1. Introduction
An important point in investigating the solution behavior of polyelectrolytes is the relation between structure and properties. Very appropriate samples for such studies are those with the same chemical structure but different spatial orientation of functional groups along the chain, such as the three stereoregular forms of poly(methacrylic acid), PMA [
1]: isotactic, syndiotactic, and the usual heterotactic (or atactic) PMA: iPMA, sPMA, and aPMA, respectively (for structures see
Scheme S1 in the Supplementary Materials). The interest in PMA, a weak polyelectrolyte, was in the past related to another property,
i.e., the conformational transition of the chain induced by ionization of carboxyl groups [
2], which places PMA side by side with biopolymers. The conformational transition entails a cooperative change of the shape of the polymer chain from a compact to a more open (extended) form. This feature has been and still is a focus of extensive investigations with both natural and synthetic polymers. The compact conformation is of a particular interest because the native conformations of biological macromolecules (e.g., globular proteins) are compact,
i.e., tightly packed. Such compact coils or globules, as they are often called, offer sites for incorporation of smaller nonpolar (hydrophobic) solutes like drug molecules. When a change in conformation is induced by some environmental factor (most often pH or temperature) the polymer shape changes to a more open one and the nonpolar solute is released into the aqueous medium [
3]. In order to direct and control such processes, the nature of the compact conformation and the mechanism of the chain unfolding have to be characterized.
One of the prerequisites for the occurrence of a cooperative change in conformation is the presence of hydrophobic side groups on the chain and/or the ability to form hydrogen (H) bonds. In the case of PMA, the hydrophobic groups responsible for the conformational transition are the methyl ones. In addition, hydrogen bonds can form between carboxyl, COOH, groups intra- and also intermolecularly, the latter leading to association/aggregation between chains. The ability to form intermolecular associates between aPMA chains was mostly excluded in the past. However, it has been demonstrated by light scattering measurements [
4,
5,
6] that this option should not be completely ignored, in particular when aPMA chains are essentially uncharged,
i.e., at low pH. With iPMA, intermolecular association is clearly very important and reflected in several properties [
2,
5,
6,
7,
8,
9,
10,
11,
12,
13]: (i) iPMA is insoluble in water below a certain critical degree of neutralization, α
N, of carboxyl groups (α
N < 0.2); (ii) it is a weaker acid in comparison with aPMA over the whole region of α
N values; (iii) the conformational transition of the isotactic chain is irreversible, denoting that charging (neutralization) and discharging (protonation) of the chain proceed along different paths (intermediate states); and, last but not least, (iv) iPMA chains are strongly associated/aggregated even at nonzero α
N values,
i.e., little above the solubility limit at α
N ≈ 0.2 [
5,
6]. The most obvious reason on the molecular level for this clearly different affinity of PMAs for water is the stereoregularity of iPMA or,
vice versa, the random structure of aPMA.
Our purpose in this contribution is to explore the molecular reasons for these well manifested differences between iPMA and aPMA by using the classical thermodynamic approach. We perform potentiometric and calorimetric measurements to determine the standard free energy, Δ
GtrӨ, and enthalpy, Δ
Htr, changes and therefrom the entropy, Δ
Str, change associated with the transition of the chain from the uncharged to the (completely) charged state. These measurements are performed in the presence of different alkali chlorides, XCl (X = Li, Na, Cs). In addition, measurements are carried out at different temperatures (15, 25, 45 °C) in order to evaluate also the heat capacity change, (Δ
cp)
tr, of the transition. Thermal properties, in particular the sign of (Δ
cp)
tr, are namely the fingerprint of the hydrophobic effect [
14,
15,
16,
17], which drives many complex processes in chemistry and biology.
4. Discussion
In addition to Δ
G and Δ
H, a complete thermodynamic analysis of a certain process also comprises the entropy, Δ
S, and the heat capacity, Δ
cp, values. Δ
S and Δ
cp were estimated from the measured Δ
GtrӨ and Δ
Htr values. To estimate the entropy change accompanying the transition, Δ
Str, at a specified temperature the standard thermodynamic equation
was used. One has to keep in mind that Δ
GtrӨ values determined from
Kapp are the standard ones (applying to concentrations/activities of 1 mol/L) whereas the Δ
Htr was measured at
cp = 0.01 mol/L. However, Crescenzi
et al. [
23] have shown that Δ
Htr for aPMA is practically independent of the concentration. A value measured at any other concentration is approximately equal to the standard value. This is in agreement with our results as well (see the comparison of Δ
Htr values above) and justifies the calculation of Δ
Str from Δ
GtrӨ and Δ
Htr.
