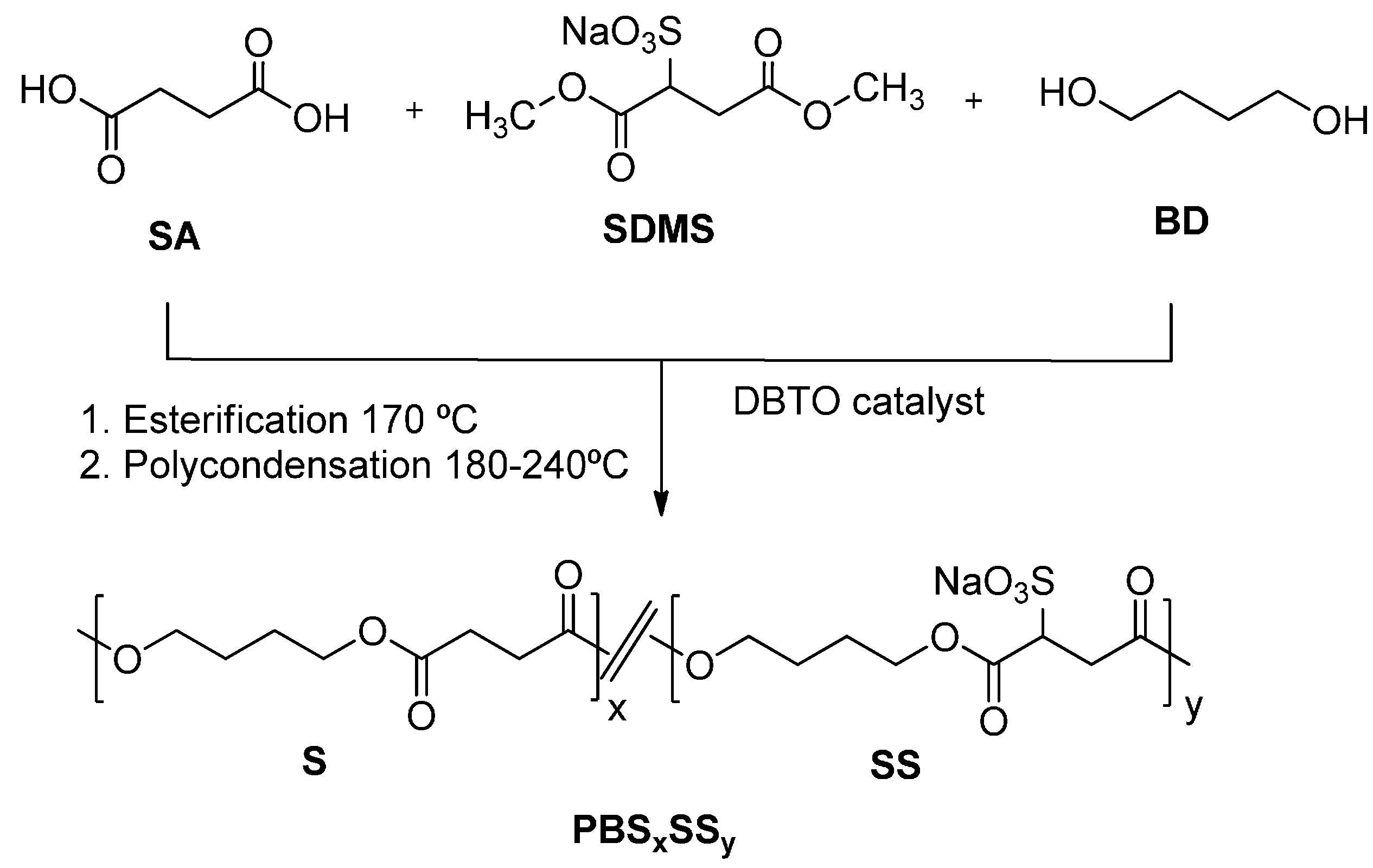
3.1. Synthesis and Chemical Structure of PBSxSSy Copolyesters
The synthesis pathway leading to the PBS
xSS
y ionomers is shown in
Scheme 1. The procedure consists of two successive steps, the first one is an esterification/transesterification reaction leading to low molecular weight oligomers, and the second one is the polycondensation of the oligomers formed in the previous step to render the final copolyesters. The first step was carried out at 170 °C with removal of the released water and methanol and the second one was carried out at higher temperatures under high vacuum to accelerate the transesterification reaction and to unbalance the equilibrium towards the formation of high molecular weight polymers by releasing of the excess of BD. The maximum temperature applied was in all cases 190 °C because the β-elimination reaction of SS units with releasing of NaHSO
3 and concomitant generation of fumarate units started to happen above this temperature.
Scheme 1.
Polymerization reactions leading to homopolyesters and PBSxSSy copolyesters.
Scheme 1.
Polymerization reactions leading to homopolyesters and PBSxSSy copolyesters.
It was observed that the melt viscosity attained by the reaction mass increased gradually with the content of SS units as it should be expected for ionomers; it is widely known that sulfonate groups can interact to generate ionic aggregates or clusters which act as thermoreversible crosslinks [
25,
26]. Dibutyl tin oxide (DBTO) was the catalyst of choice. Initially, the synthesis of the copolymers was done with titanium (IV) tetrabutoxide (TBT), but the result was not satisfactory due to the decomposition of SS units and formation of copolyesters with very low intrinsic viscosities. DBTO allowed the reaction to proceed at lower temperatures and copolymers with significantly higher intrinsic viscosities could be then attained. All the copolyesters were obtained in high yields (higher than 90%) and molecular weights fell with increasing content in SS units (
Table 1). It should be noted that GPC measurements could be affected by ionic aggregations. Although one can expect that the treatment of samples with TFA previous to injection and the use of HFIP as the mobile phase will minimize the occurrence of ionic interactions, their complete absence cannot be fully ascertained. However results afforded by GPC were very consistent with intrinsic viscosities measured in dichloroacetic acid (DCA), which ranged between 0.74 and 1.18 dL·g
−1 with values also decreasing as the content of the copolyester in SS units increased. Nevertheless, the clear conclusion that can be drawn from these data is that the polycondensation reaction is hindered in the presence of the dimethyl sulfonated succinate comonomer. One factor motivating the decrease in molecular weight is the limited temperature at which polycondensation has to be conducted in order to avoid the β-elimination reaction of sulfonate groups. A second factor is the increase in melt viscosity of the mass reaction with the content of the forming copolyester in SS units, which hinders the volatilization of BD that is released in the polycondensation step.
Table 1.
Composition and molecular weights of poly(butylene succinate) (PBS) homopolyesters and poly(butylene succinate-co-butylene sulfonated succinate) (PBSxSSy) copolyesters.
Table 1.
Composition and molecular weights of poly(butylene succinate) (PBS) homopolyesters and poly(butylene succinate-co-butylene sulfonated succinate) (PBSxSSy) copolyesters.
