Intrinsically Disordered Proteins: Where Computation Meets Experiment

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
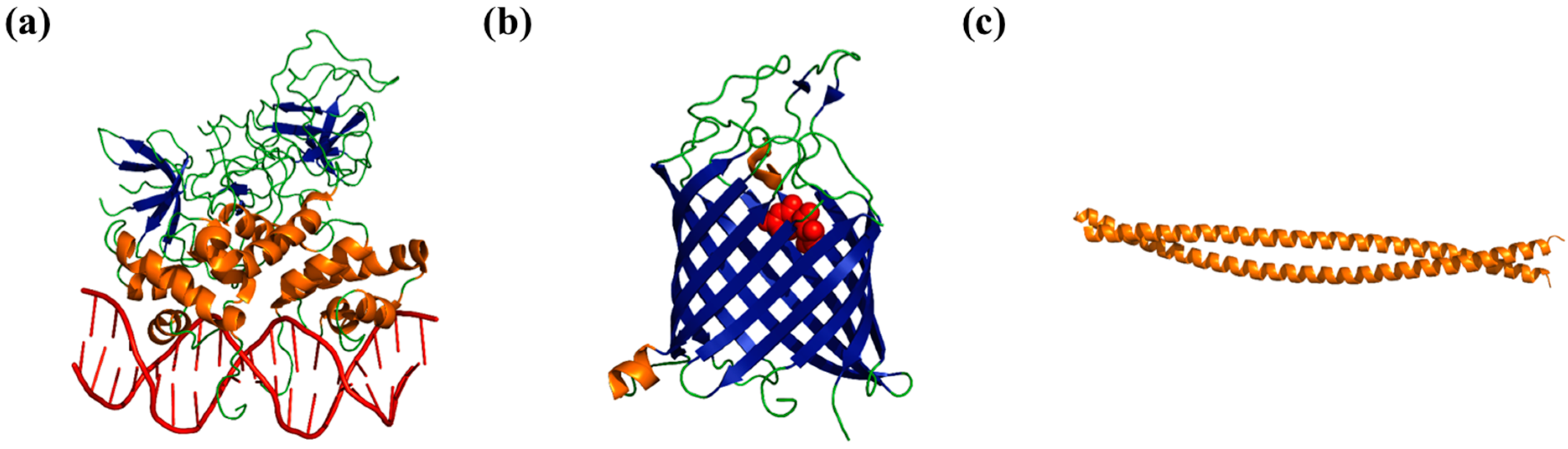
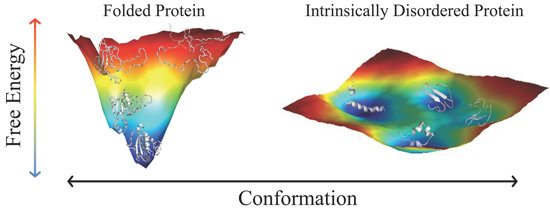
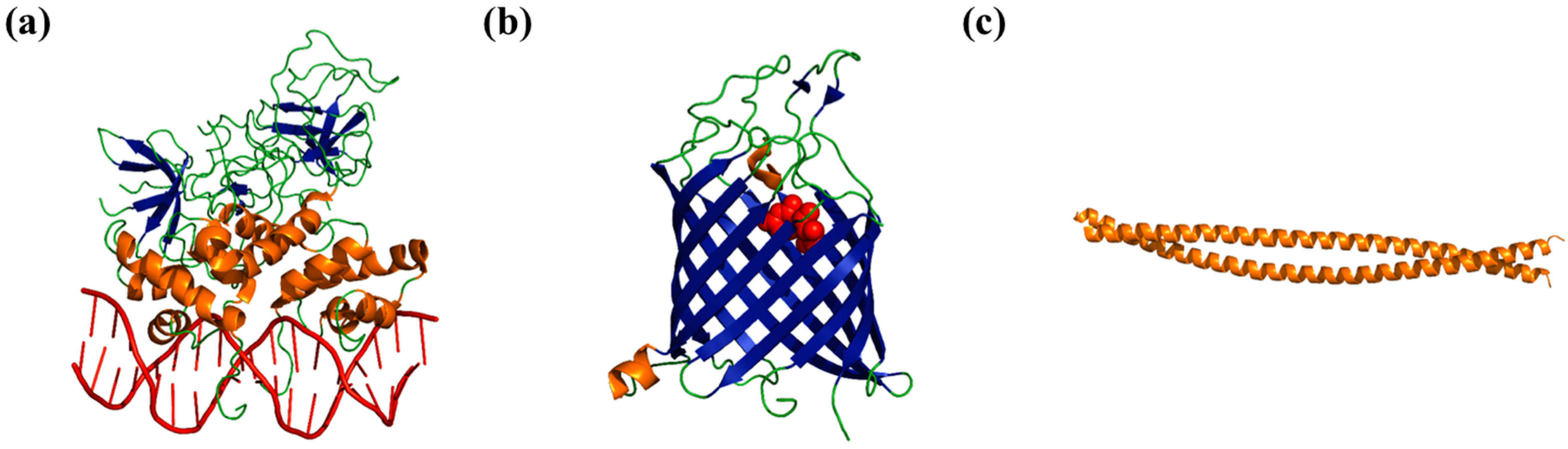
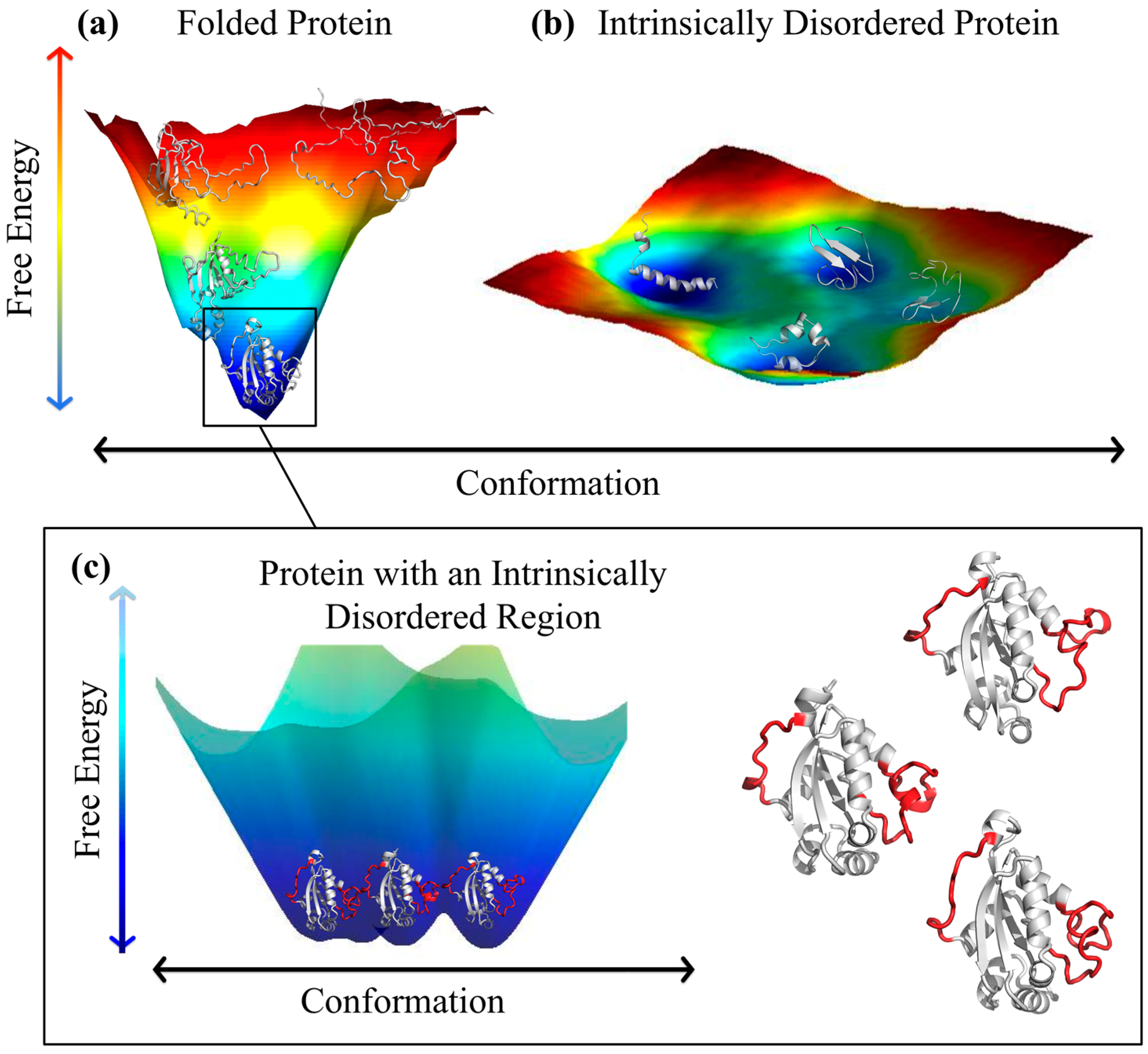
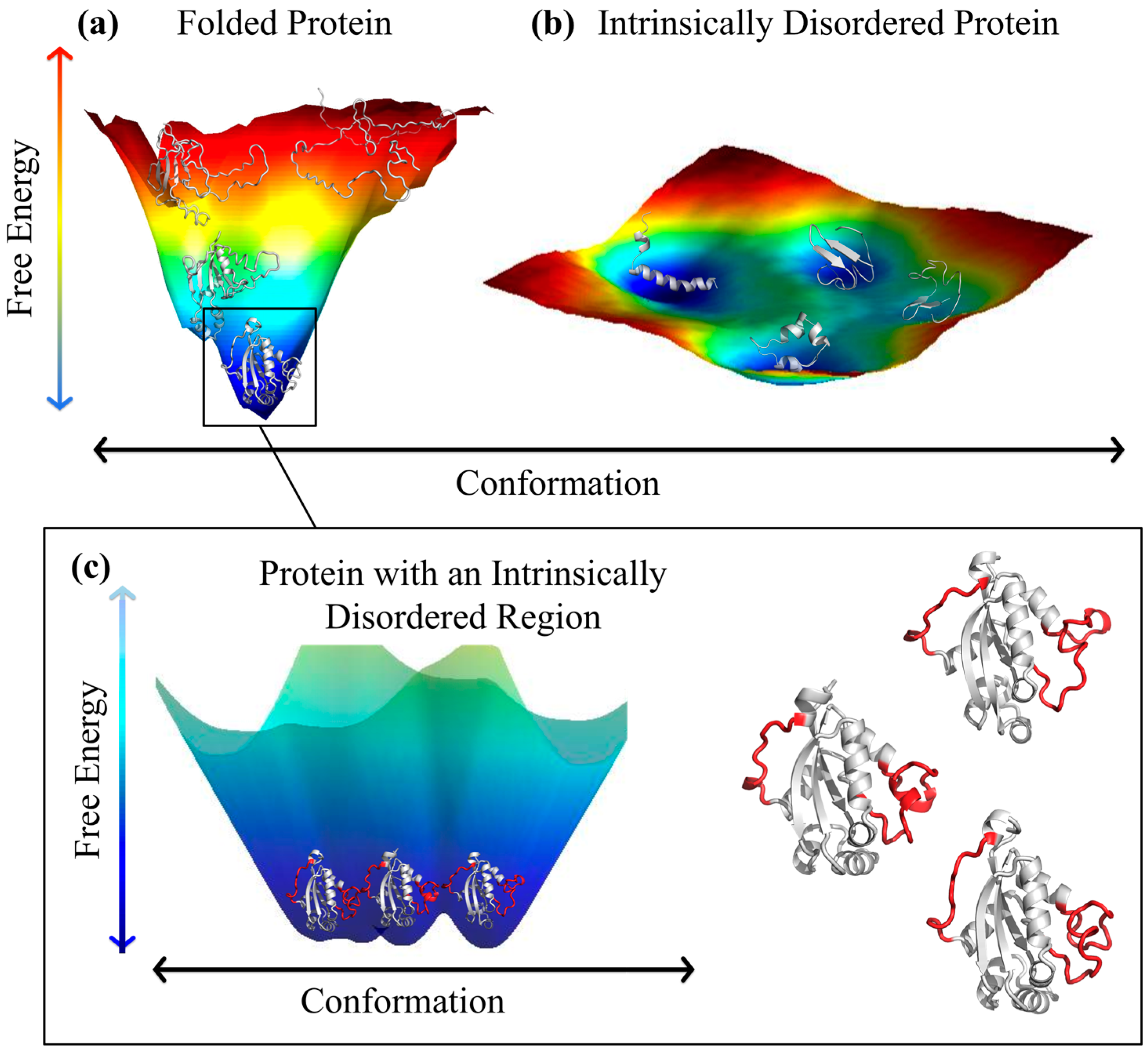
2. Folded Proteins versus Disordered Proteins—A Comparison
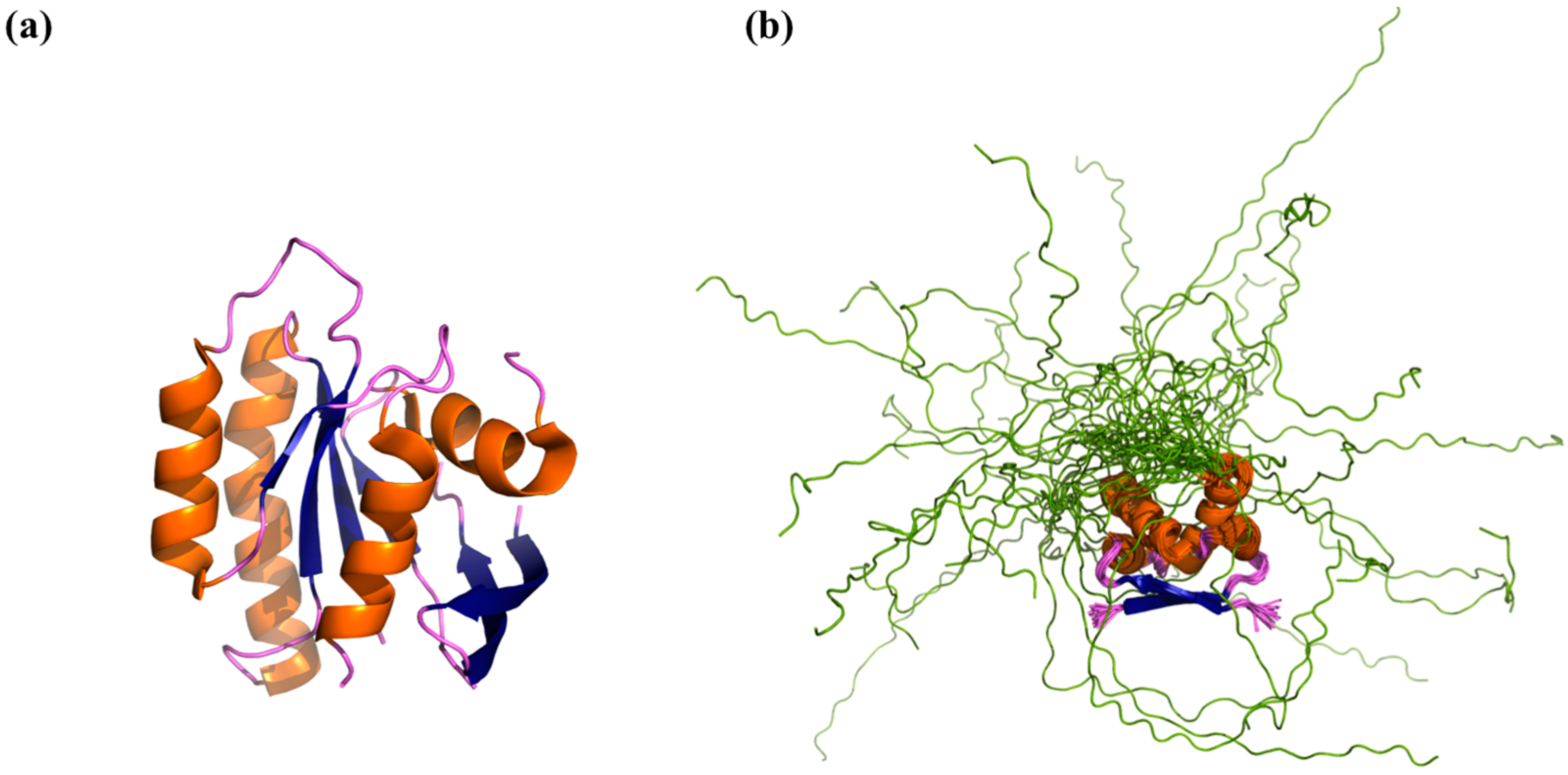
3. Experimental Studies of IDP “Structure”
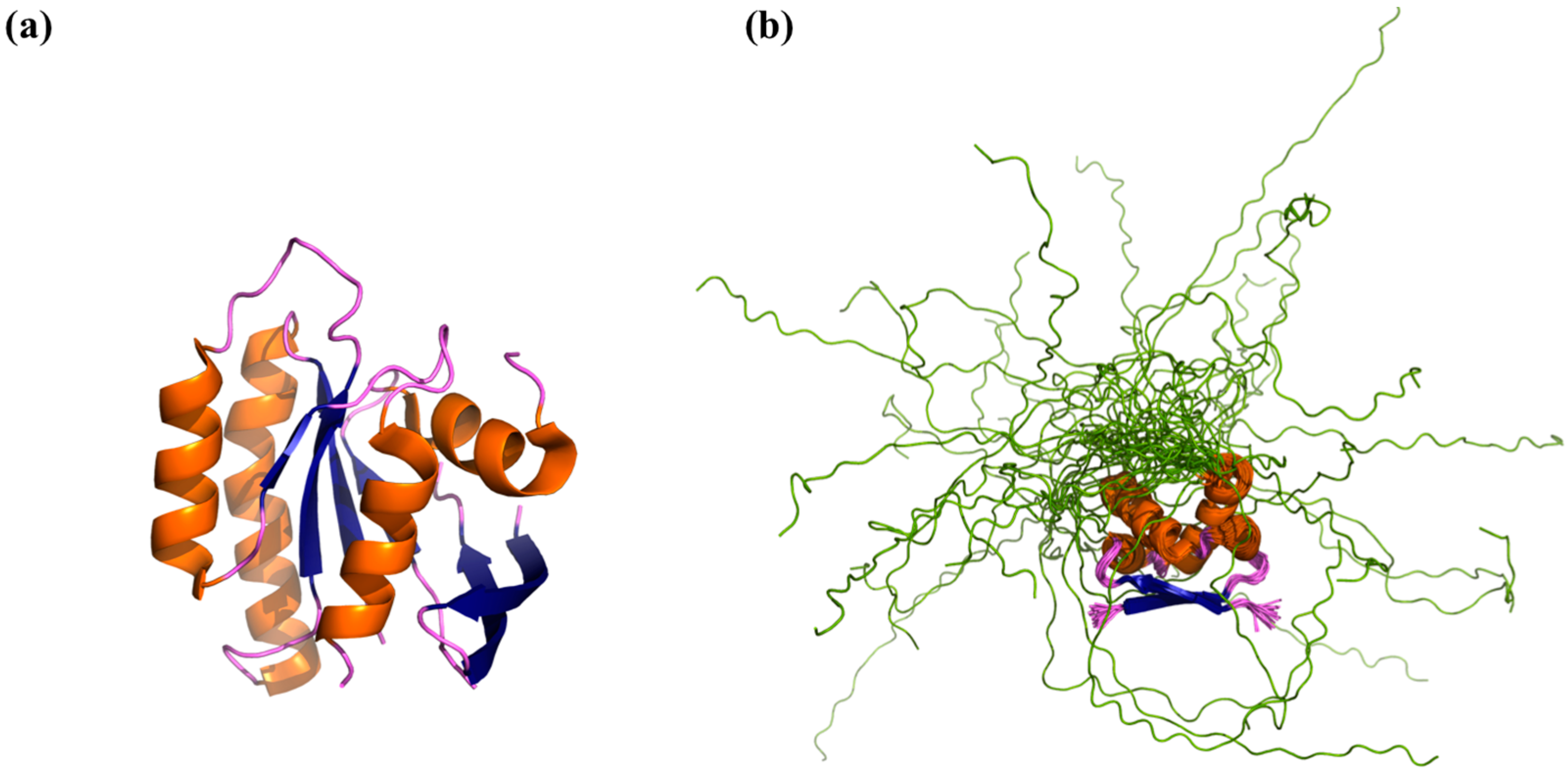
4. Computational Methods for Describing IDP Ensembles
