
Polysarcosine-Based Lipids: From Lipopolypeptoid Micelles to Stealth-Like Lipids in Langmuir Blodgett Monolayers



Abstract
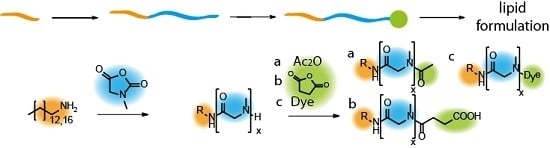
:
1. Introduction
2. Materials and Methods
3. Results
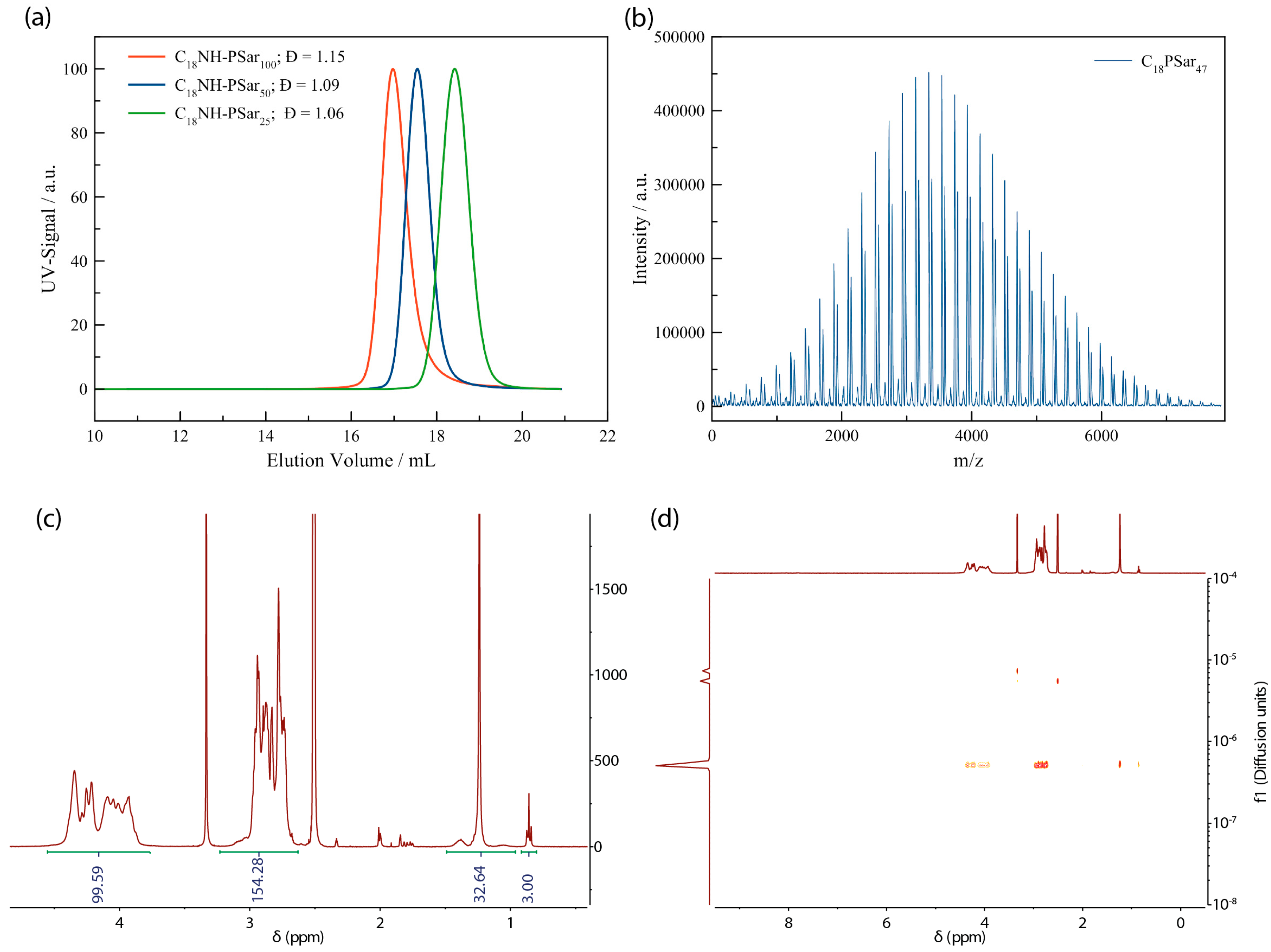
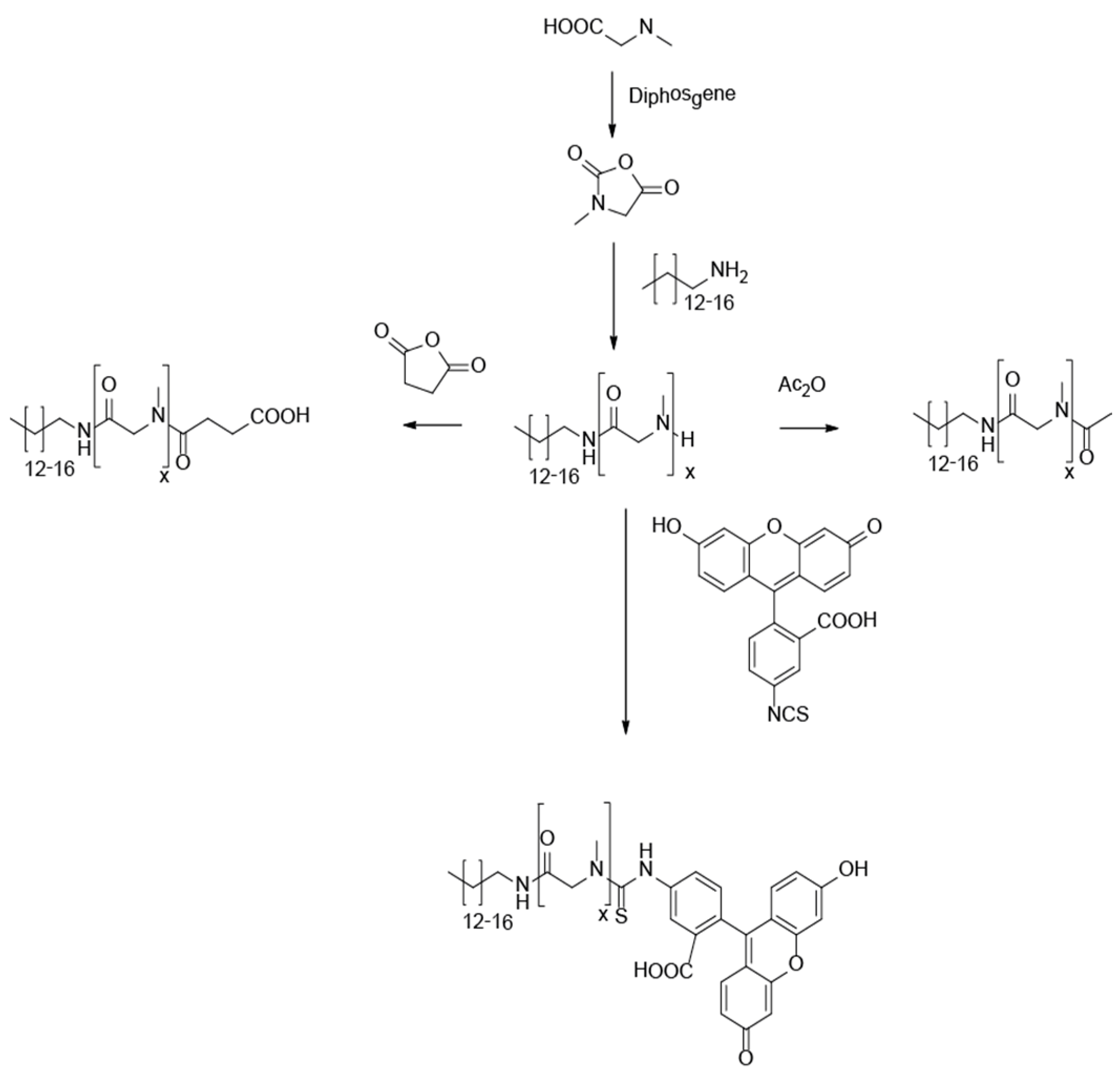
3.1. Synthesis of Lipopolypeptoids
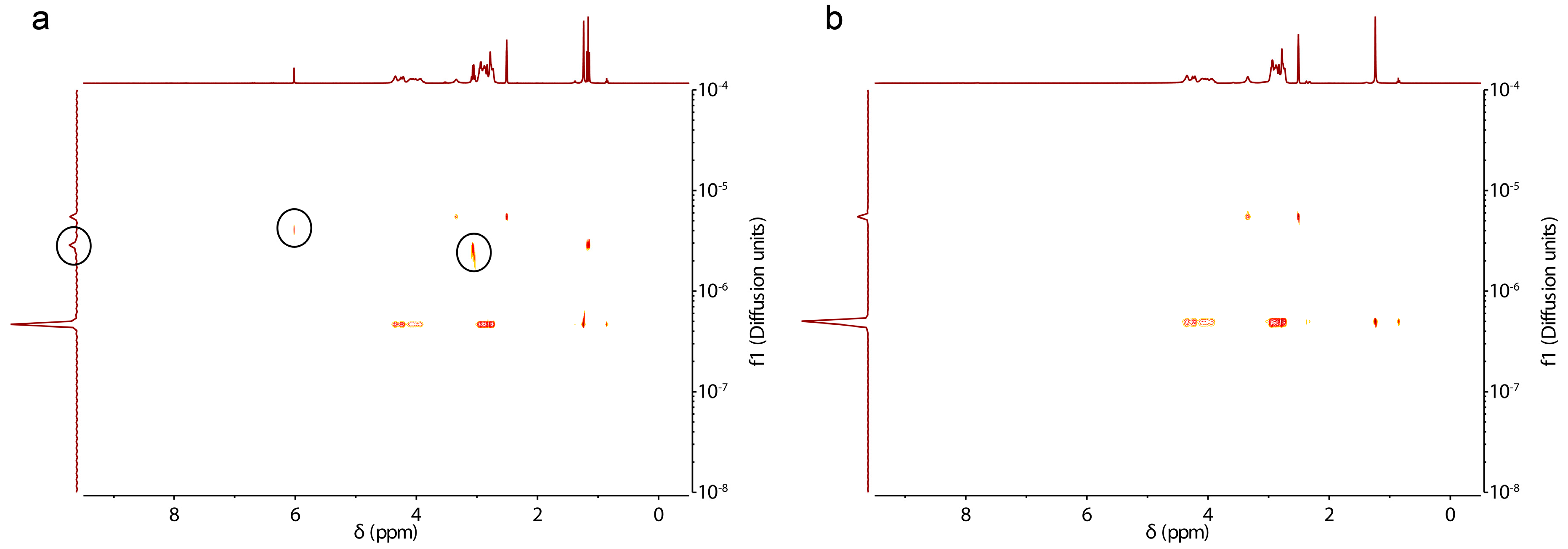
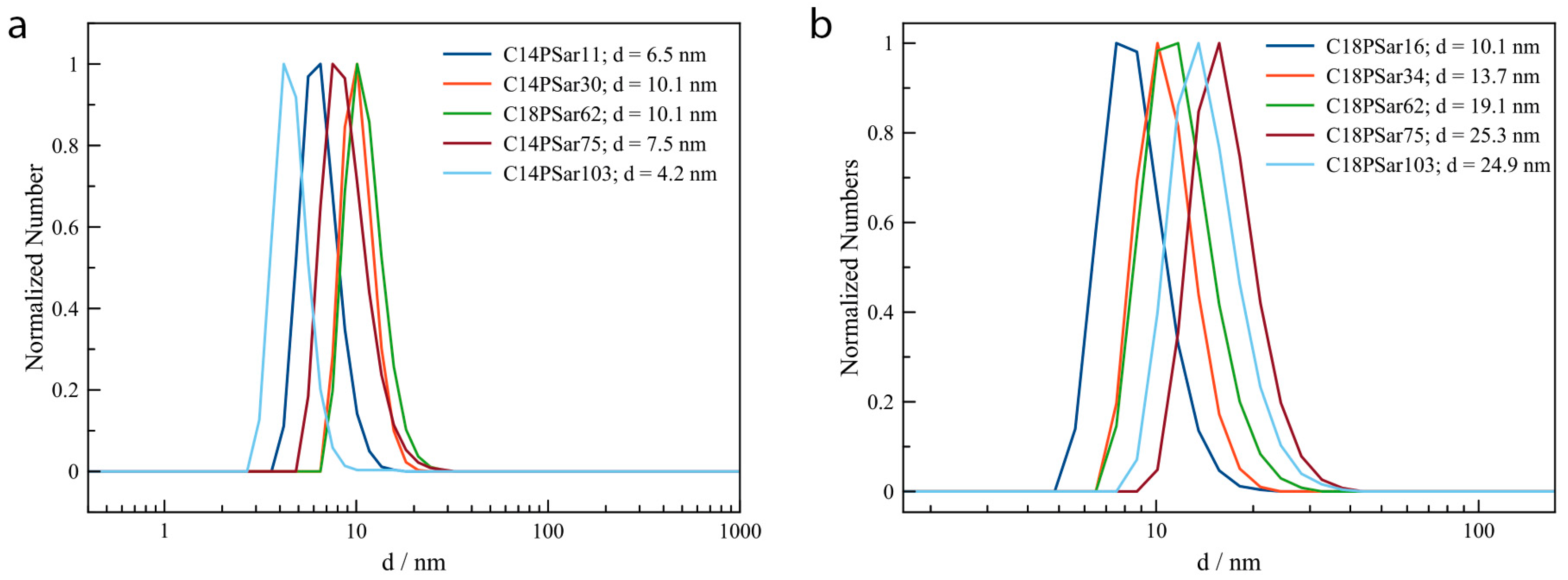
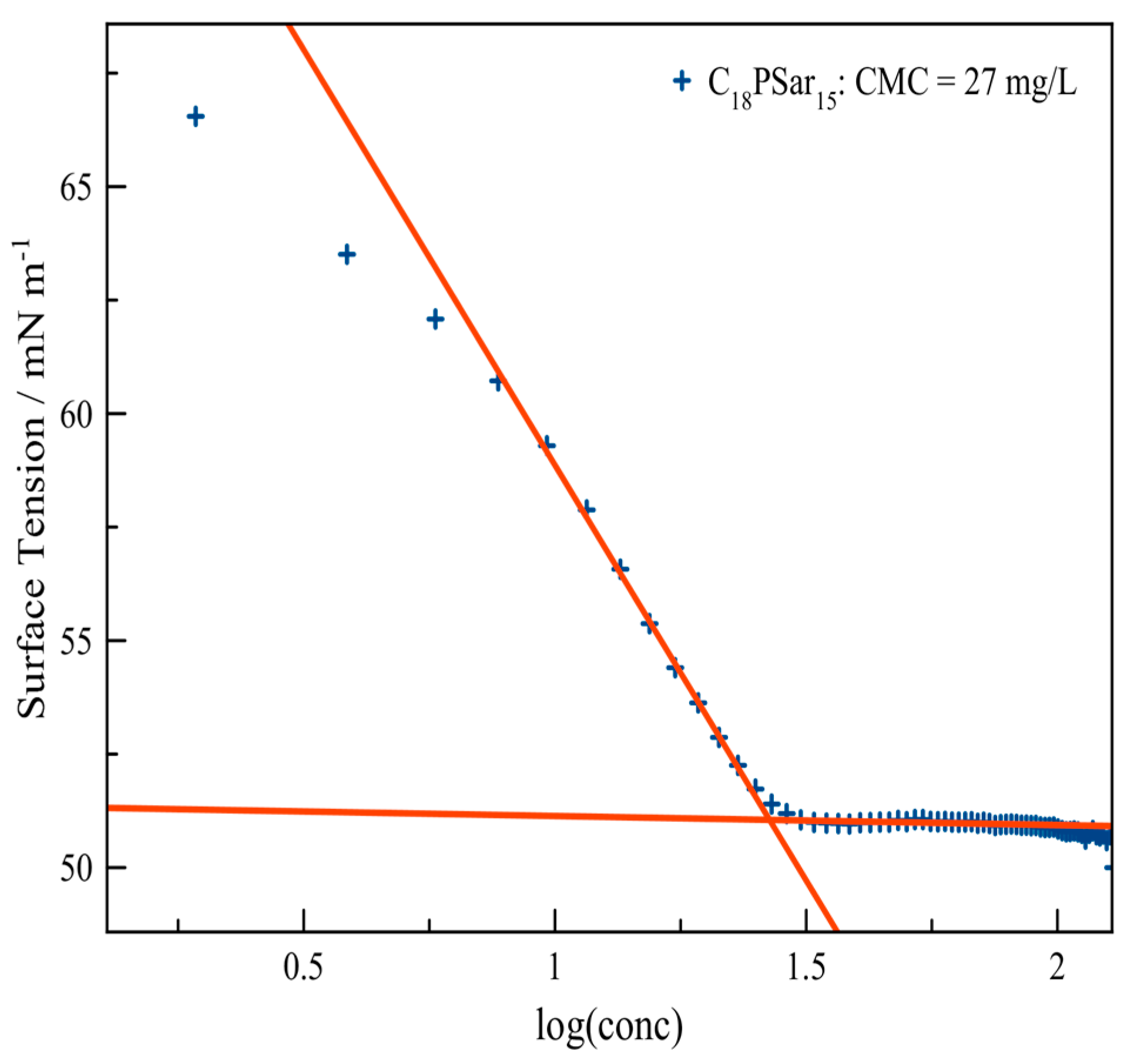
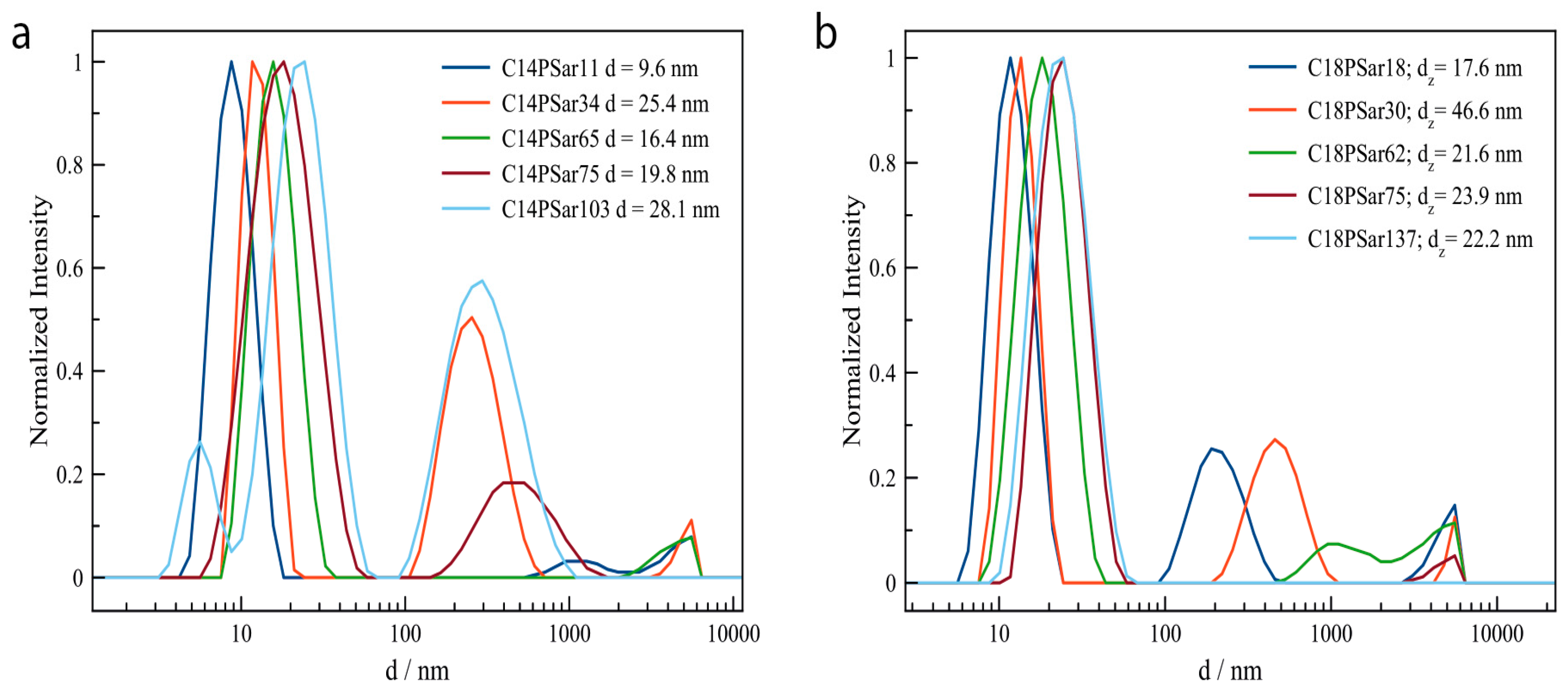
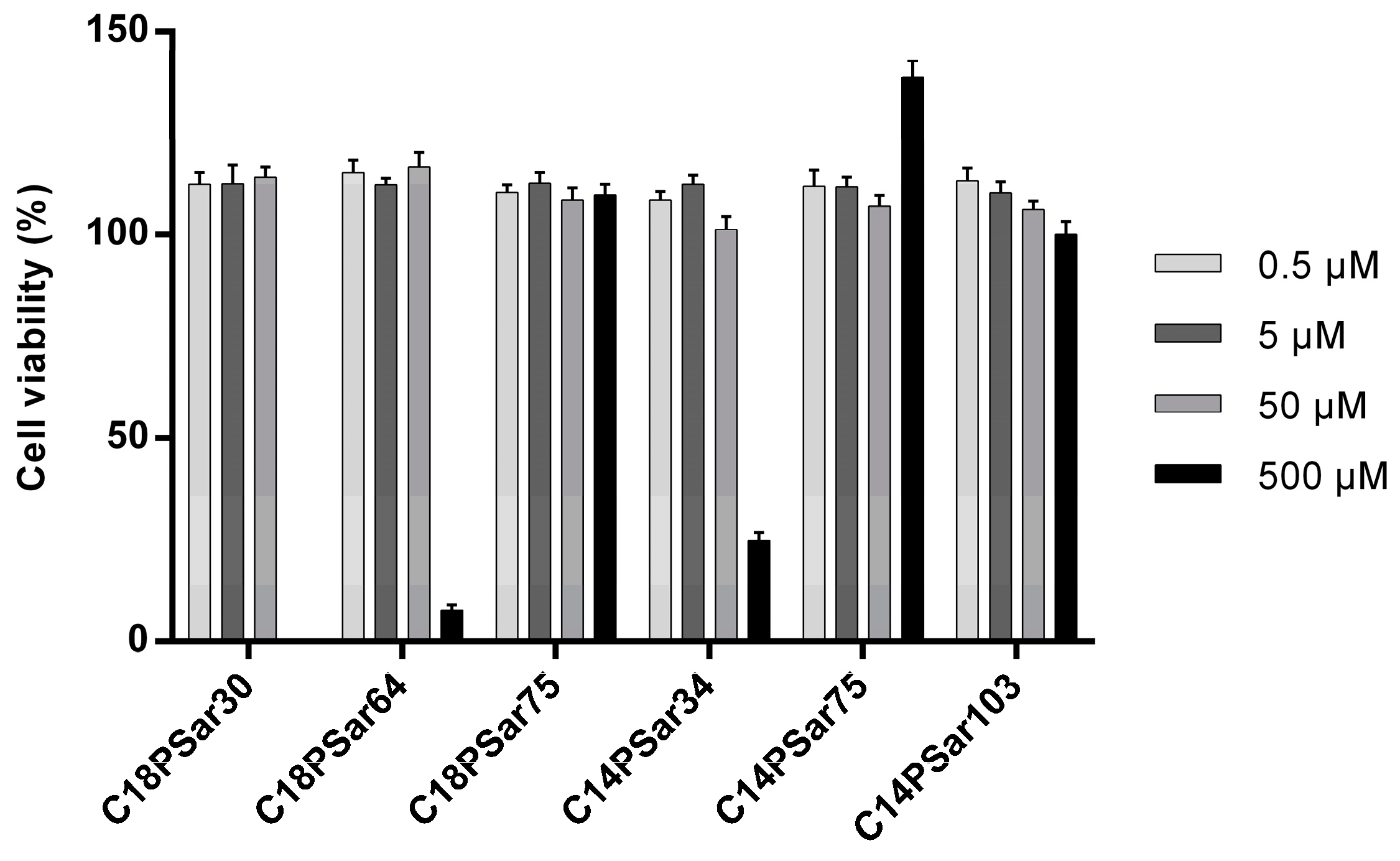
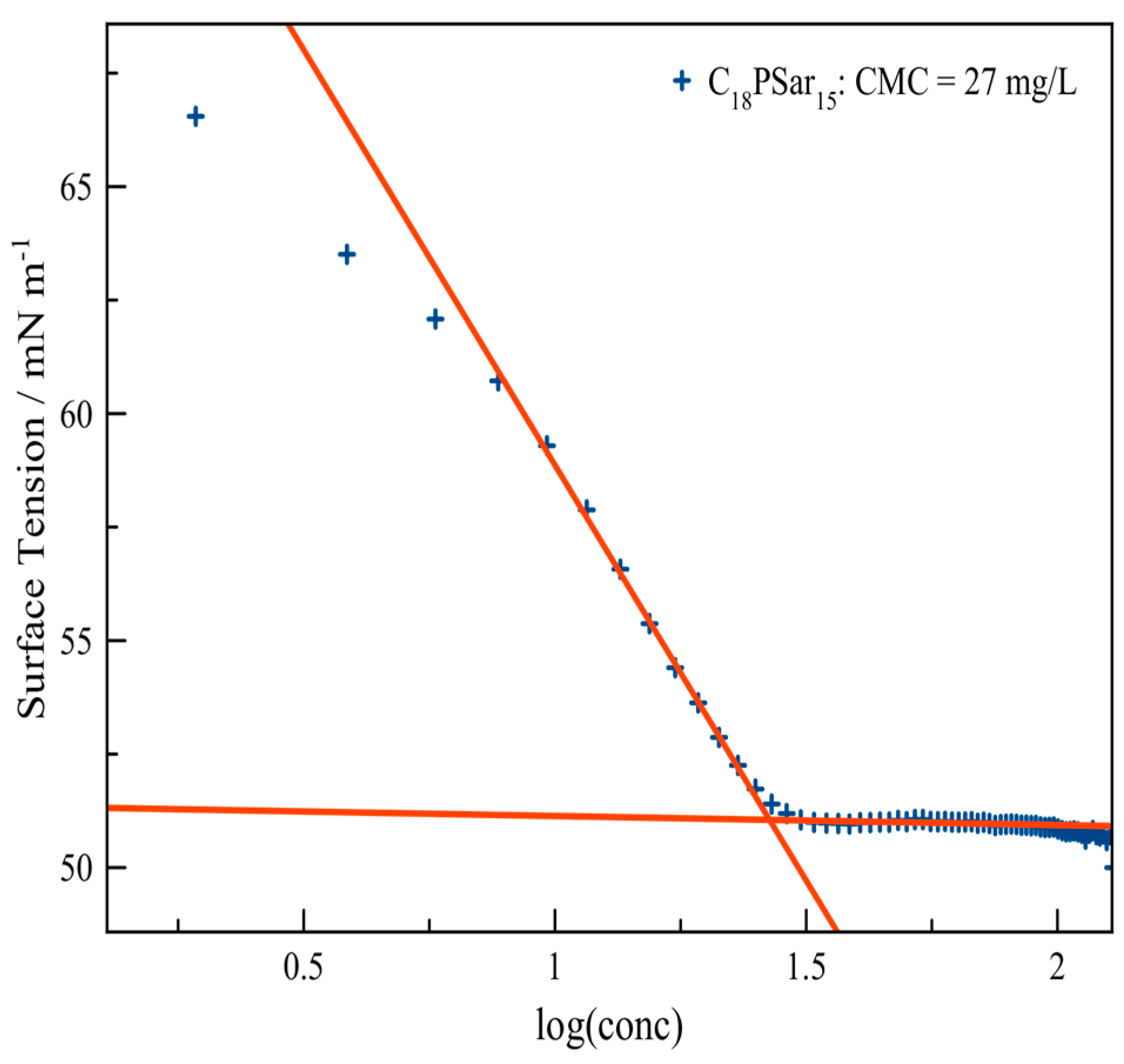
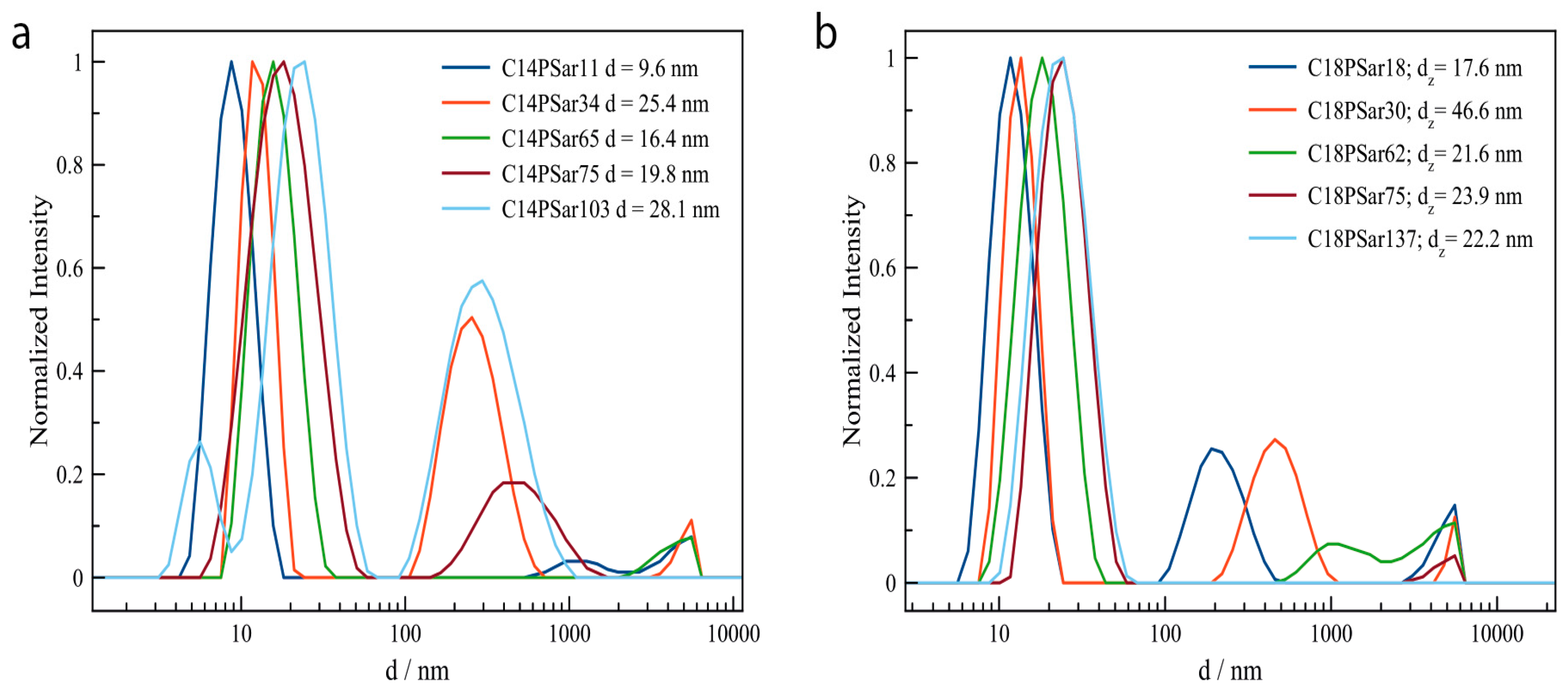
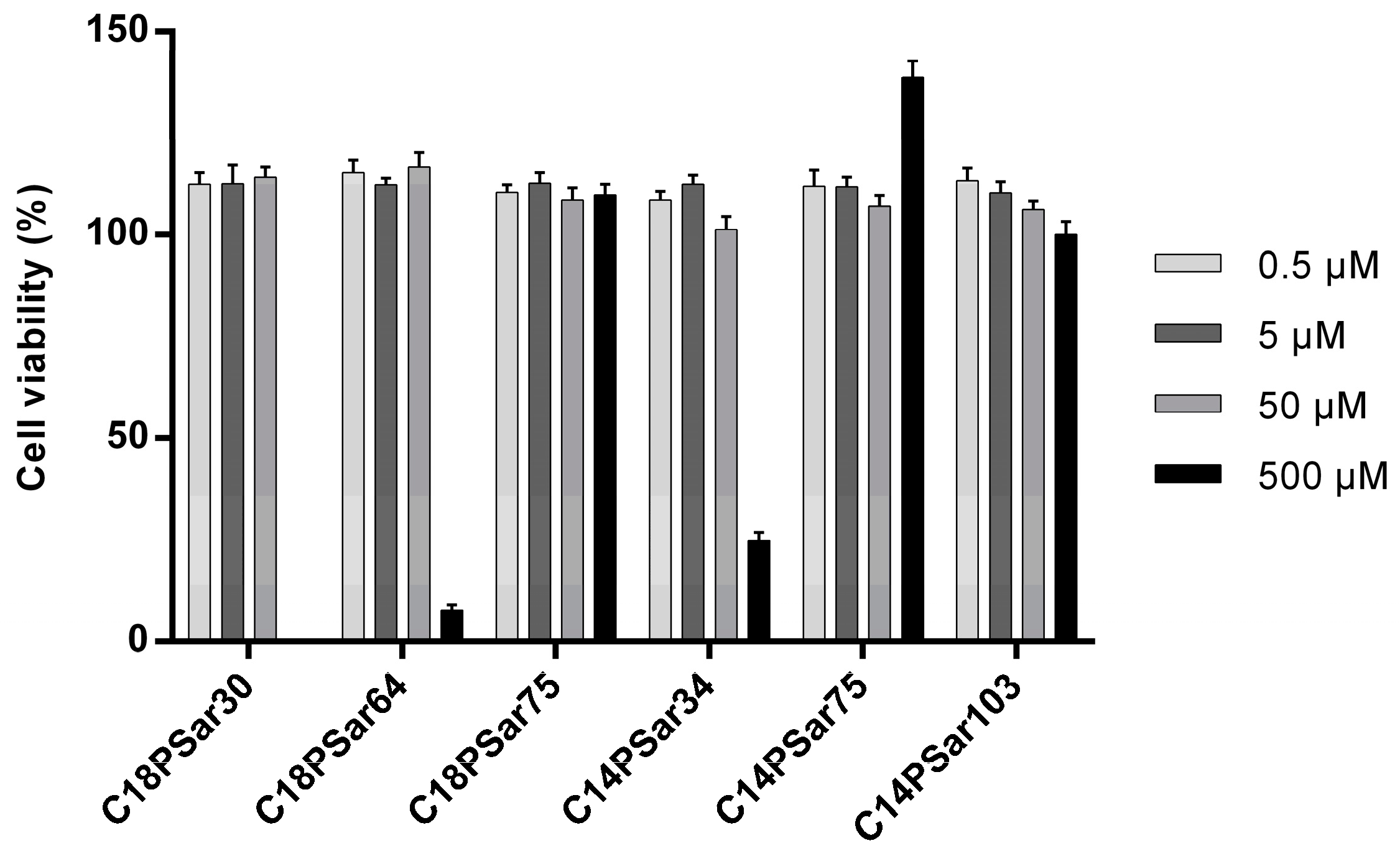
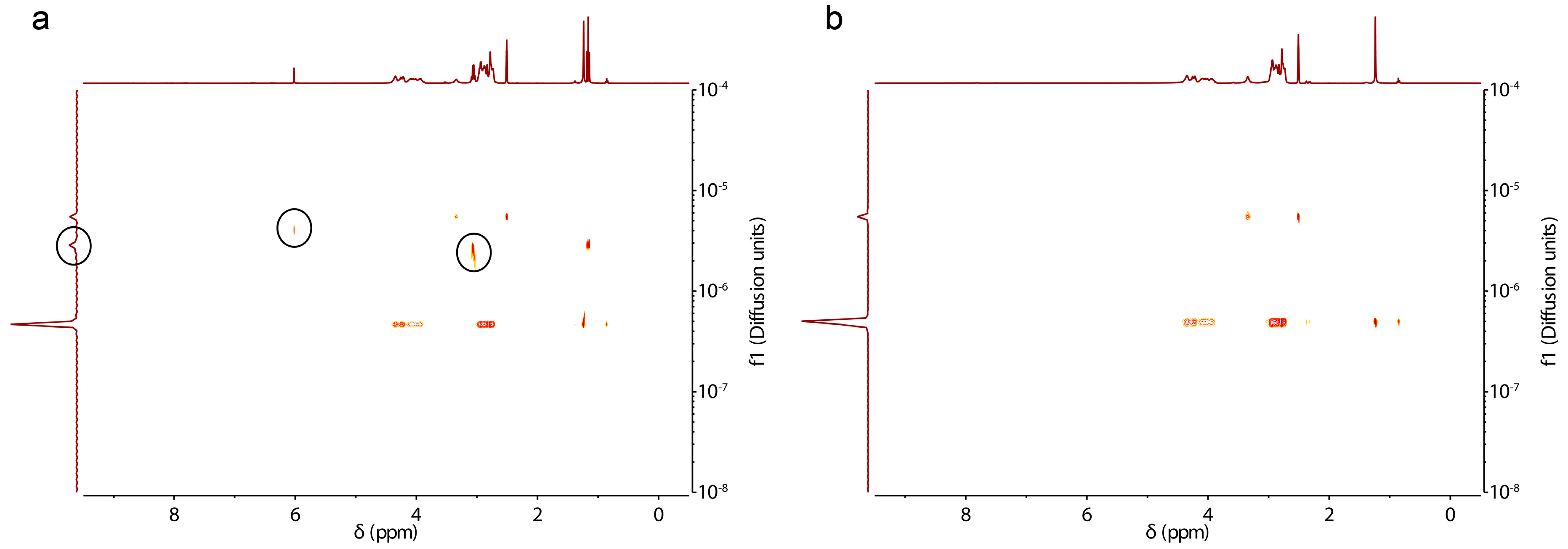
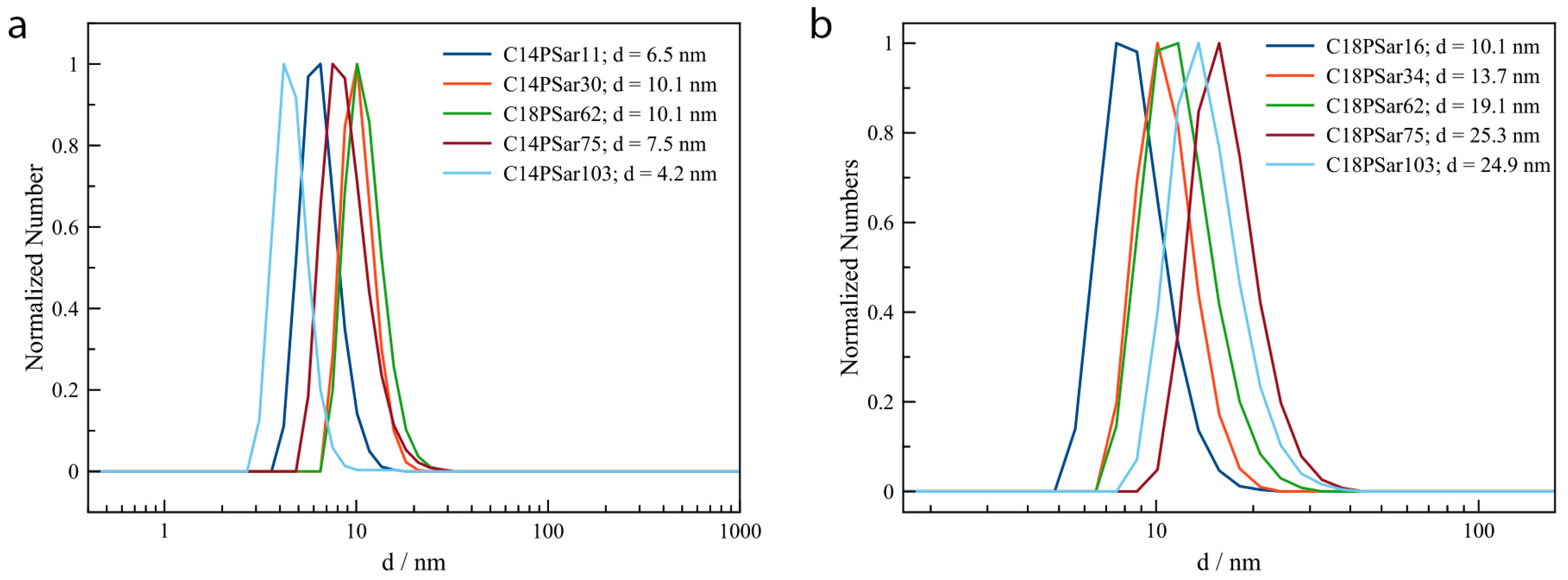
3.2. Characterization, Solution Properties, and Cellular Toxicity of Lipopolypeptoids
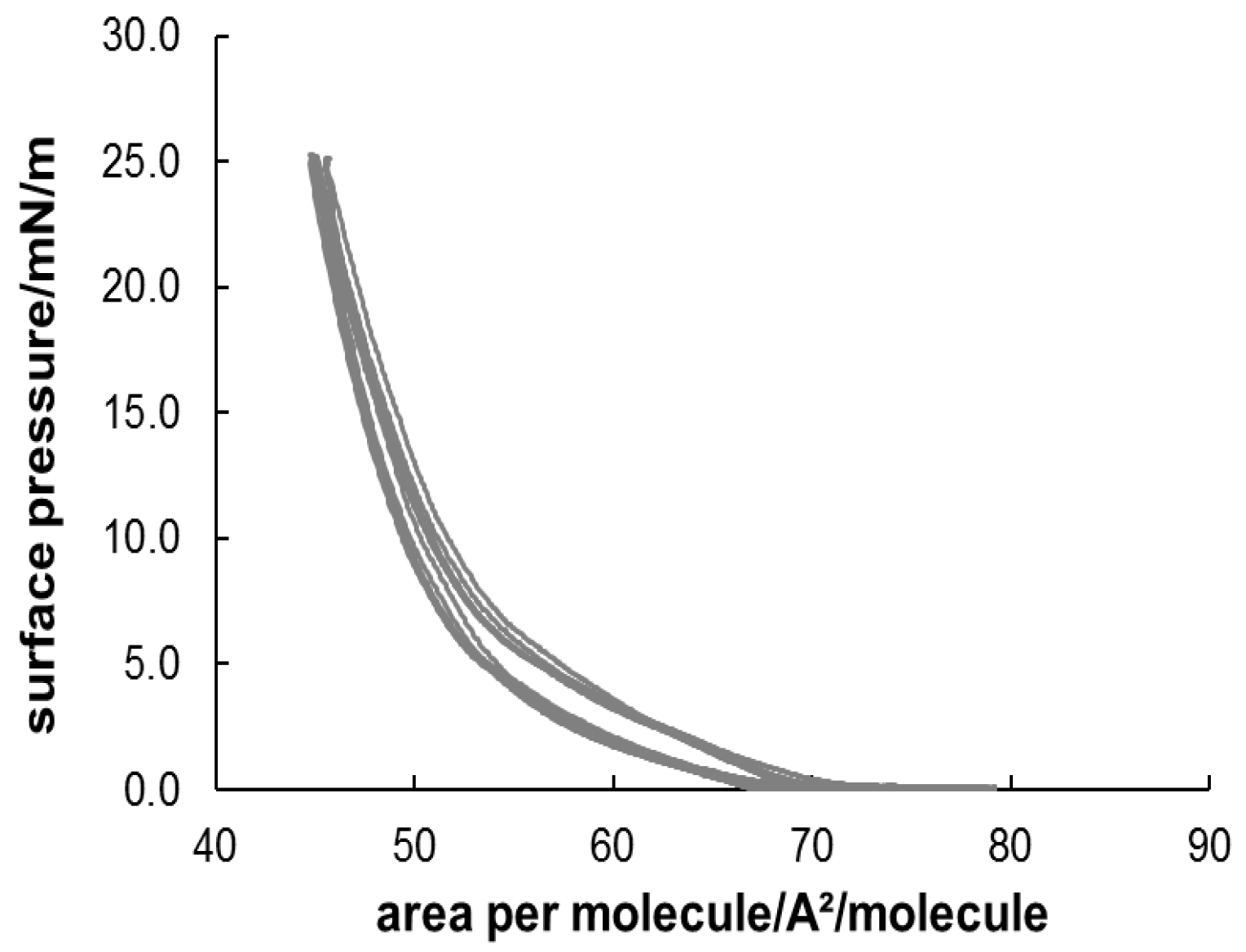
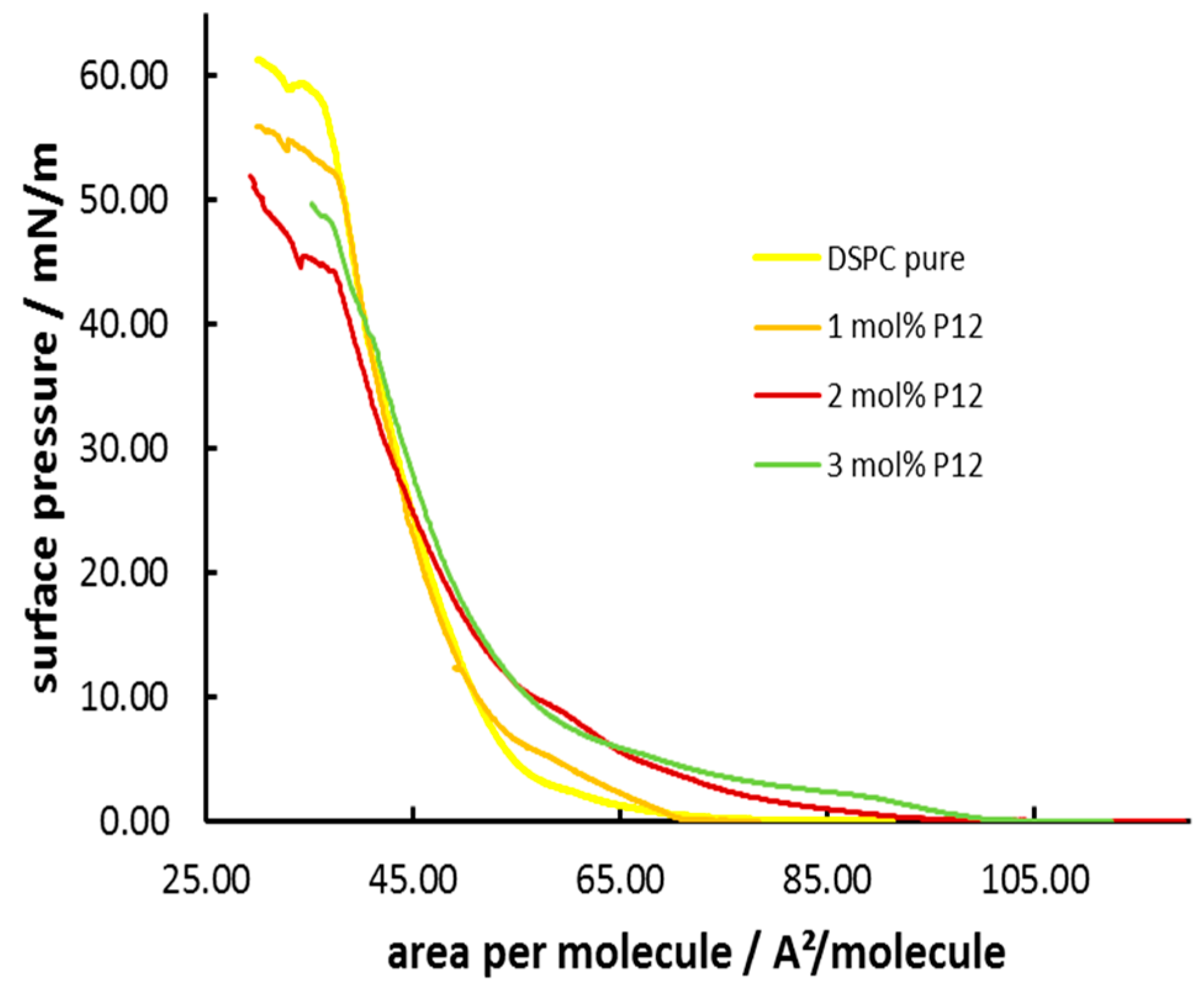
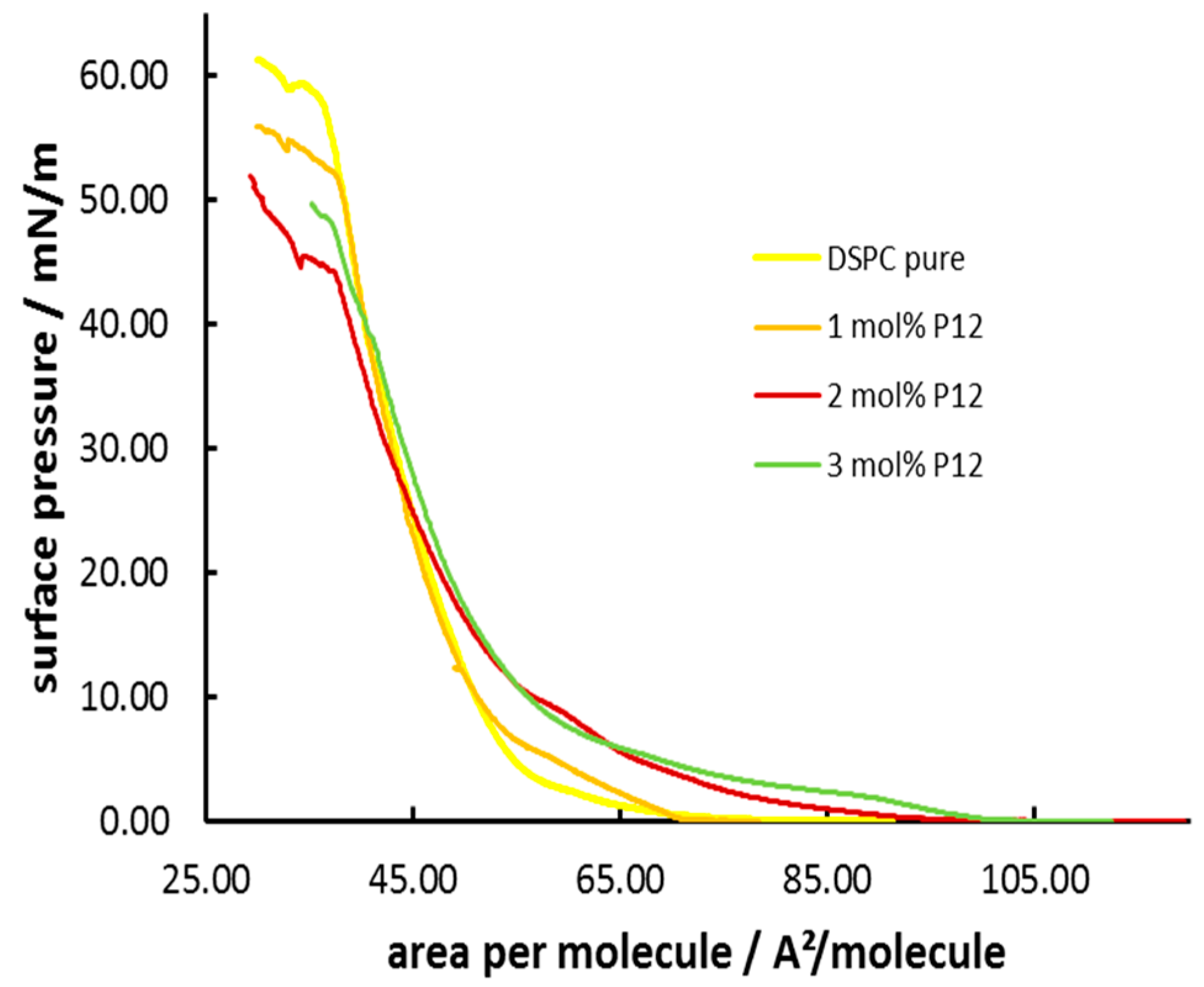
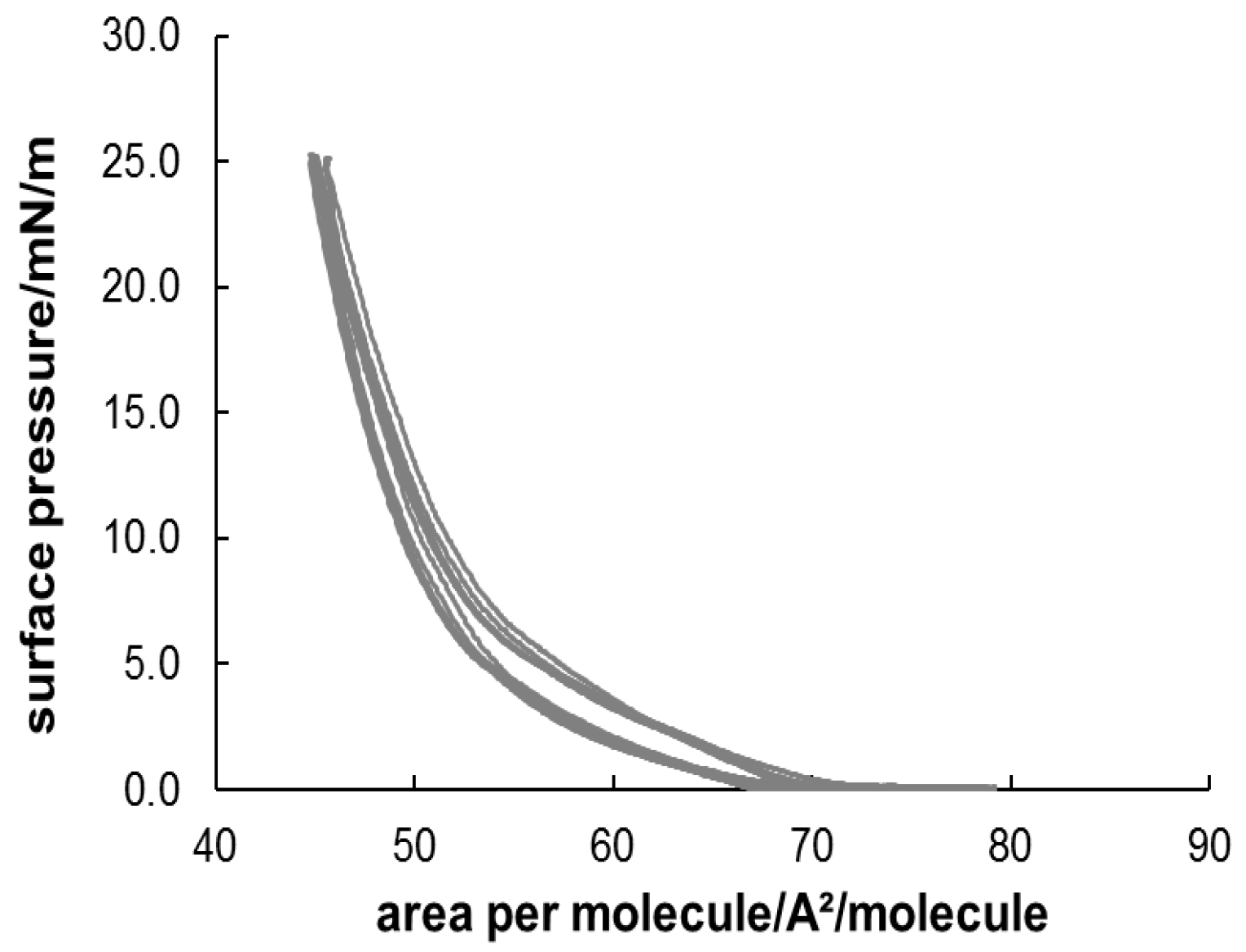
3.3. Formation and Characterization of Lipopolypeptoid Containing Langmuir-Blodgett Monolayers
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A
References
- Kosaric, N. Biosurfactants: Production, Properties, Applications; CRC Press: New York, NY, USA, 1993. [Google Scholar]
- Holmberg, K. Natural surfactants. Curr. Opin. Colloid Interface Sci. 2001, 6, 148–159. [Google Scholar] [CrossRef]
- Raffa, P.; Wever, D.A.Z.; Picchioni, F.; Broekhuis, A.A. Polymeric surfactants: Synthesis, properties, and links to applications. Chem. Rev. 2015, 115, 8504–8563. [Google Scholar] [CrossRef] [PubMed]