| Copolyester | Copolyester composition a | Copolyester composition b | Molecular weights |
|---|
| S/SS | S/SS | [η] c | Mw d | Ð d |
|---|
| PBS1 e | 100/0 | 100/0 | 1.33 | 112,000 | 2.2 |
| PBS2 f | 100/0 | 100/0 | 1.0 | 45,600 | 2.5 |
| PBS97SS3 | 97/3 | 97.9/2.1 | 1.18 | 72,000 | 2.5 |
| PBS95SS5 | 95/5 | 96.1/3.9 | 1.03 | 39,000 | 2.4 |
| PBS92SS8 | 92/8 | 92.8/7.2 | 1.02 | 38,000 | 2.4 |
| PBS90SS10 | 90/10 | 90.9/9.1 | 0.94 | 35,000 | 2.2 |
| PBS85SS15 | 85/15 | 86.2/13.8 | 0.74 | 33,000 | 2.1 |
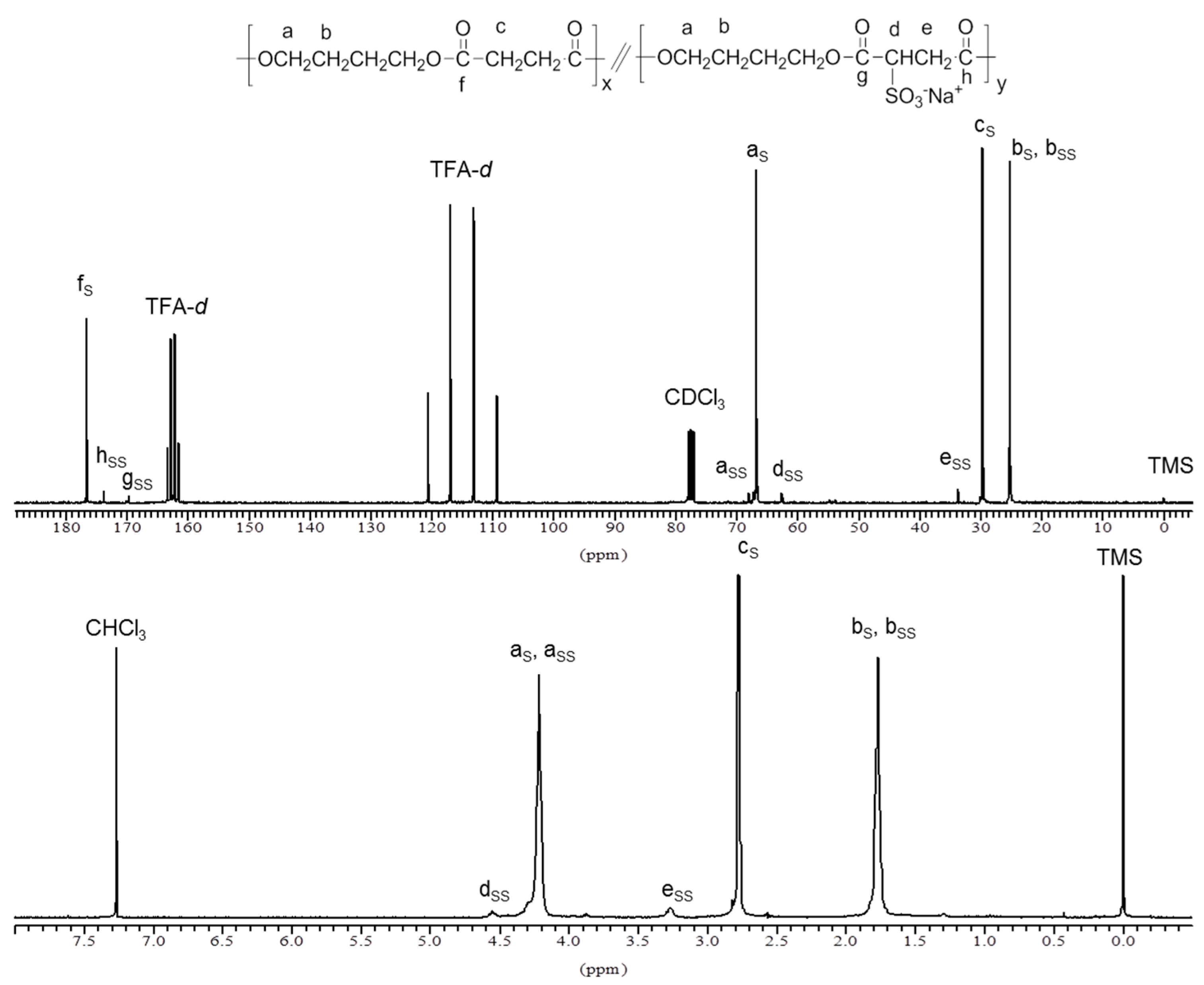
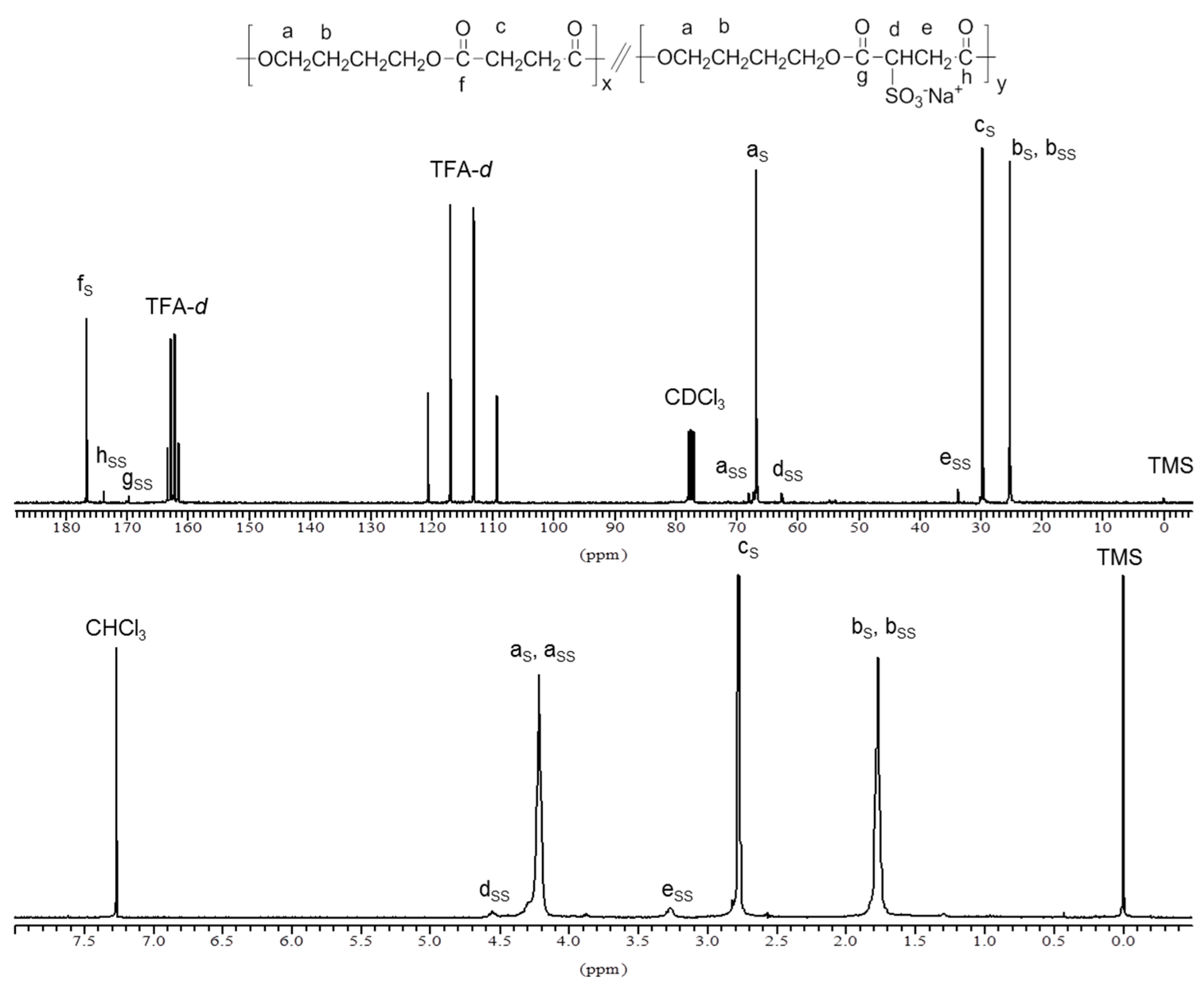
Figure 1.
1H NMR (bottom) and 13C NMR (top) of PBS85SS15 copolyester.
Figure 1.
1H NMR (bottom) and 13C NMR (top) of PBS85SS15 copolyester.
The chemical structure and composition of PBS
xSS
y copolyesters was ascertained by NMR. The
1H and
13C NMR spectra of PBS
85SS
15 are shown in
Figure 1 as representative of the series. The content in SS units was determined by the ratio of the integrated NMR signals at 3.25 ppm for the CH
2 in the SS unit (signal e) and at 2.76 ppm for the two CH
2 in the succinate unit (signal c). Results are listed in
Table 1 indicating that the content of copolyesters in sulfonated units was lower than in the feed. Differences are however small, around 10% in overall, and they must be attributed to the β-elimination reaction of sulfonated groups happening in the SS units. This side reaction was observed to be enhanced at higher temperatures [
22]; as it is shown in the
supplementary information file (
Figure S2), the peak corresponding to fumarate protons becomes clearly observed in the
1H NMR spectra of a copolyester heated at 210 °C for 30 min.