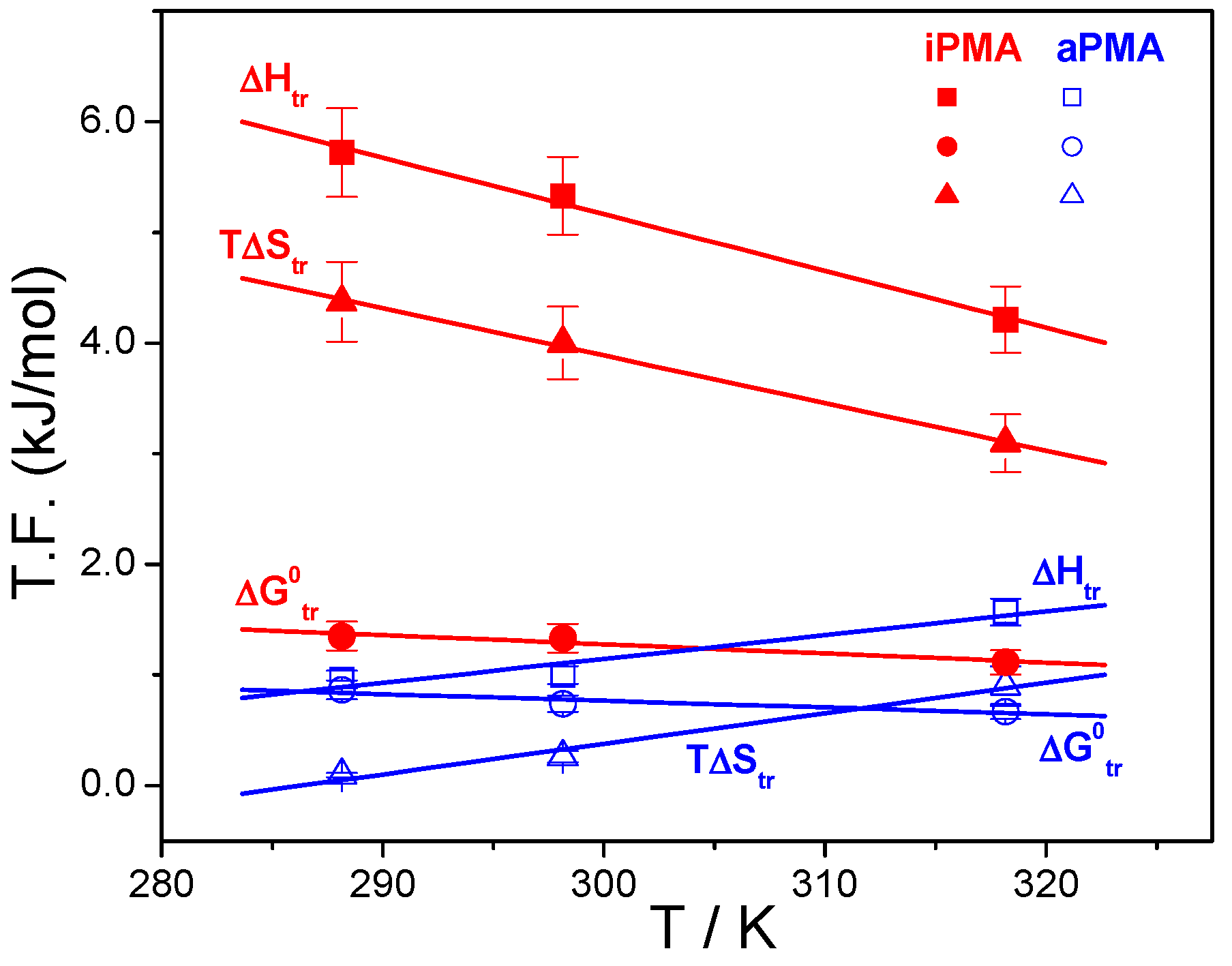
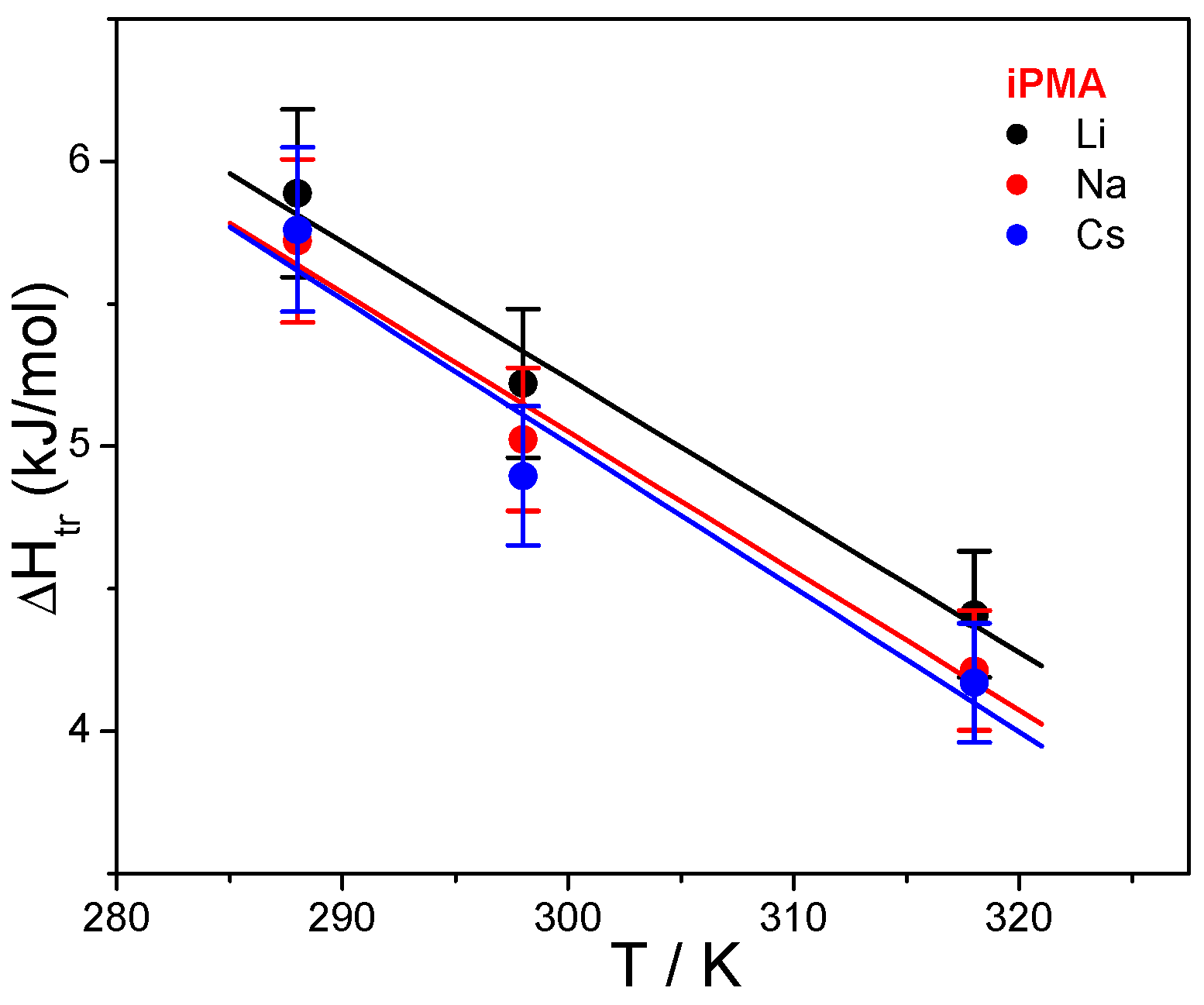
Results of this calculation are reported in
Table 2 and plotted as a function of temperature in
Figure 7, together with Δ
GtrӨ and Δ
Htr. All Δ
Str, so as Δ
GtrӨ and Δ
Htr, values are positive and depend on temperature and also on the added XCl. Our data for aPMA (Δ
Str = 0.85 and 2.84 J/(Kmol) at 25 and 45 °C, respectively, in 0.01 M NaCl) can again be compared with the literature values [
23]: at 25 °C (45 °C) the reported Δ
Str values are 0.84 (2.23) and 0.71 (2.72) J/(Kmol) for aPMA and sPMA, respectively. It is well established that aPMA and sPMA display similar solution behavior. We therefore conclude that the agreement is good. The Δ
Str values for iPMA are reported in this study for the first time, preventing any comparison with the published data.