- Lutz, J.F. Solution self-assembly of tailor-made macromolecular building blocks prepared by controlled radical polymerization techniques. Polym. Int. 2006, 55, 979–993. [Google Scholar] [CrossRef]
- Laschewsky, A. Molecular concepts, self-organisation and properties of polysoaps. In Polysoaps/Stabilizers/Nitrogen-15 NMR; Springer: Berlin/Heidelberg, Germany, 1995; pp. 1–86. [Google Scholar]
- Charalambopoulou, A.; Bokias, G.; Staikos, G. Template copolymerisation of N-isopropylacrylamide with a cationic monomer: Influence of the template on the solution properties of the product. Polymer 2002, 43, 2637–2643. [Google Scholar] [CrossRef]
- Clapés, P.; Infante, M.R. Amino acid-based surfactants: Enzymatic synthesis, properties and potential applications. Biocatal. Biotransform. 2002, 20, 215–233. [Google Scholar] [CrossRef]
- Yokota, H.; Sagawa, K.; Eguchi, C.; Takehara, M. New amphoteric surfactants derived from lysine. I. Preparation and properties of Nε-acyllysine derivatives-acyllysine derivatives. J. Am. Oil Chem. Soc. 1985, 62, 1716–1719. [Google Scholar]
- Baschang, G.; Hartmann, A.; Wacker, O. Lipopeptides Having Antitumor Activity. U.S. Patent 4666886 A, 19 May 1987. [Google Scholar]
- Braun, D. Developments with lipoamino acids and their salts. Cosmet. Toilet. 1989, 104, 87–96. [Google Scholar]
- Xia, J. Protein-Based Surfactants: Synthesis: Physicochemical Properties, and Applications; CRC Press: Boca Raton, FL, USA, 2001. [Google Scholar]