3.2. Thermal and Mechanical Properties
The effects exerted by the incorporation of SS units on the thermal properties of PBS were evaluated by DSC and TGA. The DSC data obtained for the whole PBS
xSS
y series examined in this work are collected in
Table 2. The two samples of PBS showed practically the same
Tg value of −37 °C, which was found to increase up to −32 °C with the content in SS units. Such slight but significant increase in
Tg is supposedly caused by the ionic interactions taking place between the sulfonate groups, which are known to act as physical crosslinks that restrict the chain mobility. Eisenberg
et al. [
27,
28], reported a similar behavior for styrene-based ionomers, which contain ionic multiplets, each of them surrounded by a shell of restricted chain mobility. The restricted mobility region surrounding an isolated multiplet would be too small as to have a noticeable effect on the overall
Tg of the polymer, but the multiplet itself would increase the
Tg of the polymer by acting as a large cross-link. A similar effect has been reported by Im
et al. [
22,
23] for the same copolyesters with low content of SS units and attributed to the restricted chain mobility caused by the strong intermolecular association of ionic groups.
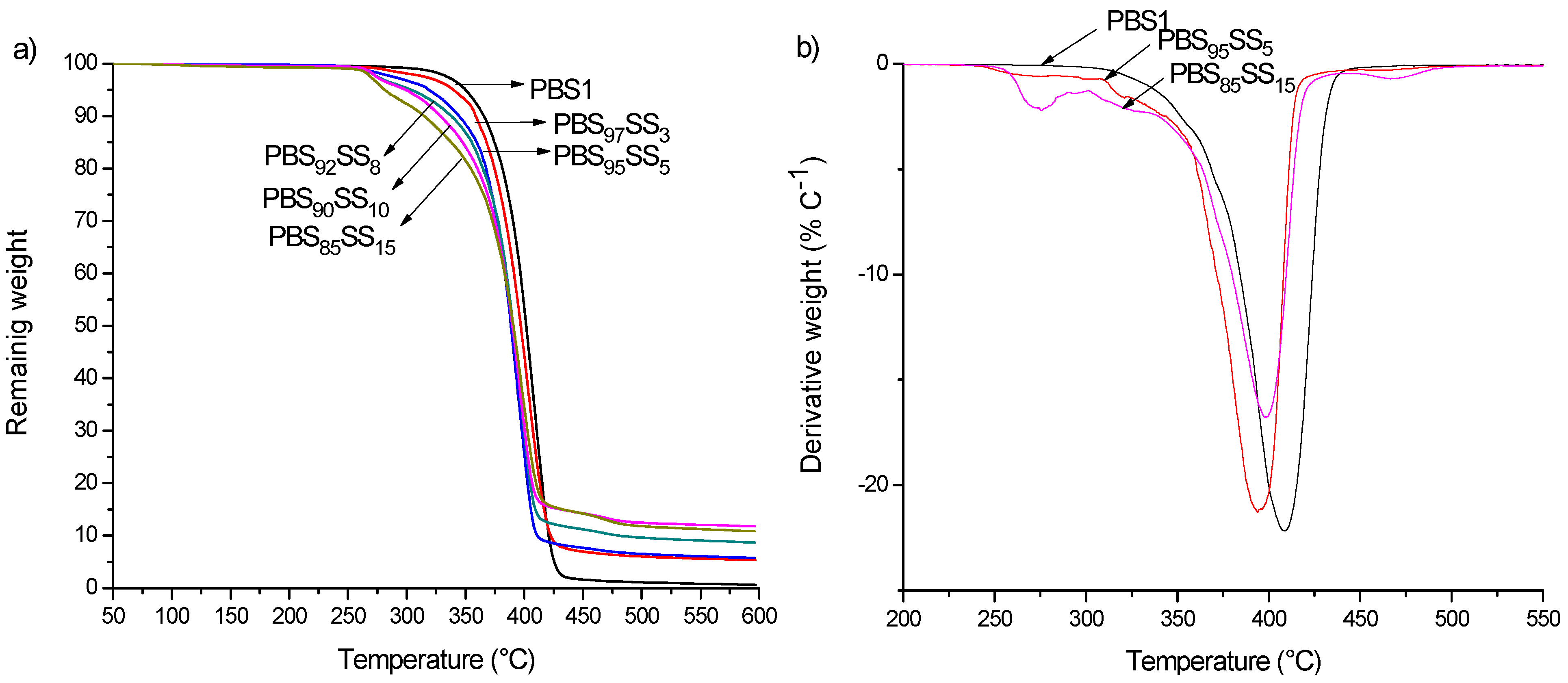
To evaluate the thermal stability of the polyesters, TGA measurements were carried out under a nitrogen atmosphere in the 30–600 °C interval. The TGA traces of PBS
xSS
y copolyesters are shown in
Figure 2a where the trace for PBS1 has been included for comparison, and data obtained therefrom are summarized in
Table 2, which shows that the thermal stability of copolyesters decreased with the content in SS units. Thermal decomposition of PBS occurs in a single stage with maximum rate at about 405 °C with a 1%–2% of residual weight left upon heating at 600 °C. The insertion of SS units in PBS not only tends to decrease the decomposition temperature but also makes the process more complex. Up to four decomposition steps are detected in the PBS
xSS
y copolyesters with that happening at 390−400 °C being much more prominent than the others (
Figure 2b). The remaining weight after the thermal degradation at 600 °C increased continuously with the content in SS units, which may be accounted for by the metallic residue left in the decomposition of the SS units (
Table 2).
Figure 2.
(a) TGA traces of PBS1 and PBSxSSy; (b) Derivative curves of PBS1 and a selection of PBSxSSy copolyesters.
Figure 2.
(a) TGA traces of PBS1 and PBSxSSy; (b) Derivative curves of PBS1 and a selection of PBSxSSy copolyesters.
Table 2.
Thermal and mechanical properties of PBS homopolymer and PBSxSSy copolyesters.
Table 2.
Thermal and mechanical properties of PBS homopolymer and PBSxSSy copolyesters.
