3.1. Protein Recovery from Emulsions
Table 1 summarizes the percent recovery of insulin in the supernatant aqueous phase after the homogenization step. Insulin recovery was significantly (
p < 0.05) affected by homogenization speed and methylene chloride with and without PLGA. In the presence of PLGA in methylene chloride, about 65% initial content of insulin was recovered after homogenization at 5000 rpm and decreased to 60% when the homogenization speed was increased to 10,000 rpm. This data suggests that the increased homogenization speed enhanced the amount of aggregates formed at the interfaces resulting in less soluble insulin in the supernatant. It has been suggested that emulsification by the homogenizer not only increases the total surface area of the organic solvent/water interface that serves as a hydrophobic adsorbent, but also generates turbulent flow for the diffusion of native protein molecules to the interface [
8]. It can be assumed here that increasing the homogenization speed from 5000 to 10,000 rpm further reduces the emulsion droplet size, increases the surface area, and diffusion of native protein to the interface leading to increased aggregation. A single extraction step was found to extract all the soluble insulin and subsequent extractions performed did not show any significant extraction of insulin from the interfaces. A 100% recovery would only be possible with a solvent having a high partition coefficient given there are no major interactions between insulin and methylene chloride. However, no solvent has been found or reported to recover proteins completely from the primary emulsion or interfaces.
Table 1.
Insulin recovered (%) from the primary emulsion with and without poly(lactic-co-glycolic acid) (PLGA) in methylene chloride (n = 4).
Table 1.
Insulin recovered (%) from the primary emulsion with and without poly(lactic-co-glycolic acid) (PLGA) in methylene chloride (n = 4).
| Sample | Homogenization Speed (rpm) | With PLGA (% ± SD) | Without PLGA (% ± SD) |
|---|
| Insulin | 5,000 | 65.1 ± 2.1 | 71.4 ± 1.4 |
| Insulin | 10,000 | 60.4 ± 1.7 | 68.2 ± 1.9 |
| Insulin + Zn2+ | 5,000 | 70.3 ± 0.8 | 74.2 ± 0.3 |
| Insulin + Zn2+ | 10,000 | 67.5 ± 1.1 | 70.6 ± 1.7 |
Recovery values increased significantly (
p < 0.05) in the absence of water-insoluble PLGA in methylene chloride. Presence of PLGA in methylene chloride enhances the aggregation rate of insulin, which is attributed to the adsorption of insulin to the hydrophobic PLGA surfaces and a decrease in the o/w interfacial tension as compared to methylene chloride without PLGA [
10]. With the addition of zinc, significantly (
p < 0.05) improved recovery values were observed at both homogenization speeds, irrespective of the presence of PLGA in methylene chloride. Under all the experimental conditions, a fraction of water-insoluble insulin precipitates resided at water–methylene chloride interfaces. Since the formed aggregates are water insoluble, low recovery values were observed.
The interfaces formed with water–methylene chloride are due to steep concentration gradients of the solvents and in our case methylene chloride is high in concentration making the interfaces highly hydrophobic. The interfaces formed are said to be hydrophobic because: (a) Methylene chloride is a highly non-polar/hydrophobic solvent and (b) The volume of methylene chloride far surpasses the volume of water used in the primary emulsification step (
i.e., 2 mL
vs. 0.1 mL). With this being said, the stability of a protein at w/o interfaces can be determined by placing an aqueous solution of the protein in an organic phase without any polymer [