- Saadati, R.; Dadashzadeh, S.; Abbasian, Z.; Soleimanjahi, H. Accelerated blood clearance of PEGylated PLGA nanoparticles following repeated injections: Effects of polymer dose, PEG coating, and encapsulated anticancer drug. Pharm. Res. 2013, 30, 985–995. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Harada, M.; Wang, X.Y.; Ichihara, M.; Irimura, K.; Kiwada, H. Accelerated blood clearance of PEGylated liposomes following preceding liposome injection: Effects of lipid dose and PEG surface-density and chain length of the first-dose liposomes. J. Control. Release 2005, 105, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Hara, E.; Makino, A.; Kurihara, K.; Yamamoto, F.; Ozeki, E.; Kimura, S. Pharmacokinetic change of nanoparticulate formulation “Lactosome” on multiple administrations. Int. Immunopharmacol. 2012, 14, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Ulbricht, J.; Jordan, R.; Luxenhofer, R. On the biodegradability of polyethylene glycol, polypeptoids and poly(2-oxazoline)s. Biomaterials 2014, 35, 4848–4861. [Google Scholar] [CrossRef] [PubMed]
- Barz, M.; Luxenhofer, R.; Zentel, R.; Vicent, M.J. Overcoming the PEG-addiction: Well-defined alternatives to PEG, from structure–property relationships to better defined therapeutics. Polym. Chem. 2011, 2, 1900–1918. [Google Scholar] [CrossRef]
- Gangloff, N.; Ulbricht, J.; Lorson, T.; Schlaad, H.; Luxenhofer, R. Peptoids and polypeptoids at the frontier of supra- and macromolecular engineering. Chem. Rev. 2015, 116, 1753–1802. [Google Scholar] [CrossRef] [PubMed]
- Luxenhofer, R.; Fetsch, C.; Grossmann, A. Polypeptoids: A perfect match for molecular definition and macromolecular engineering? J. Polym. Sci. Part A 2013, 51, 2731–2752. [Google Scholar] [CrossRef]