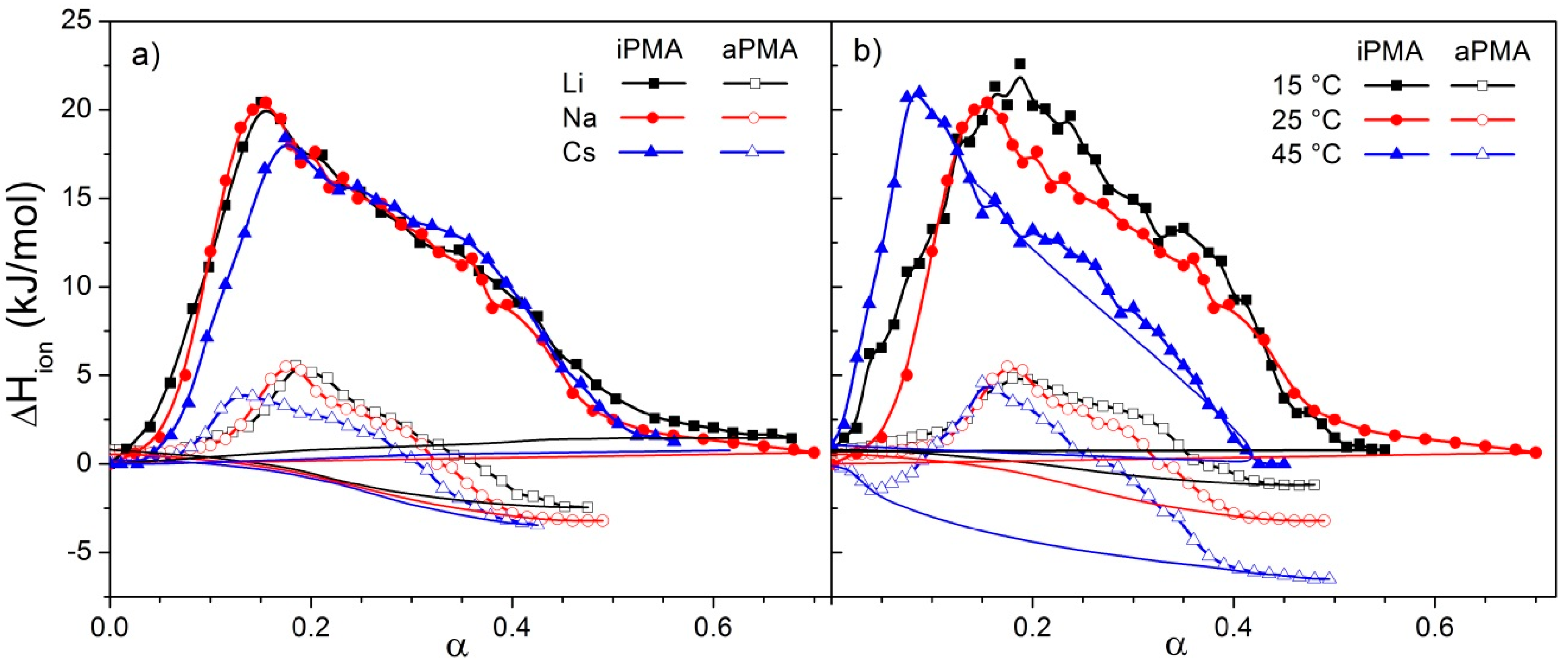
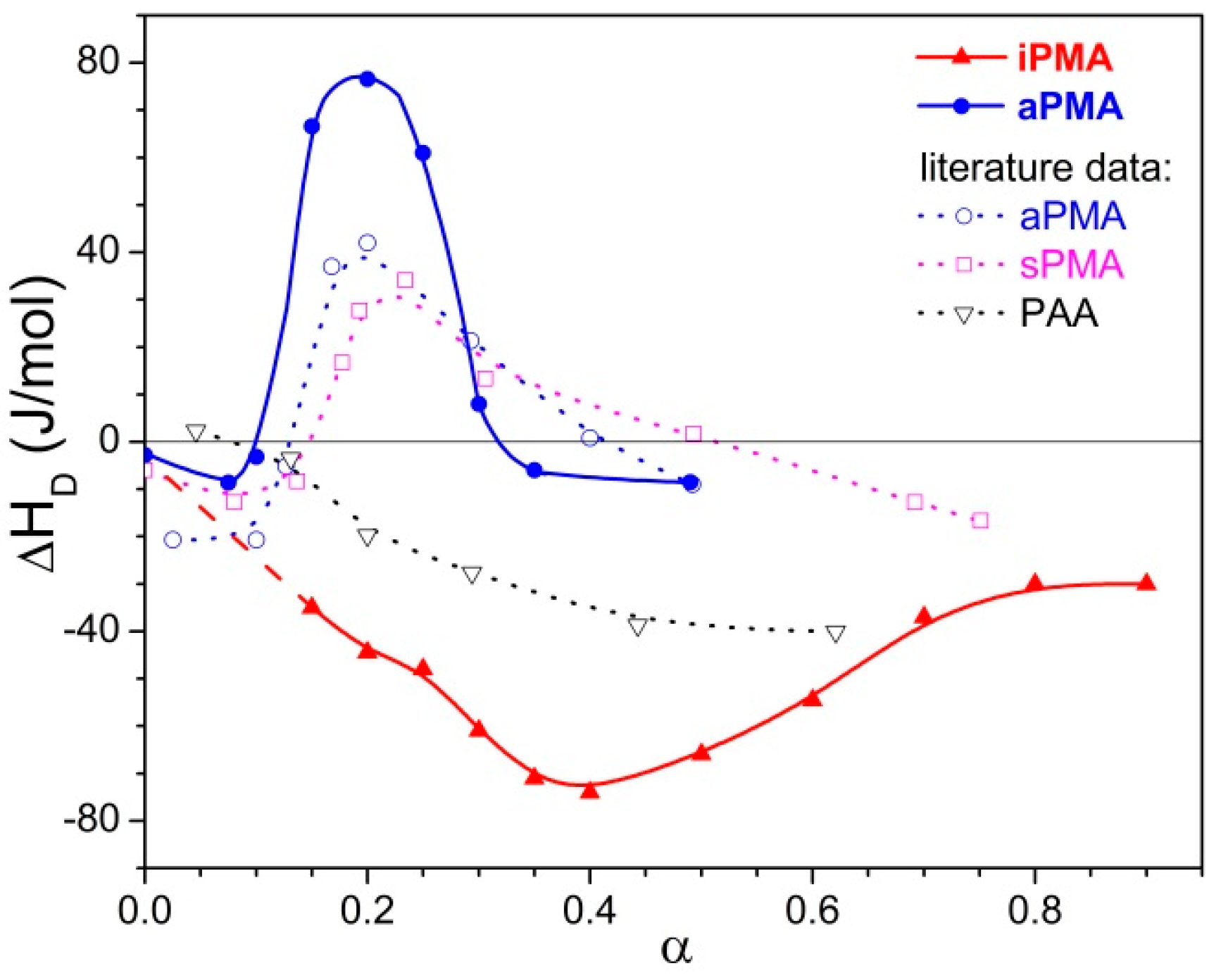
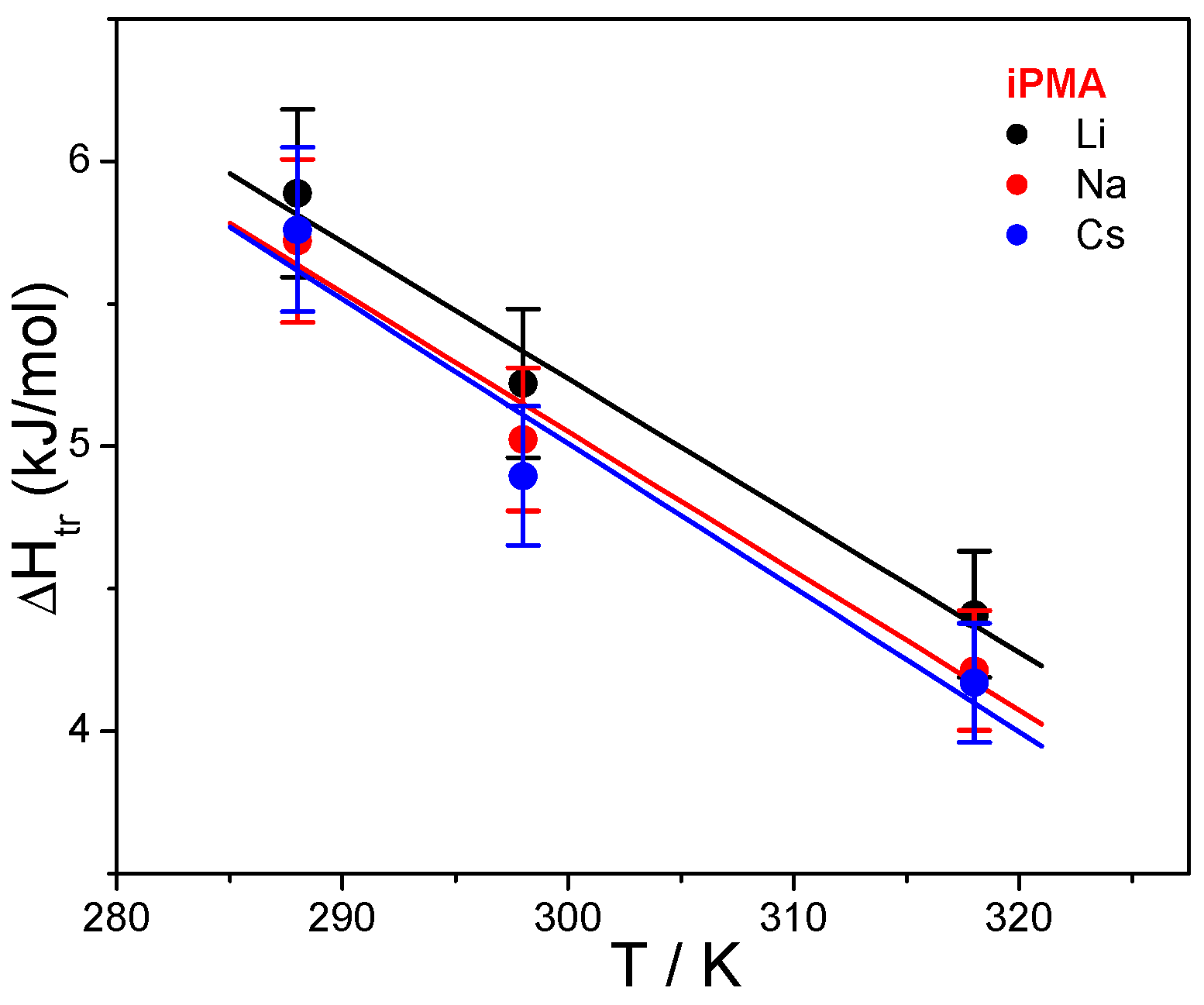
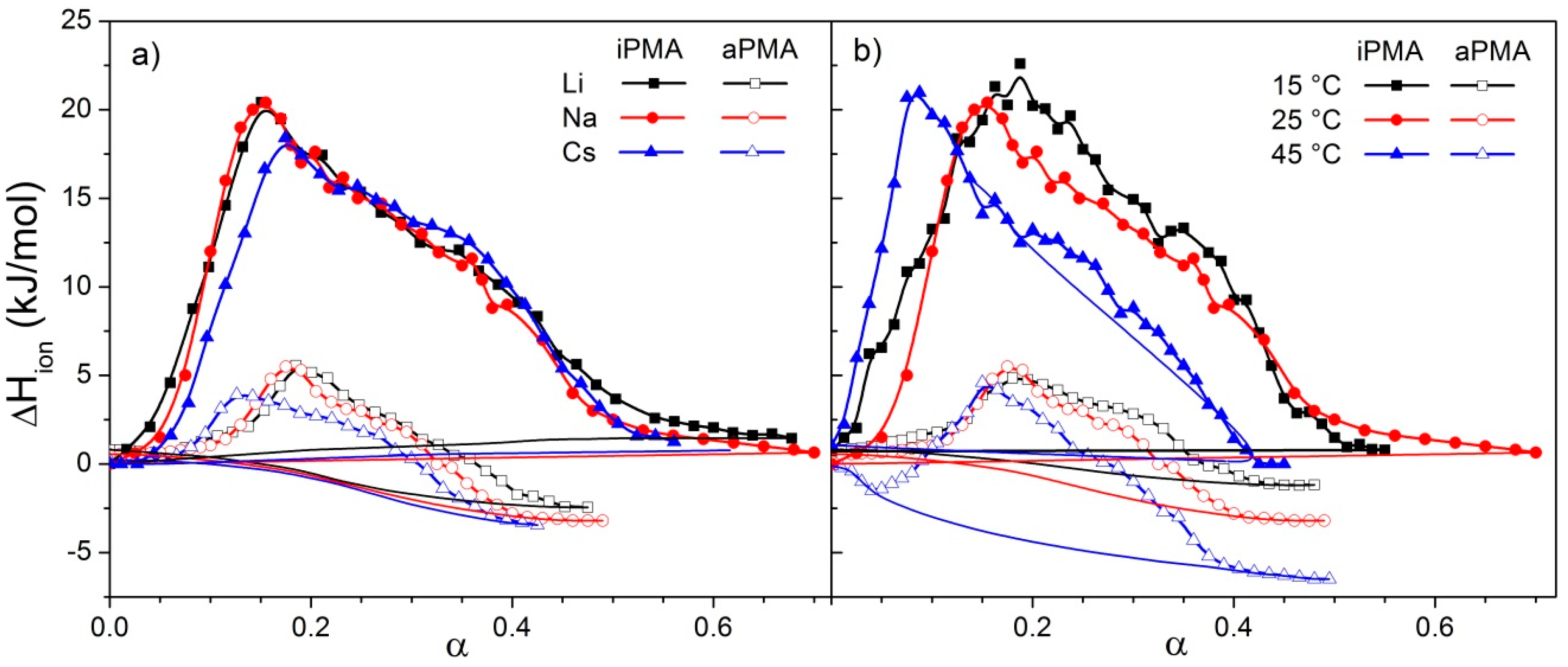
Let us focus first on the effect of ions. For both PMAs, Δ
Htr and Δ
Str are the lowest in CsCl solutions, whereas the effect of LiCl and NaCl may be regarded as comparable. This effect of ions can be accounted for by the so-called law of matching water affinities, LMWA [
25,
26,
27,
28], which is related to the hydration radii and enthalpies of the ions. LMWA was originally proposed to explain the well-known Hofmeister series, which is related to the specific effect that ions have on the precipitation of proteins from solutions. However, it was recently demonstrated that these effects are important not only in so-called ion-pairing phenomena but also in the interaction of ions with uncharged/neutral surfaces [
29,
30], for example with the acidic groups on macromolecules [
30] such as the protonated carboxyl group on PMA.
The Li
+ ion, with the smallest atomic radius, is more strongly hydrated (coordinated with more water molecules) than the Cs
+ ion, which has a larger atomic radius. The PMA’s carboxyl, COO
−, group is also strongly hydrated. Its hydration enthalpy is actually the closest to that of Na