4,
5,
19]. Nonetheless, the presence of a polymer in the organic phase cannot be neglected when studying issues confronted during microsphere preparation. Breaking emulsions in the presence of a polymer becomes difficult due to the high viscosity of the emulsion. In addition, polymers such as PLGA tend to precipitate out once the aqueous buffer is added in excess into the emulsion, and the encapsulated protein may precipitate together with the polymer [
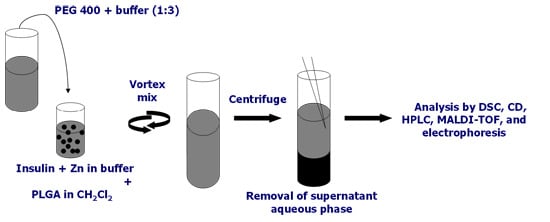
11]. To overcome these obstacles, extraction of protein using PEG 400 will completely break the emulsion; thereby more protein encapsulated in the emulsion droplets is released. This extraction method allows us to study the behavior of insulin and other proteins at the w/o interface in the presence of polymer in the organic phase.
3.2. Conformational Stability of Insulin
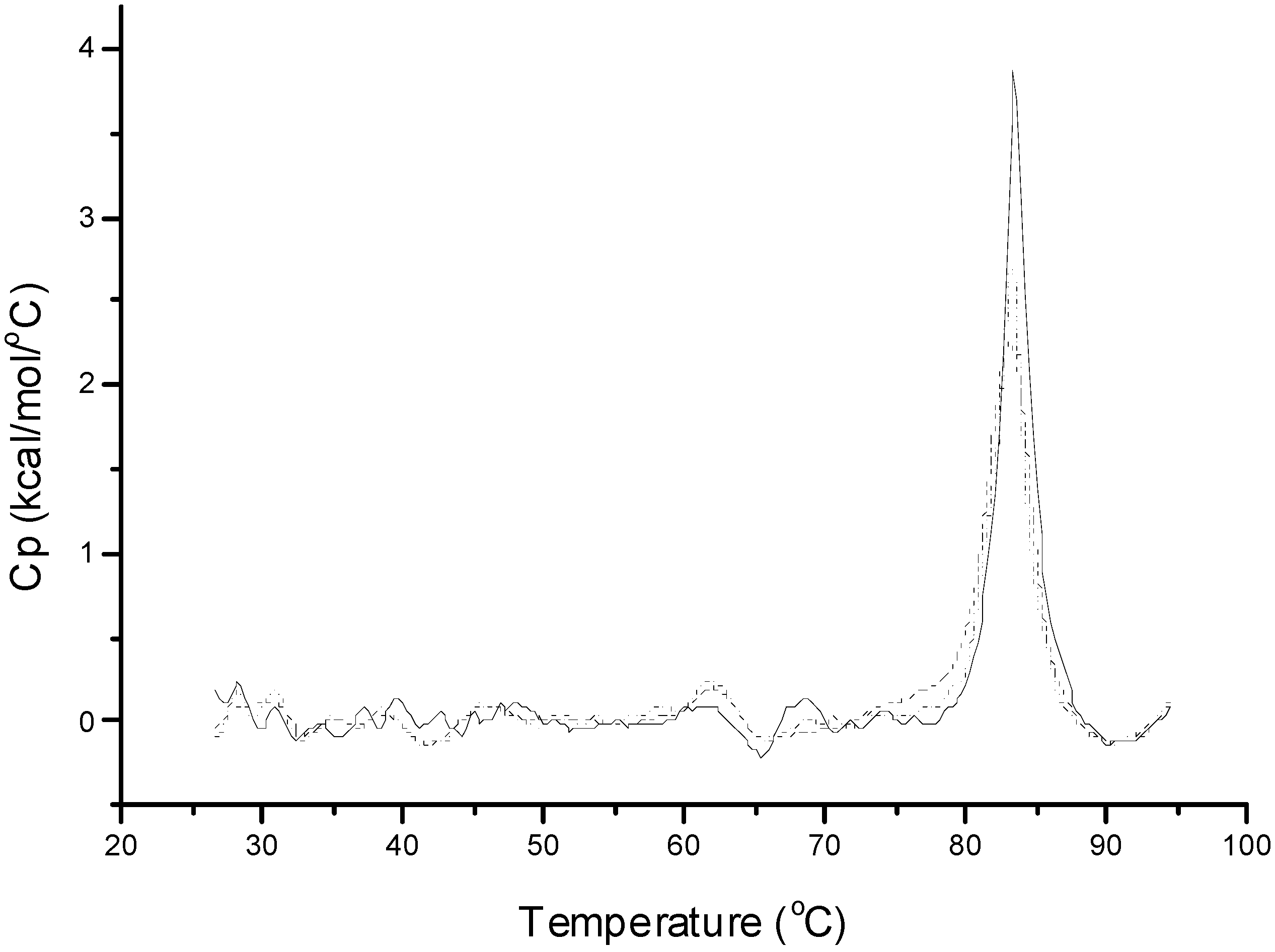
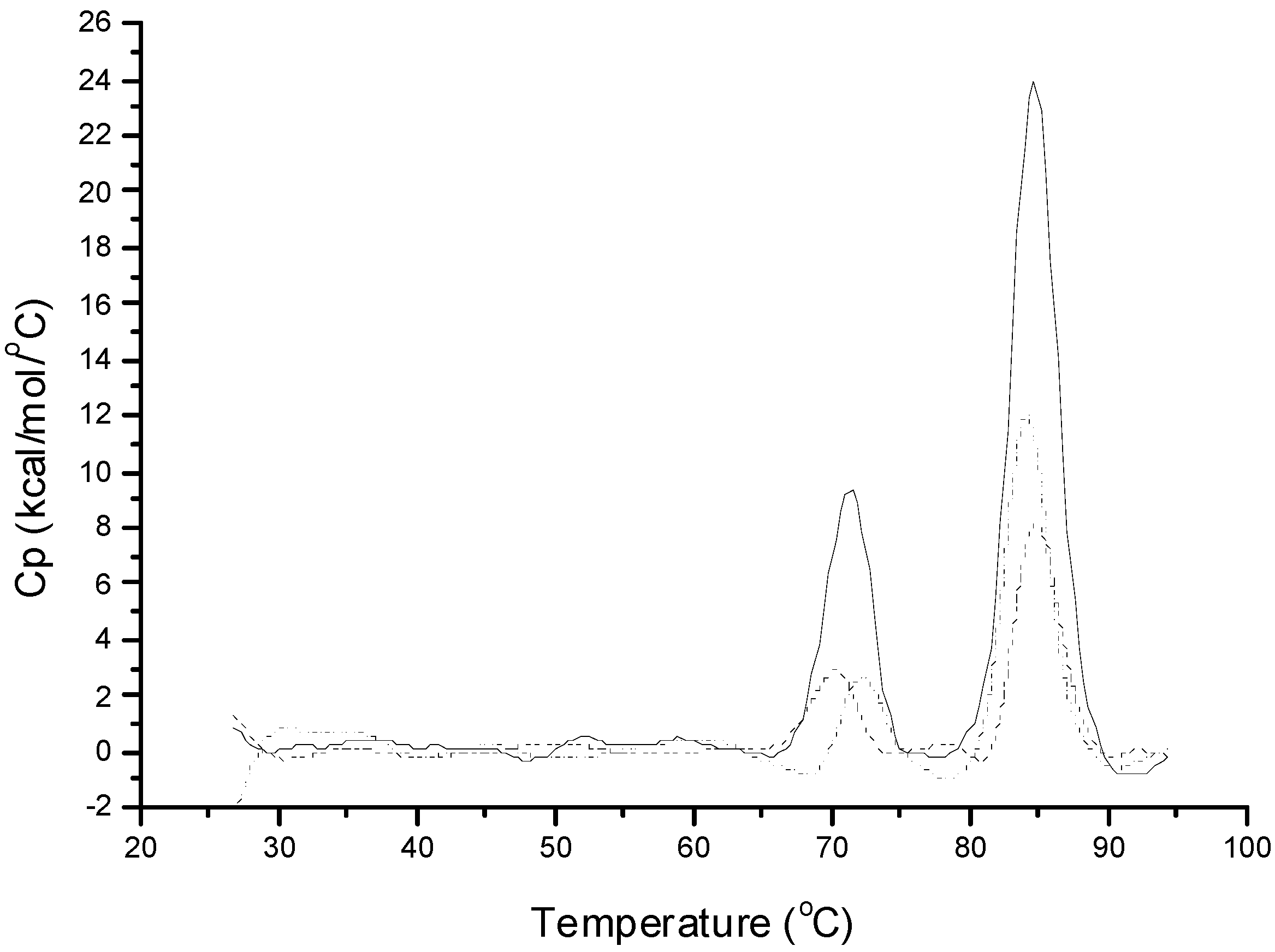
Figure 1 shows the DSC thermogram of native insulin as obtained from the supplier in 10 mM phosphate buffer (pH 7.4). Two distinct endothermic transition peaks were observed with
Tm’s at 72.1 and 84.8 °C. Based on previously published reports, the first and second transition peaks represent dimer and hexamer denaturation, respectively [
20]. Insulin recovered in the aqueous phase after emulsifying with methylene chloride in the presence of PLGA at 5000 and 10,000 rpm also showed two transition peaks, but with decreased magnitudes. To assess the retention of insulin native conformation, Δ
H was used since it provides valuable information on protein conformation [
21]. A decreased Δ
H was observed for the extracted insulin when compared to native insulin (
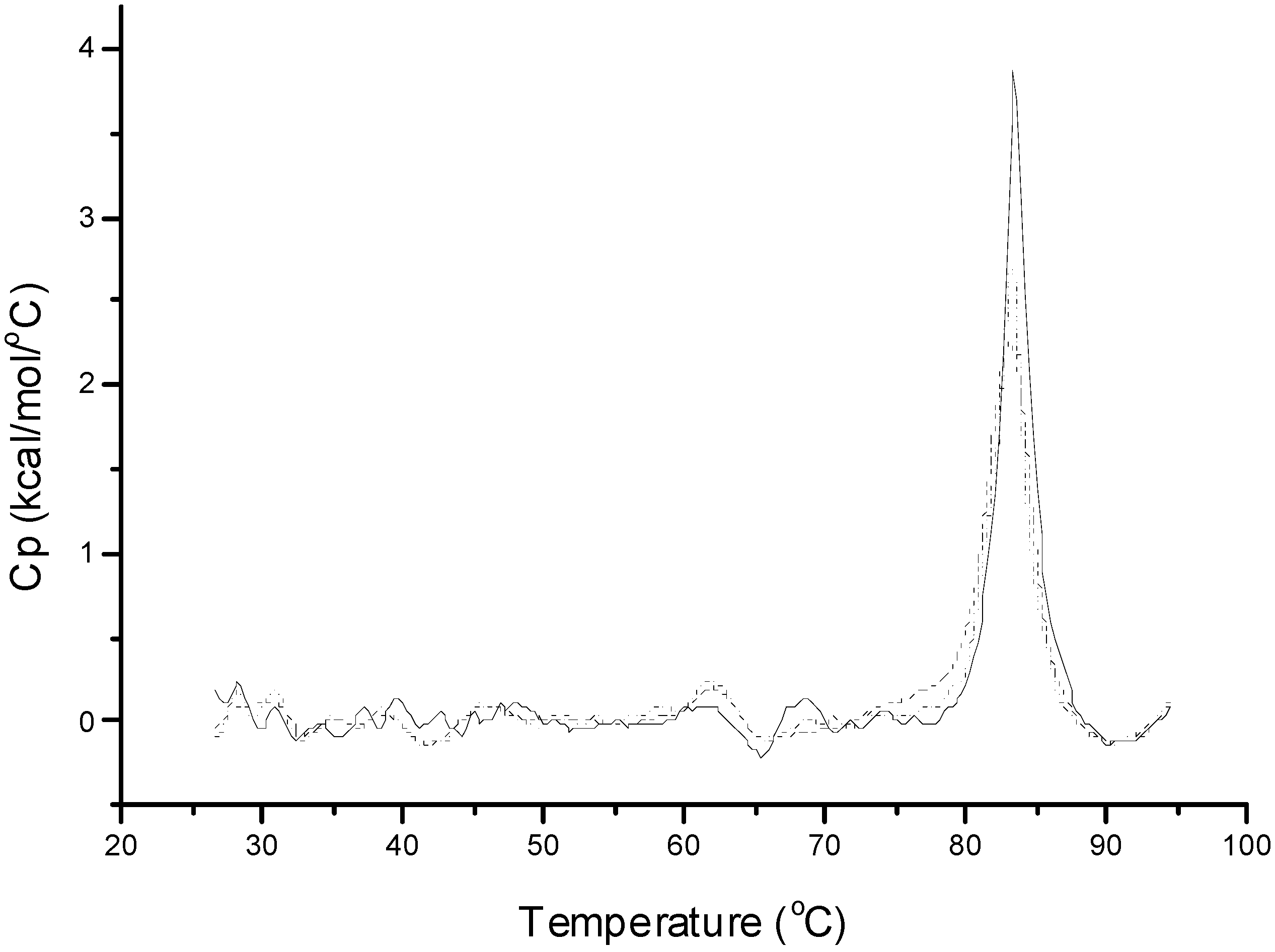
Table 2), indicating a partially denatured conformation. This decreased conformational stability can be due to the adsorption of insulin at the interfaces and to the hydrophobic PLGA surfaces. With the addition of zinc to insulin at a molar ratio of 5:6, a single endothermic transition at 83 °C for hexamer denaturation was observed (
Figure 2) which was in well accorded with the previously reported data [
20]. Similar transition peaks for insulin emulsified with the addition of zinc at different homogenization speeds was observed and their Δ
H values were close to that of hexameric insulin indicating the preservation of conformational structure. The strategy of stabilizing insulin during emulsification by converting insulin monomers to hexamers prevents contacts between the protein and the water–organic solvent interface and protein unfolding. This strategy is supported by previous reports that the hexamer conformational stability prevents denaturation at hydrophobic surfaces [
22,
23,
24].
Figure 1.
Differential scanning calorimetry (DSC) thermograms of 0.6 mM insulin in phosphate buffer (pH 7.4, 10 mM) and insulin extracted after homogenization. Full line, native insulin solution; dashed line, insulin extracted after homogenization at 5000 rpm; and dotted line, insulin extracted after homogenization at 10,000 rpm.
Figure 1.
Differential scanning calorimetry (DSC) thermograms of 0.6 mM insulin in phosphate buffer (pH 7.4, 10 mM) and insulin extracted after homogenization. Full line, native insulin solution; dashed line, insulin extracted after homogenization at 5000 rpm; and dotted line, insulin extracted after homogenization at 10,000 rpm.
Table 2.
Thermal parameters of recovered insulin after emulsification with methylene chloride in the presence of PLGA (n = 3).