- Sisido, M.; Imanishi, Y.; Higashimura, T. Nuclear magnetic resonance spectra of poly(N-alkylamino acid)s. Biopolymers 1972, 11, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Klinker, K.; Barz, M. Polypept(o)ides: Hybrid systems based on polypeptides and polypeptoids. Macromol. Rapid Commun. 2015, 36, 1943–1957. [Google Scholar] [CrossRef] [PubMed]
- Fetsch, C.; Grossmann, A.; Holz, L.; Nawroth, J.F.; Luxenhofer, R. Polypeptoids from N-substituted glycine N-carboxyanhydrides: Hydrophilic, hydrophobic, and amphiphilic polymers with poisson distribution. Macromolecules 2011, 44, 6746–6758. [Google Scholar] [CrossRef]
- Gangloff, N.; Fetsch, C.; Luxenhofer, R. Polypeptoids by living ring-opening polymerization of N-substituted N-carboxyanhydrides from solid supports. Macromol. Rapid Commun. 2013, 34, 997–1001. [Google Scholar] [CrossRef] [PubMed]
- Chapman, R.G.; Ostuni, E.; Liang, M.N.; Meluleni, G.; Kim, E.; Yan, L.; Pier, G.; Warren, H.S.; Whitesides, G.M. Polymeric thin films that resist the adsorption of proteins and the adhesion of bacteria. Langmuir 2001, 17, 1225–1233. [Google Scholar] [CrossRef]
- Lau, K.H.A.; Ren, C.; Park, S.H.; Szleifer, I.; Messersmith, P.B. An experimental-theoretical analysis of protein adsorption on peptidomimetic polymer brushes. Langmuir 2012, 28, 2288–2298. [Google Scholar] [CrossRef] [PubMed]
- Secker, C.; Brosnan, S.M.; Luxenhofer, R.; Schlaad, H. Poly(α-peptoid)s revisited: Synthesis, properties, and use as biomaterial. Macromol. Biosci. 2015, 15, 881–891. [Google Scholar] [CrossRef] [PubMed]
