Abstract
Protein aggregation and protein self-assembly is an important occurrence in natural systems, and is in some form or other dictated by biopolymers. Very obvious influences of biopolymers on protein assemblies are, e.g., virus particles. Viruses are a multi-protein assembly of which the morphology is dictated by poly-nucleotides namely RNA or DNA. This “biopolymer” directs the proteins and imposes limitations on the structure like the length or diameter of the particle. Not only do these bionanoparticles use polymer-directed self-assembly, also processes like amyloid formation are in a way a result of directed protein assembly by partial unfolded/misfolded biopolymers namely, polypeptides. The combination of proteins and synthetic polymers, inspired by the natural processes, are therefore regarded as a highly promising area of research. Directed protein assembly is versatile with respect to the possible interactions which brings together the protein and polymer, e.g., electrostatic, v.d. Waals forces or covalent conjugation, and possible combinations are numerous due to the large amounts of different polymers and proteins available. The protein-polymer interacting behavior and overall morphology is envisioned to aid in clarifying protein-protein interactions and are thought to entail some interesting new functions and properties which will ultimately lead to novel bio-hybrid materials.
1. Introduction
In nature, self-association and -assembly is a key factor for it to function. Whether it is phospholipids forming the membranes of cells and organelles, proteins assembled within the lipid bilayer for regulating trans-membrane transport, polynucleotides like RNA in combination with proteins forming the ribosomes or the microtubules which are built from a protein dimer aggregating up to 20–25 µm in length with a width of only 25 nm, and not only provide strength to the cell but also act as an intra-cellular highway along which transport occurs. In many of the processes, directed protein assembly is pivotal and hence, understanding the assembly of proteins is an important aspect to elucidate. Not only is this important in proper functioning biological systems, also many life-threatening disorders are the result of improper or undesired aggregation of proteins or polypeptides. In neurodegenerative diseases like Parkinson’s, Alzheimer’s and Huntington’s disease the common mutual cause is the atypical aggregation of proteins [1]. While the proteins by themselves do not necessarily aggregate, it is often associated with partial denaturation or from partial cleavage creating a polypeptide, which induces the aggregation, or because of a polypeptide mutation (polyglutamine) [2,3]. One can say that a biopolymeric species induces proteins to aggregate creating such disorders. Other systems where a biopolymer dictates protein aggregation are, e.g., virus particles, which are used in many approaches for new materials synthesis [4,5]. A virus consists of small capsids, protein structures, which self-assemble around a polynucleotide (RNA or DNA). The size, shape and stability of these virus particles is dictated by the presence of the biopolymer, and though without its presence some viruses are still able to self-assemble; the overall structure or morphology is affected because of the absence of it [6].
Studying protein assembly behavior and especially the ability to direct it, as mentioned above, is not only important for understanding nature and developing new treatments for protein aggregation related disorders but also for the development of new types of (bio-hybrid) materials. In nature, many of the processes are dictated by polymeric species (oligo- and polypeptides/polynucleotides) and therefore it is important to highlight the progress made in synthetic polymer induced and polymer directed protein assembly with respect to the development of new systems and approaches. In this review, the directed assembly of proteins with synthetic polymers is discussed, ranging from non-covalent systems where coordination and interaction between protein and polymer is facilitated via electrostatic interactions, and other non-covalent interactions like hydrogen bonding and hydrophobic interactions. Additionally, many systems are based on combining the protein and the polymer covalently [7]. Covalently connecting the polymer and protein alters the behavior, response and stability of the protein and increases compatibility with other polymeric systems, e.g., in phase-separated block-copolymer system [8,9].
Using polymers for directed protein assembly is very attractive because of the highly diverse character of synthetic polymers. At present time there are so many different types of polymers available, each with its own properties and responsiveness. Charged polymers are very useful because proteins are charged and electrostatic interactions are a very convenient approach for assembly (Figure 1a). The charges on proteins originate from the amino acids within the polypeptide structure and the amino acids also possess various reactivities in the form –NH2 (lysine), COOH (glutamic acid, aspartic acid), Aryl–OH (tyrosine) or –SH (cysteine). These reactive amino acids can be used as a tool for functionalization either to attach polymers directly or be modified in order to be able to grow the polymers directly from the protein surface (Figure 1c) [10,11,12]. Also possible is a combination of the two approaches, while some proteins would not display any affinity for certain polymers or oligomers, when modified with a small molecular component or receptor, specific and direct assembly can be induced which combines very well with many supramolecular polymeric hydrogel systems [13]. In such coordinated systems, charge interactions with specific metal ligand residues is possible, but also hydrophobic interactions or hydrogen-bonding can be used (Figure 1b). In the following sections, the mentioned approaches will be addressed according to the most recently performed studies and system developments.
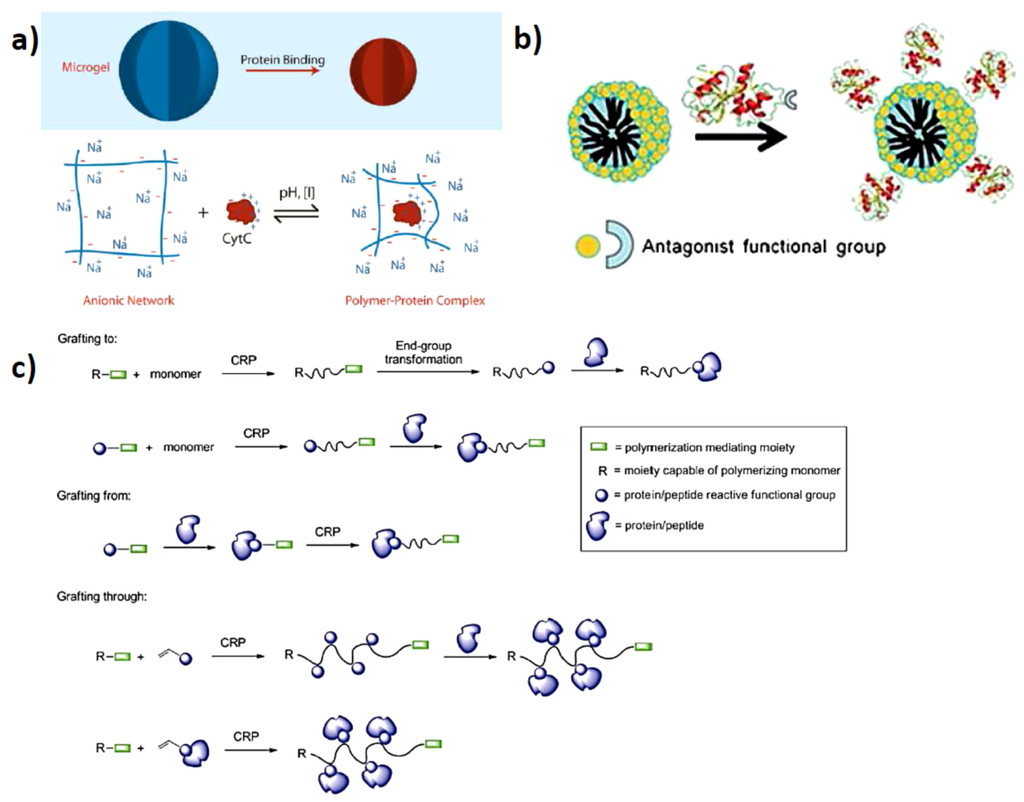
Figure 1.
Schematics of (a) polymer protein interactions by opposite charges (reprinted with permission from [14]); (b) Covalent functionalization of the protein to incorporate specific receptor-like interactions with polymeric systems (reprinted with permission from [15]); (c) Covalent attachment to- and growing from the protein surfaces (reprinted with permission from [7]).
Figure 1.
Schematics of (a) polymer protein interactions by opposite charges (reprinted with permission from [14]); (b) Covalent functionalization of the protein to incorporate specific receptor-like interactions with polymeric systems (reprinted with permission from [15]); (c) Covalent attachment to- and growing from the protein surfaces (reprinted with permission from [7]).
2. Electrostatically Directed Protein-Polymer Assembly
Electrostatic interactions are one of the most frequently used approaches for combining polymers and proteins mainly because the different components can be easily used in aqueous solutions under ambient conditions which is convenient for proteins. Also the surface charge of proteins can be adjusted by controlling the pH. The available lysine, arginine and histidine residues provide positive charges while negative charges reside on the aspartic acid, glutamic acid and tyrosine residues. Each has a certain pKa and thus can be switched between charged and neutral depending on the pH. This allows for controlled interactions with polyelectrolyte polymers and provides the possibility to release the protein or to disassemble a complex structure [14,16]. This has been used to create electrostatically assembled protein-polymer structures in solution as well as on surfaces [17]. In some systems, a natural polyanionic structure is involved in the assembly process namely, virus particles. The virus capsids (viral protein subunits) can be redirected by removing the nucleotide strand and replacing it with a synthetic polymer to alter the morphology. The native nucleotide can also be used to direct the assembly in a non-natural fashion. While in principle the particle morphology is maintained when the capsids aggregate around it, when e.g., a surface, is modified with the RNA of the respective virus in a brush-like manner, surface directed growth can be initiated [18].
2.1. Solution Assembled Structures via Electrostatics
Electrostatic interactions between proteins and polymers are important in many biological settings. An important class of biopolymers which naturally have electrostatic interactions with proteins are glycosaminoglycans. These are flexible polysaccharides, which are decorated with carboxylic acid residues and sulfate groups. Glycosaminoglycans are found on the cell surface and in connective tissue where it functions as an important component of the extracellular matrix [19]. One class in particular which has many different functions are the “heparinoids”, which regulate cell -adhesion, -differentiation, -signaling and anti-coagulation [20]. Because of the anti-coagulation properties in blood, it is also used as a post-operative medicine to prevent thrombosis, although nowadays a synthetic variant of lower molecular weight is used to decrease side-effects. Just by looking at the processes of these natural occurring polyelectrolytes (PE) one can recognize that there are many possibilities to transform this concept into a synthetic system and expand upon it to create hybrid structures, which are useful not only for medical applications but also for industry as a way to introduce enzymes into environments that would normally not allow for enzymatic activity. A recently extensive review by Kayitmazer et al. provides an in-depth analysis of the physical properties and interactions in polyelectrolyte-protein systems [21].