Table 2.
Thermal parameters of recovered insulin after emulsification with methylene chloride in the presence of PLGA (n = 3).
| Sample | Homogenization Speed (rpm) | Tm1 (°C ± SD) | Tm2 (°C ± SD) | ΔHtot (Kcal/mol ± SD) |
|---|
| Insulin control | – | 72.1 ± 0.1 | 84.8 ± 0.3 | 248 ± 3.2 |
| Insulin | 5,000 | 69.9 ± 0.4 | 84.2 ± 0.1 | 189 ± 0.9 |
| Insulin | 10,000 | 72.7 ± 0.2 | 85.0 ± 0.2 | 172 ± 2.8 |
| Insulin + Zn2+ | – | – | 83.2 ± 0.4 | 193 ± 1.8 |
| Insulin + Zn2+ | 5,000 | – | 82.9 ± 0.1 | 182 ± 0.6 |
| Insulin + Zn2+ | 10,000 | – | 83.1 ± 0.2 | 177 ± 2.1 |
Figure 2.
DSC thermograms of 0.6 mM insulin with 0.3 mM zinc acetate in phosphate buffer (pH 7.4, 10 mM) and insulin extracted after homogenization. Full line, native insulin solution with zinc; dotted line, insulin extracted after homogenization at 5000 rpm; and dashed line, insulin extracted after homogenization at 10,000 rpm.
Figure 2.
DSC thermograms of 0.6 mM insulin with 0.3 mM zinc acetate in phosphate buffer (pH 7.4, 10 mM) and insulin extracted after homogenization. Full line, native insulin solution with zinc; dotted line, insulin extracted after homogenization at 5000 rpm; and dashed line, insulin extracted after homogenization at 10,000 rpm.
Irrespective of the presence of zinc, emulsions formed at a homogenization speed of 10,000 rpm showed significantly (
p < 0.05) decreased Δ
H values. The
Tm and Δ
H values for insulin in the absence of PLGA during emulsification are compiled in
Table 3. The
Tm values were found to remain close to the native insulin. Significantly (
p < 0.05) increased Δ
H values were observed for insulin homogenized in the absence of PLGA when compared to insulin homogenized in the presence of PLGA. The use of PEG 400 to break the emulsion and extract insulin can affect the
Tm values. As reported previously, the
Tm values for lysozyme were found to decrease due to the addition of PEG 400 [
11]. However, a marked decrease in the
Tm for insulin due to the addition of PEG 400 was not noticed in this study.
Table 3.
Thermal parameters of recovered insulin after emulsification with methylene chloride in the absence of PLGA (n = 3)
Table 3.
Thermal parameters of recovered insulin after emulsification with methylene chloride in the absence of PLGA (n = 3)
| Sample | Homogenization Speed (rpm) | Tm1 (°C ± SD) | Tm2 (°C ± SD) | ΔHtot (Cal/mol ± SD) |
|---|
| Insulin control | – | 71.5 ± 0.4 | 82.6 ± 0.8 | 245 ± 4.0 |
| Insulin | 5,000 | 70.3 ± 0.5 | 83.1 ± 1.2 | 201 ± 2.7 |
| Insulin | 10,000 | 71.2 ± 0.7 | 82.8 ± 0.5 | 183 ± 1.9 |
| Insulin + Zn2+ | – | – | 83.8 ± 0.8 | 187 ± 0.2 |
| Insulin + Zn2+ | 5,000 | – | 81.4 ± 0.8 | 182 ± 1.7 |
| Insulin + Zn2+ | 10,000 | – | 82.6 ± 1.5 | 176 ± 2.3 |
3.3. Stability of Insulin
One of the major concerns for the encapsulation of proteins into biodegradable microspheres prepared by the w/o/w technique is the denaturation of proteins during the fabrication process. Addition of the aqueous protein solution to the organic polymer solution followed by shear stress makes the organic solvent molecules diffuse into the aqueous phase. The organic solvent can bind to the protein directly by hydrophobic interaction or alter the ionic strength inside the aqueous medium, fostering the destabilization of protein molecules [
25]. It was previously shown that the destabilizing effect of the organic solvents depends mainly on how the protein is incorporated (aqueous or solid form) and the organic solvent chosen for the dissolution of the polymer [
26].