- Kidchob, T.; Kimura, S.; Imanishi, Y. Amphiphilic poly(Ala)-b-poly(Sar) microspheres loaded with hydrophobic drug. J. Control. Release 1998, 51, 241–248. [Google Scholar] [CrossRef]
- Yamamoto, F.; Yamahara, R.; Makino, A.; Kurihara, K.; Tsukada, H.; Hara, E.; Hara, I.; Kizaka-Kondoh, S.; Ohkubo, Y.; Ozeki, E.; et al. Radiosynthesis and initial evaluation of (18)F labeled nanocarrier composed of poly(l-lactic acid)-block-poly(sarcosine) amphiphilic polydepsipeptide. Nucl. Med. Biol. 2013, 40, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Gallot, B. Liposarcosine-Based Polymerizable and Polymeric Surfactants; American Chemical Society: Washington, DC, USA, 1991; pp. 103–113. [Google Scholar]
- Trimpin, S.; Rouhanipour, A.; Az, R.; Räder, H.J.; Müllen, K. New aspects in matrix-assisted laser desorption/ionization time-of-flight mass spectrometry: A universal solvent-free sample preparation. Rapid Commun. Mass Spectrom. 2001, 15, 1364–1373. [Google Scholar] [CrossRef] [PubMed]
- Griffin, W.C. Classification of surface-active agents by “HLB”. J. Soc. Cosmet. Chem. 1949, 1, 311–326. [Google Scholar]
- Naught, A.D.M.; Wilkinson, A. IUPAC Gold Book, 2nd ed.; Blackwell Scientific Publication: Oxford, UK, 1997. [Google Scholar]
- Strickley, R.G. Solubilizing excipients in oral and injectable formulations. Pharm. Res. 2004, 21, 201–230. [Google Scholar] [CrossRef] [PubMed]
- Douy, A.; Gallot, B. New amphipathic lipopeptides, 1. Synthesis and mesomorphic structures of lipopeptides with polysarcosine peptidic chains. Die Macromol. Chem. 1986, 187, 465–483. [Google Scholar]