+ ions. LMWA states that the closer the hydration enthalpy of ions the stronger their mutual interaction (
i.e., the formation of an ion pair between X
+ and COO
−), through which ions lose their hydration shell and form so-called contact ion pairs (CIP). An opposite case is the solvent shared ion pair (SIP) characteristic of the combination of two large ions that do not dehydrate (an example is the pair Cs
+/F
−). Thus, Li
+ and Na
+ ions bind more strongly to the PMA’s carboxyl groups than the Cs
+ ones, and form CIPs. The attraction of larger Cs
+ ions with the carboxyl group is less pronounced since it involves a shared solvation shell (SIP). How does this affect the Δ
Htr values? Stronger binding facilitates formation of a more compact coil, which requires more energy input to stretch the chain upon ionization and leads to higher Δ
Htr values in LiCl (iPMA) or NaCl (aPMA) in comparison with CsCl solutions. The same conclusion on the effect of ions was obtained previously from light scattering studies of intermolecular association of both PMA isomers in aqueous XCl solutions at low α [
6]. In that case, a reversed order of Na
+ and Li
+ in relation to the size of microgel-like aggregates between iPMA or aPMA chains was observed, in agreement with the present findings. Note that the aggregation refers to the low α state of PMAs. Although PMA chains at low α are only weakly charged, the LMWA seems to apply fairly well, in agreement with recent computer simulations involving a neutral protein [
29] or an acidic surface with carboxyl groups [
30]. The neutral protein followed the same series with respect to ion specificity as did the charged proteins [
29]. It was therefore argued by the authors [
29] that the cation specificity is determined mainly by the properties of the cation’s hydration water, in particular their ability to donate hydrogen bonds to carboxylate groups. Pronounced ion-specific effects were observed also in the case of acidic carboxyl groups [
30], irrespective of whether they were protonated (uncharged) or not (
i.e., charged). Upon increasing pH, a pH-dependent reversal of the Hofmeister series was observed in that case, which was attributed to the change in affinities of cations for the protonated and deprotonated carboxyl groups [
30]. The authors suggested that these effects arose from direct ion-surface (charged or uncharged) and indirect hydration-related interactions. To conclude, the phenomena taking place during titration of PMA in relation to ion-specificity are highly complex and may present a challenge for a separate study.