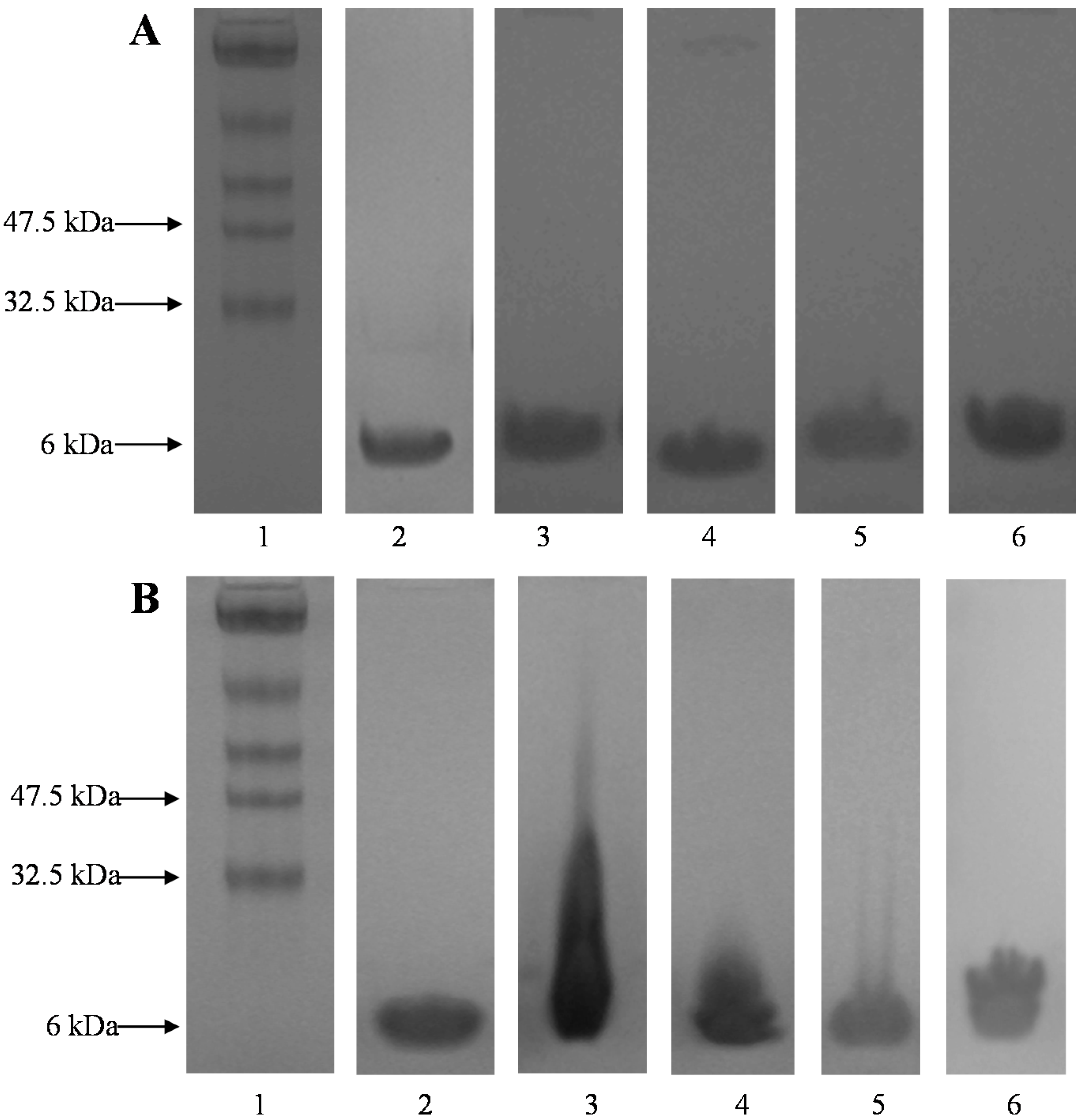
The primary structure of the extracted insulin was assessed by SDS-PAGE.
Figure 3A,B show the SDS-PAGE gel patterns of insulin extracted into the aqueous phase and insulin obtained from w/o interfaces after various emulsification conditions, respectively. All the experiments were carried out in the presence of PLGA in methylene chloride. Analysis of the supernatant aqueous phase containing insulin under all the emulsifications conditions showed a single intense band around 6 kDa, suggesting the preservation of primary structure (
Figure 3A). After the de-emulsification and extraction step, water insoluble white protein precipitates were observed at the w/o interfaces. The precipitates were carefully collected and analyzed to observe the nature of aggregation taking place at the interfaces. Native insulin showed a single intense band at 6 kDa and insulin obtained from the interfaces also showed a main band around 6 kDa (
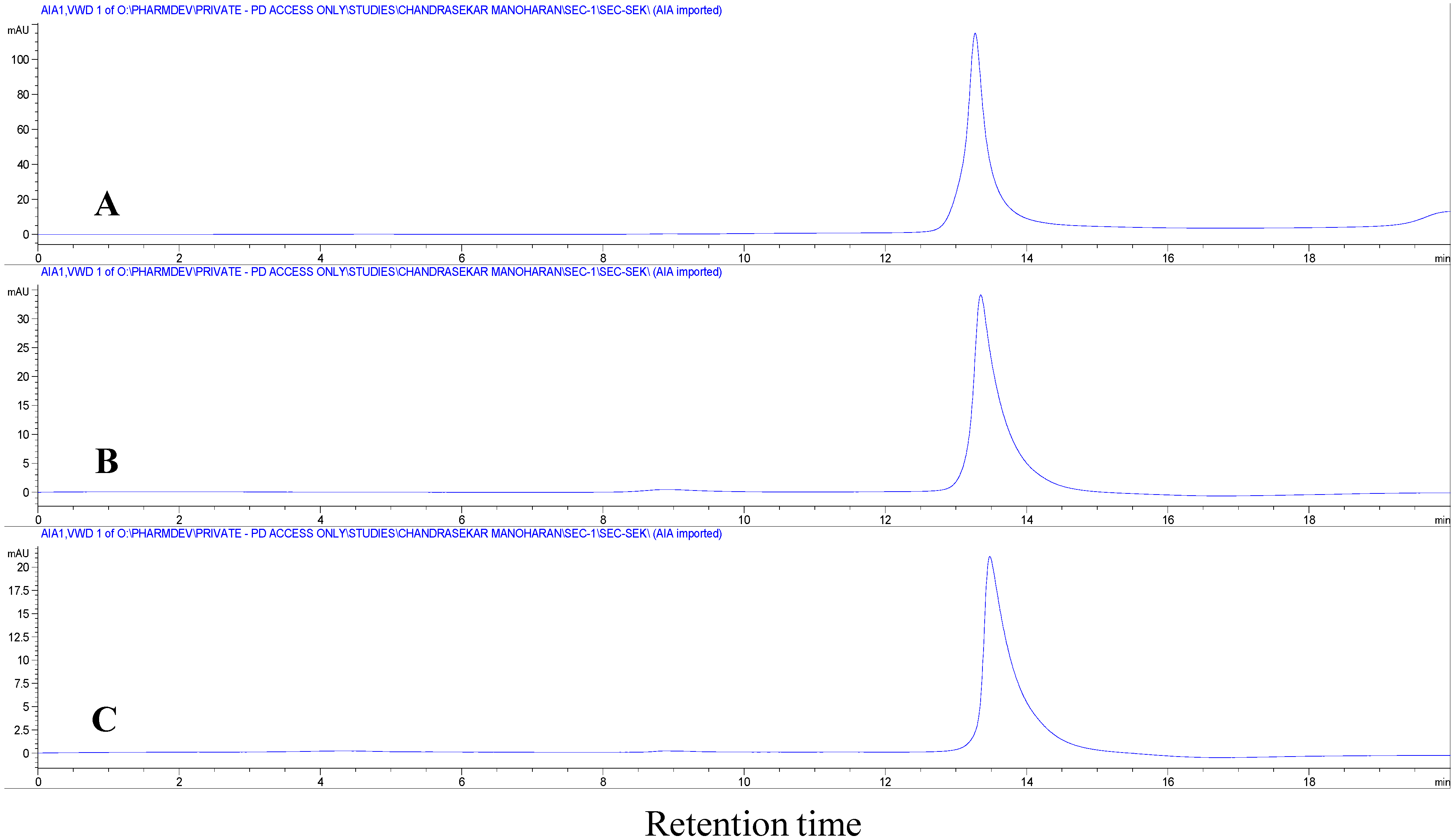
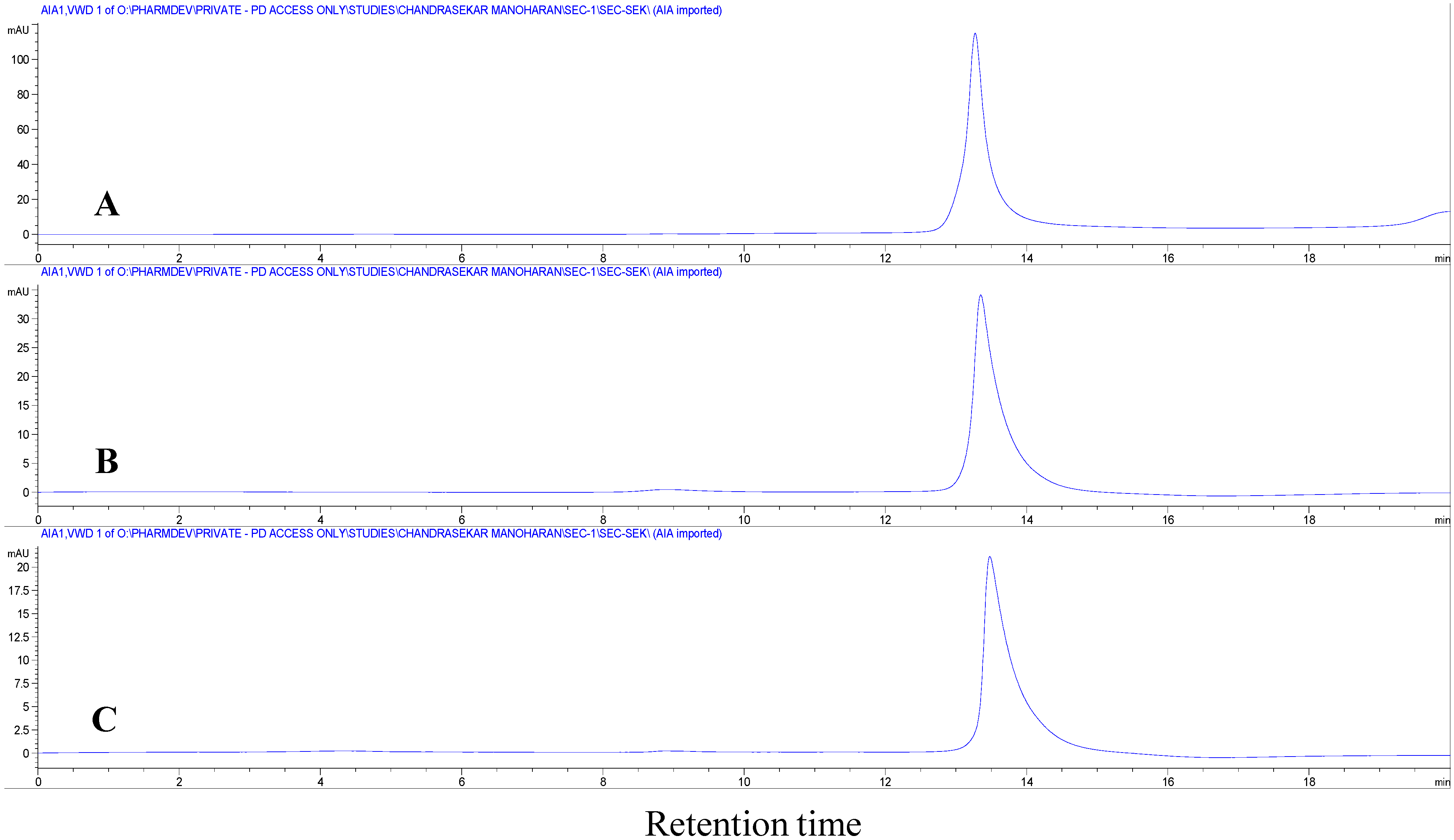
Figure 3B). However, the bands were not precise and they appeared as a smear. This clearly shows that the precipitates formed at the interfaces are insoluble insulin aggregates and not un-dissolved insulin. In addition, if there is any un-dissolved insulin, it should be dissolved during the extraction process carried out with 3 mL of phosphate buffer and 1 mL of PEG 400 leaving no insulin precipitates at the interfaces. The obtained precipitates dissolved completely when treated with 0.5% SDS containing aqueous solution, suggesting that the water-insoluble precipitates were aggregates of non-covalent nature. To further confirm the non-covalent nature of these aggregates, the precipitates dissolved in 0.5% SDS containing aqueous solution was subjected to SE-HPLC. In the SE-HPLC chromatograms, insulin extracted after homogenization at the highest speed (10,000 rpm) showed a single peak for monomeric insulin similar to native insulin (
Figure 4). Absence of peaks for higher molecular weight aggregates (
i.e., covalent dimers) confirmed the non-covalent nature of aggregation occurring between insulin molecules at the w/o interfaces.
Figure 3.
(A) Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) of insulin extracted into the aqueous phase; and (B) SDS-PAGE of insulin obtained from the interfaces after various emulsification conditions. Lane 1: standard protein markers, lane 2: native insulin, lane 3: insulin obtained after homogenization at 5000 rpm, lane 4: insulin obtained after homogenization at 10,000 rpm, lane 5: insulin with zinc after homogenization at 5000 rpm, and lane 6: insulin with zinc after homogenization at 10,000 rpm.
Figure 3.