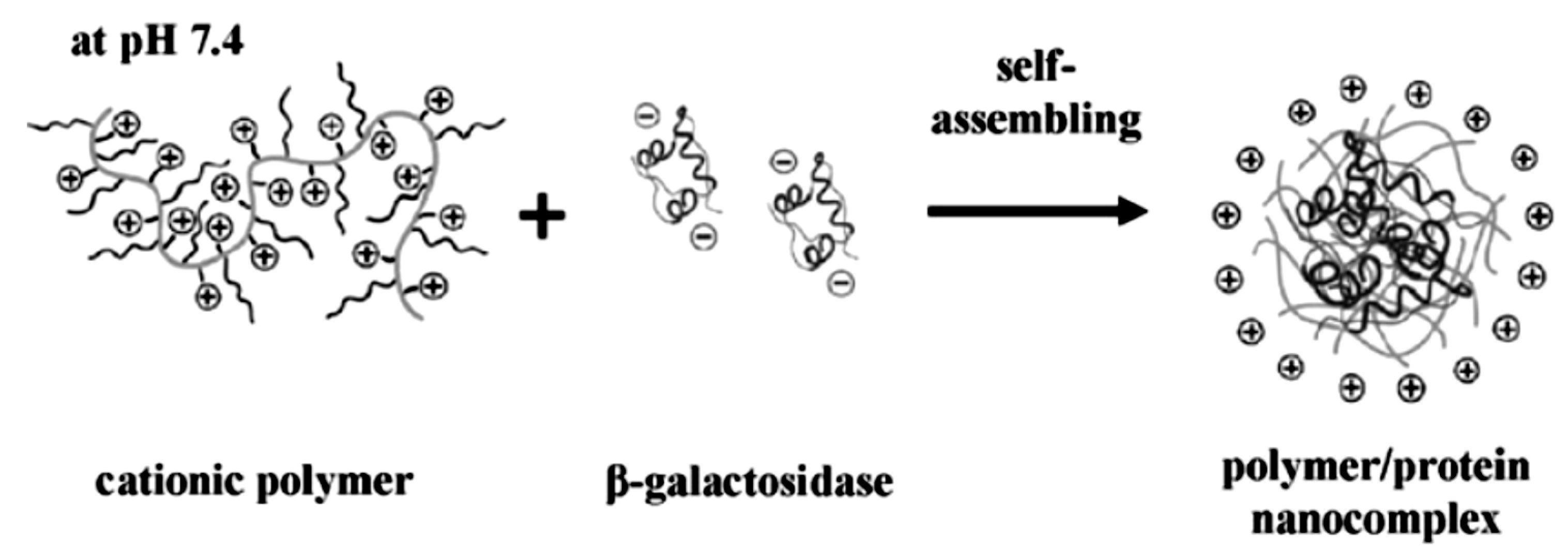
The polyelectrolyte-protein interaction and the nano-sized structures that are produced are regarded as a promising way for intra-cellular delivery of proteins. There are two main approaches for the fabrication of the electrostatically derived polymer-protein complexes. The charged polymer and the protein can be mixed and it aggregates into a hybrid soft nanoparticle-like structure or a predetermined charged soft polymeric nanoparticle structure is prepared, so called microgels, which acts as a porous hydrogel particle capable of binding oppositely charged species [14]. Simple mixing provides a convenient way of adjusting the system without too much synthetic effort, and it is easy to mix in various other types of charged species or attempt a layer-by-layer type of assembly. Because the charge of the protein, and also often that of the polymer, can be adjusted by simply changing the pH, it is a convenient way to induce disassembly. This has been done for various combinations of negatively charged as well as positively charged proteins. For this purpose, linear poly(amidoamine)s were synthesized via a poly-addition via a Michael-type reaction using histamine which provides the positive charges at neutral pH. This positively charged polyelectrolyte was mixed with β-galactosidase which is overall negatively charged at neutral pH. Mixing the two components resulted in the formation of hybrid nanoparticles with a size of 100–150 nm in diameter (Figure 2). The protein up-take was determined to be about 95% and could be released via acidification of the medium or via the reduction of a build-in disulfide bridge in the linear polymeric array [22]. It is important to have more than one way of releasing the protein contents from the overall structure because in this particular case the pH needs to be lowered to pH 5 and is not always physiologically feasible. Although pH 5 is still viable in some cases, other examples where e.g., the polymer, is targeted by protonating carboxylic acid moieties, the pH needs to be even lower which is not often found in vivo. For the in vitro study of intra-cellular uptake of the cationic-PE/β-galactosidase complex into COS-7 cells, it was found that internalization of up to 94% of the cells was achieved with absence of any cell toxicity.
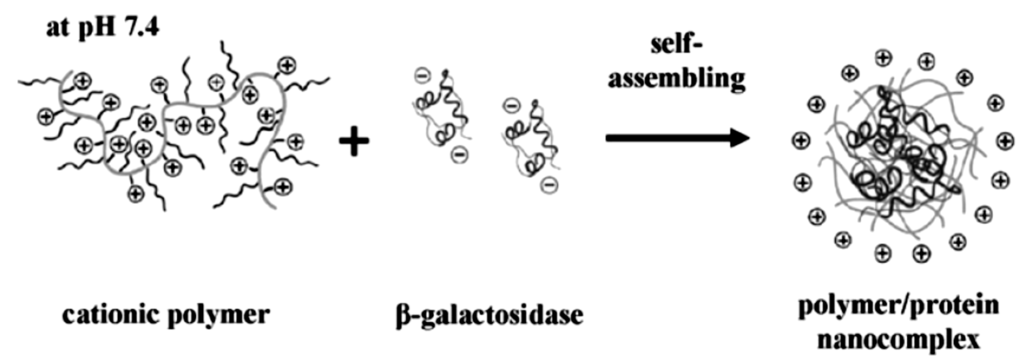
Figure 2.
Schematics of the cationic polyelectrolyte in combination with the negatively charged protein (β-galactosidase) producing hybrid protein-polymer nanostructures (reprinted with permission from [22]).
Figure 2.
Schematics of the cationic polyelectrolyte in combination with the negatively charged protein (β-galactosidase) producing hybrid protein-polymer nanostructures (reprinted with permission from [22]).
A similar approach was used to create complexes with insulin. For this a charged oligo-peptide in the form of an oligo-lysine and oligo-arginine was connected to a linear polyethylene glycol (PEG). Upon mixing of a fixed concentration of insulin with varying amounts of the cationic oligo-peptide-PEG structure, differently sized structures were obtained in the range between ~100–1800 nm with increasing amounts of cationic polymer [23]. This increase in size was associated with a reversal of surface charge going from negative for pure insulin to positive upon addition of the cationic oligo-peptide structure, which indicates coordinating and enclosing the protein. The release was studied by introducing the protein-polymer complex into simulated intestine fluid at pH 6.8 and displaying a controlled release, which was influenced by the type of oligo-peptide used and the length of the PEG-chain. In addition to having a controlled release system, the assembled structure can also provide stability or protection against denaturing or inactivating conditions. While insulin without complexation to the cationic oligo-peptide displayed a very fast degradation in the presence of trypsin and α-chymotripsin (within ~60 min), the complex displayed prolonged stability and had a much slower degradation rate.
While in some cases the protein is protected by the polymer, one needs to take care that only coordination occurs and no other effect will influence or harm the protein structure or function. This was demonstrated by Kurinomaru et al. who encapsulated/complexed α-amylase and β-galactosidase with poly-allylamine [24]. In order to recover the protein as a “free” structure, poly-acrylic acid was added to interact with the cationic poly-allylamine and thereby setting free the protein. The protein was found to be deactivated with respect to catalytic activity, which was caused by the loss of secondary structure and was confirmed by circular dichroism. So, although there is reversible complexation, in this case it is not useful since the protein does not function properly anymore after release. To prevent this, they designed a new polyelectrolyte block-copolymer in the form of poly(N,N-diethylaminoethyl methacrylate)-block-poly(ethylene glycol). This also displayed complexation of the protein but in a more controlled fashion. The aggregates not only remained small, two orders of magnitude smaller than for the poly-allylamine, but also after the addition of poly-acrylic acid the liberated protein still displays catalytic activity. This is a clear example which demonstrates that just complexation-release studies is not enough to obtain interesting hybrid protein-polymer structures but also that one needs to take care to ensure that the function of the protein is not compromised during the process. The described process here deactivates the protein during mixing with the cationic polyelectrolyte and activity is regained after removal. Also possible is that activity is retained during complexation and in that case it can be regarded as a protected activated enzymatic container allowing for the use of the enzyme in otherwise impossible reaction conditions, e.g., in organic solvents. This would be very convenient for industrial applications since enzymatic catalysis offers high selectivity but is limited to mild aqueous conditions. Wilson and coworkers co-aggregated penicillin G acylase with polyionic polymers polyethyleneimine and dextran sulfate, creating a protein-polymer ammonium sulfate like complex which afforded complexes which were tolerant to organic cosolvents (dioxane) up to 60% without too much inactivation of the enzymatic activity. The created micro-environment assures stability by rigidity of the matrix surrounding it and hence denaturing effects of the organic cosolvent is minimized [25].
2.2. Surface Assembled Electrostatic Structures
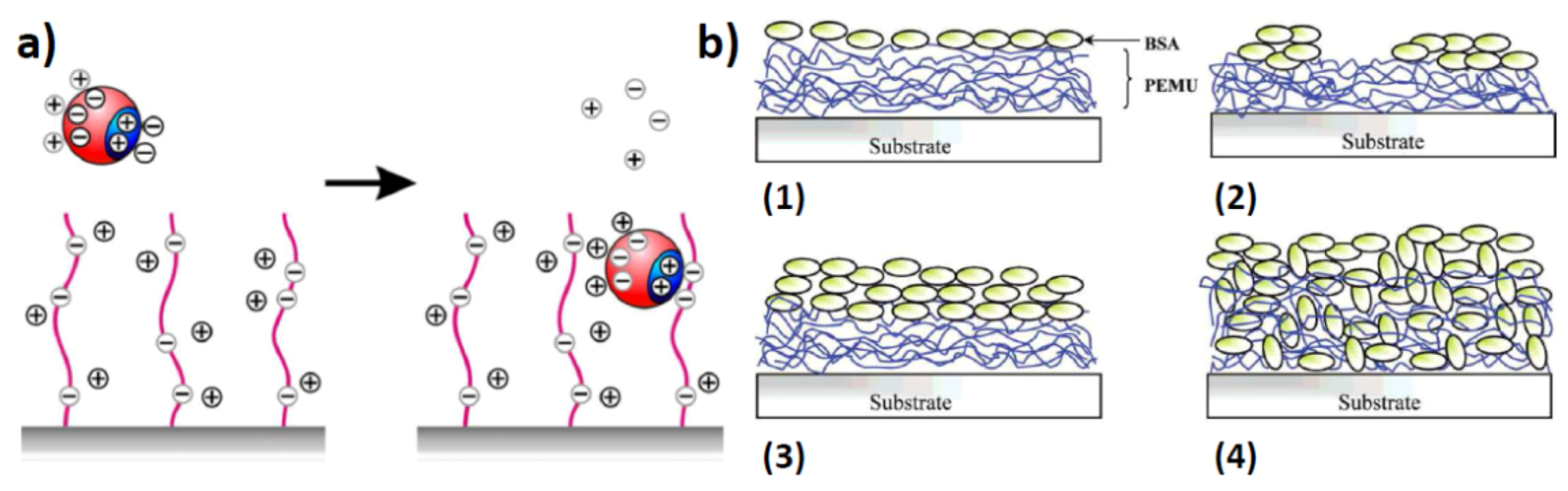
The formation of solution-based structured complexes can also be translated to surface-confined structures. By using a similar approach of electrostatic adsorption of proteins onto a charged surface [26] will allow for a controlled surface structuring of proteins and polymers in various combinations. However, on surfaces one needs to take particular care of denaturation at the surface after binding since adsorption can result in (partial) denaturation of the protein structure, especially on hard surfaces [27]. While in general brush-like and hydrogel-like layers, e.g., polyethylene glycols, attached to the surface will repel proteins [28,29], using charged polymers will result in attraction when the appropriate charge is being used. The main driving force is the replacement of several counterions from the brush-layer into the solution medium upon complexation of the protein which makes it entropically favorable while the multiple opposite charges ensure strong binding (Figure 3a) [30,31]. In order to remove the proteins from the surface, either changing the pH to remove any charge interactions is needed or extremely high concentrations of strongly coordinating counterions to overcome the entropic penalty for protein removal. When attracting proteins onto a charged surface, one can rely on the so-called layer-by-layer deposition, which is commonly used for the controlled fabrication of layered surfaces using oppositely charged polyelectrolytes [32]. These layered structures can also be formed when exchanging one of the polyelectrolytes for a similarly charged protein. In this case, an alternating sequence of polymer-protein-polymer-protein is created by alternatingly dipping the surface in the polymer and protein solution. These hybrid layers are regarded as useful for coating materials, biodegradable coatings, drug delivery layers and biocompatible films. One such system has recently been developed which displays multilayers of insulin with negatively charged polyelectrolytes namely, polystyrene sulfonic acid and hyaluronic acid [17]. First poly(diallyldimethylammonium chloride) was adsorbed onto a negatively charged silicon oxide substrate. This was then alternatingly subjected to a solution of polystyrene sulfonic acid and insulin or hyaluronic acid and insulin. A steady increase in thickness was observed after every dipping procedure. This approach with similar interactions towards polyelectrolytes can be generalized for other combinations as well which offers the potential of creating complex films with potentially different protein sequences internalized inside the film (Figure 3b) [33].