- Birke, A.; Huesmann, D.; Kelsch, A.; Weilbächer, M.; Xie, J.; Bros, M.; Bopp, T.; Becker, C.; Landfester, K.; Barz, M. Polypeptoid-block-polypeptide copolymers: Synthesis, characterization, and application of amphiphilic block Copolypept(o)ides in drug formulations and miniemulsion techniques. Biomacromolecules 2014, 15, 548–557. [Google Scholar] [CrossRef] [PubMed]
- Heller, P.; Weber, B.; Birke, A.; Barz, M. Synthesis and sequential deprotection of triblock copolypept(o)ides using orthogonal protective group chemistry. Macromol. Rapid Commun. 2015, 36, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Holm, R.; Klinker, K.; Weber, B.; Barz, M. Synthesis of Amphiphilic Block Copolypept(o)ides by Bifunctional Initiators: Making PeptoMicelles Redox Sensitive. Macromol. Rapid Commun. 2015, 36, 2083–2091. [Google Scholar] [CrossRef] [PubMed]
- Kessel, D. Properties of cremophor EL micelles probed by fluorescence. Photochem. Photobiol. 1992, 56, 447–451. [Google Scholar] [CrossRef] [PubMed]
- El-Azab, W.I.M.; Aiad, I.; Azzam, E.M.S.; Gad, E.A.M. Molecular, Surface, and thermodynamic properties of nonionic surfactants based on castor oil. J. Dispers. Sci. Technol. 2010, 31, 1150–1156. [Google Scholar] [CrossRef]
- Huesmann, D.; Sevenich, A.; Weber, B.; Barz, M. A head-to-head comparison of poly(sarcosine) and poly(ethylene glycol) in peptidic, amphiphilic block copolymers. Polymer 2015, 67, 240–248. [Google Scholar] [CrossRef]