The effect of ion size/hydration on ΔStr is more difficult to explain due to several, sometimes contradicting, contributions. However, the loss of hydration water upon binding (see above) definitely makes an important contribution to the positive ΔStr value. When the PMA chain is gradually charged along the titration path, X+ starts to strongly interact with COO−, i.e., they bind electrostatically to the negatively charged chain. Because Li+ and Na+, together with COO−, lose more water molecules through this interaction than does Cs+, the entropy increase due to dehydration is largest in the presence of LiCl and NaCl. It is frequently the case that the contribution of hydration/dehydration prevails over other effects. Another important effect is the electrostatic binding of X+ to the polyion, which leads to a decrease in the number of particles in solution and makes a negative contribution to the overall ΔS change.
Let us recall at the end that the total ΔGtrӨ values show an opposite trend when comparing iPMA and aPMA: they increase for iPMA but decrease for aPMA in the direction Li → Na → Cs and are higher for iPMA. Because precipitation presents a considerable contribution to ΔGtrӨ for iPMA, further discussion on this point would be too speculative. However, it is clear that PMA solutions display a typical case where the high, and positive, ΔHtr and ΔStr values compensate for each other and result in a rather weak dependence of ΔGtrӨ on temperature. ΔGtrӨ values alone can therefore not be used to appraise the pronounced differences in the solution behavior of iPMA and aPMA, which was sometimes attempted in the past.
The dependence of Δ
Htr and Δ
Str on temperature points to differences in the interaction of iPMA and aPMA with water and reflects a dissimilar state of isotactic and atactic chains in aqueous solutions, in particular at low α. The exact opposite temperature dependence of Δ
Htr and Δ
Str suggests that they have different affinity for water. It is generally accepted that the temperature dependence of Δ
Htr and Δ
Str is a fingerprint of the hydrophobic effect [
14,
15,
28]. The latter is most clearly reflected in the sign of the heat capacity change, Δ
cp, accompanying a certain process. In the present case, this is the transition of a weak polyelectrolyte chain from an uncharged to a charged state, leading to a designation (Δ
cp)
tr for the heat capacity change of the transition.
The (Δ
cp)
tr is determined by the temperature dependence of Δ
Htr (or Δ
Str):
Here, (Δ
cp)
tr values were calculated from the slope of the Δ
Htr =
f(
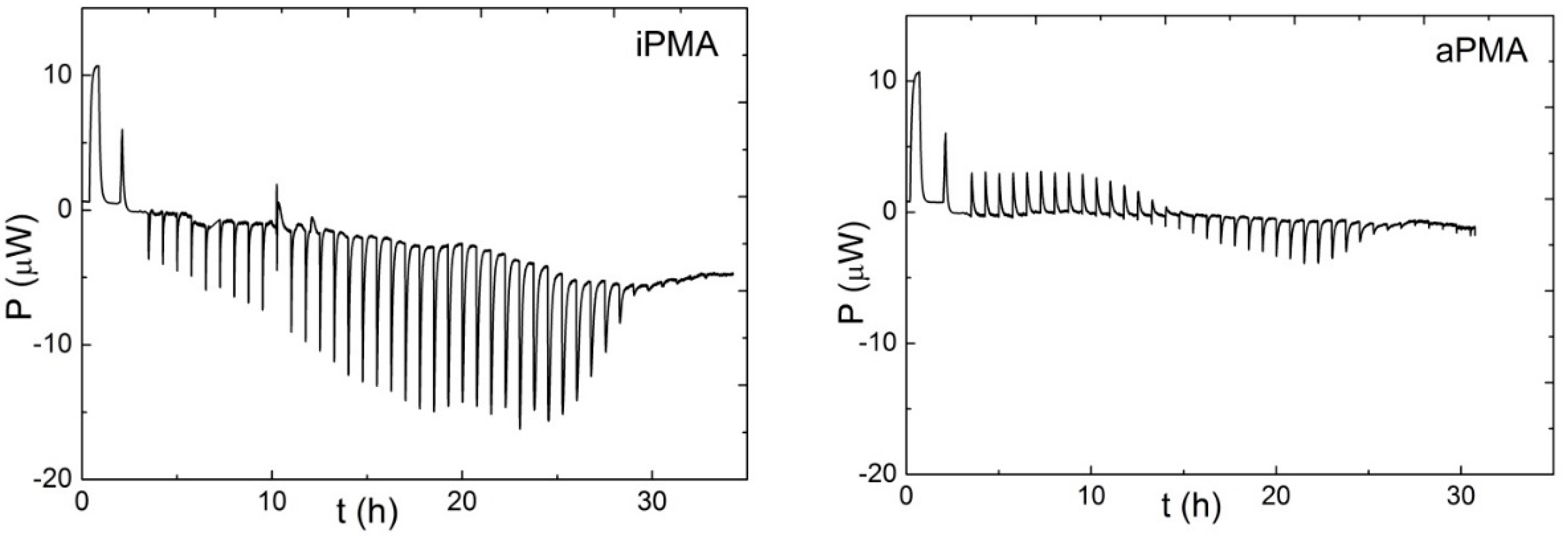
T) plots. These plots can be appreciated for both PMAs in the presence of 0.01 M NaCl in
Figure 7. Temperature dependencies of Δ
Htr for iPMA in the presence of all 0.01 M XCls are shown in
Figure 8. For aPMA, (Δ
cp)
tr is positive (around +20 J/Kmol), whereas it is negative for iPMA and approximately equal for all three XCls (around −50 J/Kmol). It may be noted that the (Δ
cp)
tr values obtained from the temperature dependence of Δ
Str are close to these,
i.e., +28 and −43 J/Kmol for aPMA and iPMA, respectively.
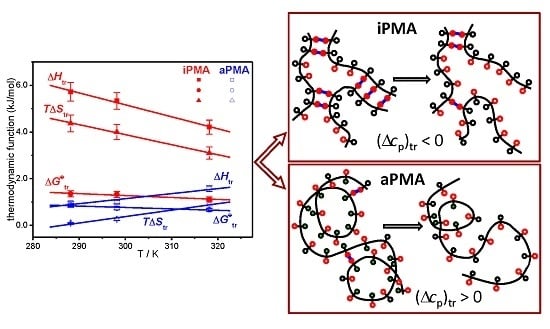
Let us recall at this point that the dilution enthalpies also show an opposite sign: they are negative for iPMA and positive for aPMA. The reason for the opposite sign of (Δcp)tr and ΔHD of PMA isomers can be explained by proposing a different “structure” for the compact coil of iPMA and aPMA in water at low α, which entails different hydration, i.e., interaction with water. In the following, the features of the compact state of iPMA and aPMA in aqueous solutions are discussed in detail.