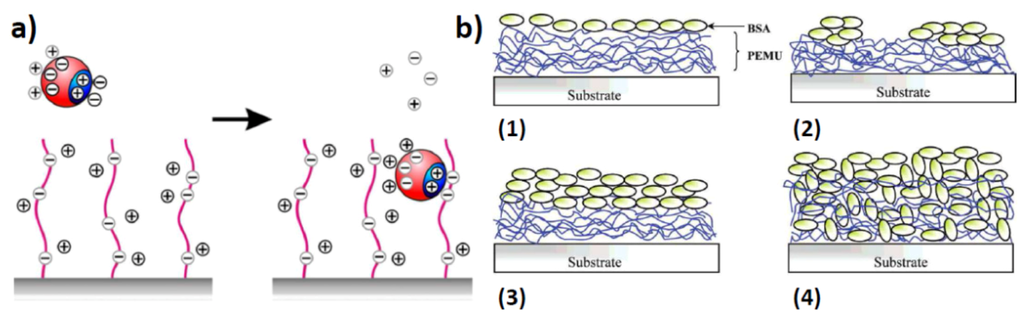
Figure 3.
(a) Uptake of proteins by polyelectrolyte brushes. A positively charged patch on a protein can replace the counterions in a negatively charged polymer brush, despite the fact that the overall charge of the protein is negative. The counterions from the surface of the protein, and the counterions from the polymer brush are released into the bulk of the solution. The increase in entropy upon counterion release is the main driving force for protein adsorption on the “wrong side” of the isoelectric point, i.e., when the net charge of the protein globule has the same sign as the chains in the brush (reprinted with permission from [31]); (b) Schematic of protein adsorption onto/into polyelectrolyte multilayers. (1 and 2) Apparent monolayers. (3 and 4) Apparent multilayers (reprinted with permission from [33]).
Figure 3.
(a) Uptake of proteins by polyelectrolyte brushes. A positively charged patch on a protein can replace the counterions in a negatively charged polymer brush, despite the fact that the overall charge of the protein is negative. The counterions from the surface of the protein, and the counterions from the polymer brush are released into the bulk of the solution. The increase in entropy upon counterion release is the main driving force for protein adsorption on the “wrong side” of the isoelectric point, i.e., when the net charge of the protein globule has the same sign as the chains in the brush (reprinted with permission from [31]); (b) Schematic of protein adsorption onto/into polyelectrolyte multilayers. (1 and 2) Apparent monolayers. (3 and 4) Apparent multilayers (reprinted with permission from [33]).
As mentioned, polyethylene glycol will repel proteins from the surface while charged polymers may attract them. This notion allowed for the development of more complex films where there is a combination of attractiveness and repulsion in a single system. Delcroix et al. used a mixed brush system in which both polyethylene glycols as well as poly-acrylic acid was grafted onto the surface. Both polymer-types can be made to collapse or extend depending on the stimuli applied. Ethylene glycol generally collapses with increasing temperature due to the lower critical solution temperature while acrylic acid tends to “precipitate” when decreasing the pH or change conformation with deviating ionic strength [34]. This approach resulted in a mixed combination of an attractive and repelling surface where initially, in the mixed phase where both polyethylene glycol and polyacrylic acid are extended, albumin is adsorbed by the favorable interactions with the negatively charged acrylic acid. Lowering the pH or increasing the ionic strength leads to collapse and reduced attraction between the acrylic acid and the albumin. Additionally, the repulsive effect of the polyethylene glycol ensures fast release of the protein from the surface. These “smart” surfaces are expected to be developed further in the near future as potential bio-sensing platforms or anti-fouling surfaces. This “smart” triggered approach was also demonstrated by using a block-copolymer system based on poly(N-isopropylacrylamide)-block-2-(dimethylamino)-ethyl acrylate) (PND) [35]. In solution, the anionic fluorescent protein mCherry coordinates to the micelle forming PND via electrostatic interactions with the positively charged dimethylamino-ethyl acrylate. Either by lowering the pH or increasing the temperature the dimethylamino)-ethyl acrylate and N-isopropylacrylamide are affected, respectively. Both situations induce release of mCherry. This approach not only functioned in solution but was also transferred to thin phase-separated block-copolymer films.
2.3. Artificial Assembly of Virus Particles
In the above described section, the binding of the proteins to the polyelectrolytes resulted in fairly unspecific structures without a high degree of internal structural control or specific overall morphology. These mixed systems are in general not as defined as natural systems. One protein system in particular interacts very specifically with polyelectrolytes, and that is viruses. These particles are composed of sometimes several thousand protein subunits, and they assemble in a highly defined fashion around the negatively charged biopolymer namely, RNA or DNA [6]. The oligonucleotide initiates the assembly process and also dictates specific morphology and size. The controlled assembly of the viral coat proteins around the natural RNA-sequence can be used to target, e.g., surfaces, when the RNA is attached by one side to a planar or particle surface, the same directed protein assembly can be used to structure the surface with coat proteins. This has been shown where the native RNA was attached via a streptavidin-biotin label onto the surface of silicon oxide on which the viral assembly process was initiated and resulted in reconstituting the morphology from the bottom-up [18]. While this results in a surface modification/structuring via the natural assembly process, alternatively it is also possible to initiate aggregation in solution by exchanging the natural anionic biopolymer (RNA) for a synthetic polyelectrolyte. When the viral coat proteins are separated from the RNA and isolated, they often still are able to re-assemble, but in a different structure/morphology or with an altered symmetry. Cowpea chlorotic mottle virus (CCMV), which normally assembles around a single stranded RNA structure was mixed with a double stranded DNA as an anionic biopolymer template [36]. It was found that this extended polynucleotide, which is more rigid than the virus’s original single stranded RNA, allows for assembly into rigid rod-like structures. By offering an alternative template, the assembly process is redirected and completely new structures arise. This approach was also taken using a synthetic fluorescent polymer consisting of a rhodamine B labeled polystyrene sulfonate [37]. Depending on the size of the polyelectrolyte, differently sized virus-like particles (VLPs) were formed. With a molecular weight of about 1 MDa and lower, the VLPs formed are about 22–23 nm in diameter which is consistent with a T = 2 triangulation number consisting of 120 protein units organized into 12 pentamers of dimers. When the molecular weight is increased to 2–3.4 MDa, the VLPs displayed a diameter of 27–28 nm, which indicates a T = 3 structure containing 180 subunits arranged in 12 pentamers and 20 hexamers [38]. Similar rod-like and globular particle structures with ccmv were obtained when mixed with a semi-conducting polymer poly-2-methoxy-5-propyloxy sulfonate phenylenevinylene (MPS-PPV). By controlling the polymer conformation using different ionic strengths, going from low to high ionic strength, the morphology changed from rod-like to mixed rod-like/globular to finally completely globular. Additionally, the fluorescent properties also changed with the polymer conformation obtaining not only different overall particle morphologies, but also with different optical and electronic properties [39]. Future research will produce more of these biohybrid materials since new morphologies with new applications will emerge and the strength of combining electrostatic interactions with synthetic and biological structures is a straightforward and easy way of fabrication and allows also for large-scale fabrication under mild aqueous conditions. Not only for the directed intra-viral self-assembly but also for the directed aggregation of inter-viral assemblies where particles are directed by the surface charge and complementary charged polymers of different architectures and controlled/reversed by factors like ionic strength, pH or temperature [40].
3. Directed Assembly of Polymer-Protein Conjugates
When directing the assembly of proteins, non-covalent forces are not always sufficient for coordination or to direct the protein in a specific way. In recent years, covalently connecting polymer and protein has become an important research area and the protein-polymer conjugates produce new classes of materials, which are not accessible using non-covalent interactions alone. There are various methods for achieving the covalent connection between a protein and a polymer and the topic is a much reported one. Therefore, the reader is directed to one of the recent reviews on the matter which offer a broad perspective on synthetic approaches for protein modifications and in particular using different polymerization techniques, e.g., grafting to and grafting from which the latter is performed via controlled radical polymerizations (ATRP or RAFT) [10,15,41].
By covalently connecting the polymer and protein, new bio-block-copolymers emerge either in the form of one linear synthetic polymer attached to one side of the protein which results in a semi-structured block-copolymer but also when several polymers are attached to a central protein structure, a star-block-copolymer is formed which in some ways can be regarded as a core-shell hybrid microgel-like structure. These types of structures have been used in different settings, forming various novel materials in thin films and by solution-based aggregation or interface directed self-assembly. Additional directional assembly and environmental responsiveness is incorporated when responsive polymers are used of which N-isopropylacrylamide (NIPAAm) is probably the most popular one.
3.1. Protein-Polymer Block-Copolymers
Block-copolymers have been known for many years and are able to phase-separate into various structures in bulk and thin films like micellar/cylindrical/lamellar but also in solution various morphologies and architectures can be achieved in the form of micelles, elongated micelles and bilayer structures [42]. The concept of block-copolymer behavior has been extended towards protein-polymer structures in which the protein is considered as one of the blocks. In this way, depending on the difference in polarity between the protein and polymer used as well as the relative lengths, one can predict more or less where the protein will end-up.