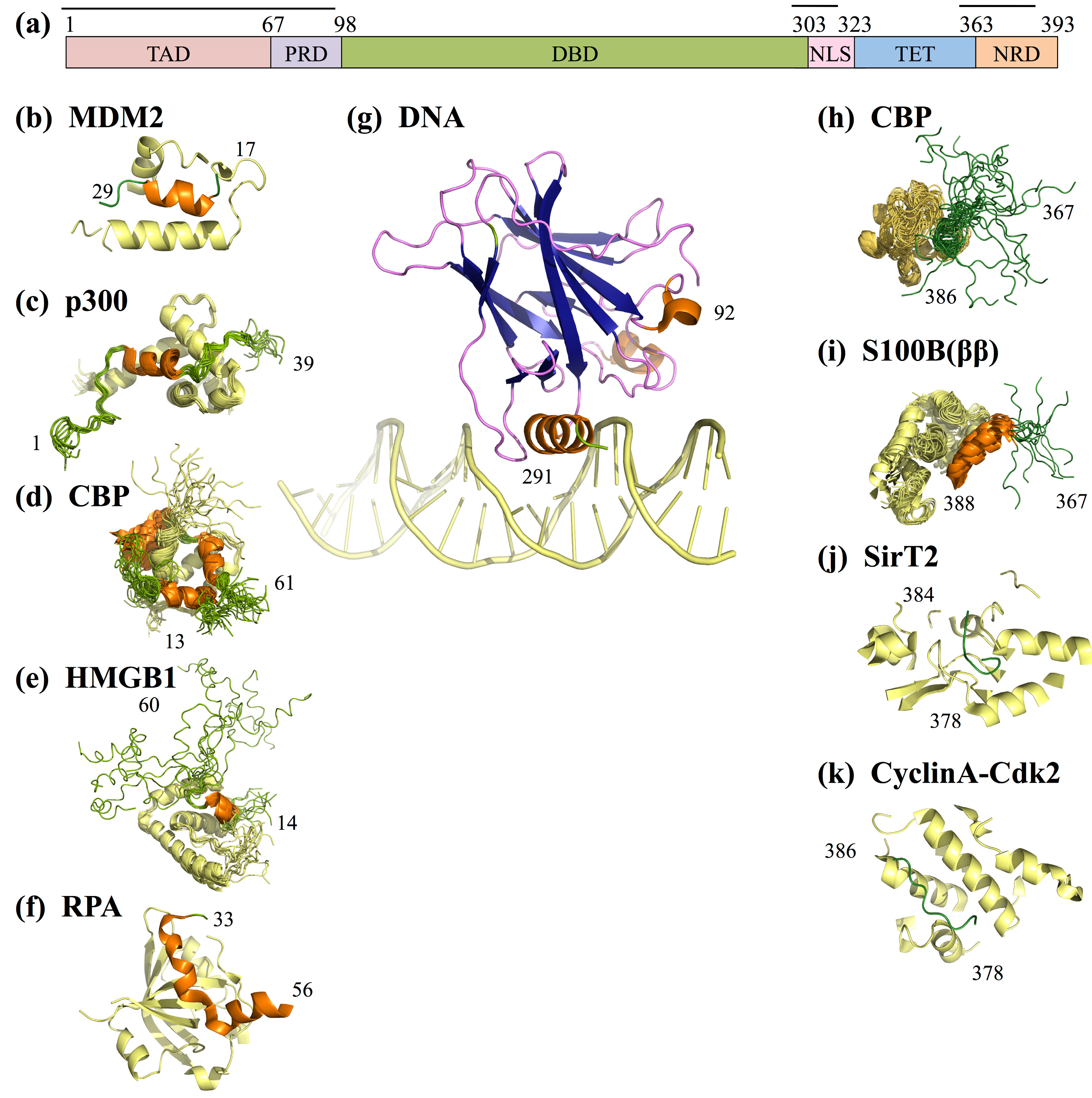
5. p53
5.1. p53 Transcription Activation Domain (TAD)
- In the absence of cellular stress, p53 TAD is bound by its inhibitor MDM2 (mouse double minute 2 homolog), which both tags p53 for degradation and inhibits the binding site of p53 to transcriptional co-activator proteins (Figure 4b) [112,113]. The TAD region is phosphorylated in response to cellular stress; phosphorylation of the TAD disrupts the interaction between p53 and MDM2, thereby allowing p53 to act as a transcriptional regulator [114].