- Fetsch, C.; Gaitzsch, J.; Messager, L.; Battaglia, G.; Luxenhofer, R. Self-assembly of amphiphilic block copolypeptoids—Micelles, worms and polymersomes. Sci. Rep. 2016, 6, 33491. [Google Scholar] [CrossRef] [PubMed]
- Kiss, L.; Walter, F.R.; Bocsik, A.; Veszelka, S.; Ózsvári, B.; Puskás, L.G.; Szabó-Révész, P.; Deli, M.A. Kinetic analysis of the toxicity of pharmaceutical excipients cremophor EL and RH40 on endothelial and epithelial cells. J. Pharm. Sci. 2013, 102, 1173–1181. [Google Scholar] [CrossRef] [PubMed]
- Hao, C.; Liu, Q.; Li, Q.; Zhang, J.; Sun, R. Thermodynamic and structural studies of DMPC and DSPC with DOTAP mixed monolayers at the air–water interface 1. Russ. J. Phys. Chem. A 2016, 90, 214–219. [Google Scholar] [CrossRef]
- Tanwir, K.; Tsoukanova, V. Lateral distribution of a poly(ethylene glycol)-grafted phospholipid in phosphocholine monolayers studied by epifluorescence microscopy. Langmuir 2008, 24, 14078–14087. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | Xn (Calculated) | Xn (NMR) | Xn (MALDI) | Mn (GPC) | Đ |
|---|---|---|---|---|---|
| C18PSar12 | 10 | 12 | 5232 | 1.06 | |
| C18PSar30 | 30 | 30 | 7619 | 1.06 | |
| C18PSar45 | 50 | 45 | 47 | 13,933 | 1.07 |
| C18PSar64 | 70 | 64 | 15,090 | 1.08 | |
| C18PSar117 | 100 | 117 | 24,890 | 1.13 | |
| C14PSar11 | 10 | 11 | 3960 | 1.05 | |
| C14PSar34 | 30 | 34 | 9035 | 1.07 | |
| C14PSar53 | 50 | 53 | 49 | 14,718 | 1.08 |
| C14PSar75 | 70 | 75 | 18,220 | 1.12 | |
| C14PSar103 | 100 | 103 | 23,210 | 1.11 |
| Polymer | HLB value 1 | CMC2/mg·L−1 | Diameter of main peak (DLS/nm) | Distribution |
|---|---|---|---|---|