3.1.1. Self-Assembly in Phase-Separating Thin Films
In order to achieve surface confined protein structures with a distinct arrangement, surface patterning via contact printing of protein structures or covalent connecting proteins to a predetermined reactive surface pattern is usually the preferred method [43,44]. However, the features obtained by contact printing are limited in size and coordination, or adsorption of proteins onto block-copolymer thin films offer a two-step approach of first, film formation and second, protein adsorption, to achieve nano-sized protein features on surfaces. This approach has been performed using polystyrene-block-poly(methyl methacrylate) as well as polystyrene-block-polyvinylpyridine, in combination with immunoglobulin G, protein G, mushroom tyrosinase and horseradish peroxidase [45,46]. It was found that one of the phases in the phase-separated amphiphilic block-copolymer film could be used to bind the protein and therefore direct the assembly of the proteins and create well-ordered nanoscale protein features.
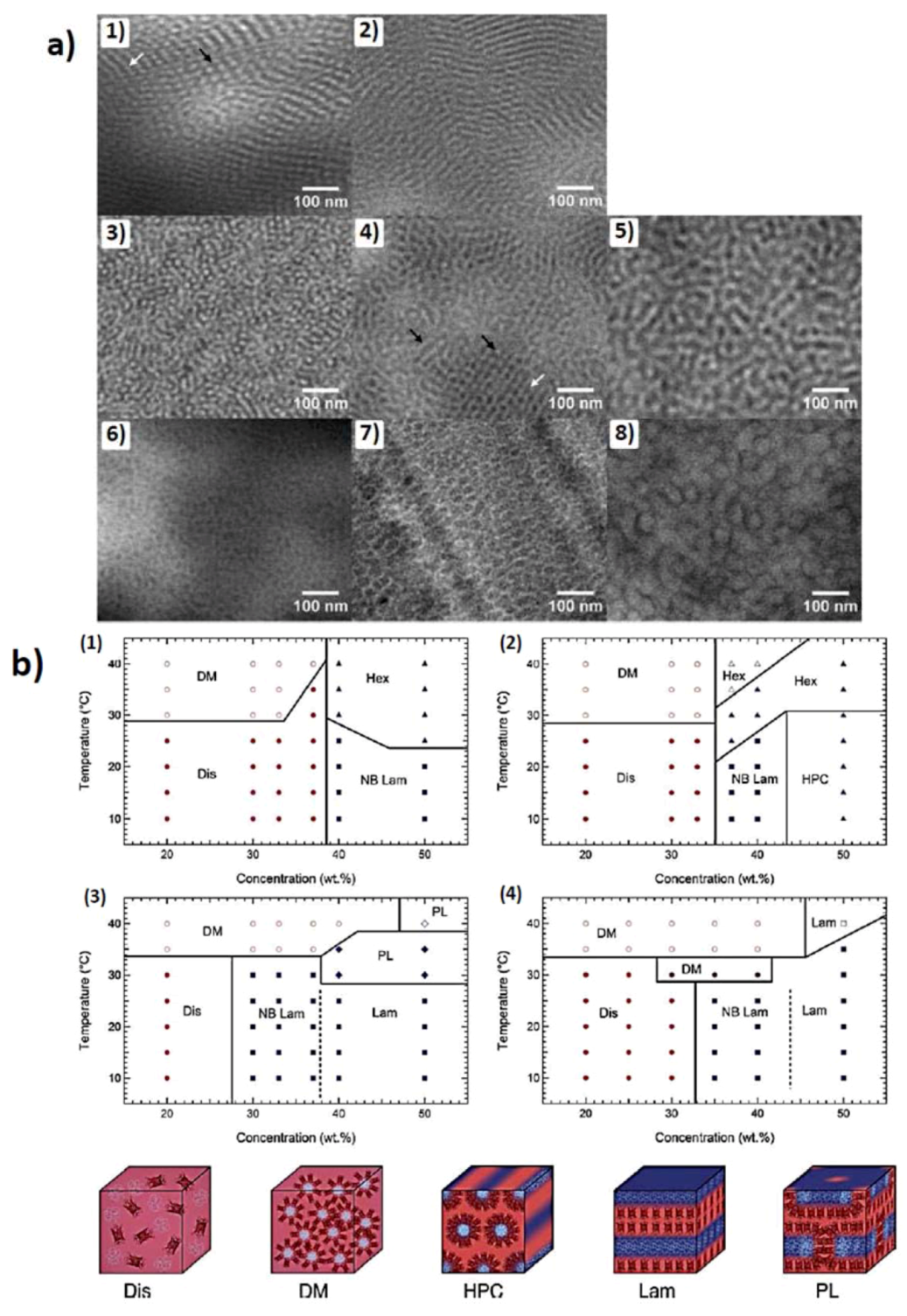
The two-step method has the advantage that the film morphology can be controlled and since protein adsorption is in a separate step, it is possible to use other proteins as well with either different phase-preference or with other intrinsic protein properties (catalysis, storage, fluorescence, specific binding affinities etc.). However, to expand the scope a one-step approach has been developed as well. Here the second block is composed of the protein structure and a single polymer chain is attached. Olsen and coworkers used an engineered red fluorescent protein, mCherry, which was modified to contain one exposed cysteine residue making a thiol-functionality available for covalent attachment of a single polymer. Onto the thiol, a pre-synthesized poly-NIPAAm chain was attached which has on one end a maleimide-functionality which is able to conjugate to a thiol-group under mild conditions [47]. The use of pNIPAAm is a clever choice since it allows for water to be used as a non-selective as well as a selective solvent, which is normally a way to influence assembly behavior in block-copolymer films. Above the lower critical solution temperature (LCST, ~32–37 °C) pNIPAAm is non-compatible with water due to dehydration and therefore water is only selective towards the protein phase. At room temperature pNIPAAm is fully hydrated and water soluble, making room temperature water a non-selective solvent for the system. The use of water at moderate temperatures ensures that the secondary structure of the protein is not compromised. It was found that the newly developed block-copolymer structures displayed similar thin film morphologies as conventional block-copolymers and depending on the treatment and temperature various structures were obtained like lamellae and hexagonally perforated lamellae (Figure 4). The assembly behavior of these protein-polymer conjugates were found to be dependent on the relative block-size just like conventional block-copolymer. Since the protein structure cannot be expanded, the size of the pNIPAAm-block was varied [48]. Depending on the length of the pNIPAAm in combination with the conjugate contents (wt %) and solvent quality which is controlled by the temperature, a complete phase diagram was established which displayed many of the commonly seen morphologies ranging from disordered, micellar, cylindrical to lamellar and perforated lamellar (Figure 4) [49].
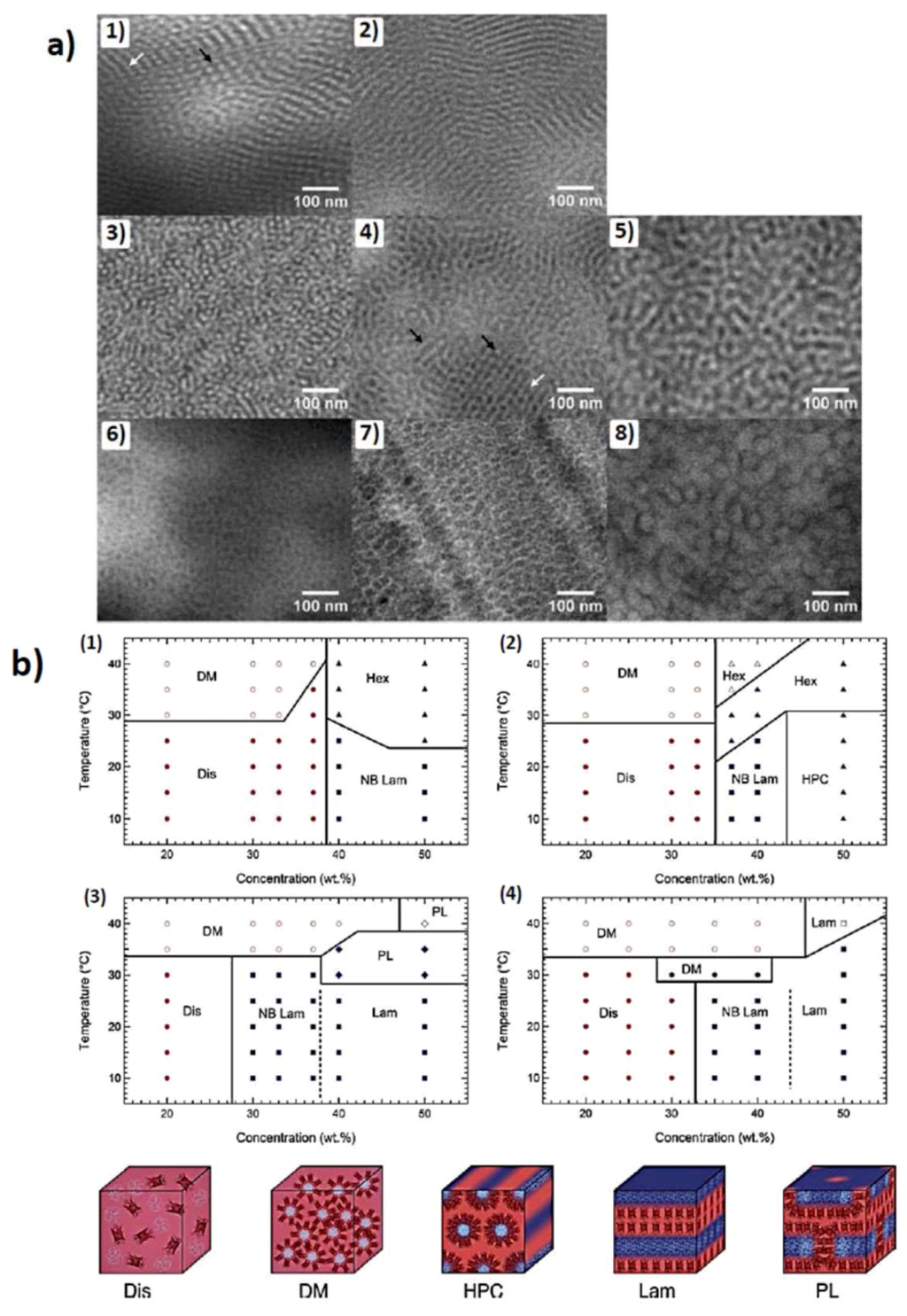
Figure 4.
(a) TEM images for as-cast mCherry-PNIPAM block copolymers. (1) Cylinders cast from a polymer-selective solvent. The white arrow shows an example of cylinders parallel to the field of view, and the black arrow points out perpendicular cylinders. (2) Lamellae cast from a polymer-selective solvent. (3) Cylinders cast from a nonselective solvent. (4) Nanostructure cast from a nonselective solvent. The white arrow designates a region of perforated lamellae, and the black arrows show side-on and edge-on cylindrical nanostructures. (5) Disordered lamellae cast from a nonselective solvent. (6) Disordered micelles cast from a protein selective solvent. (7) Hexagonally packed micelles cast from a protein-selective solvent. (8) Hexagonally packed micelles cast from a protein selective solvent (reprinted with permission from [48]); (b) Phase diagrams of (1) mChP8 (2) mChP17(3) mChP30 (4) mChP57 (P plus number depicts Mn of pNIPAAm in kD) as a function of temperature and concentration. The various phases are denoted as disordered (Dis), disordered micellar (DM), non-birefringent lamellar (NB Lam), lamellar (Lam), nonbirefringent hexagonal (Hex), hexagonally packed cylinders (HPC), and perforated lamellar (PL). Open symbols represent regions where macrophase separation between a conjugate rich ordered phase and a water-rich phase is observed. (reprinted with permission from [49]).