- In response to cellular stress signals, the TAD domain binds to the transcriptional co-activators CBP (CREB-binding protein; CREB is the cAMP-response element-binding protein) and p300, which function as scaffolds for assembling transcription factors on DNA that regulate genes for stress response pathways (Figure 4b). CBP and p53 additionally bind the NRD and perform post-translational modifications of NRD residues, leading to increased stabilization of the p53-DNA complex [113].
- P53 is also activated as a transcription factor through its interaction with High Mobility Group Protein B1 (HGMB1)), which forms part of the transcription machinery on DNA (Figure 4b). HGMB1 has two binding domains: one domain binds to p53 and the other domain binds and bends DNA, most likely into a more suitable conformation for binding of the p53 DBD [115].
- p53 TAD also interacts with its inhibitor Replication Protein A (RPA), which preferentially binds single-stranded DNA (ssDNA) (Figure 4b) [116]. If damaged DNA results in an increase in ssDNA, RPA will instead bind ssDNA, freeing p53 to activate transcription of stress response genes [116]. Hyperphosphorylation of RPA through UV radiation also disrupts the interaction between p53 and RPA, allowing p53 to initiate repair of DNA damaged by the UV radiation [116].
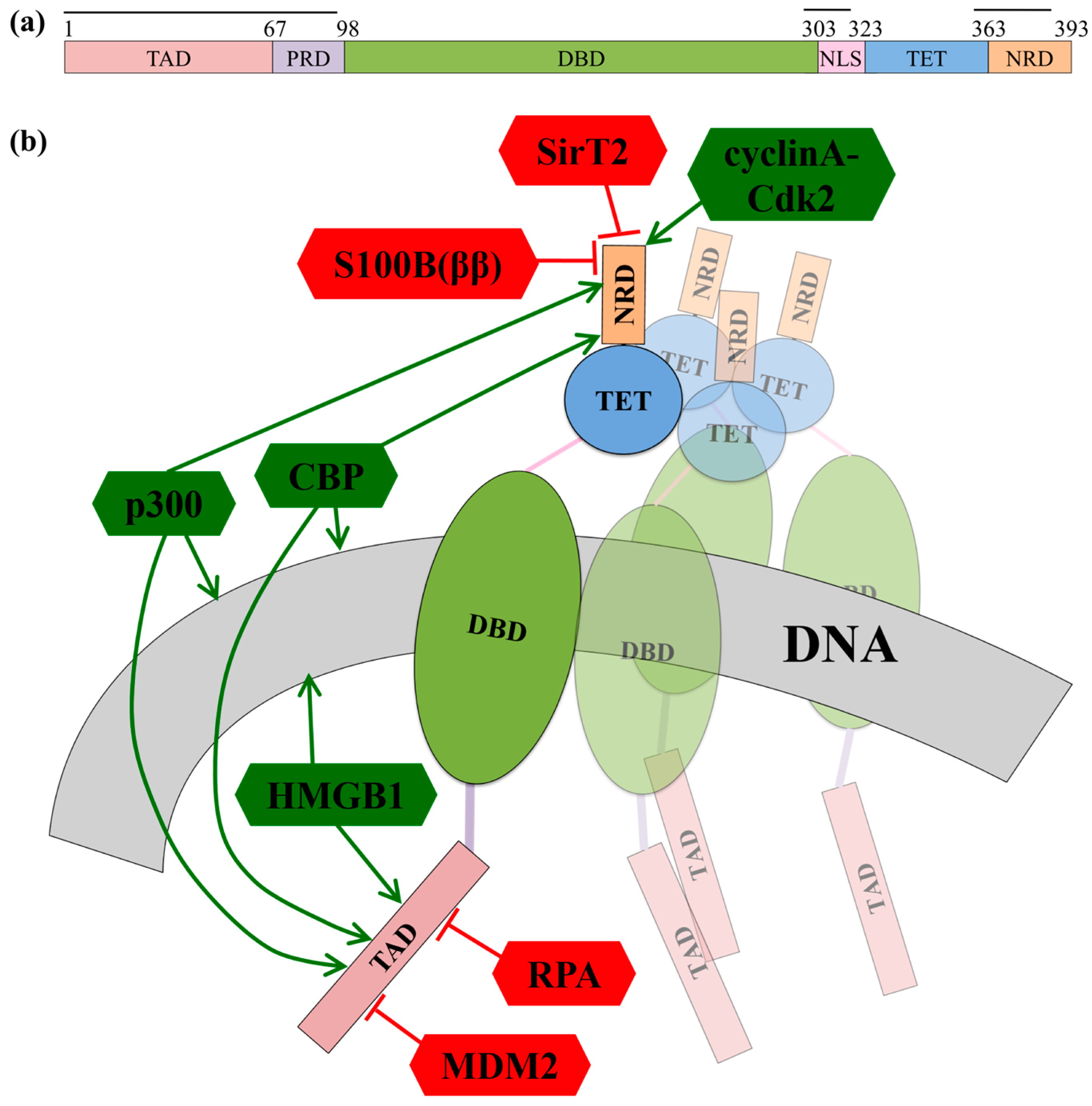
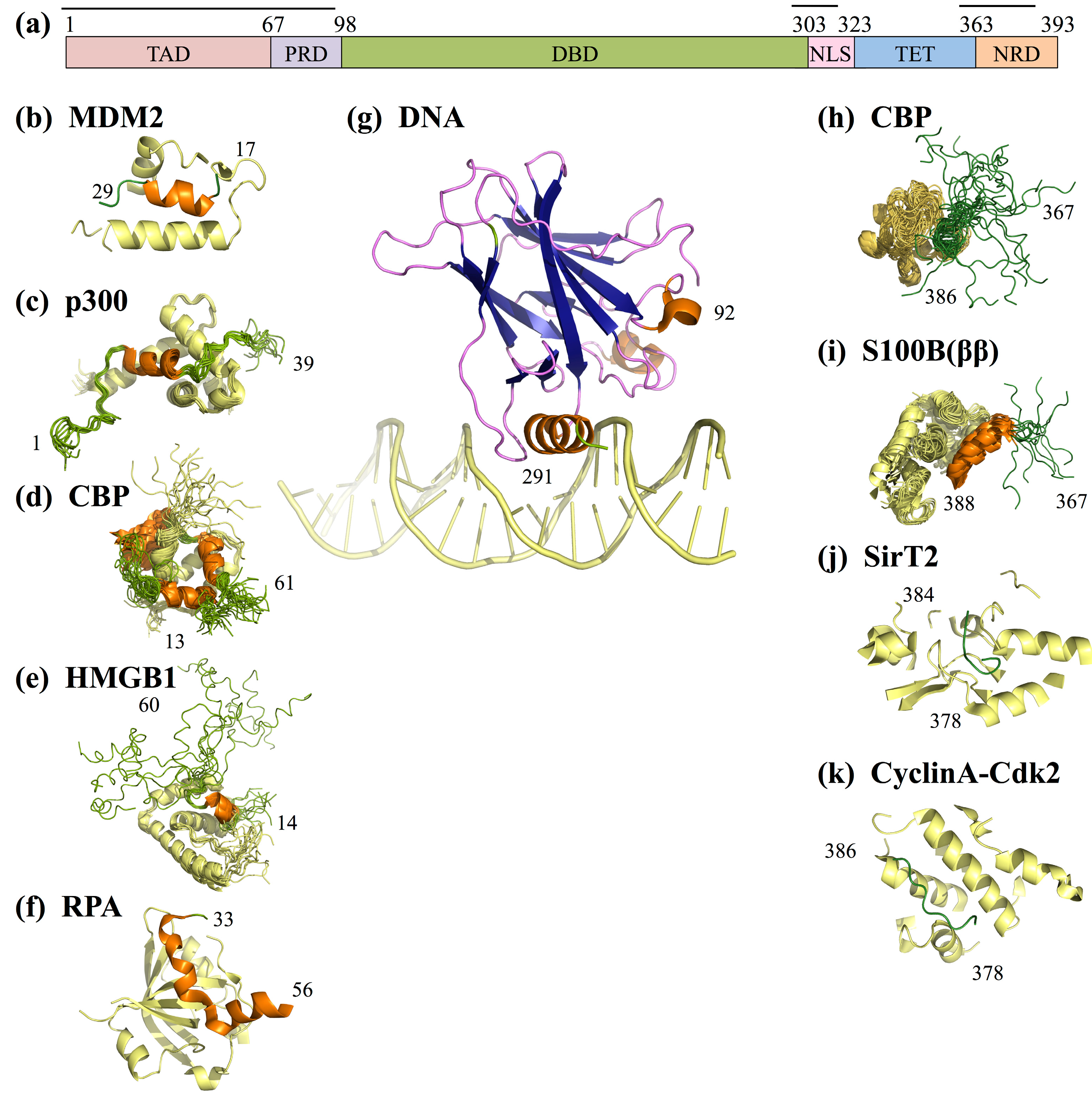
5.2. p53 Negative Regulatory Domain (NRD)
- Acetylation of p53NRD at Lysine 382 facilitates binding by the bromodomain of CBP (Figure 4b). This leads to recruitment of transcriptional co-activators essential for p53 to activate transcription of genes involved in cell cycle arrest [121]. Cell cycle arrest prevents division of the damaged cell, providing time for p53-initiated damage control pathways to repair the cell’s DNA before cell division is reinitiated.
- Cyclin A-CDk2 binds p53 NRD and phosphorylates Serine 315 after irradiation damage (Figure 4b), leading to activation of p53 [135]. Interestingly, both SirT2 and Cyclin A can regulate p53 in more roles than described here, alternately inhibiting or promoting p53’s transcriptional activation functions [137,138].