| C18PSar12 | 16.0 | 27 | 10.1 | bimodal |
| C18PSar30 | 17.8 | 62 | 13.7 | bimodal |
| C18PSar45 | 18.4 | 94 | 19.1 | monomodal |
| C18PSar65 | 19.0 | 181 | 25.2 | monomodal |
| C18PSar117 | 19.5 | 1181 | 24.9 | monomodal |
| C14PSar11 | 15.7 | 213 | 9.1 | monomodal |
| C14PSar34 | 18.4 | - | 12.7 | bimodal |
| C14PSar53 | 18.9 | - | 16.4 | monomodal |
| C14PSar75 | 19.1 | - | 19.2 | bimodal |
| C14PSar103 | 19.4 | - | 24.4 | trimodal |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weber, B.; Seidl, C.; Schwiertz, D.; Scherer, M.; Bleher, S.; Süss, R.; Barz, M. Polysarcosine-Based Lipids: From Lipopolypeptoid Micelles to Stealth-Like Lipids in Langmuir Blodgett Monolayers. Polymers 2016, 8, 427. https://doi.org/10.3390/polym8120427
Weber B, Seidl C, Schwiertz D, Scherer M, Bleher S, Süss R, Barz M. Polysarcosine-Based Lipids: From Lipopolypeptoid Micelles to Stealth-Like Lipids in Langmuir Blodgett Monolayers. Polymers. 2016; 8(12):427. https://doi.org/10.3390/polym8120427
Chicago/Turabian StyleWeber, Benjamin, Christine Seidl, David Schwiertz, Martin Scherer, Stefan Bleher, Regine Süss, and Matthias Barz. 2016. "Polysarcosine-Based Lipids: From Lipopolypeptoid Micelles to Stealth-Like Lipids in Langmuir Blodgett Monolayers" Polymers 8, no. 12: 427. https://doi.org/10.3390/polym8120427
APA StyleWeber, B., Seidl, C., Schwiertz, D., Scherer, M., Bleher, S., Süss, R., & Barz, M. (2016). Polysarcosine-Based Lipids: From Lipopolypeptoid Micelles to Stealth-Like Lipids in Langmuir Blodgett Monolayers. Polymers, 8(12), 427. https://doi.org/10.3390/polym8120427