| Copolyester | TGA | DSC | Stress-strain Parameters |
|---|
| Td a (°C) | maxTd b (°C) | RW c (%) | Tg d (°C) | Tm e (°C) | ΔHm e (J g−1) | Tc f (°C) | ΔHc f (J g−1) | E g (Mpa) | σmax h (Mpa) | εmax i (%) |
|---|
| PBS1 | 363 | 408 | 1 | −37 | 115 (114) | 70 (74) | 75 | 66 | 440 ± 5 | 35 ± 1 | 282 ± 10 |
| PBS2 | 356 | 405 | 3 | −37 | 113 (114) | 77 (67) | 78 | 62 | 403 ± 3 | 26 ± 1 | 10 ± 1 |
| PBS97SS3 | 359 | 403 | 5 | −36 | 113 (111) | 90 (69) | 71 | 65 | 480 ± 10 | 36 ± 2 | 121 ± 3 |
| PBS95SS5 | 344 | 399 | 6 | −36 | 113 (111) | 71 (66) | 63 | 63 | 515 ± 10 | 39 ± 1 | 21 ± 3 |
| PBS92SS8 | 338 | 397 | 9 | −34 | 111 (110) | 61 (60) | 54 | 57 | 860 ± 5 | 42 ± 1 | 9 ± 1 |
| PBS90SS10 | 328 | 396 | 12 | −33 | 110 (108) | 43 (56) | 53 | 50 | 930 ± 7 | 38 ± 1 | 6 ± 1 |
| PBS85SS15 | 315 | 393 | 13 | −32 | 106 (105) | 53(46) | 36 | 25 | 1050 ± 8 | 28 ± 2 | 3 ± 1 |
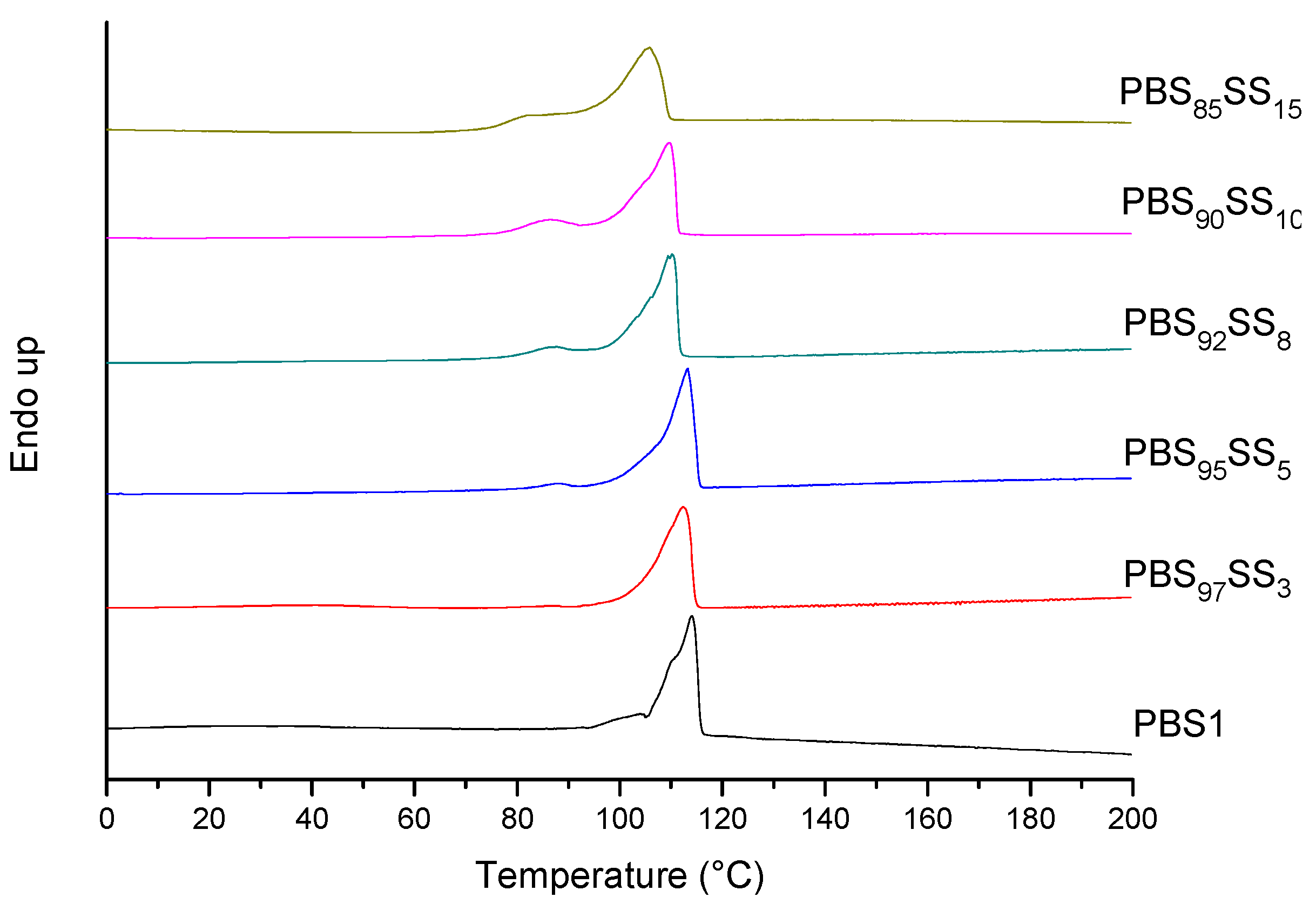
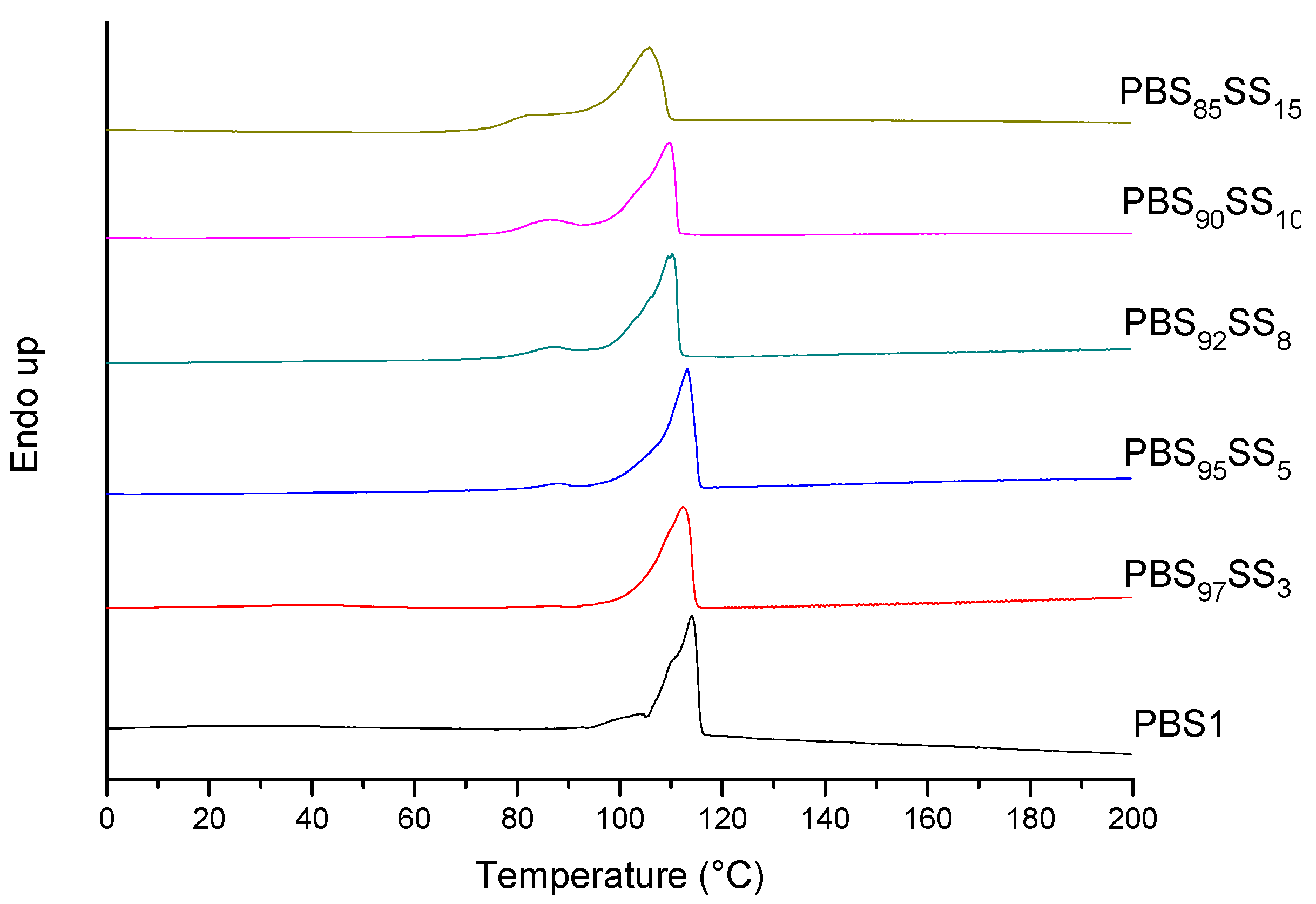
All PBS
xSS
y copolyesteres are semicrystalline with melting temperatures and enthalpies slightly decreasing with the content in SS units. Since the random insertion of the second comonomer will diminish the chain regularity of PBS, a decrease in both crystallite size and crystallinity should be expected for the copolyesters. Nevertheless the effect is really moderate with a decay of
Tm in less than 10 °C for a content of 15 mol% in SS units. The thermograms registered at the first heating from copolyester samples prepared by precipitation from CHCl
3 with methanol are shown in
Figure 3. All of them display well-defined endothermic peaks characteristic of melting. Moreover, all of the copolyesters were able to crystallize from the melt upon cooling at 10 °C·min
−1 with a good reproduction of melting at the second heating (
Figure S3). Samples obtained by precipitation from solution are expected to be well crystallized and to therefore display high melting temperature and enthalpy. Samples used for the second heating were crystallized under much more unfavorable conditions. The small differences observed in both
Tm and Δ
Hm between first and second heating traces (
Table 2) are indicative of the ability of these copolyesters to crystallize from the melt. In principle, it could be thought that ionic clusters that have not been destroyed after melting may act as nucleating agents favoring the crystallization of the molten material, as it was reported for these copolyesters with low content of SS units [
22]. In the present case, however, the large supercooling observed for crystallization, which attains up to near 70 °C, makes such interpretation less probable.