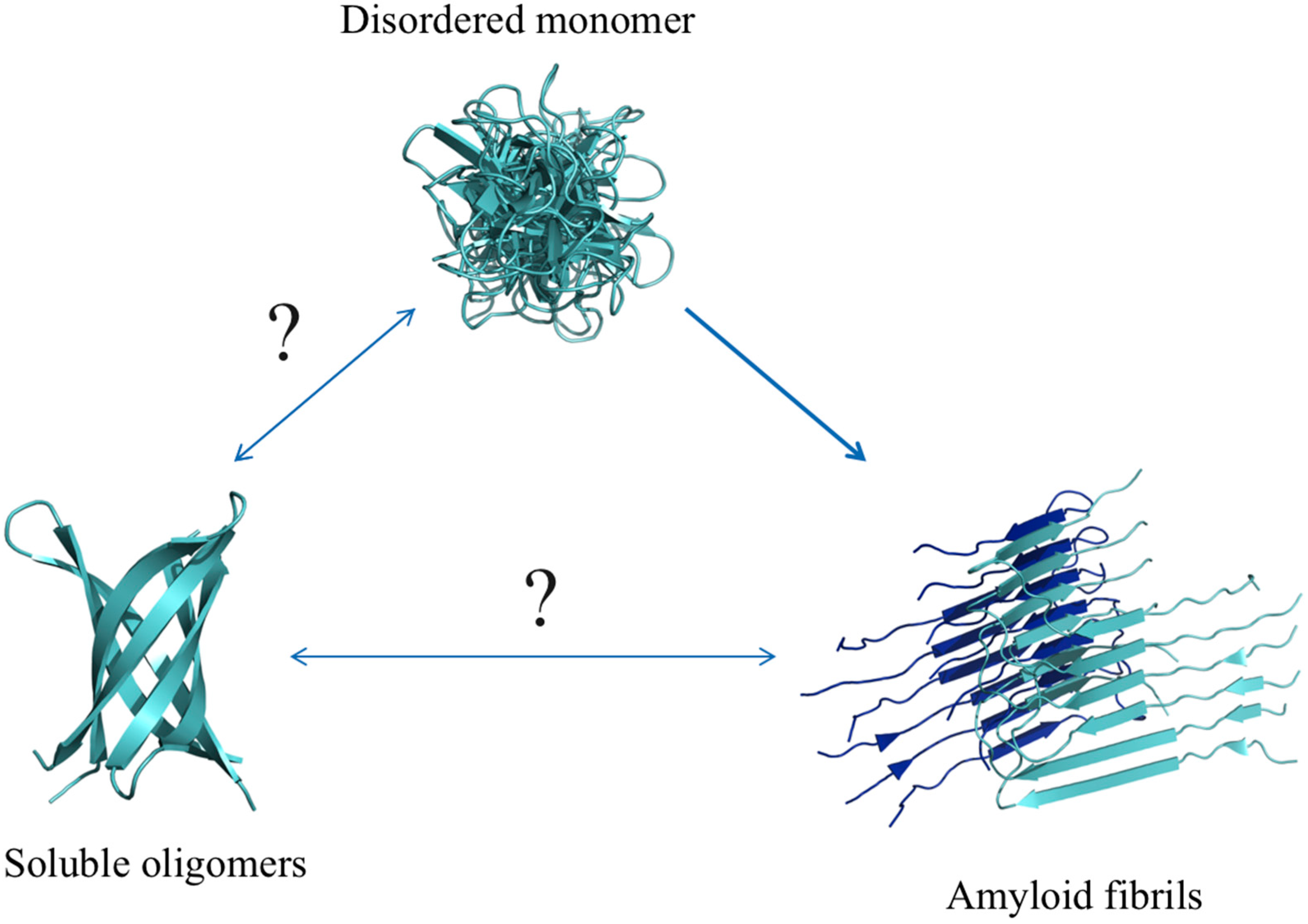
6. Aβ
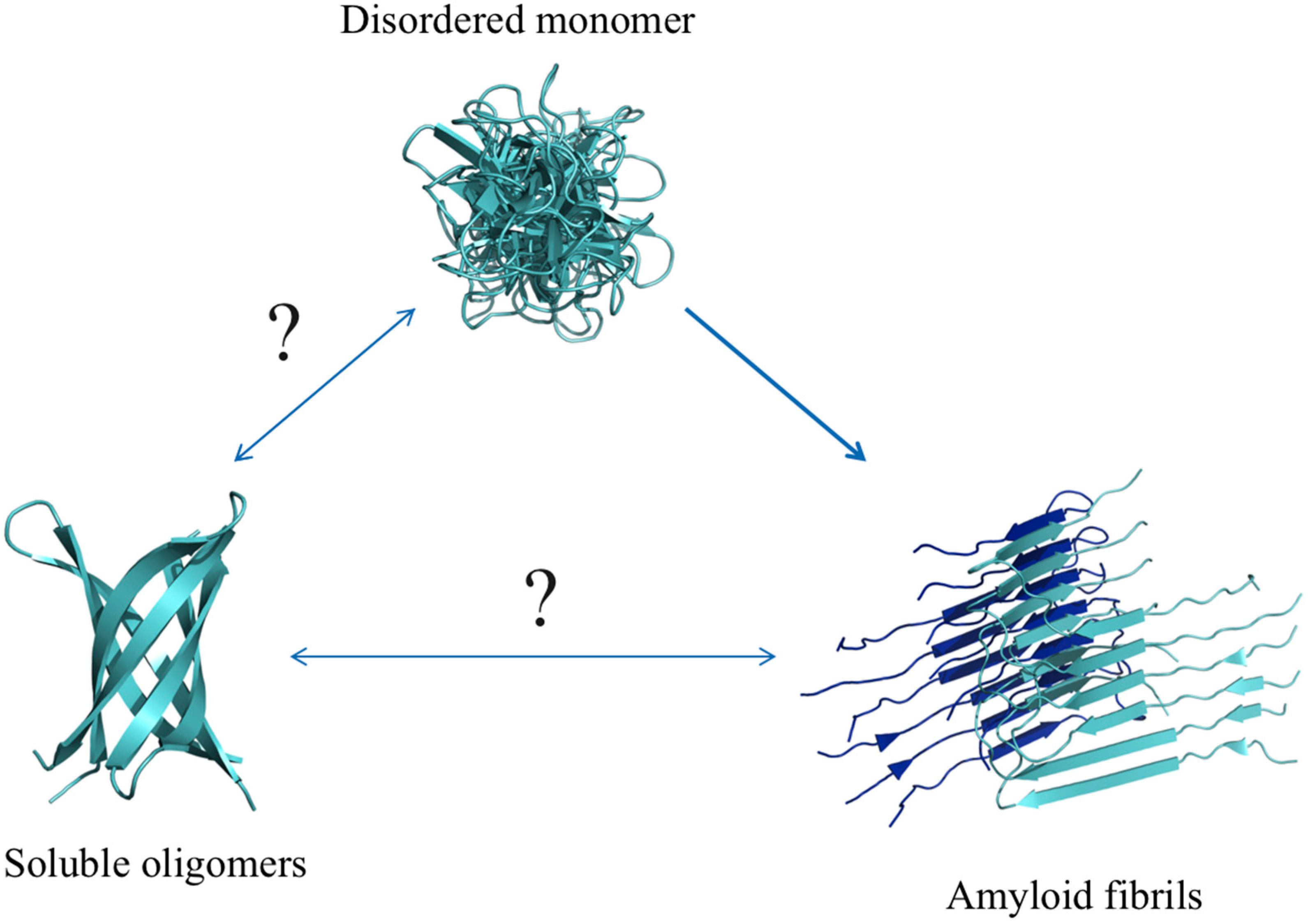
6.1. Aβ Mutations and Aggregates
6.2. Aβ Oligomers
6.3. Aβ Fibrils
6.4. Insight into Aβ Structure and Its Aggregation Mechanism through Computation
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Petsko, G.A.; Ringe, D. Protein Structure and Function; New Science Press: London, UK, 2004. [Google Scholar]
- Richardson, J.S. The anatomy and taxonomy of protein structure. Adv. Protein Chem. 1981, 34, 167–339. [Google Scholar]
- Wang, Z.; Moult, J. SNPs, protein structure, and disease. Hum. Mutat. 2001, 17, 263–270. [Google Scholar] [CrossRef]
- Stayrook, S.; Jaru-Ampornpan, P.; Ni, J.; Hochschild, A.; Lewis, M. Crystal structure of the lambda repressor and a model for pairwise cooperative operator binding. Nature 2008, 452, 1022–1025. [Google Scholar] [CrossRef]
- Ye, J.; van den Berg, B. Crystal structure of the bacterial nucleoside transporter Tsx. EMBO J. 2004, 23, 3187–3195. [Google Scholar] [CrossRef]
- Lee, C.H.; Kim, M.S.; Chung, B.M.; Leahy, D.J.; Coulombe, P.A. Structural basis for heteromeric assembly and perinuclear organization of keratin filaments. Nat. Struct. Mol. Biol. 2012, 19, 707–715. [Google Scholar] [CrossRef]
- Vihinen, M. Relationship of protein flexibility to thermostability. Protein Eng. 1987, 1, 477–480. [Google Scholar] [CrossRef]
- Falke, J.J. A moving story. Science 2002, 295, 1480–1481. [Google Scholar] [CrossRef]
- Fisher, C.K.; Stultz, C.M. Protein structure along the order–disorder continuum. J. Am. Chem. Soc. 2011, 133, 10022–10025. [Google Scholar] [CrossRef]
- Oldfield, C.J.; Cheng, Y.; Cortese, M.S.; Brown, C.J.; Uversky, V.N.; Dunker, A.K. Comparing and combining predictors of mostly disordered proteins. Biochemistry 2005, 44, 1989–2000. [Google Scholar] [CrossRef]
- Uversky, V.N.; Dunker, A.K. Understanding protein non-folding. Biochim. Biophys. Acta 2010, 1804, 1231–1264. [Google Scholar] [CrossRef]
- Chouard, T. Structural biology: Breaking the protein rules. Nature 2011, 471, 151–153. [Google Scholar] [CrossRef]
- Conway, K.A.; Lee, S.-J.; Rochet, J.-C.; Ding, T.T.; Williamson, R.E.; Lansbury, P.T. Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson’s disease: Implications for pathogenesis and therapy. Proc. Natl. Acad. Sci. USA 2000, 97, 571–576. [Google Scholar] [CrossRef]
- Perutz, M.F. Glutamine repeats and neurodegenerative diseases: Molecular aspects. Trends Biochem. Sci. 1999, 24, 58–63. [Google Scholar] [CrossRef]
- Romero, P.; Obradovic, Z.; Li, X.; Garner, E.C.; Brown, C.J.; Dunker, A.K. Sequence complexity of disordered protein. Proteins Struct. Funct. Bioinf. 2001, 42, 38–48. [Google Scholar] [CrossRef]
- Crick, S.; Jayaraman, M.; Frieden, C.; Wetzel, R.; Pappu, R. Fluorescence correlation spectroscopy shows that monomeric polyglutamine molecules form collapsed structures in aqueous solutions. Proc. Natl. Acad. Sci. USA 2006, 103, 16764–16769. [Google Scholar] [CrossRef]
- Vitalis, A.; Lyle, N.; Pappu, R.V. Thermodynamics of β-sheet formation in polyglutamine. Biophys. J. 2009, 97, 303–311. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef]
- Pitschke, M.; Prior, R.; Haupt, M.; Riesner, D. Detection of single amyloid beta-protein aggregates in the cerebrospinal fluid of Alzheimer’s patients by fluorescence correlation spectroscopy. Nat. Med. 1998, 4, 832–834. [Google Scholar] [CrossRef]
- El-Agnaf, O.M.A.; Mahil, D.S.; Patel, B.P.; Austen, B.M. Oligomerization and toxicity of β-amyloid-42 implicated in Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2000, 273, 1003–1007. [Google Scholar]
- Zhang, S.; Iwata, K.; Lachenmann, M.J.; Peng, J.W.; Li, S.; Stimson, E.R.; Lu, Y.A.; Felix, A.M.; Maggio, J.E.; Lee, J.P. The Alzheimer’s peptide Aβ adopts a collapsed coil structure in water. J. Struct. Biol. 2000, 130, 130–141. [Google Scholar] [CrossRef]
- Schweers, O.; Schönbrunn-Hanebeck, E.; Marx, A.; Mandelkow, E. Structural studies of tau protein and Alzheimer paired helical filaments show no evidence for beta-structure. J. Biol. Chem. 1994, 269, 24290–24297. [Google Scholar]
- Iakoucheva, L.M.; Brown, C.J.; Lawson, J.D.; Obradovic, Z.; Dunker, A.K. Intrinsic disorder in cell-signaling and cancer-associated proteins. J. Mol. Biol. 2002, 323, 573–584. [Google Scholar] [CrossRef]
- Bell, S.; Klein, C.; Muller, L.; Hansen, S.; Buchner, J. p53 contains large unstructured regions in its native state. J. Mol. Biol. 2002, 322, 917–927. [Google Scholar] [CrossRef]
- Joerger, A.C.; Fersht, A.R. The tumor suppressor p53: From structures to drug discovery. Cold Spring Harb. Perspect. Biol. 2010, 2. [Google Scholar] [CrossRef]
- Lasky, T.; Silbergeld, E. P53 mutations associated with breast, colorectal, liver, lung, and ovarian cancers. Environ. Health Perspect. 1996, 104, 1324–1331. [Google Scholar] [CrossRef]
- Bogler, O.; Huang, H.J.; Kleihues, P.; Cavenee, W.K. The p53 gene and its role in human brain tumors. Glia 1995, 15, 308–327. [Google Scholar] [CrossRef]