Figure 4.
(a) TEM images for as-cast mCherry-PNIPAM block copolymers. (1) Cylinders cast from a polymer-selective solvent. The white arrow shows an example of cylinders parallel to the field of view, and the black arrow points out perpendicular cylinders. (2) Lamellae cast from a polymer-selective solvent. (3) Cylinders cast from a nonselective solvent. (4) Nanostructure cast from a nonselective solvent. The white arrow designates a region of perforated lamellae, and the black arrows show side-on and edge-on cylindrical nanostructures. (5) Disordered lamellae cast from a nonselective solvent. (6) Disordered micelles cast from a protein selective solvent. (7) Hexagonally packed micelles cast from a protein-selective solvent. (8) Hexagonally packed micelles cast from a protein selective solvent (reprinted with permission from [48]); (b) Phase diagrams of (1) mChP8 (2) mChP17(3) mChP30 (4) mChP57 (P plus number depicts Mn of pNIPAAm in kD) as a function of temperature and concentration. The various phases are denoted as disordered (Dis), disordered micellar (DM), non-birefringent lamellar (NB Lam), lamellar (Lam), nonbirefringent hexagonal (Hex), hexagonally packed cylinders (HPC), and perforated lamellar (PL). Open symbols represent regions where macrophase separation between a conjugate rich ordered phase and a water-rich phase is observed. (reprinted with permission from [49]).
3.1.2. Self-Assembly in Solution
Self-assembly of protein-polymer block-copolymers is not limited to bulk/thin films. The protein is obviously hydrophilic and when a polymer is attached which is incompatible with water or becomes incompatible with water upon applying special conditions like increased temperature (pNIPAAm) or a change in pH (pAAc for low pH, pEI for high pH), an amphiphilic system is created which is able to self-assemble in very much the same way as conventional surfactants or amphiphilic synthetic block-copolymers do [50].
One of the first examples of such a system came from Nolte and coworkers which used lipase B from candida antarctica (CALB) onto which a polystyrene chain was attached via a maleimide-thiol conjugation. Only one chain of ~40 styrene-units per protein was attached and dispersing the bioconjugate in water resulted in micrometer long rods and bundles of rods with a diameter between 25 and 30 nm which is in good agreement with twice the conjugate-size [51]. Since then the available structures has expanded and also vesicles as well as micellar aggregates have been developed (Figure 5) creating the same self-assembled morphologies as one would expect from conventional surfactants and amphiphilic block-copolymers but now with locally arranged protein structures on the surface of the aggregates [50].
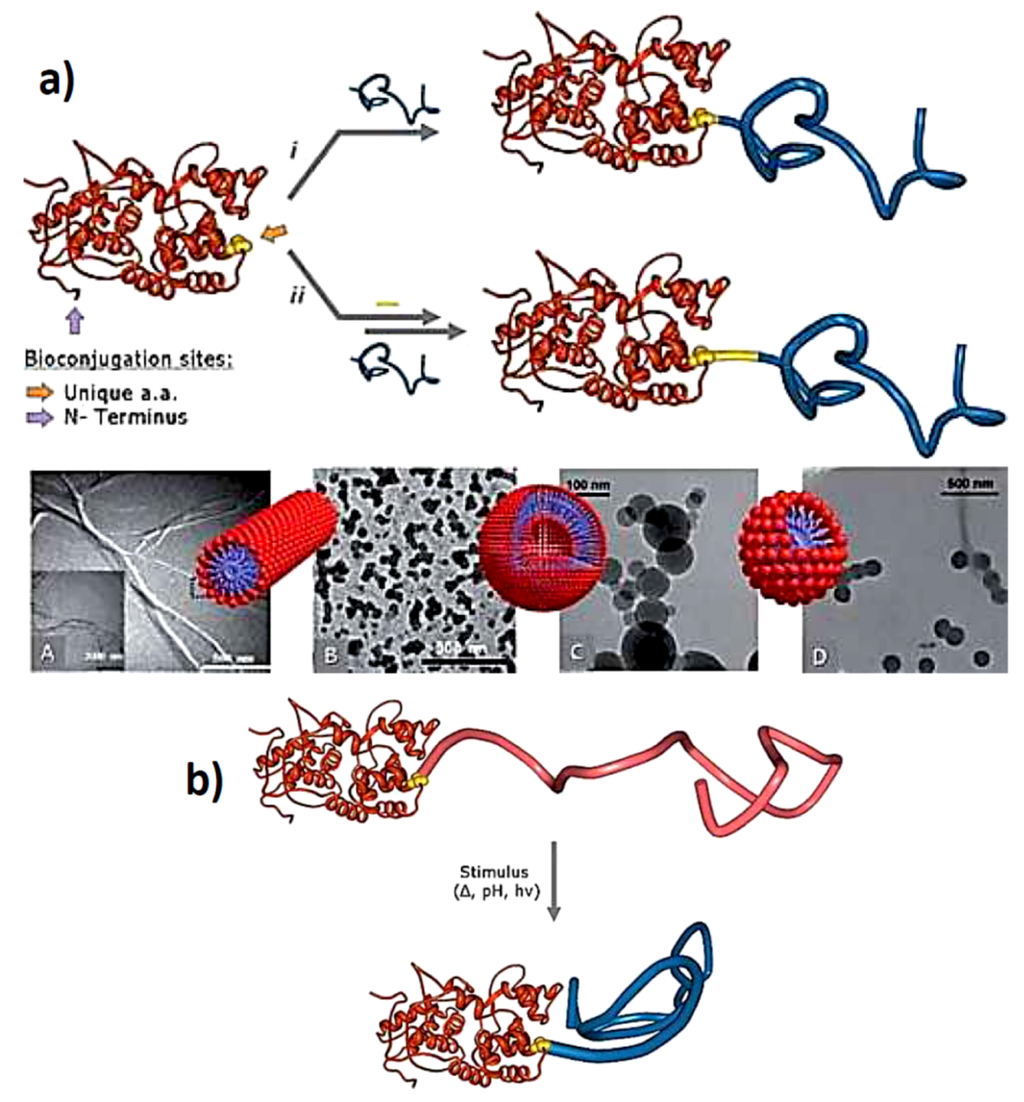
Figure 5.
(a) (A) schematic representation of the ‘‘grafting to’’ synthesis of Giant Amphiphiles through the i direct and ii indirect bioconjugation approach. Morphologies of Giant amphiphiles: (A) CALB-PS micellar rods, (B) BSAPS vesicles, (C) BSA-PA@Bz and (D) BSA-PA@C10 spherical structures, (reprinted with permission from [50]); (b) Representation of stimuli responsive “smart” giant amphiphile (reprinted with permission from [50]).
Figure 5.
(a) (A) schematic representation of the ‘‘grafting to’’ synthesis of Giant Amphiphiles through the i direct and ii indirect bioconjugation approach. Morphologies of Giant amphiphiles: (A) CALB-PS micellar rods, (B) BSAPS vesicles, (C) BSA-PA@Bz and (D) BSA-PA@C10 spherical structures, (reprinted with permission from [50]); (b) Representation of stimuli responsive “smart” giant amphiphile (reprinted with permission from [50]).
Similar approaches can be taken as for the previously described thin film systems. When taking a water soluble polymer, which is able to change the preference towards water upon applying a stimulus, controlled association and dissociation is possible. Also this allows for a more convenient synthetic approach since the complete synthesis can be performed in water instead of using a cosolvent as was needed in the case for CALB in combination with polystyrene. This also opens the gate towards less stable and more delicate proteins. In that respect, onto EGFP (enhanced green fluorescent protein) different random copolymers of ethylene glycol methyl ether acrylate (EGMEA) and methoxy ethoxy ethyl acrylate (MEEA) were conjugated with different relative ratios [52]. The created polymer is temperature responsive and with increasing temperature it becomes dehydrated and precipitates. The different ratios affect the temperature where the dehydration occurs and hence, more control is obtained on the aggregation behavior and hence, application. It was found that the aggregate size could be affected in a reversible manner by increasing and decreasing the temperature, which increases and decreases the size of the aggregate, respectively. The protein-polymer block-copolymer concept has even been extended towards triblock-copolymers with additional possibilities with post-modifications of the polymer via “click”-reactions [53,54]. The attachment of a single polymer chain onto a single protein can also be done by using the protein-cofactor combination. The cofactor can be reversibly removed and reconstituted, so when the cofactor is modified with the polymer and introduced to the “empty” protein without cofactor (Apo-variant) in situ, the protein-polymer conjugate structure is then formed [55]. This approach was performed using horseradish peroxidase and myoglobin which both use protoporphyrin IX as a cofactor. This resulted also in a giant amphiphile formation as well as a triple hybrid block-copolymer structure [56]. In this case the structure formation and assembly is reversible since the cofactor can be removed via oxidation or coordination of additional ligands to the heme-like group. Also linear polymeric protein-polymer structures were constructed with this method were a single polymer with a heme-group attached on one end was covalently attached to the protein with the other end of the polymer. Upon reconstituting the cofactor, the protein-polymer structures formed a linear array of chemically-controlled supramolecular protein polymer structures [57].
3.2. Protein Centralized Multiple Polymer Grafts
In many cases, the protein has more than one reactive functionality, and hence during modification more than one polymer is attached giving it more the character of a protein-core/polymer-shell soft microgel-like particle. One of the earlier approaches was performed on Bovine Serum Albumin (BSA), which was reduced to have more reactive cysteine-groups exposed and was subsequently functionalized with a radical initiator used in ATRP-reactions and polymers were grown in a controlled fashion [11]. This led to many sequential studies on how to design other protein-multiple polymer systems as well as investigating their behavior with respect to remaining catalytic activity and also directed self-assembly [58]. One such directed assembly is towards interfaces, which is facilitated by the polymer that surrounds the protein structure.
Proteins by themselves as stated earlier can be regarded as bionanoparticles in the sense that in a certain way they behave like particles. Colloidal particles are able to interact with interfaces and tend to assemble at these because of minimization of surface interaction energies and this allows colloidal particles to stabilize polar-apolar interfaces [59]. In addition to the conventional “hard” colloids also “soft” (polymeric hydrogel) colloids are also able to stabilize interfaces and hence proteins, which can also be regarded as “soft” deformable colloids, stabilize interfaces as well [59]. The ability of interface stabilization by protein structures has been shown for small proteins, globular virus particles as well as tubular particles [60,61]. The adsorption of proteins onto polar-apolar interfaces is not restricted to liquid-liquid interfaces but also applies to liquid-gas and liquid-solid. One of the drawbacks of proteins at interfaces is that they tend do denature up to a certain extend [62]. Surrounding the protein with a polymer shell prevents such denaturation and also is able to direct the assembly. While PEGylated surfaces, the attachment of polyethylene glycol chains, is regarded as a way to prevent bio-fouling and protein adsorption, when applying the PEG-based polymer onto the protein surface also a reduced adsorption tendency and a more reversible adsorption is created [63]. Russell and coworkers also used a glycol-based polymers attached onto ferritin [64]. Here, instead of a linear PEG, a PEG-methacylate was used and was grafted from the ferritin surface by first functionalization of the surface with an ATRP-initiator via a maleimide-coupling followed by controlled polymerization using ATRP. While normally PEGylation would prevent surface assembly, it was used to direct the assembly in phase separated polystyrene-block-polyethylene glycol thin films. Due to compatible interactions, the protein-polymer conjugate selectively incorporated into the polar phase.