(A) Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) of insulin extracted into the aqueous phase; and (B) SDS-PAGE of insulin obtained from the interfaces after various emulsification conditions. Lane 1: standard protein markers, lane 2: native insulin, lane 3: insulin obtained after homogenization at 5000 rpm, lane 4: insulin obtained after homogenization at 10,000 rpm, lane 5: insulin with zinc after homogenization at 5000 rpm, and lane 6: insulin with zinc after homogenization at 10,000 rpm.
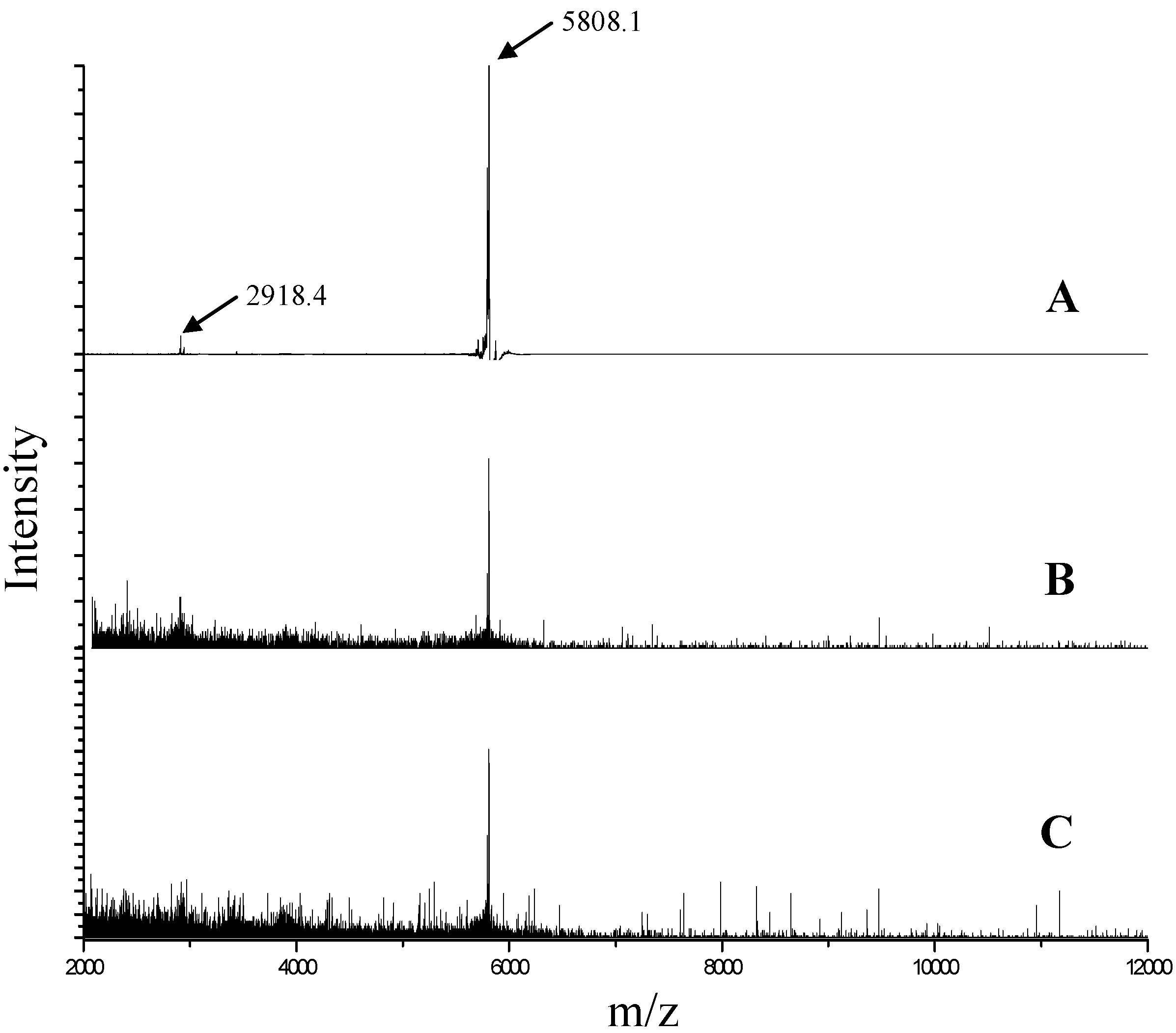
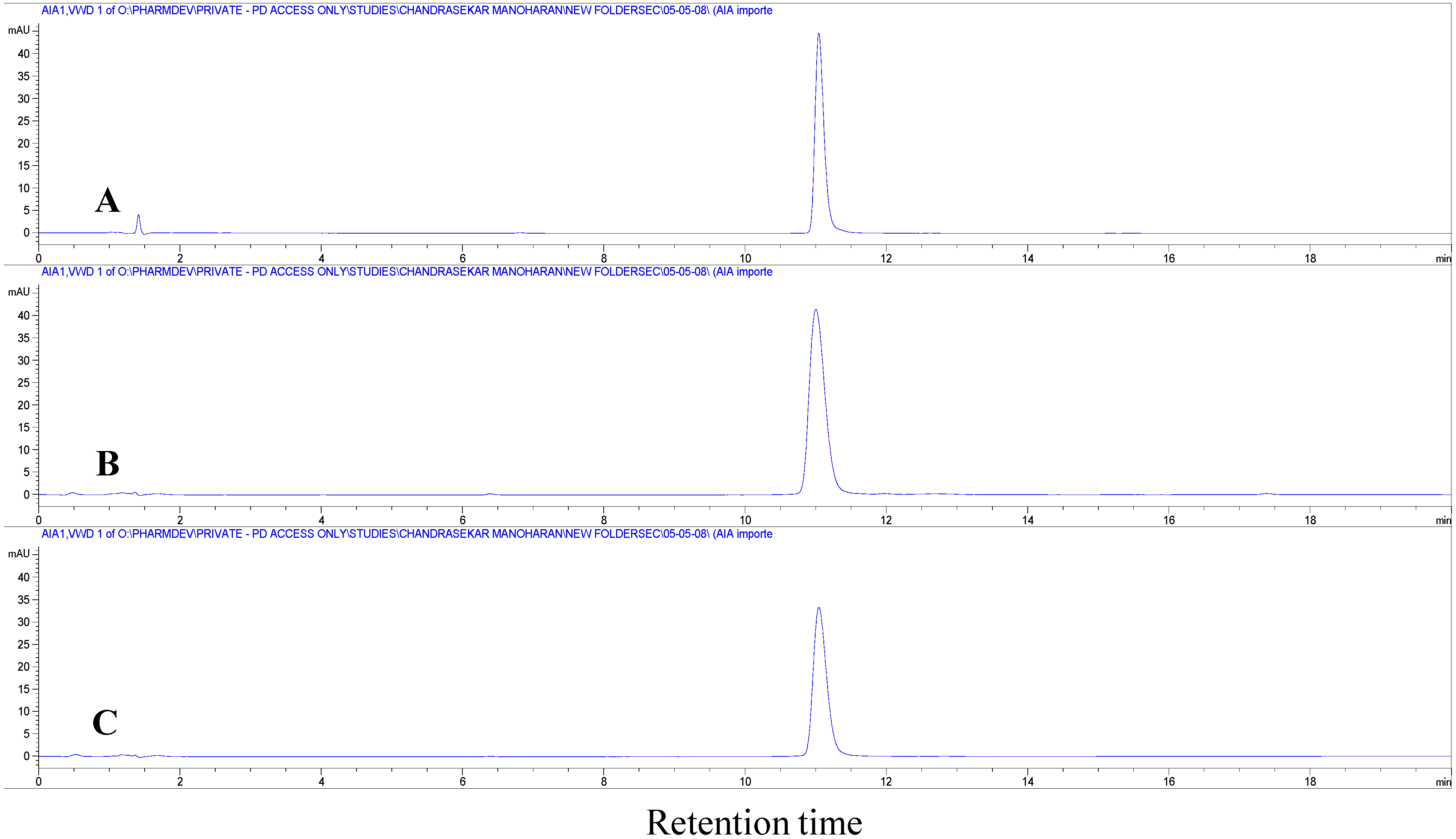
The extent of insulin degradation during the primary emulsification step that was carried out at 10,000 rpm in the presence of PLGA in methylene chloride was assessed by HPLC and MALDI-TOF MS. Comparison of chromatograms of insulin recovered in the supernatant aqueous phase with the chromatogram of native insulin indicated that insulin did not degrade after homogenization irrespective of the presence of zinc (
Figure 5). All the chromatograms showed an insulin monomer peak at a retention time around 11 min. No additional peaks were observed which provided evidence that insulin did not degrade into products of different chemical nature such as the A21 desamido-insulin. Similar results were obtained from MALDI-TOF MS experiments. Analysis of the extracted insulin by MALDI-TOF MS is shown in
Figure 6. In the mass spectrum of native insulin, a high intensity peak at m/z 5808 corresponds to the [M + H]
+ ion, and a low intensity peak at
m/
z 2918 corresponds to the [M + H]
2+ ion. The spectra of insulin and insulin with added zinc extracted after homogenization at 10,000 rpm showed peaks identical to native insulin.