- Koudinov, A.R.; Berezov, T.T. Alzheimer’s amyloid-beta (a beta) is an essential synaptic protein, not neurotoxic junk. Acta Neurobiol. Exp. 2004, 64, 71–79. [Google Scholar]
- Anfinsen, C.B. Principles that govern the folding of protein chains. Science 1973, 181, 223–230. [Google Scholar] [CrossRef]
- Bahar, I.; Lezon, T.R.; Yang, L.W.; Eyal, E. Global dynamics of proteins: Bridging between structure and function. Annu. Rev. Biophys. 2010, 39, 23–42. [Google Scholar] [CrossRef]
- Huang, A.; Stultz, C.M. Finding order within disorder: Elucidating the structure of proteins associated with neurodegenerative disease. Fut. Med. Chem. 2009, 1, 467–482. [Google Scholar] [CrossRef]
- Leopold, P.E.; Montal, M.; Onuchic, J.N. Protein folding funnels: A kinetic approach to the sequence-structure relationship. Proc. Natl. Acad. Sci. USA 1992, 89, 8721–8725. [Google Scholar] [CrossRef]
- Onuchic, J.N.; Wolynes, P.G. Theory of protein folding. Curr. Opin. Struct. Biol. 2004, 14, 70–75. [Google Scholar] [CrossRef]
- Chen, J. Towards the physical basis of how intrinsic disorder mediates protein function. Arch. Biochem. Biophys. 2012, 524, 123–131. [Google Scholar] [CrossRef]
- Tompa, P. Unstructural biology coming of age. Curr. Opin. Struct. Biol. 2011, 3, 419–425. [Google Scholar] [CrossRef]
- Uversky, V.N. Natively unfolded proteins: A point where biology waits for physics. Protein Sci. 2002, 11, 739–756. [Google Scholar] [CrossRef]
- Dunker, A.; Cortese, M.; Romero, P.; Iakoucheva, L.; Uversky, V. Flexible nets. The roles of intrinsic disorder in protein interaction networks. FEBS J. 2005, 272, 5129–5148. [Google Scholar] [CrossRef]
- Tompa, P.; Szasz, C.; Buday, L. Structural disorder throws new light on moonlighting. Trends Biochem. Sci. 2005, 30, 484–489. [Google Scholar] [CrossRef]
- Oldfield, C.; Meng, J.; Yang, J.; Yang, M.; Uversky, V.; Dunker, A. Flexible nets: Disorder and induced fit in the associations of p53 and 14-3-3 with their partners. BMC Genomics 2008, 9, S1. [Google Scholar] [CrossRef]
- Wright, P.E.; Dyson, H.J. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J. Mol. Biol. 1999, 293, 321–331. [Google Scholar]
- Gsponer, J.; Futschik, M.E.; Teichmann, S.A.; Babu, M.M. Tight regulation of unstructured proteins: From transcript synthesis to protein degradation. Science 2008, 322, 1365–1368. [Google Scholar] [CrossRef]
- Tompa, P.; Prilusky, J.; Silman, I.; Sussman, J.L. Structural disorder serves as a weak signal for intracellular protein degradation. Proteins 2008, 71, 903–909. [Google Scholar] [CrossRef]
- Gross, M. Anarchy in the proteome. Chem. World 2011, 8, 42–45. [Google Scholar]
- Plaxco, K.W.; Gross, M. Cell biology. The importance of being unfolded. Nature 1997, 386, 657–659. [Google Scholar] [CrossRef]
- Webb, P.A.; Perisic, O.; Mendola, C.E.; Backer, J.M.; Williams, R.L. The crystal structure of a human nucleoside diphosphate kinase, NM23-H2. J. Mol. Biol. 1995, 251, 574–587. [Google Scholar] [CrossRef]
- Drobnak, I.; De Jonge, N.; Haesaerts, S.; Vesnaver, G.; Loris, R.; Lah, J. Energetic basis of uncoupling folding from binding for an intrinsically disordered protein. J. Am. Chem. Soc. 2013, 135, 1288–1294. [Google Scholar] [CrossRef]
- Brooks, B.R.; Bruccoleri, R.E.; Olafson, B.D.; States, D.J.; Swaminathan, S.; Karplus, M. Charmm-A program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1983, 4, 187–217. [Google Scholar] [CrossRef]
- Brooks, B.R.; Brooks, C.L., 3rd; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef]
- Monod, J.; Wyman, J.; Changeux, J.-P. On the nature of allosteric transitions: A plausible model. J. Mol. Biol. 1965, 12, 88–118. [Google Scholar] [CrossRef]
- Koshland, D.E. Application of a theory of enzyme specificity to protein synthesis. Proc. Natl. Acad. Sci. USA 1958, 44, 98–104. [Google Scholar] [CrossRef]
- Kumar, S.; Showalter, S.A.; Noid, W.G. Native-based simulations of the binding interaction between RAP74 and the disordered FCP1 peptide. J. Phys. Chem. B 2013, 117, 1–12. [Google Scholar]
- Shoemaker, B.A.; Portman, J.J.; Wolynes, P.G. Speeding molecular recognition by using the folding funnel: The fly-casting mechanism. Proc. Natl. Acad. Sci. USA 2000, 97, 8868–8873. [Google Scholar] [CrossRef]
- Levy, Y.; Onuchic, J.N.; Wolynes, P.G. Fly-casting in protein-DNA binding: Frustration between protein folding and electrostatics facilitates target recognition. J. Am. Chem. Soc. 2007, 129, 738–739. [Google Scholar] [CrossRef]
- Huang, Y.; Liu, Z. Kinetic advantage of intrinsically disordered proteins in coupled folding-binding process: A critical assessment of the “fly-casting” mechanism. J. Mol. Biol. 2009, 393, 1143–1159. [Google Scholar] [CrossRef]
- Tompa, P.; Fuxreiter, M. Fuzzy complexes: Polymorphism and structural disorder in protein-protein interactions. Trends Biochem. Sci. 2008, 33, 2–8. [Google Scholar] [CrossRef]
- Anand, K.; Parret, A.; Denayer, E.; Petrova, B.; Legius, E.; Scheffzek, K. Crystal structure of the activating H-Ras I163F mutant in costello syndrome, bound to Mg-Gdp. Available online: http://www.rcsb.org/pdb/explore/explore.do?pdbId=2X1V (accessed on 17 August 2014).