Figure 3.
DSC thermograms (first heating) of PBSxSSy copolyesters and PBS1.
Figure 3.
DSC thermograms (first heating) of PBSxSSy copolyesters and PBS1.
The basic mechanical parameters of the copolyesters registered in tensile essays are presented in
Table 2 together with those of PBS1 and PBS2 that have been included for comparison. The stress-strain curves experimentally obtained are depicted in
Figure S4 of the
Supplementary Informaiton file. Large differences in the mechanical parameters are observed between PBS1 and PBS2 according to what should be expected from their greatly different molecular weights. In fact, a drastic decay in extensibility in addition to slight reductions in the elastic modulus and maximum stress were observed for PBS when the molecular weight decreased from ~100,000 to ~45,000. Mechanical data for the PBS
xSS
y series can be then analyzed as a function of composition by taking as reference PBS2, which has a molecular weight acceptably close to those of copolyesters. Sample PBS
97SS
3 displays, however, an exceptional behavior due to its higher molecular weight and crystallinity; in this case, the three analyzed mechanical parameters are enhanced with respect to those of PBS2, as should be reasonably expected. The trend observed for the Young’s modulus along the whole series is very clear:
E increased steadily with the content in SS units to reach near three-times the PBS value for a SS units content of about 15 mol%. Such a variation is in agreement with the increasing value of
Tg. The elongation at break was found to follow an opposite trend which is consistent with the variation observed for the elastic modulus. On the other hand, the influence of composition on the maximum stress is such that σ
max differences for copolyesters in the 5–10 mol% range are less than 10% and may be considered just as due to uncontrolled experimental factors. The exceptional low value observed for PBS
85SS
15 can be explained by the lower crystallinity displayed by this sample. In spite of the lack of an apparent systematic variation in the measured parameters, it can be preliminarily concluded from this analysis that the insertion of ionic sulfonated units makes the polyester stiffer but also more brittle because of the high reduction undergone in the elongation to break. This behavior is in accordance with that displayed by other ionomers containing sulfonated units that have been reported by us in previous works [
2,
29].
3.3. Hydrolytic Degradation
Polyester recycling is in the focus of current polymer research concerning sustainability. Chemical recycling leading to monomer (or oligomer) recovery for subsequent valorization is an actual alternative method to physical processes, and PBS is a good candidate for hydrolysis recycling with regeneration of succinic acid [
30]. Therefore, to know the influence of comonomers on novel PBS copolyesters is a matter of unquestionable importance.