Figure 4.
Size exclusion high performance liquid chromatography (SE-HPLC) chromatograms of (A) native insulin; (B) insulin precipitates obtained after homogenization at 10,000 rpm; and (C) insulin precipitates with added zinc after homogenization at 10,000 rpm. The precipitates were dissolved in 0.5% sodium dodecyl sulfate (SDS) aqueous solution prior to analysis.
Figure 4.
Size exclusion high performance liquid chromatography (SE-HPLC) chromatograms of (A) native insulin; (B) insulin precipitates obtained after homogenization at 10,000 rpm; and (C) insulin precipitates with added zinc after homogenization at 10,000 rpm. The precipitates were dissolved in 0.5% sodium dodecyl sulfate (SDS) aqueous solution prior to analysis.
Figure 5.
High performance liquid chromatography (HPLC) chromatograms of (A) native insulin; (B) insulin extracted after homogenization at 10,000 rpm; and (C) insulin with zinc extracted after homogenization at 10,000 rpm.
Figure 5.
High performance liquid chromatography (HPLC) chromatograms of (A) native insulin; (B) insulin extracted after homogenization at 10,000 rpm; and (C) insulin with zinc extracted after homogenization at 10,000 rpm.
Figure 6.
Matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrum of (A) native insulin; (B) insulin extracted after homogenization at 10,000 rpm; and (C) insulin with zinc extracted after homogenization at 10,000 rpm.
Figure 6.
Matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrum of (A) native insulin; (B) insulin extracted after homogenization at 10,000 rpm; and (C) insulin with zinc extracted after homogenization at 10,000 rpm.
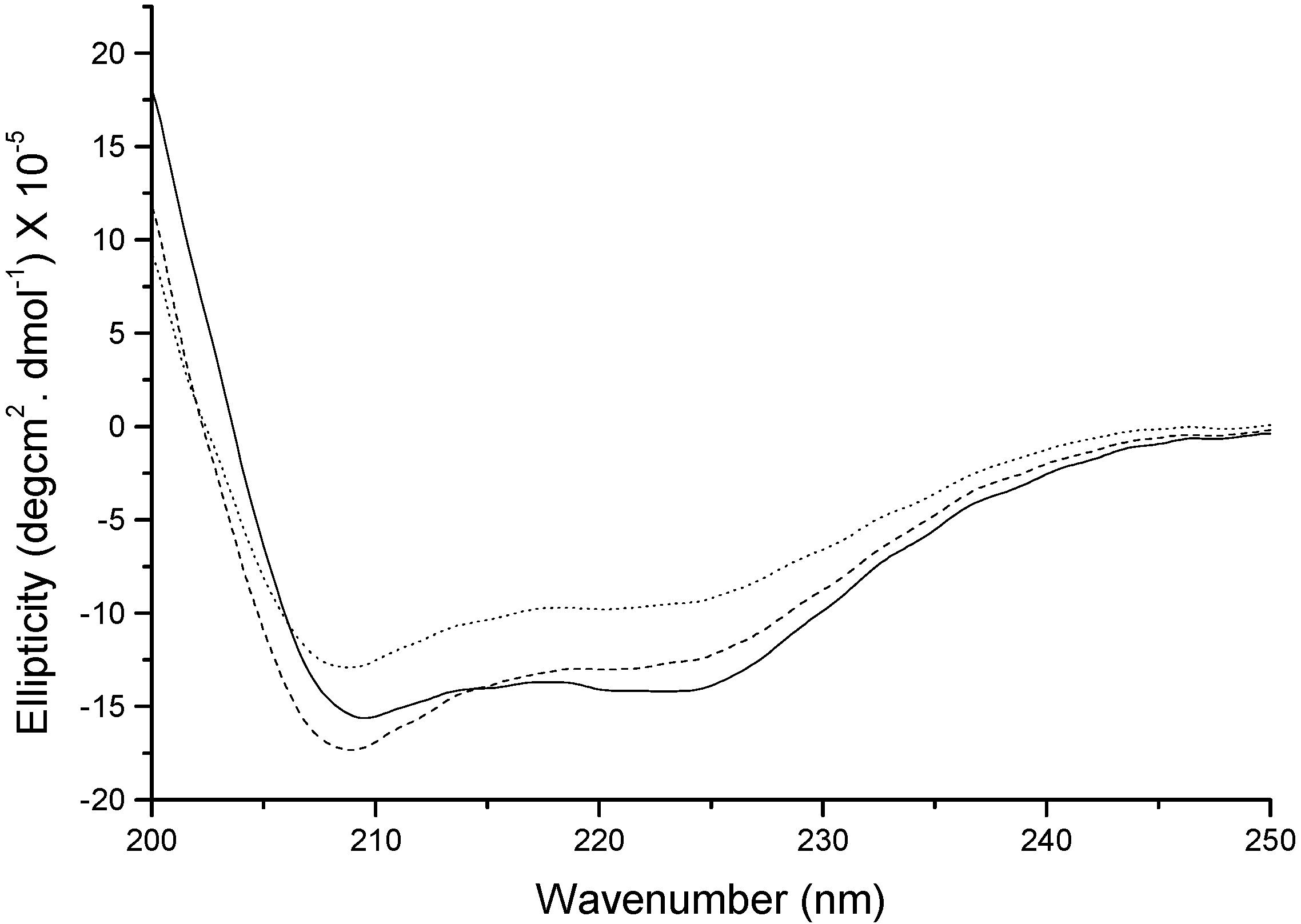
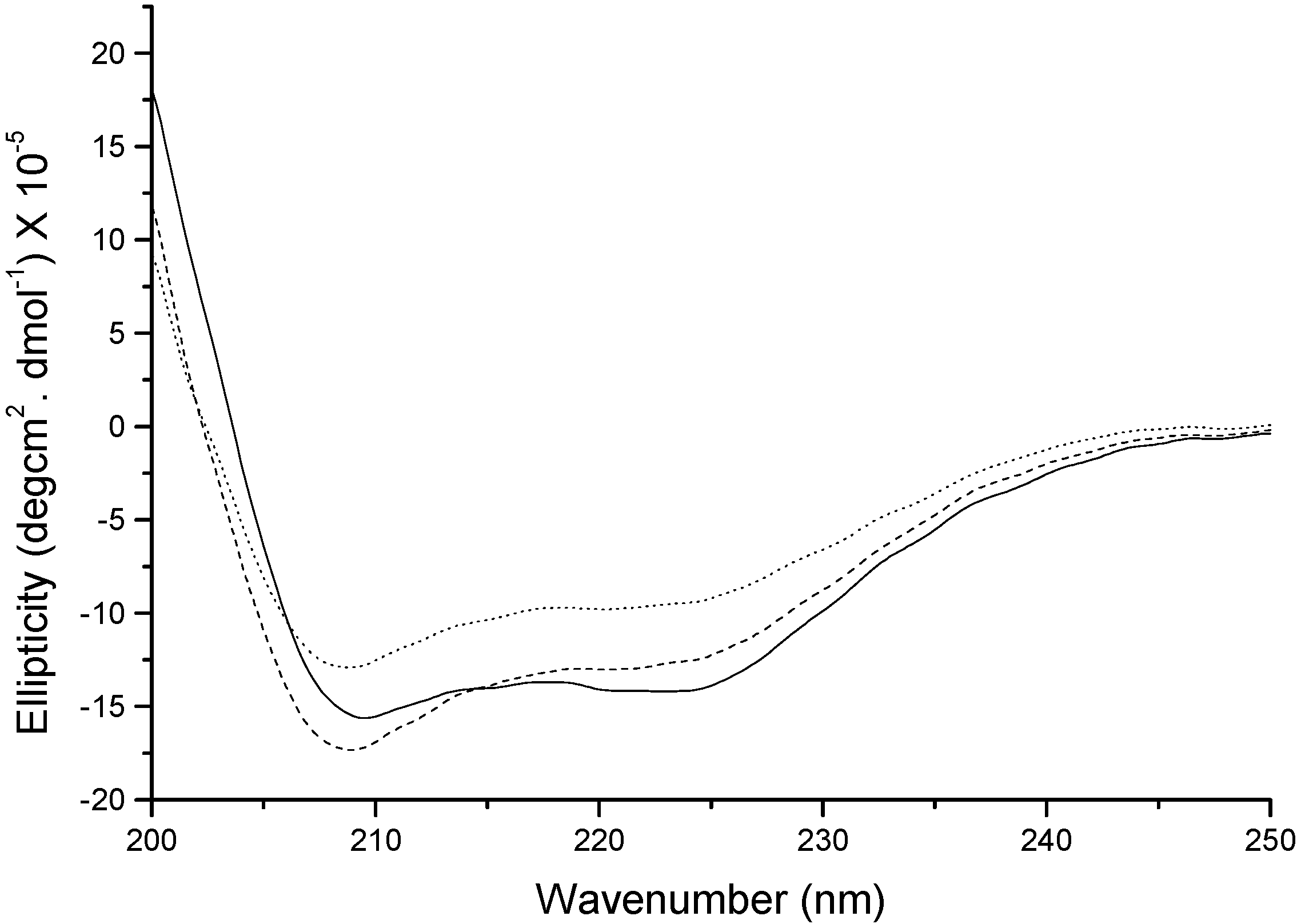
Figure 7 shows the far UV-CD spectrum of native insulin and insulin extracted after homogenization at 10,000 rpm with PLGA in methylene chloride. Native insulin showed two minima for α-helix at 208 and 223 nm. The spectrum of insulin homogenized at 10,000 rpm was found to be different from the native insulin spectrum. The spectrum showed characteristic α-helix minima peak at 208 and 223 with decreased magnitude indicating a partial loss in the secondary structure. With the addition of zinc at 10,000 rpm homogenization speed, insulin spectrum resembled closely to that of native insulin demonstrating the structural stabilizing effect of zinc.
Figure 7.
Far UV-circular dichroism (CD) of insulin extracted in the aqueous phase. Solid line: native insulin, dotted line: insulin extracted after homogenization at 10,000 rpm, and dashed line: insulin with zinc extracted after homogenization at 10,000 rpm.
Figure 7.
Far UV-circular dichroism (CD) of insulin extracted in the aqueous phase. Solid line: native insulin, dotted line: insulin extracted after homogenization at 10,000 rpm, and dashed line: insulin with zinc extracted after homogenization at 10,000 rpm.