- Madl, T.; van Melderen, L.; Mine, N.; Respondek, M.; Oberer, M.; Keller, W.; Khatai, L.; Zangger, K. Structural basis for nucleic acid and toxin recognition of the bacterial antitoxin CcdA. J. Mol. Biol. 2006, 364, 170–185. [Google Scholar] [CrossRef]
- MacArthur, M.W.; Driscoll, P.C.; Thornton, J.M. NMR and crystallography—Complementary approaches to structure determination. Trends Biotechnol. 1994, 12, 149–153. [Google Scholar] [CrossRef]
- Redfield, C. Using nuclear magnetic resonance spectroscopy to study molten globule states of proteins. Methods 2004, 34, 121–132. [Google Scholar] [CrossRef]
- Kosol, S.; Contreras-Martos, S.; Cedeno, C.; Tompa, P. Structural characterization of intrinsically disordered proteins by NMR spectroscopy. Molecules 2013, 18, 10802–10828. [Google Scholar] [CrossRef]
- Gillespie, J.R.; Shortle, D. Characterization of long-range structure in the denatured state of staphylococcal nuclease. I. Paramagnetic relaxation enhancement by nitroxide spin labels. J. Mol. Biol. 1997, 268, 158–169. [Google Scholar]
- Tugarinov, V.; Choy, W.Y.; Orekhov, V.Y.; Kay, L.E. Solution NMR-derived global fold of a monomeric 82-kda enzyme. Proc. Natl. Acad. Sci. USA 2005, 102, 622–627. [Google Scholar] [CrossRef]
- Dedmon, M.M.; Lindorff-Larsen, K.; Christodoulou, J.; Vendruscolo, M.; Dobson, C.M. Mapping long-range interactions in alpha-synuclein using spin-label NMR and ensemble molecular dynamics simulations. J. Am. Chem. Soc. 2005, 127, 476–477. [Google Scholar] [CrossRef]
- Eliezer, D. Biophysical characterization of intrinsically disordered proteins. Curr. Opin. Struct. Biol. 2009, 19, 23–30. [Google Scholar] [CrossRef]
- Tjandra, N.; Bax, A. Direct measurement of distances and angles in biomolecules by NMR in a dilute liquid crystalline medium. Science 1997, 278, 1111–1114. [Google Scholar] [CrossRef]
- Jensen, M.R.; Markwick, P.R.; Meier, S.; Griesinger, C.; Zweckstetter, M.; Grzesiek, S.; Bernado, P.; Blackledge, M. Quantitative determination of the conformational properties of partially folded and intrinsically disordered proteins using NMR dipolar couplings. Structure 2009, 17, 1169–1185. [Google Scholar] [CrossRef]
- Bernado, P.; Svergun, D.I. Structural analysis of intrinsically disordered proteins by small-angle X-ray scattering. Mol. Biosyst. 2012, 8, 151–167. [Google Scholar] [CrossRef]
- Ando, T. High-speed atomic force microscopy. Microscopy 2013, 62, 81–93. [Google Scholar] [CrossRef]
- Katan, A.J.; Dekker, C. High-speed AFM reveals the dynamics of single biomolecules at the nanometer scale. Cell 2011, 147, 979–982. [Google Scholar] [CrossRef]
- Miyagi, A.; Tsunaka, Y.; Uchihashi, T.; Mayanagi, K.; Hirose, S.; Morikawa, K.; Ando, T. Visualization of intrinsically disordered regions of proteins by high-speed atomic force microscopy. Chemphyschem 2008, 9, 1859–1866. [Google Scholar] [CrossRef]
- Allison, J.R.; Varnai, P.; Dobson, C.M.; Vendruscolo, M. Determination of the free energy landscape of α-synuclein using spin label nuclear magnetic resonance measurements. J. Am. Chem. Soc. 2009, 131, 18314–18326. [Google Scholar] [CrossRef]
- Schwalbe, M.; Ozenne, V.; Bibow, S.; Jaremko, M.; Jaremko, L.; Gajda, M.; Jensen, M.R.; Biernat, J.; Becker, S.; Mandelkow, E.; et al. Predictive atomic resolution descriptions of intrinsically disordered htau40 and alpha-synuclein in solution from NMR and small angle scattering. Structure 2014, 22, 238–249. [Google Scholar] [CrossRef]
- Tompa, P.; Varadi, M. Predicting the predictive power of IDP ensembles. Structure 2014, 22, 177–178. [Google Scholar] [CrossRef]
- Rauscher, S.; Pomès, R. Molecular simulations of protein disorder. Biochem. Cell Biol. 2010, 88, 269–290. [Google Scholar] [CrossRef]
- Tuckerman, M.E. Statistical Mechanics: Theory and Molecular Simulation; Oxford University Press: Oxford, UK & New York, NY, USA, 2010. [Google Scholar]
- Karplus, M.; McCammon, J.A. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 2002, 9, 646–652. [Google Scholar] [CrossRef]
- Leach, A.R. Molecular Modelling: Principles and Applications, 2nd ed.; Prentice Hall: Harlow, UK & New York, NY, USA, 2001. [Google Scholar]
- Vitalis, A.; Pappu, R.V. Absinth: A new continuum solvation model for simulations of polypeptides in aqueous solutions. J. Comput. Chem. 2009, 30, 673–699. [Google Scholar] [CrossRef]
- Vitalis, A.; Caflisch, A. Micelle-like architecture of the monomer ensemble of Alzheimer’s amyloid-beta peptide in aqueous solution and its implications for Abeta aggregation. J. Mol. Biol. 2010, 403, 148–165. [Google Scholar] [CrossRef]
- Meng, W.; Lyle, N.; Luan, B.; Raleigh, D.P.; Pappu, R.V. Experiments and simulations show how long-range contacts can form in expanded unfolded proteins with negligible secondary structure. Proc. Natl. Acad. Sci. USA 2013, 110, 2123–2128. [Google Scholar] [CrossRef]
- Wuttke, R.; Hofmann, H.; Nettels, D.; Borgia, M.B.; Mittal, J.; Best, R.B.; Schuler, B. Temperature-dependent solvation modulates the dimensions of disordered proteins. Proc. Natl. Acad. Sci. USA 2014, 111, 5213–5218. [Google Scholar] [CrossRef]
- Bottaro, S.; Lindorff-Larsen, K.; Best, R.B. Variational optimization of an all-atom implicit solvent force field to match explicit solvent simulation data. J. Chem. Theory Comput. 2013, 9, 5641–5652. [Google Scholar] [CrossRef]
- Jha, A.K.; Colubri, A.; Freed, K.F.; Sosnick, T.R. Statistical coil model of the unfolded state: Resolving the reconciliation problem. Proc. Natl. Acad. Sci. USA 2005, 102, 13099–13104. [Google Scholar] [CrossRef]
- Ozenne, V.; Bauer, F.; Salmon, L.; Huang, J.-R.; Jensen, M.R.; Segard, S.; Bernadó, P.; Charavay, C.; Blackledge, M. Flexible-meccano: A tool for the generation of explicit ensemble descriptions of intrinsically disordered proteins and their associated experimental observables. Bioinformatics 2012, 28, 1463–1470. [Google Scholar] [CrossRef]
- Bernstein, F.C.; Koetzle, T.F.; Williams, G.J.B.; Meyer, E.F., Jr.; Brice, M.D.; Rodgers, J.R.; Kennard, O.; Shimanouchi, T.; Tasumi, M. The protein data bank: A computer-based archival file for macromolecular structures. J. Mol. Biol. 1977, 112, 535–542. [Google Scholar] [CrossRef]
- Vendruscolo, M. Determination of conformationally heterogeneous states of proteins. Curr. Opin. Struct. Biol. 2007, 17, 15–20. [Google Scholar] [CrossRef]
- Daughdrill, G.W.; Kashtanov, S.; Stancik, A.; Hill, S.E.; Helms, G.; Muschol, M.; Receveur-Brechot, V.; Ytreberg, F.M. Understanding the structural ensembles of a highly extended disordered protein. Mol. Biosyst. 2012, 8, 308–319. [Google Scholar] [CrossRef]
- Fisher, C.K.; Stultz, C.M. Constructing ensembles for intrinsically disordered proteins. Curr. Opin. Struct. Biol. 2011, 21, 426–431. [Google Scholar] [CrossRef]
- Krzeminski, M.; Marsh, J.A.; Neale, C.; Choy, W.-Y.; Forman-Kay, J.D. Characterization of disordered proteins with ENSEMBLE. Bioinformatics 2013, 29, 398–399. [Google Scholar] [CrossRef]
- Jensen, M.R.; Salmon, L.; Nodet, G.; Blackledge, M. Defining conformational ensembles of intrinsically disordered and partially folded proteins directly from chemical shifts. J. Am. Chem. Soc. 2010, 132, 1270–1272. [Google Scholar] [CrossRef]
- Varadi, M.; Kosol, S.; Lebrun, P.; Valentini, E.; Blackledge, M.; Dunker, A.K.; Felli, I.C.; Forman-Kay, J.D.; Kriwacki, R.W.; Pierattelli, R.; et al. Pe-db: A database of structural ensembles of intrinsically disordered and of unfolded proteins. Nucleic Acids Res. 2014, 42, D326–D335. [Google Scholar] [CrossRef]
- Fisher, C.K.; Huang, A.; Stultz, C.M. Modeling intrinsically disordered proteins with bayesian statistics. J. Am. Chem. Soc. 2010, 132, 14919–14927. [Google Scholar] [CrossRef]
- Marsh, J.A.; Forman-Kay, J.D. Structure and disorder in an unfolded state under nondenaturing conditions from ensemble models consistent with a large number of experimental restraints. J. Mol. Biol. 2009, 391, 359–374. [Google Scholar] [CrossRef]
- Ganguly, D.; Chen, J.H. Structural interpretation of paramagnetic relaxation enhancement-derived distances for disordered protein states. J. Mol. Biol. 2009, 390, 467–477. [Google Scholar] [CrossRef]
- Huang, A.; Stultz, C.M. The effect of a Delta K280 mutation on the unfolded state of a microtubule-binding repeat in tau. PLoS Comput. Biol. 2008, 4, e1000155. [Google Scholar] [CrossRef]
- Pitera, J.W.; Chodera, J.D. On the use of experimental observations to bias simulated ensembles. J. Chem. Theory Comput. 2012, 8, 3445–3451. [Google Scholar] [CrossRef]
- Boomsma, W.; Ferkinghoff-Borg, J.; Lindorff-Larsen, K. Combining experiments and simulations using the maximum entropy principle. PLoS Comput. Biol. 2014, 10, e1003406. [Google Scholar] [CrossRef]
- Lane, J.L.; Schwantes, C.R.; Beauchamp, K.A.; Pande, V.S. Efficient inference of protein structural ensembles. Biol. Phys. 2014. 1408.0255. [Google Scholar]
- Fisher, C.K.; Ullman, O.; Stultz, C.M. Efficient construction of disordered protein ensembles in a bayesian framework with optimal selection of conformations. Pac. Symp. Biocomput. 2012, 17, 82–93. [Google Scholar]
- Gurry, T.; Ullman, O.; Fisher, C.K.; Perovic, I.; Pochapsky, T.; Stultz, C.M. The dynamic structure of alpha-synuclein multimers. J. Am. Chem. Soc. 2013, 135, 3865–3872. [Google Scholar] [CrossRef]
- Neal, S.; Nip, A.M.; Zhang, H.Y.; Wishart, D.S. Rapid and accurate calculation of protein H-1, C-13 and N-15 chemical shifts. J. Biomol. NMR 2003, 26, 215–240. [Google Scholar] [CrossRef]
- Lane, D.P. Cancer. p53, guardian of the genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef]
- Pagano, B.; Jama, A.; Martinez, P.; Akanho, E.; Bui, T.T.T.; Drake, A.F.; Fraternali, F.; Nikolova, P.V. Structure and stability insights into tumour suppressor p53 evolutionary related proteins. PLoS One 2013, 8. [Google Scholar] [CrossRef]
- Vousden, K.H.; Prives, C. Blinded by the light: The growing complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef]
- Zhao, R.; Gish, K.; Murphy, M.; Yin, Y.; Notterman, D.; Hoffman, W.H.; Tom, E.; Mack, D.H.; Levine, A.J. Analysis of p53-regulated gene expression patterns using oligonucleotide arrays. Genes Dev. 2000, 14, 981–993. [Google Scholar] [CrossRef]