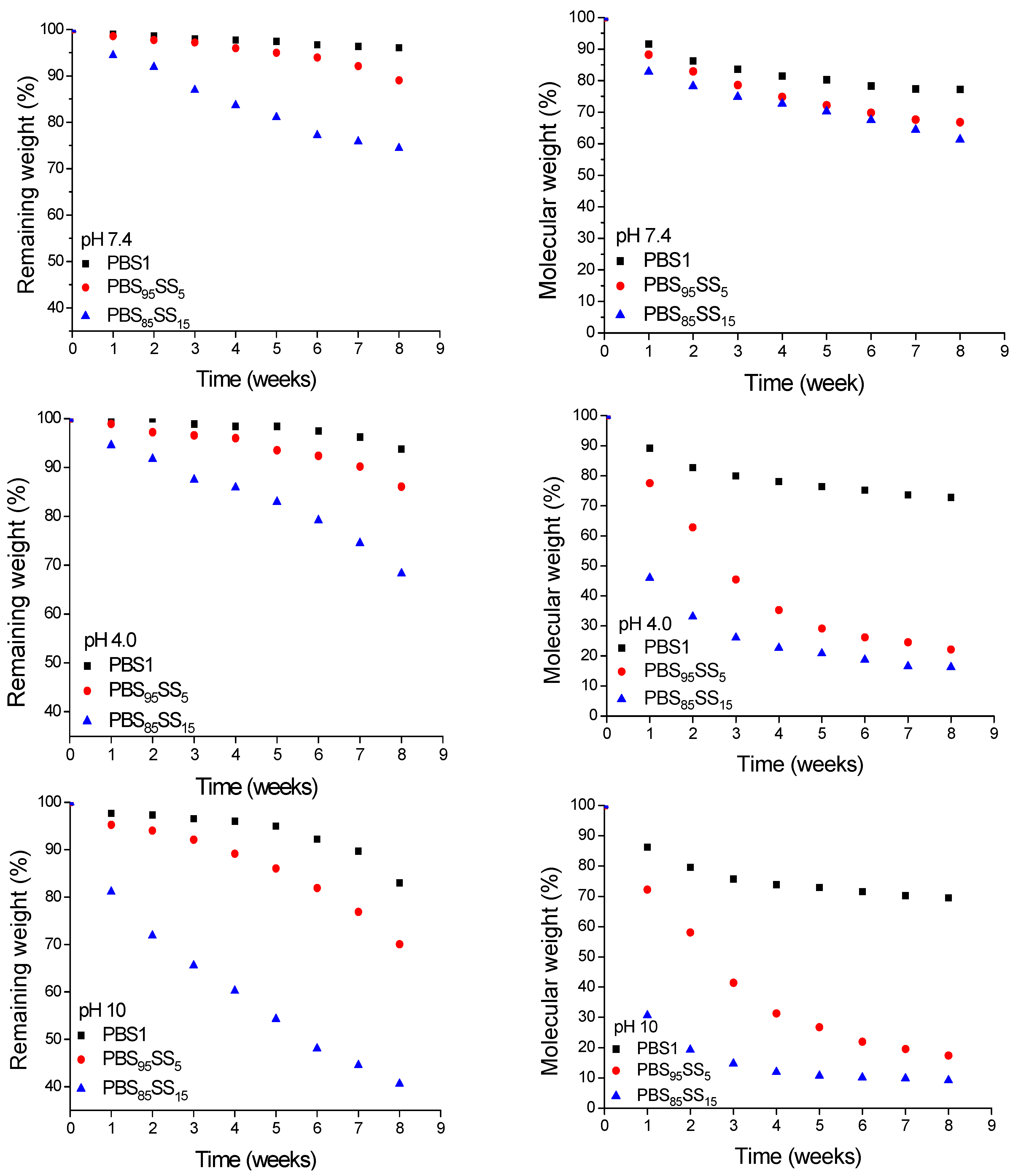
Accordingly, the effect exerted by the incorporation of SS units on the hydrolytic degradation of PBS has been evaluated under a variety of conditions. The copolyesters chosen for comparison with PBS were PBS
95SS
5 and PBS
85SS
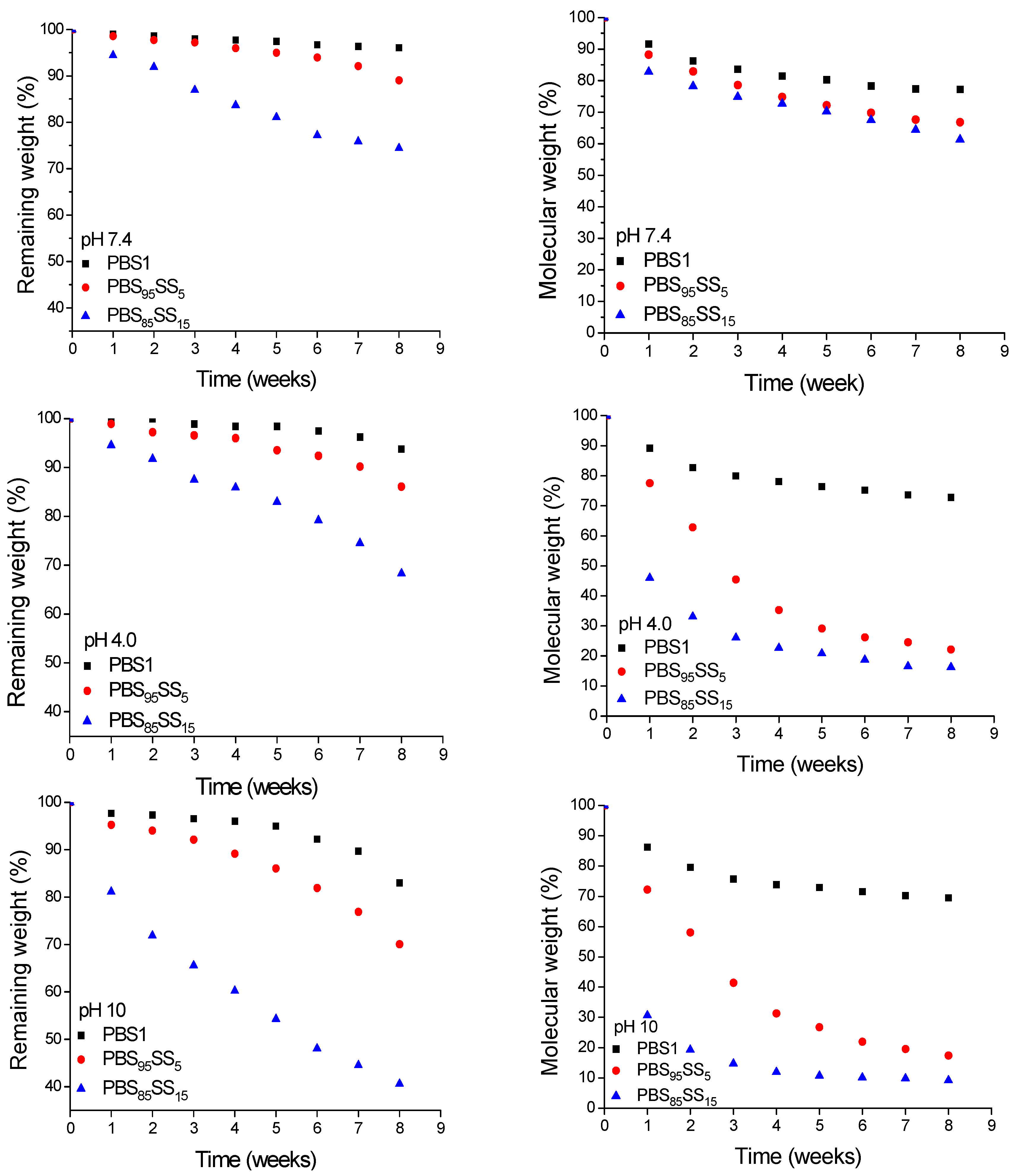
15. They were incubated in pH 7.4 buffers at 37 °C and also at pH 4.0 and 10 to estimate their susceptibility to hydrolysis under both acidic and basic conditions. The degradation progress was monitored by following the weight loss and molecular weight decay of the residual sample as a function of the incubation time. These data are presented in
Figure 4. After eight weeks of incubation at pH 7.4, the
Mw of samples with SS unit contents of 5 and 15 mol% was 33% and 39% of the initial value, respectively whereas PBS decreased only in less than 20%. It becomes clear, therefore, from these results that the hydrolytic degradation is enhanced by increasing contents in SS units.
To gain insight into the degradation of the polyester chain at the molecular level, PBS1, PBS
95SS
5 and PBS
85SS
15 polyesters were incubated at 37 °C in aqueous buffer at pH 4.0 and pH 10 and both the releasing fragments and the residual polymer were analyzed weekly over a period of two months. Changes taking place in sample weight and
Mw of the three polyesters upon hydrolysis are comparatively depicted in
Figure 4. A significant decay in both sample weight and molecular weight was observed for the PBS
xSS
y copolyesters. It was observed that the hydrolytic degradation was drastically accelerated by increasing either ionic content or pH. Similar observations have been recently reported for PBS ionenes [
31] as well as for other copolyesters containing sulfoisophthalic units [
2,
18,
29], and such increase has been attributed to the enhancement of the hydrophilicity caused by the presence of the ionic groups.
Figure 4.
Evolution of sample remaining weight (left) and weight average molecular weight (right) for PBS1, PBS95SS5 and PBS85SS15 at pH 7.4, 4.0 and 10 (top to bottom) at 37 °C, as a function of incubation time.
Figure 4.
Evolution of sample remaining weight (left) and weight average molecular weight (right) for PBS1, PBS95SS5 and PBS85SS15 at pH 7.4, 4.0 and 10 (top to bottom) at 37 °C, as a function of incubation time.
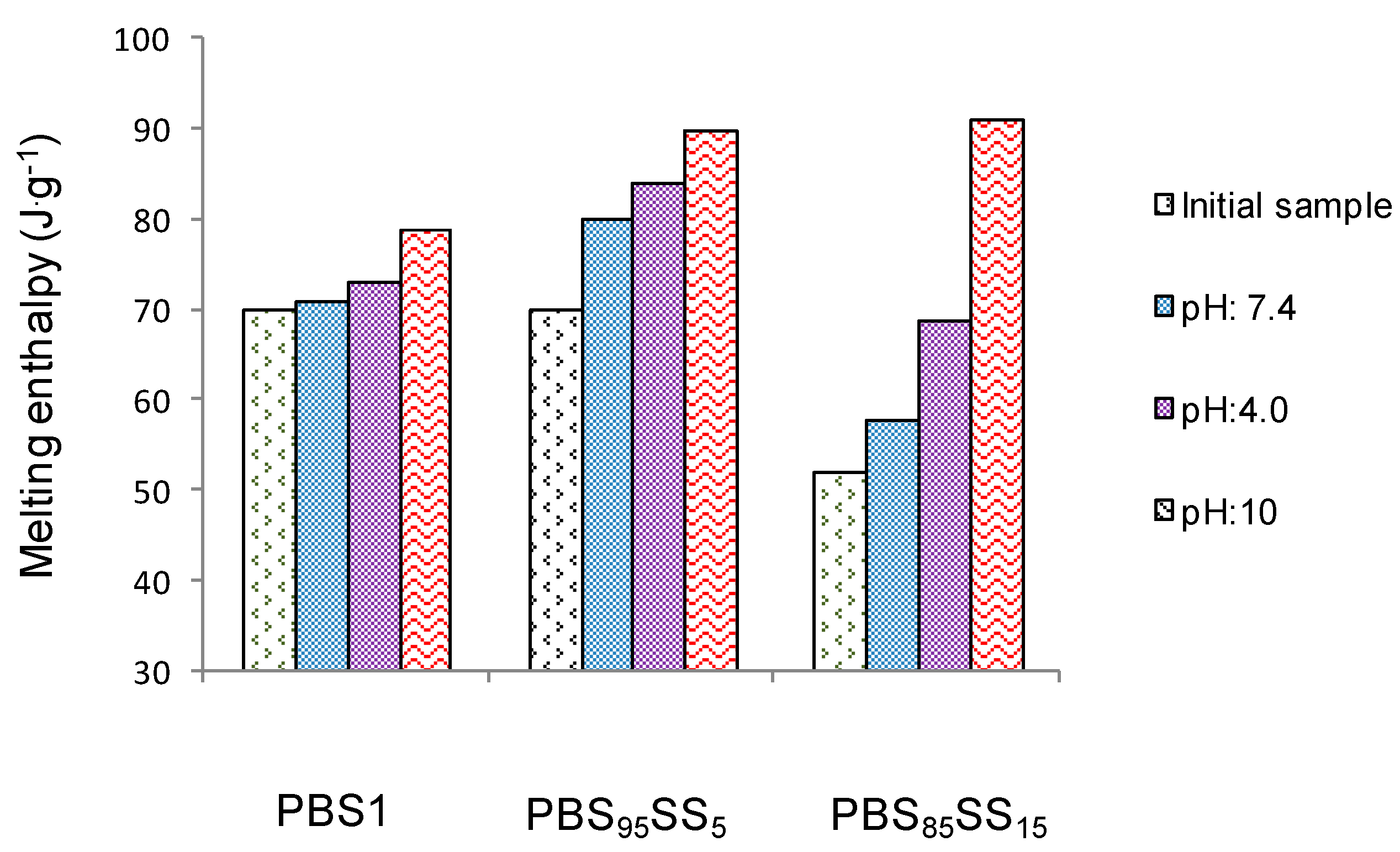
Melting enthalpies displayed by PBS1, PBS
95SS
5 and PBS
85SS
15 polyesters after incubation are compared in
Figure 5. It is noteworthy to highlight that crystallinity of the three polyesters increased as degradation proceeded, with the highest values attained when samples were incubated in basic conditions. Such increase in crystallinity is indicative that hydrolysis has taken place preferably in the more permeable amorphous phase, as it is usually observed in semicrystalline polymers [
32].
