google Scholar]
- Schwieger, C.; Blume, A. Interaction of poly(l-lysines) with negatively charged membranes: An FT-IR and DSC study. Eur. Biophys. J. Biophys. Lett. 2007, 36, 437–450. [Google Scholar] [CrossRef] [PubMed]
- Ladokhin, A.S.; White, S.H. Folding of amphipathic alpha-helices on membranes: Energetics of helix formation by melittin. J. Mol. Biol. 1999, 285, 1363–1369. [Google Scholar] [CrossRef] [PubMed]
- Bremer, A.; Wolff, M.; Thalhammer, A.; Hincha, D.K. Folding of intrinsically disordered plant LEA proteins is driven by glycerol-induced crowding and the presence of membranes. FEBS J. 2017, 284, 919–936. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, M.M.; Campelo, F.; Liska, N.; Chernomordik, L.V.; Marrink, S.J.; McMahon, H.T. Mechanisms shaping cell membranes. Curr. Opin. Cell Biol. 2014, 29, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Boucrot, E.; Pick, A.; Camdere, G.; Liska, N.; Evergren, E.; McMahon, H.T.; Kozlov, M.M. Membrane Fission Is Promoted by Insertion of Amphipathic Helices and Is Restricted by Crescent BAR Domains. Cell 2012, 149, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Krüger, D.; Ebenhan, J.; Werner, S.; Bacia, K. Measuring Protein Binding to Lipid Vesicles by Fluorescence Cross-Correlation Spectroscopy. Biophys. J. 2017, 113, 1311–1320. [Google Scholar] [CrossRef] [PubMed]















© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schwieger, C.; Meister, A.; Daum, S.; Blume, A.; Bacia, K. Binding of the GTPase Sar1 to a Lipid Membrane Monolayer: Insertion and Orientation Studied by Infrared Reflection–Absorption Spectroscopy. Polymers 2017, 9, 612. https://doi.org/10.3390/polym9110612
Schwieger C, Meister A, Daum S, Blume A, Bacia K. Binding of the GTPase Sar1 to a Lipid Membrane Monolayer: Insertion and Orientation Studied by Infrared Reflection–Absorption Spectroscopy. Polymers. 2017; 9(11):612. https://doi.org/10.3390/polym9110612
Chicago/Turabian StyleSchwieger, Christian, Annette Meister, Sebastian Daum, Alfred Blume, and Kirsten Bacia. 2017. "Binding of the GTPase Sar1 to a Lipid Membrane Monolayer: Insertion and Orientation Studied by Infrared Reflection–Absorption Spectroscopy" Polymers 9, no. 11: 612. https://doi.org/10.3390/polym9110612
APA StyleSchwieger, C., Meister, A., Daum, S., Blume, A., & Bacia, K. (2017). Binding of the GTPase Sar1 to a Lipid Membrane Monolayer: Insertion and Orientation Studied by Infrared Reflection–Absorption Spectroscopy. Polymers, 9(11), 612. https://doi.org/10.3390/polym9110612