- Braithwaite, A.W.; Del Sal, G.; Lu, X. Some p53-binding proteins that can function as arbiters of life and death. Cell Death Differ. 2006, 13, 984–993. [Google Scholar] [CrossRef]
- Maslon, M.M.; Hupp, T.R. Drug discovery and mutant p53. Trends Cell Biol. 2010, 20, 542–555. [Google Scholar] [CrossRef]
- Greenblatt, M.S.; Bennett, W.P.; Hollstein, M.; Harris, C.C. Mutations in the p53 tumor suppressor gene: Clues to cancer etiology and molecular pathogenesis. Cancer Res. 1994, 54, 4855–4878. [Google Scholar]
- Okorokov, A.L.; Orlova, E.V. Structural biology of the p53 tumour suppressor. Curr. Opin. Struct. Biol. 2009, 19, 197–202. [Google Scholar] [CrossRef]
- Silva, J.L.; Gallo, C.V.D.M.; Costa, D.C.F.; Rangel, L.P. Prion-like aggregation of mutant p53 in cancer. Trends Biochem. Sci. 2014, 39, 260–267. [Google Scholar] [CrossRef]
- Merrill, G.F.; Dowell, P.; Pearson, G.D. The human p53 negative regulatory domain mediates inhibition of reporter gene transactivation in yeast lacking thioredoxin reductase. Cancer Res. 1999, 59, 3175–3179. [Google Scholar]
- Moll, U.M.; Petrenko, O. The MDM2–p53 interaction. Mol. Cancer Res. MCR 2003, 1, 1001–1008. [Google Scholar]
- Lee, C.W.; Martinez-Yamout, M.A.; Dyson, H.J.; Wright, P.E. Structure of the p53 transactivation domain in complex with the nuclear receptor coactivator binding domain of CREB binding protein. Biochemistry 2010, 49, 9964–9971. [Google Scholar] [CrossRef]
- Lambert, P.F.; Kashanchi, F.; Radonovich, M.F.; Shiekhattar, R.; Brady, J.N. Phosphorylation of p53 serine 15 increases interaction with CBP. J. Biol. Chem. 1998, 273, 33048–33053. [Google Scholar] [CrossRef]
- Rowell, J.P.; Simpson, K.L.; Stott, K.; Watson, M.; Thomas, J.O. HMGB1-facilitated p53 DNA binding occurs via HMG-Box/p53 transactivation domain interaction, regulated by the acidic tail. Structure 2012, 20, 2014–2024. [Google Scholar] [CrossRef]
- Bochkareva, E.; Kaustov, L.; Ayed, A.; Yi, G.S.; Lu, Y.; Pineda-Lucena, A.; Liao, J.C.; Okorokov, A.L.; Milner, J.; Arrowsmith, C.H.; et al. Single-stranded DNA mimicry in the p53 transactivation domain interaction with replication protein A. Proc. Natl. Acad. Sci. USA 2005, 102, 15412–15417. [Google Scholar] [CrossRef]
- Chang, J.; Kim, D.H.; Lee, S.W.; Choi, K.Y.; Sung, Y.C. Transactivation ability of p53 transcriptional activation domain is directly related to the binding affinity to tata-binding protein. J. Biol. Chem. 1995, 270, 25014–25019. [Google Scholar] [CrossRef]
- Kussie, P.H.; Gorina, S.; Marechal, V.; Elenbaas, B.; Moreau, J.; Levine, A.J.; Pavletich, N.P. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 1996, 274, 948–953. [Google Scholar] [CrossRef]
- Feng, H.; Jenkins, L.M.; Durell, S.R.; Hayashi, R.; Mazur, S.J.; Cherry, S.; Tropea, J.E.; Miller, M.; Wlodawer, A.; Appella, E.; et al. Structural basis for p300 Taz2-p53 TAD1 binding and modulation by phosphorylation. Structure 2009, 17, 202–210. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, X.; Dantas Machado, A.C.; Ding, Y.; Chen, Z.; Qin, P.Z.; Rohs, R.; Chen, L. Structure of p53 binding to the BAX response element reveals DNA unwinding and compression to accommodate base-pair insertion. Nucleic Acids Res. 2013, 41, 8368–8376. [Google Scholar] [CrossRef]
- Mujtaba, S.; He, Y.; Zeng, L.; Yan, S.; Plotnikova, O.; Sachchidanand; Sanchez, R.; Zeleznik-Le, N.J.; Ronai, Z.; Zhou, M.M. Structural mechanism of the bromodomain of the coactivator CBP in p53 transcriptional activation. Mol. Cell 2004, 13, 251–263. [Google Scholar] [CrossRef]
- Rustandi, R.R.; Baldisseri, D.M.; Weber, D.J. Structure of the negative regulatory domain of p53 bound to S100B(betabeta). Nat. Struct. Biol. 2000, 7, 570–574. [Google Scholar] [CrossRef]
- Cosgrove, M.S.; Bever, K.; Avalos, J.L.; Muhammad, S.; Zhang, X.; Wolberger, C. The structural basis of sirtuin substrate affinity. Biochemistry 2006, 45, 7511–7521. [Google Scholar] [CrossRef]
- Lowe, E.D.; Tews, I.; Cheng, K.Y.; Brown, N.R.; Gul, S.; Noble, M.E.; Gamblin, S.J.; Johnson, L.N. Specificity determinants of recruitment peptides bound to phospho-CDK2/cyclin A. Biochemistry 2002, 41, 15625–15634. [Google Scholar] [CrossRef]
- Xiong, K.; Zwier, M.C.; Myshakina, N.S.; Burger, V.M.; Asher, S.A.; Chong, L.T. Direct observations of conformational distributions of intrinsically disordered p53 peptides using UV Raman and explicit solvent simulations. J. Phys. Chem. A 2011, 115, 9520–9527. [Google Scholar] [CrossRef]
- Lee, H.; Mok, K.H.; Muhandiram, R.; Park, K.H.; Suk, J.E.; Kim, D.H.; Chang, J.; Sung, Y.C.; Choi, K.Y.; Han, K.H. Local structural elements in the mostly unstructured transcriptional activation domain of human p53. J. Biol. Chem. 2000, 275, 29426–29432. [Google Scholar] [CrossRef]
- Zondlo, S.C.; Lee, A.E.; Zondlo, N.J. Determinants of specificity of MDM2 for the activation domains of p53 and p65: Proline27 disrupts the MDM2-binding motif of p53. Biochemistry 2006, 45, 11945–11957. [Google Scholar] [CrossRef]
- Fuxreiter, M.; Simon, I.; Friedrich, P.; Tompa, P. Preformed structural elements feature in partner recognition by intrinsically unstructured proteins. J. Mol. Biol. 2004, 338, 1015–1026. [Google Scholar] [CrossRef]
- Lee, S.H.; Kim, D.H.; Han, J.J.; Cha, E.J.; Lim, J.E.; Cho, Y.J.; Lee, C.; Han, K.H. Understanding pre-structured motifs (PreSMos) in intrinsically unfolded proteins. Curr. Protein Peptide Sci. 2012, 13, 34–54. [Google Scholar] [CrossRef]
- Lee, C.; Kalmar, L.; Xue, B.; Tompa, P.; Daughdrill, G.W.; Uversky, V.N.; Han, K.-H. Contribution of proline to the pre-structuring tendency of transient helical secondary structure elements in intrinsically disordered proteins. Biochim. Biophys. Acta 2014, 1840, 993–1003. [Google Scholar] [CrossRef]
- Szöllősi, D.; Horváth, T.; Han, K.-H.; Dokholyan, N.V.; Tompa, P.; Kalmar, L.; Hegedűs, T. Discrete molecular dynamics can predict helical prestructured motifs in disordered proteins. PLoS One 2014, 9. [Google Scholar] [CrossRef]
- Huang, Y.; Liu, Z. Anchoring intrinsically disordered proteins to multiple targets: Lessons from N-terminus of the p53 protein. Int. J. Mol. Sci. 2011, 12, 1410–1430. [Google Scholar] [CrossRef]
- Dawson, R.; Muller, L.; Dehner, A.; Klein, C.; Kessler, H.; Buchner, J. The N-terminal domain of p53 is natively unfolded. J. Mol. Biol. 2003, 332, 1131–1141. [Google Scholar] [CrossRef]
- Schon, O.; Friedler, A.; Bycroft, M.; Freund, S.M.; Fersht, A.R. Molecular mechanism of the interaction between MDM2 and p53. J. Mol. Biol. 2002, 323, 491–501. [Google Scholar] [CrossRef]
- Luciani, M.G.; Hutchins, J.R.; Zheleva, D.; Hupp, T.R. The C-terminal regulatory domain of p53 contains a functional docking site for cyclin A. J. Mol. Biol. 2000, 300, 503–518. [Google Scholar] [CrossRef]
- Hoffmann, G.; Breitenbucher, F.; Schuler, M.; Ehrenhofer-Murray, A.E. A novel sirtuin 2 (SIRT2) inhibitor with p53-dependent pro-apoptotic activity in non-small cell lung cancer. J. Biol. Chem. 2014, 289, 5208–5216. [Google Scholar] [CrossRef]
- Bosch-Presegue, L.; Vaquero, A. The dual role of sirtuins in cancer. Genes Cancer 2011, 2, 648–662. [Google Scholar] [CrossRef]
- Hsieh, J.K.; Yap, D.; O’Connor, D.J.; Fogal, V.; Fallis, L.; Chan, F.; Zhong, S.; Lu, X. Novel function of the cyclin A binding site of E2F in regulating p53-induced apoptosis in response to DNA damage. Mol. Cell. Biol. 2002, 22, 78–93. [Google Scholar] [CrossRef]
- Chen, J. Intrinsically disordered p53 extreme C-terminus binds to s100b(ββ) through “fly-casting”. J. Am. Chem. Soc. 2009, 131, 2088–2089. [Google Scholar] [CrossRef]
- Ross, C.A.; Poirier, M.A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004, 10, S10–S17. [Google Scholar] [CrossRef]
- Cohen, S.I.A.; Linse, S.; Luheshi, L.M.; Hellstrand, E.; White, D.A.; Rajah, L.; Otzen, D.E.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P.J. Proliferation of amyloid-β42 aggregates occurs through a secondary nucleation mechanism. Proc. Natl. Acad. Sci. USA 2013, 110, 9758–9763. [Google Scholar] [CrossRef]
- Kayed, R.; Head, E.; Thompson, J.L.; McIntire, T.M.; Milton, S.C.; Cotman, C.W.; Glabe, C.G. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003, 300, 486–489. [Google Scholar] [CrossRef]
- Walsh, D.M.; Selkoe, D.J. Abeta oligomers—A decade of discovery. J. Neurochem. 2007, 101, 1172–1184. [Google Scholar] [CrossRef]
- Periquet, M.; Fulga, T.; Myllykangas, L.; Schlossmacher, M.G.; Feany, M.B. Aggregated alpha-synuclein mediates dopaminergic neurotoxicity in vivo. J. Neurosci. 2007, 27, 3338–3346. [Google Scholar] [CrossRef]
- Bernstein, S.L.; Dupuis, N.F.; Lazo, N.D.; Wyttenbach, T.; Condron, M.M.; Bitan, G.; Teplow, D.B.; Shea, J.-E.; Ruotolo, B.T.; Robinson, C.V.; et al. Amyloid-beta protein oligomerization and the importance of tetramers and dodecamers in the aetiology of Alzheimer’s disease. Nat. Chem. 2009, 1, 326–331. [Google Scholar] [CrossRef]
- Petkova, A.T.; Yau, W.-M.; Tycko, R. Experimental constraints on quaternary structure in Alzheimer’s beta-amyloid fibrils. Biochemistry 2005, 45, 498–512. [Google Scholar] [CrossRef]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890. [Google Scholar] [CrossRef]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid beta-peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef]
- Haass, C. Take five—BACE and the gamma-secretase quartet conduct Alzheimer’s amyloid beta-peptide generation. EMBO J. 2004, 23, 483–488. [Google Scholar] [CrossRef]
- Burdick, D.; Soreghan, B.; Kwon, M.; Kosmoski, J.; Knauer, M.; Henschen, A.; Yates, J.; Cotman, C.; Glabe, C. Assembly and aggregation properties of synthetic Alzheimer’s Abeta amyloid peptide analogs. J. Biol. Chem. 1992, 267, 546–554. [Google Scholar]
- Citron, M.; Oltersdorf, T.; Haass, C.; McConlogue, L.; Hung, A.Y.; Seubert, P.; Vigo-Pelfrey, C.; Lieberburg, I.; Selkoe, D.J. Mutation of the beta-amyloid precursor protein in familial Alzheimer’s disease increases beta-protein production. Nature 1992, 360, 672–674. [Google Scholar] [CrossRef]