Water is a highly structured solvent due to hydrogen (H)–bonds between water molecules. When a solute is dissolved in water, the H-bond network of pure water is perturbed. If the solute is polar, new H–bonds between water and the solute are formed, which is energetically favorable (the hydration enthalpies are usually strongly negative—exothermic). However, if the solute is non-polar (hydrophobic) it does not form H-bonds with water, but its presence gives rise to enhanced H–bond formation between water molecules themselves. This is known as the hydrophobic effect (also hydrophobic hydration). The structuring of water around a simple hydrophobic solute is sometimes described as the iceberg model. As a consequence, the transferring of simple hydrophobic solutes into water usually results in positive Δ
H and Δ
G values (consumption of energy due to the breaking of H-bonds). However, it has been shown [
14,
15,
16,
17,
28] that the sign of Δ
cp, which is determined by the temperature dependence of Δ
H (or Δ
S), is actually the most important factor in appraising the hydrophobic effect and hydrophobic hydration. The hydration of a non-polar solute (or its transfer into water) produces a positive and the hydration of a polar solute a negative Δ
cp value. This rule applies, for example, to protein folding/unfolding [
16]. The thermal features of protein unfolding resemble those of transferring a small nonpolar solute into water.
The positive (Δ
cp)
tr accompanying the process of charging the aPMA chain is thus parallel to the transfer of a hydrocarbon (non-polar solute) into water [
14,
31] or to protein unfolding [
16,
31]. The Δ
cp values for protein unfolding are somewhat higher, in the range of 0.1–0.2 cal/K per gram of polymer [
17] (in the same units, the (Δ
cp)
tr for aPMA determined in this work is 0.055–0.078 cal/K per gram of the acid form of aPMA). It can be easily visualized for aPMA that due to charging and unfolding of the chain, the methyl groups that are buried inside the compact aPMA core are exposed to water, similar to the case of protein unfolding. In the latter case, it is the nonpolar amino acids that are exposed. On the other hand, the negative (Δ
cp)
tr in the iPMA case agrees with the hydration of a typical polar solute. For example, the dissociation of acetic acid is accompanied by a strongly negative (Δ
cp)
tr (=−142 J/Kmol [
14]). Negative (Δ
cp)
tr values stem from strong hydration of ions, which results in the loss of entropy due to the more ordered water structure in the hydration shell. It was recently demonstrated by molecular dynamics simulations [
31] that the overall negative heat capacity changes (the Δ
cp values) of common salts are caused by the anion (F
− in that case [
31]), while the cation (Na
+ in that case [
31]) actually contributes positively. The opposite sign of Δ
cp of individual ions was attributed to strong asymmetry in the orientation of water molecules around positively and negatively charged groups. Experimentally, of course, it is the sum of heat capacities that is determined; in the present approach we have no means to determine that. To return to iPMA, the negative (Δ
cp)
tr value in this case is in apparent contradiction with the well-defined hydrophobic nature of iPMA at low α, which is reflected in its insolubility in water and in high aggregation tendency [
5,
6]. How can this contradiction be resolved? For this purpose, we propose the following model of unfolding the iPMA and aPMA chains in water, which is schematically presented in
Figure 9. The unfolding of the chains is brought about either by ionization of carboxyl groups or by dilution of the polymer at a constant α value. The ionization undoubtedly causes a more pronounced change than dilution.
Let us start with aPMA. A schematic representation of the unfolding of the aPMA chain is shown in
Figure 9a. In the compact form of aPMA, most of the hydrophobic methyl groups (open black circles) are buried inside the core, whereas the carboxyl groups (open red circles) are oriented towards the aqueous environment and thus effectively protect the methyl groups from unfavorable contact with water. Responsible for the stability of the compact form of aPMA are the short-range interactions,
i.e., van der Waals forces, the hydrophobic effect associated with the methyl groups and partly also H-bonds (all this is schematically captured in
Figure 9a). Such picture is in agreement with the solubility of aPMA in water at α = 0 and also with low polarity of the compact core, as demonstrated through pyrene polarity ratio measurements [
13]. When the aPMA chain unfolds (due to long-range repulsive electrostatic interactions), the nonpolar methyl groups from the core are exposed to the aqueous environment. This process is parallel to the transfer of a hydrophobic solute into water and therefore accompanied by a positive (Δ
cp)
tr. Note that only one aPMA chain is shown in the scheme in
Figure 9a in order to keep presentation simple. However, it has to be kept in mind that aPMA chains are intermolecular, associated at α = 0 [
4].
In the unfolding of iPMA chains, a qualitatively opposite process occurs that parallels the transfer of a polar solute into water. iPMA chains are much more strongly aggregated in aqueous solutions in comparison with the aPMA ones. The mode of aggregation is different as well. Light scattering measurements [
4] suggest the formation of microgel-like aggregates of several iPMA chains just above the α value (α ≈ 0.25) defining the limit of its solubility in water, and that in spite of a non-negligible charge of the chains (recall again that intermolecular association of aPMA chains occurs only at or close to α = 0 [
4]). Liable for this is the ordered isotactic arrangement of the functional groups, which is the basis for a very strong and cooperative hydrogen bonding between chains [
5,
6,
11]. The methyl groups play an important role in this cooperativity by reinforcing the H-bonds between carboxyl groups. It has been shown by density functional theory calculations [
32] that the strength of H-bonds is significantly increased if an electron donating group (like methyl) is bound next to the carboxyl group. In fact, such reinforcement must be expected for both aPMA and iPMA, but not for PAA. It was demonstrated by statistical mechanics [
33] that the PAA chain merely extended in solution upon ionization, whereas it swelled substantially in an abrupt way,
i.e., in a narrow pH range, in the PMA case. It can be easily visualized that the particular orientation of carboxyl groups on the isotactic chain (always on the same side of the chain) is favorable for the cooperativeness of the H-bonding. In agreement with this, a recent study on thin iPMA films [
11] showed that a high degree of order is induced when amorphous iPMA films prepared from solutions in organic solvents are immersed in water. In line with our conclusions, the strong intermolecular association was therein ascribed to cooperative hydrogen bonding between iPMA chains that could only be disturbed by solvents capable of forming strong hydrogen bonds with the solute, like DMF and DMSO.