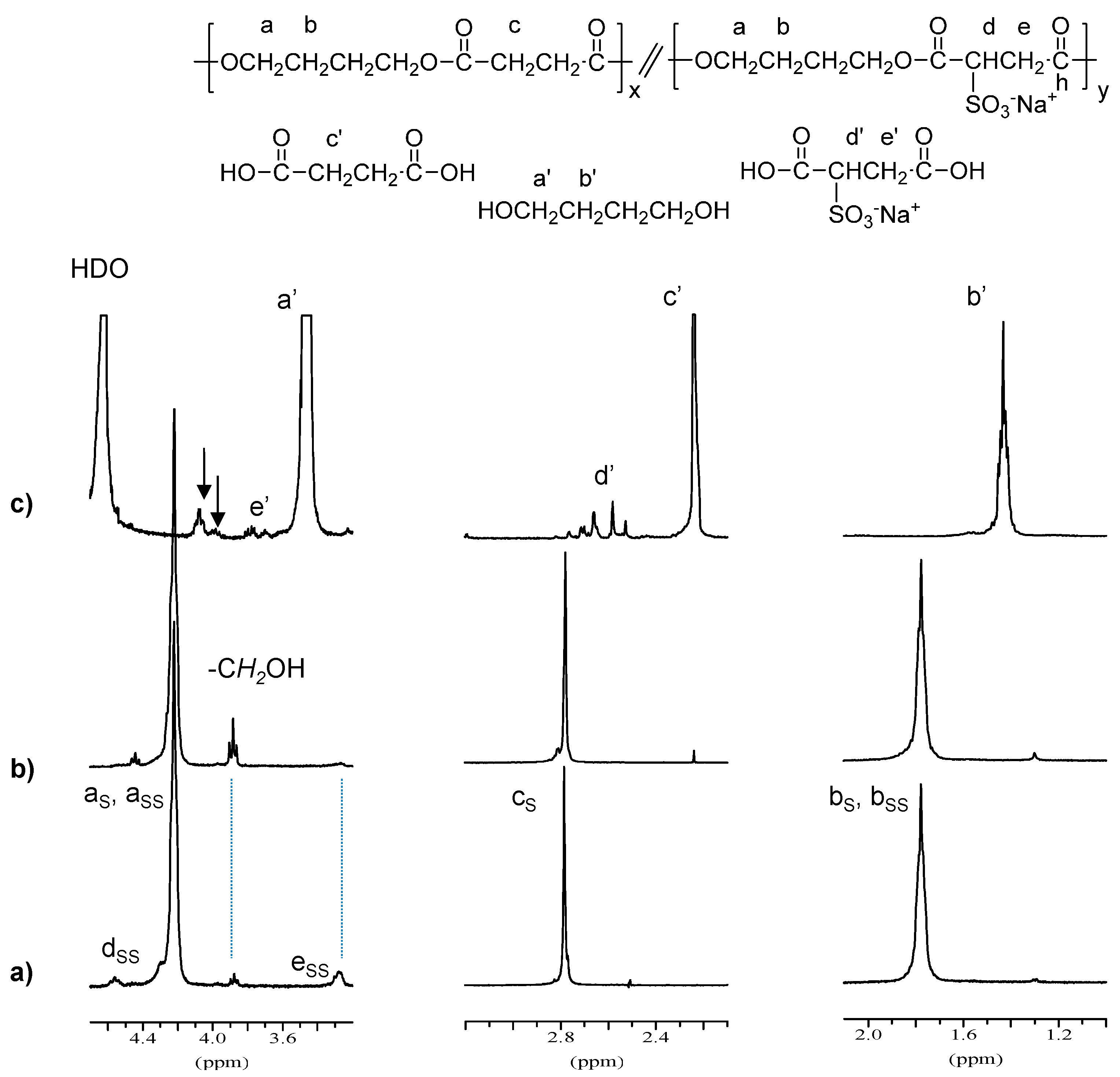
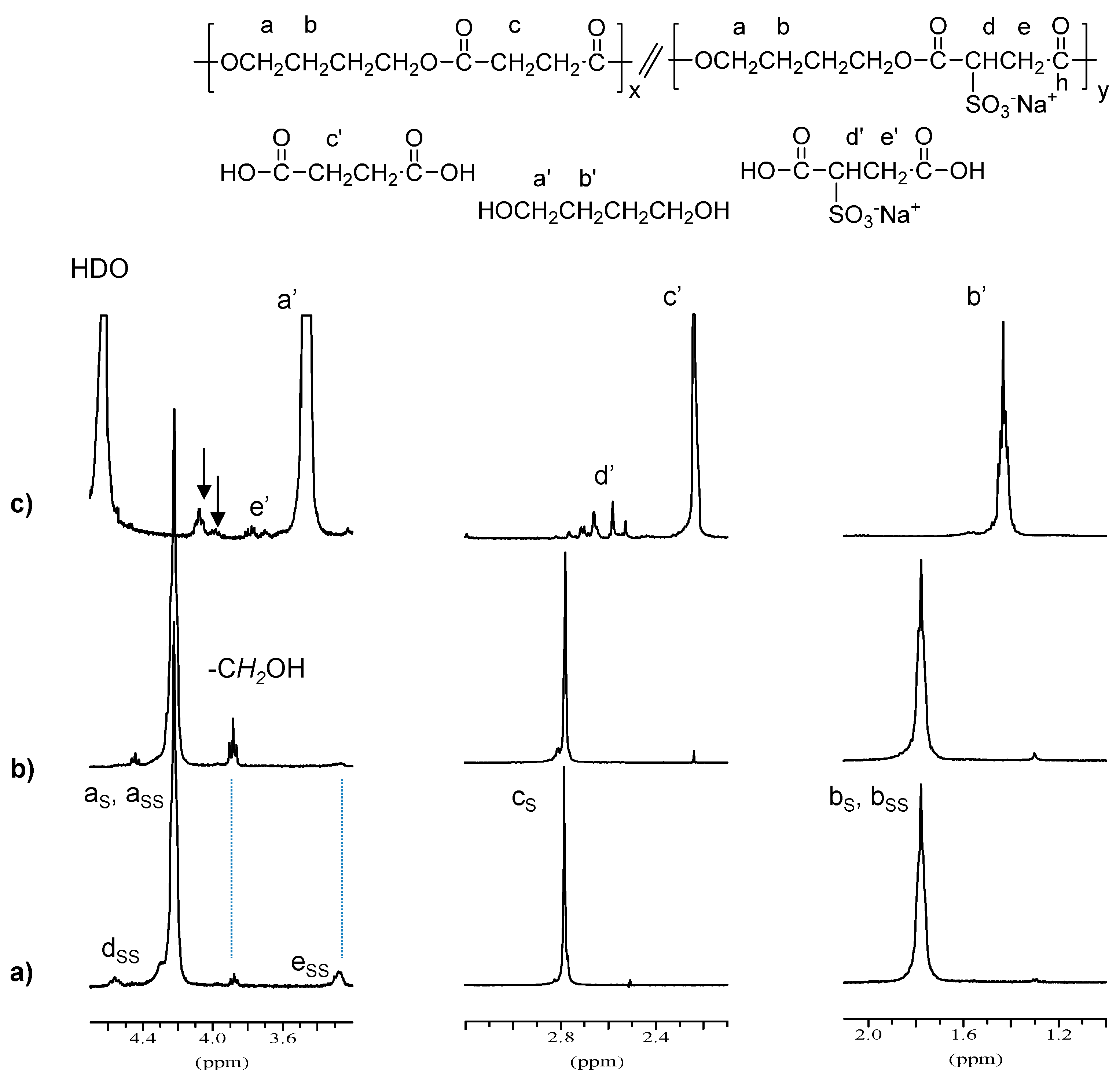
1H NMR spectra of PBS
85SS
15 residue and of the products released to the incubation medium after eight weeks of incubation are depicted in
Figure 6. It can be observed that the signals due to sulfonate units (e
SS and d
SS) almost disappeared in the degraded sample, and that signals arising from –CH
2OH end groups increased as a consequence of the hydrolysis process. The spectra of water soluble products showed signals corresponding to succinic and sulfonated succinic acids, 1,4-butanediol and short oligomers, which are more intense for samples incubated at basic pH where copolyesters underwent a more severe degradation.