- Kassler, K.; Horn, A.C.; Sticht, H. Effect of pathogenic mutations on the structure and dynamics of Alzheimer’s Aβ42-amyloid oligomers. J. Mol. Model 2010, 16, 1011–1020. [Google Scholar] [CrossRef]
- Suzuki, N.; Cheung, T.; Cai, X.; Odaka, A.; Otvos, L.; Eckman, C.; Golde, T.; Younkin, S. An increased percentage of long amyloid beta protein secreted by familial amyloid beta protein precursor (beta APP717) mutants. Science 1994, 264, 1336–1340. [Google Scholar] [CrossRef]
- Kirkitadze, M.D.; Condron, M.M.; Teplow, D.B. Identification and characterization of key kinetic intermediates in amyloid beta-protein fibrillogenesis. J. Mol. Biol. 2001, 312, 1103–1119. [Google Scholar] [CrossRef]
- Glabe, C.G. Common mechanisms of amyloid oligomer pathogenesis in degenerative disease. Neurobiol. Aging 2006, 27, 570–575. [Google Scholar] [CrossRef]
- Selkoe, D.J. Folding proteins in fatal ways. Nature 2003, 426, 900–904. [Google Scholar] [CrossRef]
- Bartels, T.; Choi, J.G.; Selkoe, D.J. Alpha-synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 2011, 477, 107–110. [Google Scholar] [CrossRef]
- Lashuel, H.A.; Hartley, D.; Petre, B.M.; Walz, T.; Lansbury, P.T. Neurodegenerative disease: Amyloid pores from pathogenic mutations. Nature 2002, 418. [Google Scholar] [CrossRef]
- Demuro, A.; Mina, E.; Kayed, R.; Milton, S.C.; Parker, I.; Glabe, C.G. Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J. Biol. Chem. 2005, 280, 17294–17300. [Google Scholar] [CrossRef]
- Silvia, C.; Benedetta, M.; Mariagioia, Z.; Anna, P.; Claudia, P.; Elisa, E.; Annalisa, R.; Massimo, S.; Christopher, M.D.; Cristina, C.; et al. A causative link between the structure of aberrant protein oligomers and their toxicity. Nat. Chem. Biol. 2010, 6, 140–147. [Google Scholar] [CrossRef]
- Sandberg, A.; Luheshi, L.M.; Söllvander, S.; Pereira de Barros, T.; Macao, B.; Knowles, T.P.J.; Biverstål, H.; Lendel, C.; Ekholm-Petterson, F.; Dubnovitsky, A.; et al. Stabilization of neurotoxic Alzheimer amyloid-β oligomers by protein engineering. Proc. Natl. Acad. Sci. USA 2010, 107, 15595–15600. [Google Scholar] [CrossRef]
- Hoyer, W.; Grönwall, C.; Jonsson, A.; Ståhl, S.; Härd, T. Stabilization of a beta-hairpin in monomeric Alzheimer’s amyloid-beta peptide inhibits amyloid formation. Proc. Natl. Acad. Sci. USA 2008, 105, 5099–5104. [Google Scholar] [CrossRef]
- Ahmed, M.; Davis, J.; Aucoin, D.; Sato, T.; Ahuja, S.; Aimoto, S.; Elliott, J.I.; van Nostrand, W.E.; Smith, S.O. Structural conversion of neurotoxic amyloid-beta1–42 oligomers to fibrils. Nat. Struct. Mol. Biol. 2010, 17, 561–567. [Google Scholar] [CrossRef]
- Laganowsky, A.; Liu, C.; Sawaya, M.R.; Whitelegge, J.P.; Park, J.; Zhao, M.; Pensalfini, A.; Soriaga, A.B.; Landau, M.; Teng, P.K.; et al. Atomic view of a toxic amyloid small oligomer. Science 2012, 335, 1228–1231. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.-Y.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Karran, E.; Mercken, M.; Strooper, B.D. The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 2011, 10, 698–712. [Google Scholar] [CrossRef]
- Chiti, F.; Dobson, C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef]
- Paravastu, A.K.; Leapman, R.D.; Yau, W.-M.; Tycko, R. Molecular structural basis for polymorphism in Alzheimer’s beta-amyloid fibrils. Proc. Natl. Acad. Sci. USA 2008, 105, 18349–18354. [Google Scholar] [CrossRef]
- Lührs, T.; Ritter, C.; Adrian, M.; Riek-Loher, D.; Bohrmann, B.; Döbeli, H.; Schubert, D.; Riek, R. 3D structure of Alzheimer’s amyloid-β(1–42) fibrils. Proc. Natl. Acad. Sci. USA 2005, 102, 17342–17347. [Google Scholar] [CrossRef]
- Fitzpatrick, A.W.P.; Debelouchina, G.T.; Bayro, M.J.; Clare, D.K.; Caporini, M.A.; Bajaj, V.S.; Jaroniec, C.P.; Wang, L.; Ladizhansky, V.; Müller, S.A.; et al. Atomic structure and hierarchical assembly of a cross-beta amyloid fibril. Proc. Natl. Acad. Sci. USA 2013, 110, 5468–5473. [Google Scholar] [CrossRef]
- Rubin, N.; Perugia, E.; Goldschmidt, M.; Fridkin, M.; Addadi, L. Chirality of amyloid suprastructures. J. Am. Chem. Soc. 2008, 130, 4602–4603. [Google Scholar] [CrossRef]
- GhattyVenkataKrishna, P.K.; Uberbacher, E.C.; Cheng, X. Effect of the amyloid beta hairpin’s structure on the handedness of helices formed by its aggregates. FEBS Lett. 2013, 587, 2649–2655. [Google Scholar] [CrossRef]
- Buchete, N.-V.; Tycko, R.; Hummer, G. Molecular dynamics simulations of Alzheimer’s β-amyloid protofilaments. J. Mol. Biol. 2005, 353, 804–821. [Google Scholar] [CrossRef]
- Fawzi, N.L.; Okabe, Y.; Yap, E.-H.; Head-Gordon, T. Determining the critical nucleus and mechanism of fibril elongation of the Alzheimer’s aβ1–40 peptide. J. Mol. Biol. 2007, 365, 535–550. [Google Scholar] [CrossRef]
- Lin, Y.-S.; Bowman, G.R.; Beauchamp, K.A.; Pande, V.S. Investigating how peptide length and a pathogenic mutation modify the structural ensemble of amyloid beta monomer. Biophys. J. 2012, 102, 315–324. [Google Scholar] [CrossRef]
- Fisher, C.K.; Ullman, O.; Stultz, C.M. Comparative studies of disordered proteins with similar sequences: Application to Aβ40 and Aβ42. Biophys. J. 2013, 104, 1546–1555. [Google Scholar] [CrossRef]
- Sciarretta, K.L.; Gordon, D.J.; Petkova, A.T.; Tycko, R.; Meredith, S.C. Aβ40-lactam(d23/k28) models a conformation highly favorable for nucleation of amyloid. Biochemistry 2005, 44, 6003–6014. [Google Scholar] [CrossRef]
- Reddy, G.; Straub, J.E.; Thirumalai, D. Influence of preformed asp23−lys28 salt bridge on the conformational fluctuations of monomers and dimers of Aβ peptides with implications for rates of fibril formation. J. Phys. Chem. B 2009, 113, 1162–1172. [Google Scholar] [CrossRef]
- Tarus, B.; Straub, J.E.; Thirumalai, D. Dynamics of asp23−lys28 salt-bridge formation in aβ10–35 monomers. J. Am. Chem. Soc. 2006, 128, 16159–16168. [Google Scholar] [CrossRef]
- Nguyen, P.H.; Li, M.S.; Stock, G.; Straub, J.E.; Thirumalai, D. Monomer adds to preformed structured oligomers of a beta-peptides by a two-stage dock-lock mechanism. Proc. Natl. Acad. Sci. USA 2007, 104, 111–116. [Google Scholar] [CrossRef]
- Takeda, T.; Klimov, D.K. Probing energetics of Abeta fibril elongation by molecular dynamics simulations. Biophys. J. 2009, 96, 4428–4437. [Google Scholar] [CrossRef]
- Rojas, A.; Liwo, A.; Browne, D.; Scheraga, H.A. Mechanism of fiber assembly: Treatment of Abeta peptide aggregation with a coarse-grained united-residue force field. J. Mol. Biol. 2010, 404, 537–552. [Google Scholar] [CrossRef]
- Esler, W.P.; Stimson, E.R.; Jennings, J.M.; Vinters, H.V.; Ghilardi, J.R.; Lee, J.P.; Mantyh, P.W.; Maggio, J.E. Alzheimer’s disease amyloid propagation by a template-dependent dock-lock mechanism. Biochemistry 2000, 39, 6288–6295. [Google Scholar] [CrossRef]
- Lee, J.; Culyba, E.K.; Powers, E.T.; Kelly, J.W. Amyloid-β forms fibrils by nucleated conformational conversion of oligomers. Nat. Chem. Biol. 2011, 7, 602–609. [Google Scholar] [CrossRef]
- Urbanc, B.; Betnel, M.; Cruz, L.; Bitan, G.; Teplow, D.B. Elucidation of amyloid β-protein oligomerization mechanisms: Discrete molecular dynamics study. J. Am. Chem. Soc. 2010, 132, 4266–4280. [Google Scholar] [CrossRef]
- Teplow, D.B.; Lazo, N.D.; Bitan, G.; Bernstein, S.; Wyttenbach, T.; Bowers, M.T.; Baumketner, A.; Shea, J.-E.; Urbanc, B.; Cruz, L.; et al. Elucidating amyloid β-protein folding and assembly: A multidisciplinary approach. Acc. Chem. Res. 2006, 39, 635–645. [Google Scholar] [CrossRef]
- Bitan, G.; Kirkitadze, M.D.; Lomakin, A.; Vollers, S.S.; Benedek, G.B.; Teplow, D.B. Amyloid β-protein (Aβ) assembly: Aβ40 and Aβ42 oligomerize through distinct pathways. Proc. Natl. Acad. Sci. USA 2003, 100, 330–335. [Google Scholar] [CrossRef]
- Bitan, G.; Vollers, S.S.; Teplow, D.B. Elucidation of primary structure elements controlling early amyloid β-protein oligomerization. J. Biol. Chem. 2003, 278, 34882–34889. [Google Scholar] [CrossRef]
- Bitan, G.; Lomakin, A.; Teplow, D.B. Amyloid β-protein oligomerization: Prenucleation interactions revealed by photo-induced cross-linking of unmodified proteins. J. Biol. Chem. 2001, 276, 35176–35184. [Google Scholar] [CrossRef]
- Barz, B.; Urbanc, B. Minimal model of self-assembly: Emergence of diversity and complexity. J. Phys. Chem. B 2014, 118, 3761–3770. [Google Scholar] [CrossRef]
- Streltsov, V.A.; Varghese, J.N.; Masters, C.L.; Nuttall, S.D. Crystal structure of the amyloid-β p3 fragment provides a model for oligomer formation in Alzheimer’s disease. J. Neurosci. 2011, 31, 1419–1426. [Google Scholar] [CrossRef]
- Urbanc, B.; Cruz, L.; Yun, S.; Buldyrev, S.V.; Bitan, G.; Teplow, D.B.; Stanley, H.E. In silico study of amyloid β-protein folding and oligomerization. Proc. Natl. Acad. Sci. USA 2004, 101, 17345–17350. [Google Scholar] [CrossRef]
- Urbanc, B.; Betnel, M.; Cruz, L.; Li, H.; Fradinger, E.A.; Monien, B.H.; Bitan, G. Structural basis for aβ1–42 toxicity inhibition by Aβ c-terminal fragments: Discrete molecular dynamics study. J. Mol. Biol. 2011, 410, 316–328. [Google Scholar] [CrossRef]
- Barz, B.; Urbanc, B. Dimer formation enhances structural differences between amyloid β-protein (1–40) and (1–42): An explicit-solvent molecular dynamics study. PLoS One 2012, 7. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burger, V.M.; Gurry, T.; Stultz, C.M. Intrinsically Disordered Proteins: Where Computation Meets Experiment. Polymers 2014, 6, 2684-2719. https://doi.org/10.3390/polym6102684
Burger VM, Gurry T, Stultz CM. Intrinsically Disordered Proteins: Where Computation Meets Experiment. Polymers. 2014; 6(10):2684-2719. https://doi.org/10.3390/polym6102684
Chicago/Turabian StyleBurger, Virginia M., Thomas Gurry, and Collin M. Stultz. 2014. "Intrinsically Disordered Proteins: Where Computation Meets Experiment" Polymers 6, no. 10: 2684-2719. https://doi.org/10.3390/polym6102684
APA StyleBurger, V. M., Gurry, T., & Stultz, C. M. (2014). Intrinsically Disordered Proteins: Where Computation Meets Experiment. Polymers, 6(10), 2684-2719. https://doi.org/10.3390/polym6102684