Recently, a two-step model of the association process between iPMA chains was proposed [
6,
11]. A sketch showing a detail of such an associate/aggregate of several iPMA chains is shown in
Figure 9b, where the H-bonded COOH groups are shown by full red circles and the parallel blue lines indicate the H-bonds between them. In such an arrangement, a large proportion of the methyl groups (open black circle) is forced to face the solvent (water). Again, such a “model” is strongly supported by pyrene fluorescence measurements [
13], which pointed out that the micropolarity of the iPMA compact conformation at low α is actually higher than that of aPMA. This is due to a different “composition” of the interior of the compact form at α = 0 in comparison with aPMA; the compact conformation of iPMA is put together mainly through hydrogen bonding and thus the methyl groups are actually forced to orient toward water, whereas the core contains more COOH groups. The breaking of H-bonds between COOH groups, either due to ionization or dilution, leads to exposure of these polar groups to water. This is equivalent to the transfer of a polar solute into water and thus accompanied by a negative (Δ
cp)
tr value, as suggested above.
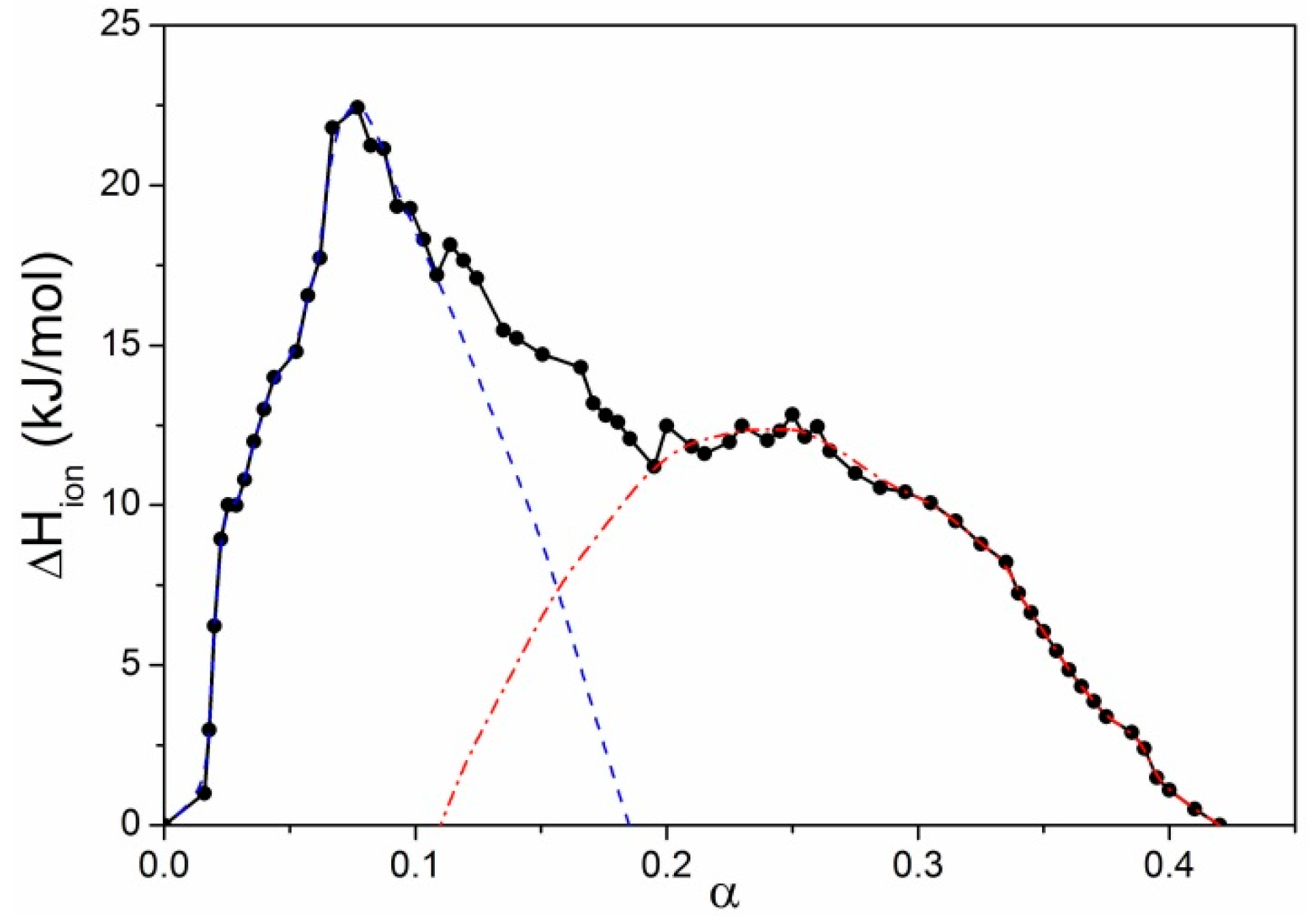
To summarize: the ionization process of both iPMA and aPMA is accompanied by positive ΔHion (and ΔHtr) values because energy is needed to pull a hydrogen ion from the chain and to unfold the compact coil. Upon unfolding of the aPMA coil, the number of unfavorable contacts of the methyl groups with water increases. These contacts become increasingly less favorable with increasing temperature, which leads to an increase of ΔHtr with temperature and to a positive (Δcp)tr. The situation is different with iPMA. In order to ionize the isotactic chain, first the H-bonds involved in intermolecular associates/aggregates have to be broken and then the hydrogen ions can be pulled from the chain. This is the reason, on the molecular level, for higher ΔHion (and ΔHtr) values in comparison with those for aPMA. Furthermore, due to ionization of carboxyl groups that are involved in hydrogen bonding between iPMA chains favorable contacts between the strongly hydrated COO− groups and water are established. ΔHtr for iPMA decreases with increasing temperature because the extent of hydration is lower at higher temperatures.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}