PEGylation prevents the protein-polymer particle to stick to the surface while the attachment of other polymers make it stick very well. It was found that attaching pNIPAAm to ferritin via the same principle, an increased preference towards polar-apolar surfaces was created [65]. Not only at liquid-liquid interfaces but also onto hydrophobized silicon oxide surfaces [66]. The enhanced assembly at the interface of oil-in-water emulsions resulted in highly stabilized emulsion systems which were transformed into stable protein-polymer capsules by additional cross-linking of the polymer matrix which displayed such a high toughness and flexibility that even anisotropic capsule-networks were formed by intense mixing via extrusion [67,68].
3.3. Virus-Polymer Conjugates
Virus particles in general are attractive protein-based particles which show great promise in bionanotechnology, as they have been used as, e.g., delivery vessel, templates, nanowires, liquid crystals [69,70,71]. It is only natural that virus particles are combined with polymers, not only in a non-covalent fashion as described above but also covalently. It has already been shown that the modification of virus particles with polymers increases their stability, and therefore, even allows them to be transferred to an organic solvent without disassembling, being more tolerant towards elevated temperature [72]. This was also shown by intra-viral capsid cross-linking using a polymer with multiple alkyne-groups attached to the backbone, which was allowed to react with a surface modified bacteriophage QB containing several azide moieties [73]. Via a Cu-catalyzed “click” reaction, the surface was coated with the polymer that covalently connected the individual viral capsids in the case when one polymer reacted with several capsids. It was found that the overall structure was maintained even until 100 °C. Using the same approach for surface functionalization only with a radical initiator attached to the azide instead of a linear polymer, various polymers were grown from the surface via ATRP as described above [74]. The interesting thing is that there are several aspects, which can be combined in a viral-polymer system. On the interior, structures can be entrapped and the outside can contain several responsive and functional polymers (repellant glycol-chains, temperature responsive, fluorescent, coordination, etc.) making it a very interesting candidate for in vivo applications.
4. Potential Applications of Polymer-Driven Protein Assembly
Combining proteins and polymers is regarded as a valuable approach to understand certain aggregation phenomena as well as developing new applications and materials. One of the main disciplines that will most likely have the best changes of using the protein-polymer combination is in biomedical sciences and biotechnology. In vivo delivery, therapeutic agents, imaging, bio-sensing, responsive biocatalysts and biodegradable systems can be envisioned with the above mentioned systems and approaches. In this section, a few approaches with directly imaginable applications will be highlighted.
4.1. Protein-Imprinting into Polymer Surfaces
Proteins can be co-assembled inside or onto a pre-polymer matrix which upon polymerization, fixates not only the protein but also the defined shape of the protein inside the matrix. Upon removal of the protein, an imprint of the protein is left behind, this process is known as protein imprinting (Figure 6) [75]. Imprinting effects confined to surfaces as well as inside hydrogel particles in which proteins are incorporated have the possibility to rebind the protein specifically after it has been removed which expands the scope considerably. Imprinted protein structures on the surface or inside of nano/micron-sized particles can be used to study protein-protein interactions in an alternative way, as an antibody analog or since there is specific binding it can also act as a scavenger particle [76,77,78]. For the incorporation one can use electrostatic interactions to coordinate the protein structure but it can also first be confined to a surface covalently after which a layer can be built in between the surface bound proteins, upon removal a void in the shape of the protein is formed [79]. The surface binding approach has recently been used to actively scavenge virus particles from solution [80]. This has a very clear application and could potentially be used in vivo to bind viral entities and excrete the fully bound structure from the body and thus aid in the struggle against viral infections.
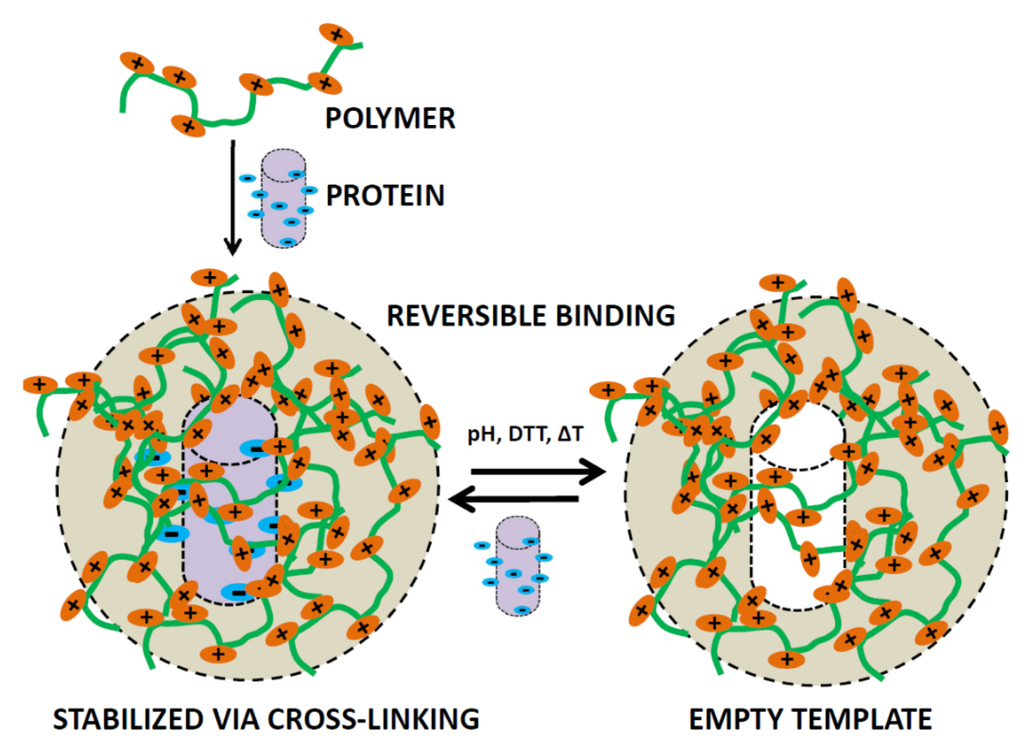
Figure 6.
Schematic representation of a polymer structure, which is able to reversibly bind protein structures using the protein-imprinted technique.
Figure 6.
Schematic representation of a polymer structure, which is able to reversibly bind protein structures using the protein-imprinted technique.
4.2. Amyloid Formation and Prevention
The formation of amyloid structures is one of the events which is present of many neurological disorders (Huntington’s, Alzheimer’s, Parkinson’s) and inhibition of the degradation or disposal of the assembled protein/peptide structures is given as one of the causes associated with these disorders [81,82]. Therefore not only polymer-induced assembly is of great importance but also polymer-induced disassembly which still focuses on protein-polymer interactions. The polymers used are often peptidic in nature with or without some additional non-natural component, the definition “synthetic” component is not correct in this context since, although natural in origin, the polypeptides are also synthetic. Proteins associated with the formation of amyloid structures are amongst others: α-synuclein (dementia, with Lewis bodies, Lewis body variant Alzheimer’s disease and Parkinson’s disease), huntingtin (Huntington’s disease), tau protein (Alzheimer’s, Prion disease). Different approaches were used in order to disassemble amyloids formed by these proteins or related model systems. The approaches range from synthetic polypeptides, non-natural polymer/polypeptide combinations and purely non-natural polymers. Cyclic polypeptides were used to counteract the aggregation of tau-based model amyloids [83]. The amino acid sequence was chosen in such a way that is maximizes the interactions with the tau aggregate regions and it is suggested that the cyclic peptide inhibits the direction of growth. In another system, a linear peptide was investigated and displayed inhibition towards the peptide structure, which accelerates α-synuclein amyloid formation and could potentially be used to prevent aggregation [84]. Also purely non-natural polymers were found to be pro-active towards amyloid disassembly. An in situ supramolecular bottle-brush formation by coordination of a sulfonic acid-capped polyethylene glycol chain onto the protein inside the aggregate, displayed disassembly properties towards positively charged β-lactoglobulin fibrils [85]. It was thought that the high coordination density causes the chains to extend, due to excluded volume interactions and the gain in free energy causes amyloid fibrils to disassemble. Both peptide and non-natural polymers have interactions with amyloid aggregates so it is only logical that also attempts were made to combine the two. One such a system is based on polymeric nanoparticles composed of poly(N-acryloyl-L-phenylalanyl-L-phenylalanine methyl ester) which contains two hydrophobic peptide moieties. The particle inhibits the fibrillation kinetics and when the hydrophilic alanyl-alanine derivative was used, the opposite effect was found [86].
4.3. Therapeutic and Smart Hybrid Bioactive Systems
The combination of proteins inside a nano-sized polymeric matrix can serve as a platform in which controlled release and targeted bioactivity are general addressed applications especially in combination with antibody labeling, fluorescence, ligand attachment and biodegradability [87]. When the polymer matrix acts as a hydrogel, the proteins are still in contact with the outside medium for small analytes and substrates because of the open porous polymeric hydrogel structure but at the same time is shielded and protected against attack by structures, which are larger than the polymer mesh-size (Figure 7). The interactions for confinement of the protein inside the polymer gel-particle can be electrostatic as well as hydrophobic interactions or covalently bound as discussed earlier [16,22,23]. Due to a change in environment, a triggered release can be induced, e.g., a lowering of pH as is seen in cancer cells, temperature in case of local inflammation or just general biodegradability by hydrolysis or via enzymatic degradation. Opposite to controlled release, retention or scavenging are also possibilities. Scavenging was addressed in protein imprinting but retention of the protein inside the polymer matrix and making use of the smart polymeric properties in combination with enzymatic and bioactivity also “on/off” systems can be created of which the bioactivity can be switched by changing the polymer hydration state surrounding the protein or giving the protein a much higher stability which not only makes it useful for biomedical applications but also potentially for the chemical industry (Figure 7) [25,88].