Figure 5.
Melting enthalpies of PBS1, PBS95SS5 and PBS85SS15 after eight weeks of incubation and initial samples.
Figure 5.
Melting enthalpies of PBS1, PBS95SS5 and PBS85SS15 after eight weeks of incubation and initial samples.
Figure 6.
1H NMR spectra of PBS85SS15 (a) residue and (b) water soluble products (c) of PBS85SS15 degraded in aqueous buffer, pH 10 for eight weeks. The arrows indicate signals arising from oligomers generated upon hydrolysis.
Figure 6.
1H NMR spectra of PBS85SS15 (a) residue and (b) water soluble products (c) of PBS85SS15 degraded in aqueous buffer, pH 10 for eight weeks. The arrows indicate signals arising from oligomers generated upon hydrolysis.
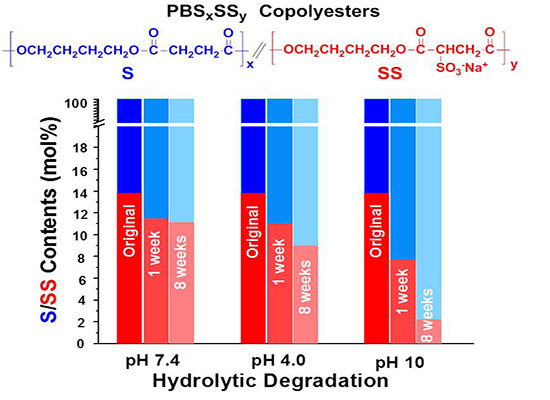
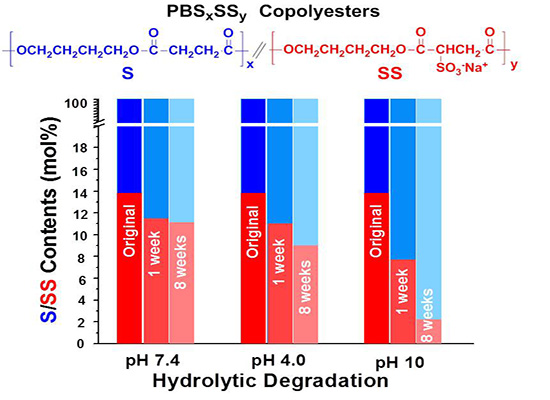
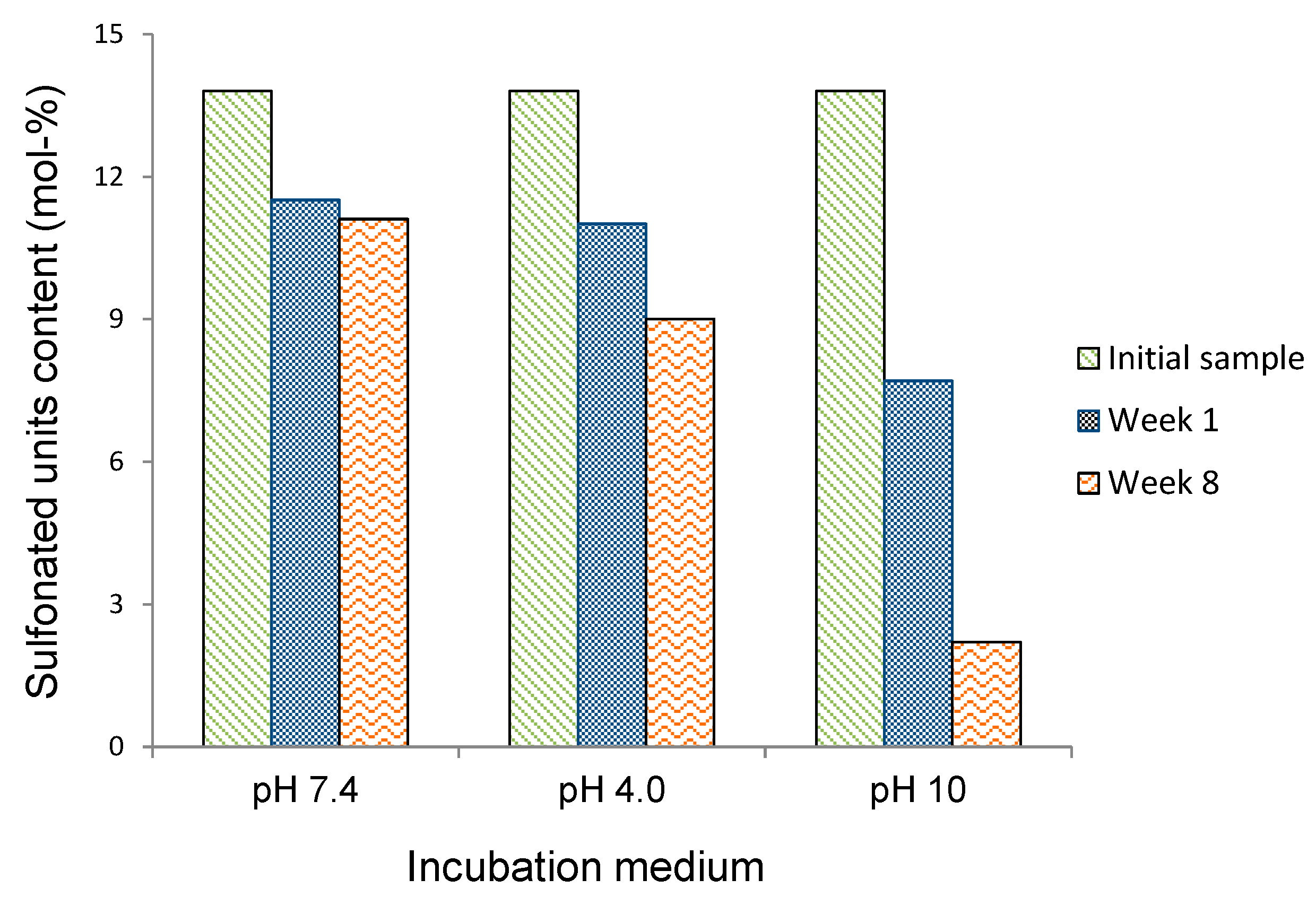
The evolution of the content in sulfonated units of the residual PBS
85SS
15 with time for an incubation period of eight weeks under the different essayed conditions is plotted in
Figure 7. These results are consistent with those obtained by sample weighing and GPC measurements and reveal that degradation involves preferably the hydrolysis of the SS units. The drastically accelerated degradation observed at pH 10 is ascribed to both the higher hydrophilicity of the copolyester and to the faster and more efficient hydrolytic degradation process that takes place in copolyesters at basic pH.
Figure 7.
Evolution of the content in sulfonated units of the residual PBS85SS15 along incubation time.
Figure 7.
Evolution of the content in sulfonated units of the residual PBS85SS15 along incubation time.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}