The exposure of insulin to water-methylene chloride interfaces, hydrophobic PLGA surfaces, and the homogenization step did not provoke the formation of covalent insulin aggregates or other degradation products as assessed by SDS-PAGE, MALDI-TOF MS, HPLC, and SE-HPLC. No significant (
p > 0.05) difference in results was seen for insulin homogenized at 5000 (data not shown) and 10,000 rpm. Two possible reasons can be attributed to this increased stability of insulin. First, insulin used in this study was not strictly zinc-free. Before the addition of extra zinc to insulin, the zinc content was about 0.4% which when calculated accounts for nearly two molecules of zinc for six molecules of insulin. At this zinc concentration, more than 75% of insulin exists in the hexameric state [
27] and the hexamer becomes the dominant species. In our case, the hexamers may have occupied the w/o interfaces and prevented the exposure of hydrophobic surfaces to the interfaces which leads to protein aggregation and denaturation. With the addition of zinc to insulin at a molar ratio of 5:6, all the insulin molecules are expected to exist in the form of hexamers and hence a greater stabilizing effect is noticed. In the absence of zinc, monomer population prevails and exposure to w/o interfaces leads to unfolding of the monomers and subsequent aggregation. It has been reported that the aggregation rate of monomeric insulin is proportional to insulin concentration [
28,
29]. Second, irrespective of the homogenization speed, the duration of homogenization was 1 min for all the experiments. The reduced homogenization time decreased the residing time for the protein molecules at the w/o interface. Kwon
et al. [
10] showed that aggregation of zinc-free insulin took place within 3 min in the presence of only methylene chloride and within ~1.5 min in the presence of 200 mg/mL PLGA in methylene chloride. In this study, it is obvious that the reduced homogenization time did not allow any degradation or aggregation to take place during the emulsification step by decreasing the residing time for the protein molecules at the w/o interface. Insulin aggregates formed at the interfaces were of non-covalent nature and the results are consistent with previous reports [
10]. No interactions between insulin and PEG were observed. Finally, evaluation of the therapeutic activity of insulin encapsulated in PLGA microspheres with zinc is critical. In a different study, insulin encapsulated into PLGA microspheres with a zinc salt was evaluated in rat models by measuring the blood glucose levels. Insulin released from the microspheres suppressed glucose levels significantly for about two weeks (data not shown) demonstrating that the therapeutic activity of insulin was preserved during the emulsification process.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}