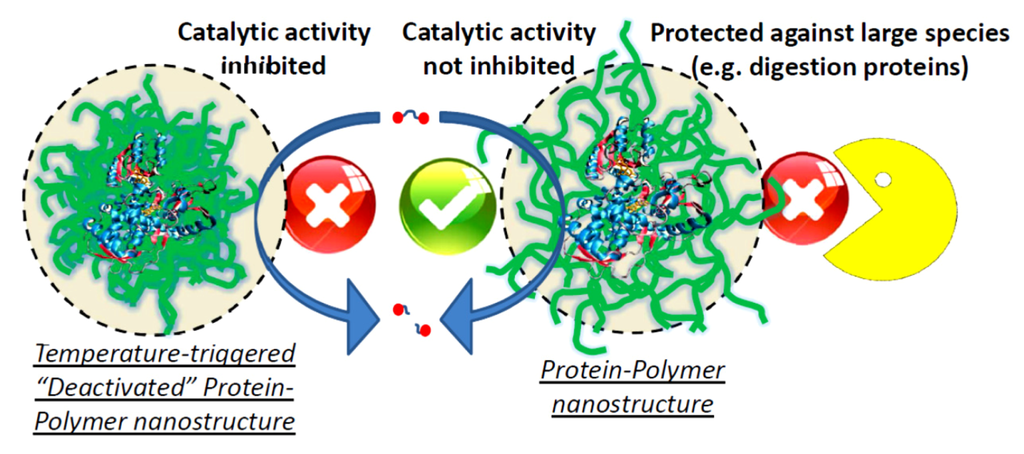
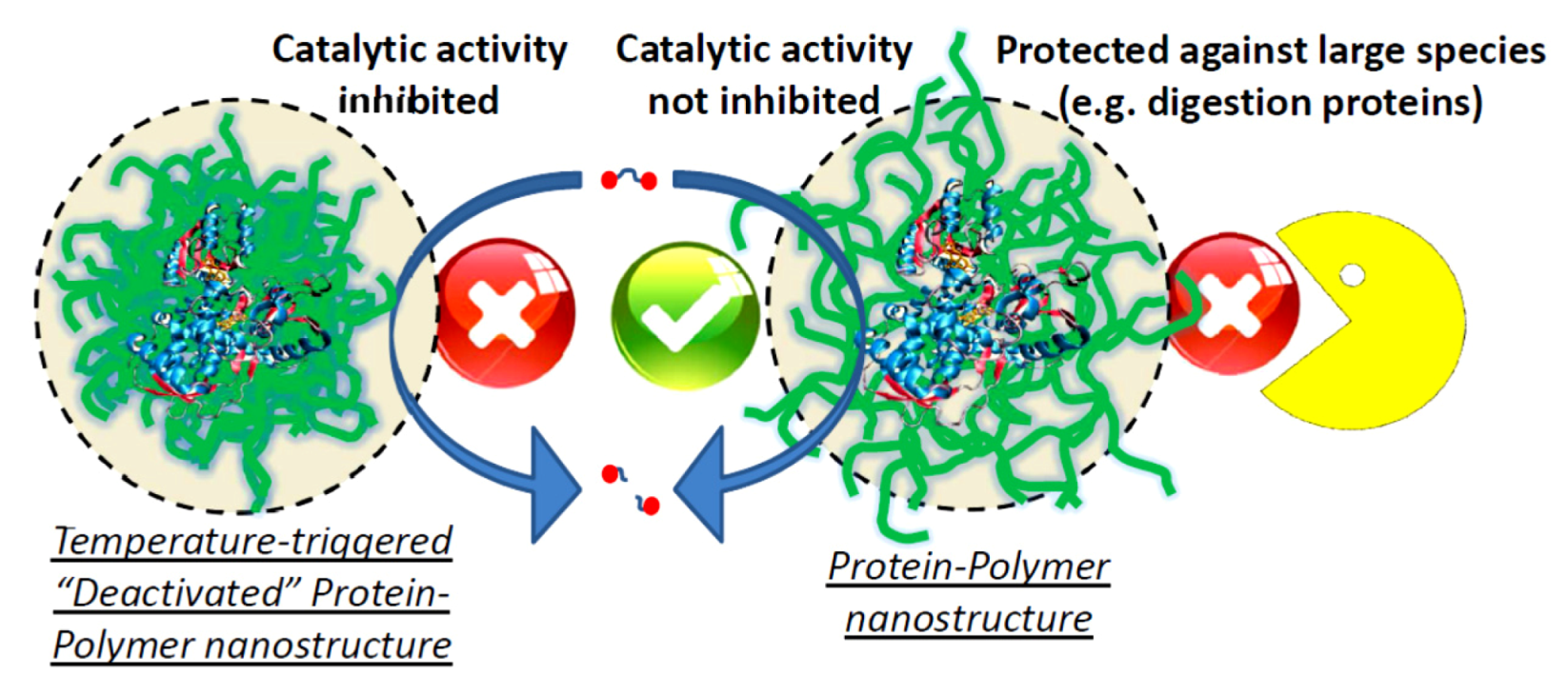
Figure 7.
Schematic representation of a protein-polymer nanostructure, which is able to use the catalytic properties of the protein, but at the same time displays protection against denaturing media or larger digestive protein structures. Additionally, the catalytic activity can be inhibited by a collapse of the surrounding polymer layer, e.g., temperature in combination with polyNIPAAm.
Figure 7.
Schematic representation of a protein-polymer nanostructure, which is able to use the catalytic properties of the protein, but at the same time displays protection against denaturing media or larger digestive protein structures. Additionally, the catalytic activity can be inhibited by a collapse of the surrounding polymer layer, e.g., temperature in combination with polyNIPAAm.
4.4. Bio-Interfaces
Cell adhesion layers and antibacterial surface properties and biofouling are important issues both in biomedical and industrial applications. For biofouling, a non-stick/repellant layer is needed which basically has non-favorable interactions with, e.g., proteins. As stated above, PEGylated surfaces are very useful for this [34]. However, to combine it with anti-bacterial properties is also possible when e.g., an anti-bacterial protein, like lysozyme, is covalently incorporated into the film [89]. Alternatively, cell adhesion is desired in the formation of scaffolds for tissue growth [90]. Here the polymer repels unwanted protein adsorption but the incorporated protein component acts as a cell binder. One can envision this to be a general approach, and since many hydrogel systems are stable enough to be formed into any shape with an integrated biodegradability, different types of tissues can potentially be addressed.
5. Conclusions
It is clear that combining proteins and polymers offer the best of both worlds and complement each other in many ways. The versatility and broad applicability originates from the diverse number of both polymers and proteins, as well as the different ways of combining them (electrostatically, vd Waals interactions, anti-body labeling, covalently bound). The systems presented here give a glimpse of what is possible, and while the polymers are getting more sophisticated, and the access to proteins and genetically modified proteins that are tailored for specific applications become easier, the designed systems become more sophisticated and applicable as well. The applications mentioned are mainly towards biomedical ones, but one can easily imagine that proteins with specific properties like redox-activity in combination with conjugated polymers, but also (inorganic-)nanoparticles, provide a platform towards electro-chemically controlled systems and bio-electronic devices/sensors. This is already in development and many interesting combinations and approaches have been presented in literature, but are beyond the scope of this review [91,92,93]. The future will be more focused on the combination of synthetic and biological systems as it not only helps in the understanding of biological phenomena and disease related disorders, but also because nature offers this tremendous source of interesting functional systems of which many are based on polymers (polypeptide and polynucleotide).
Conflict of Interest
The authors declare no conflict of interest.
References
- Rubinsztein, D.C. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 2006, 443, 780–786. [Google Scholar] [CrossRef]
- Citron, M. Alzheimer’s disease: Strategies for disease modification. Nat. Rev. Drug Discov. 2010, 9, 387–398. [Google Scholar] [CrossRef]
- Perutz, M.F.; Windle, A.H. Cause of neural death in neurodegenerative diseases attributable to expansion of glutamine repeats. Nature 2001, 412, 143–144. [Google Scholar] [CrossRef]
- Pokorski, J.K.; Steinmetz, N.F. The art of engineering viral nanoparticles. Mol. Pharmaceut. 2011, 8, 29–43. [Google Scholar] [CrossRef]
- van Rijn, P.; Böker, A. Bionanoparticles and hybrid materials: Tailored structural properties, self-assembly, materials and developments in the field. J. Mater. Chem. 2011, 21, 16735–16747. [Google Scholar] [CrossRef]
- Klug, A. The tobacco mosaic virus particle: Structure and assembly. Philos. Trans. R. Soc. B 1999, 354, 531–535. [Google Scholar] [CrossRef]
- Broyer, R.M.; Grover, G.N.; Maynard, H.D. Emerging synthetic approaches for protein-polymer conjugations. Chem. Commun. 2011, 47, 2212–2226. [Google Scholar] [CrossRef]
- Gauthier, M.A.; Klok, H.-A. Polymer-protein conjugates: An enzymatic activity perspective. Polym. Chem. 2010, 1, 1352–1373. [Google Scholar] [CrossRef]
- Vandermeulen, G.W.M.; Klok, H.-A. Peptide/protein hybrid materials: Enhanced control of structure and improved performance through conjugation of biological and synthetic polymers. Macromol. Biosci. 2004, 4, 383–398. [Google Scholar] [CrossRef]
- Theato, P. Chemical strategies for the synthesis of protein—Polymer conjugates. Adv. Polym. Sci. 2013, 253, 37–70. [Google Scholar]
- Heredia, K.L.; Bontempo, D.; Ly, T.; Byers, J.T.; Halstenberg, S.; Maynard, H.D. In situ preparation of protein-“smart” polymer conjugates with retention of bioactivity. J. Am. Chem. Soc. 2005, 127, 16955–16960. [Google Scholar]
- Boyer, C.; Bulmus, V.; Liu, J.; Davis, T.P.; Stenzel, M.H.; Barner-Kowollik, C. Well-defined protein-polymer conjugates via in situ RAFT polymerization. J. Am. Chem. Soc. 2007, 129, 7145–7154. [Google Scholar]
- Appel, E.A.; del Barrio, J.; Loh, X.J.; Scherman, O.A. Supramolecular polymeric hydrogels. Chem. Soc. Rev. 2012, 41, 6195–6214. [Google Scholar] [CrossRef]
- Smith, M.H.; Lyon, L.A. Tunable encapsulation of proteins within charged microgels. Macromolecules 2011, 44, 8154–8160. [Google Scholar] [CrossRef]
- Boyer, C.; Huang, X.; Whittaker, M.R.; Bulmus, V.; Davis, T.P. An overview of protein-polymer particles. Soft Matter 2011, 7, 1599–1614. [Google Scholar] [CrossRef]
- Morimoto, N.; Hirano, S.; Takahashi, H.; Loethen, S.; Thompson, D.H.; Akiyoshi, K. Self-assembled pH-sensitive cholesteryl pullulan nanogel as a protein delivery vehicle. Biomacromolecules 2013, 14, 56–63. [Google Scholar] [CrossRef]
- Koo, J.; Czeslik, C. Probing aggregation and fibril formation of insulin in polyelectrolyte multilayers. Colloids Surf. B 2012, 94, 80–88. [Google Scholar] [CrossRef]
- Mueller, A.; Eber, F.J.; Azucena, C.; Petershans, A.; Bittner, A.M.; Gliemann, H.; Jeske, H.; Wege, C. Inducible site-selective bottom-up assembly of virus-derived nanotube arrays on RNA-equipped wafers. ACS Nano 2011, 5, 4512–4520. [Google Scholar] [CrossRef]
- Seyrek, E.; Dubin, P. Glycosaminoglycans as polyelectrolytes. Adv. Colloid Interface Sci. 2010, 158, 119–129. [Google Scholar] [CrossRef]
- Capila, I.; Linhardt, R.J. Heparin-protein interactions. Angew. Chem. Int. Ed. 2002, 41, 391–412. [Google Scholar]
- Kayitmazer, A.B.; Seeman, D.; Minsky, B.B.; Dubin, P.L.; Xu, Y. Protein-polyelectrolyte interactions. Soft Matter 2013, 9, 2553–2583. [Google Scholar] [CrossRef]
- Coué, G.; Engbersen, J.F.J. Functionalized linear poly(amidoamine)s are efficient vectors for intracellular protein delivery. J. Control. Release 2011, 152, 90–98. [Google Scholar] [CrossRef]
- Tsiourvas, D.; Sideratou, Z.; Sterioti, N.; Papadopoulos, A.; Nounesis, G.; Paleos, C.M. Insulin complexes with PEGylated basic oligopeptides. J. Colloid Interface Sci. 2012, 384, 61–72. [Google Scholar] [CrossRef]
- Kurinomaru, T.; Tomita, S.; Kudo, S.; Ganguli, S.; Nagasaki, Y.; Shiraki, K. Improved complementary polymer pair system: Switching for enzyme activity by PEGylated polymers. Langmuir 2012, 28, 4334–4338. [Google Scholar] [CrossRef]
- Wilson, L.; Illanes, A.; Abián, O.; Pessela, B.C.C.; Fernández-Lafuente, R.; Guisán, J.M. Co-aggregation of penicillin g acylase and polyionic polymers: An easy methodology to prepare enzyme biocatalysts stable in organic media. Biomacromolecules 2004, 5, 852–857. [Google Scholar] [CrossRef]
- Kubiak-Ossowska, K.; Mulheran, P. Protein diffusion and long-term adsorption states at charged solid surfaces. Langmuir 2012, 28, 15577–15585. [Google Scholar] [CrossRef]
- Kubiak-Ossowska, K.; Mulheran, P. Mechanism of hen egg white lysozyme adsorption on a charged solid surface. Langmuir 2010, 26, 15954–15965. [Google Scholar] [CrossRef]
- Yang, Z.; Galloway, J.A.; Yu, H. Protein interactions with poly(ethylene glycol) self-assembled monolayers on glass substrates: Diffusion and adsorption. Langmuir 1999, 15, 8405–8411. [Google Scholar] [CrossRef]
- Wang, X.; Wu, G.; Lu, C.; Wang, Y.; Fan, Y.; Gao, H.; Ma, J. Synthesis of a novel zwitterionic biodegradable poly (α,β-L-aspartic acid) derivative with some L-histidine side-residues and its resistance to non-specific protein adsorption. Colloids Surf. B 2011, 86, 237–241. [Google Scholar] [CrossRef]
- Uhlmann, P.; Houbenov, N.; Brenner, N.; Grundke, K.; Burkert, S.; Stamm, M. In-situ investigation of the adsorption of globular model proteins on stimuli-responsive binary polyelectrolyte brushes. Langmuir 2007, 23, 57–64. [Google Scholar] [CrossRef]
- Becker, A.L.; Henzler, K.; Welsch, N.; Ballauff, M.; Borisov, O. Proteins and polyelectrolytes: A charged relationship. Curr. Opin. Colloid Interface Sci. 2012, 17, 90–96. [Google Scholar] [CrossRef]
- Li, Y.; Wang, X.; Sun, J. Layer-by-layer assembly for rapid fabrication of thick polymeric films. Chem. Soc. Rev. 2012, 41, 5998–6009. [Google Scholar] [CrossRef]
- Salloum, D.S.; Schlenoff, J.B. Protein adsorption modalities on polyelectrolyte multilayers. Biomacromolecules 2004, 5, 1089–1096. [Google Scholar] [CrossRef]
- Delcroix, M.F.; Huet, G.L.; Conard, T.; Demoustier-Champagne, S.; du Prez, F.E.; Landoulsi, J.; Dupont-Gillain, C.C. Design of mixed PEO/PAA brushes with switchable properties toward protein adsorption. Biomacromolecules 2013, 14, 215–225. [Google Scholar] [CrossRef]
- Kim, B.; Lam, C.N.; Olsen, B.D. Nanopatterned protein films directed by ionic complexation with water-soluble diblock copolymers. Macromolecules 2012, 45, 4572–4580. [Google Scholar] [CrossRef]
- Mukherjee, S.; Pfeifer, C.M.; Johnson, J.M.; Liu, J.; Zlotnick, A. Redirecting the coat protein of a spherical virus to assemble into tubular nanostructures. J. Am. Chem. Soc. 2006, 128, 2538–2539. [Google Scholar] [CrossRef]
- Cadena-Nava, R.D.; Hu, Y.; Garmann, R.F.; Ng, B.; Zelikin, A.N.; Knobler, C.M.; Gelbart, W.M. Exploiting fluorescent polymers to probe the self-assembly of virus-like particles. J. Phys. Chem. B 2011, 115, 2386–2391. [Google Scholar]
- Hu, Y.; Zandi, R.; Anavitarte, A.; Knobler, C.M.; Gelbart, W.M. Packaging of a polymer by a viral capsid: The interplay between polymer length and capsid size. Biophys. J. 2008, 94, 1428–1436. [Google Scholar] [CrossRef]
- Ng, B.C.; Chan, S.T.; Lin, J.; Tolbert, S.H. Using polymer conformation to control architecture in semiconducting polymer/viral capsid assemblies. ACS Nano 2011, 5, 7730–7738. [Google Scholar] [CrossRef]
- Kostiainen, M.A.; Hiekkataipale, P.; de la Torre, J.Á.; Nolte, R.J.M.; Cornelissen, J.J.L.M. Electrostatic self-assembly of virus–polymer complexes. J. Mater. Chem. 2011, 21, 2112–2117. [Google Scholar] [CrossRef]
- Grover, G.N.; Maynard, H.D. Protein-polymer conjugates: Synthetic approaches by controlled radical polymerizations and interesting applications. Curr. Opin. Chem. Biol. 2010, 14, 818–827. [Google Scholar] [CrossRef]
- Discher, D.E.; Ahmed, F. Polymersomes. Ann. Rev. Biomed. Eng. 2006, 8, 323–341. [Google Scholar] [CrossRef]
- Horn, A.; Hiltl, S.; Fery, A.; Böker, A. Ordering and printing virus arrays: A straightforward way to functionalize surfaces. Small 2010, 6, 2122–2125. [Google Scholar] [CrossRef]
- Chen, C.-H.; Yang, K.-L. Improving protein transfer efficiency and selectivity in affinity contact printing by using UV-modified surfaces. Langmuir 2011, 27, 5427–5432. [Google Scholar] [CrossRef]
- Kumar, N.; Hahm, J. Nanoscale protein patterning using self-assembled diblock copolymers. Langmuir 2005, 21, 6652–6655. [Google Scholar] [CrossRef]
- Kumar, N.; Parajuli, O.; Hahm, J.-I. Two-dimensionally self-arranged protein nanoarrays on diblock copolymer templates. J. Phys. Chem. B 2007, 111, 4581–4587. [Google Scholar] [CrossRef]
- Thomas, C.S.; Glassman, M.J.; Olsen, B.D. Solid-state nanostructured materials from self-assembly of a globular protein-polymer diblock copolymer. ACS Nano 2011, 5, 5697–5707. [Google Scholar] [CrossRef]
- Thomas, C.S.; Xu, L.; Olsen, B.D. Kinetically controlled nanostructure formation in self-assembled globular protein-polymer diblock copolymers. Biomacromolecules 2012, 13, 2781–2792. [Google Scholar] [CrossRef]
- Lam, C.N.; Olsen, B.D. Phase transitions in concentrated solution self-assembly of globular protein–polymer block copolymers. Soft Matter 2013, 9, 2393–2402. [Google Scholar] [CrossRef]
- Velonia, K. Protein-polymer amphiphilic chimeras: Recent advances and future challenges. Polym. Chem. 2010, 1, 944–952. [Google Scholar] [CrossRef]
- Velonia, K.; Rowan, A.E.; Nolte, R.J.M. Lipase polystyrene giant amphiphiles. J. Am. Chem. Soc. 2002, 124, 4224–4225. [Google Scholar] [CrossRef]
- Lavigueur, C.; García, J.G.; Hendriks, L.; Hoogenboom, R.; Cornelissen, J.J.L.M.; Nolte, R.J.M. Thermoresponsive giant biohybrid amphiphiles. Polym. Chem. 2011, 2, 333–340. [Google Scholar] [CrossRef]
- le Droumaguet, B.; Mantovani, G.; Haddleton, D.M.; Velonia, K. Formation of giant amphiphiles by post-functionalization of hydrophilic protein-polymer conjugates. J. Mater. Chem. 2007, 17, 1916–1922. [Google Scholar] [CrossRef]
- Daskalaki, E.; le Droumaguet, B.; Gérard, D.; Velonia, K. Multifunctional Giant Amphiphiles via simultaneous copper(I)-catalyzed azide-alkyne cycloaddition and living radical polymerization. Chem. Commun. 2012, 48, 1586–1588. [Google Scholar]
- Boerakker, M.J.; Hannink, J.M.; Bomans, P.H.H.; Frederik, P.M.; Nolte, R.J.M.; Meijer, E.M.; Sommerdijk, N.A.J.M. Giant amphiphiles by cofactor reconstitution. Angew. Chem. Int. Ed. 2002, 41, 4239–4241. [Google Scholar] [CrossRef]
- Reynhout, I.C.; Cornelissen, J.J.L.M.; Nolte, R.J.M. Self-assembled architectures from biohybrid triblock copolymers. J. Am. Chem. Soc. 2007, 129, 2327–2332. [Google Scholar] [CrossRef]
- Oohora, K.; Onoda, A.; Kitagishi, H.; Yamaguchi, H.; Harada, A.; Hayashi, T. A chemically-controlled supramolecular protein polymer formed by a myoglobin-based self-assembly system. Chem. Sci. 2011, 2, 1033–1038. [Google Scholar] [CrossRef]
- Yaşayan, G.; Saeed, A.O.; Fernández-Trillo, F.; Allen, S.; Davies, M.C.; Jangher, A.; Paul, A.; Thurecht, K.J.; King, S.M.; Schweins, R.; et al. Responsive hybrid block co-polymer conjugates of proteins-controlled architecture to modulate substrate specificity and solution behaviour. Polym. Chem. 2011, 2, 1567–1578. [Google Scholar] [CrossRef]
- Böker, A.; He, J.; Emrick, T.; Russell, T.P. Self-assembly of nanoparticles at interfaces. Soft Matter 2007, 3, 1231–1248. [Google Scholar] [CrossRef]
- Kaur, G.; He, J.; Xu, J.; Pingali, S.; Jutz, G.; Böker, A.; Niu, Z.; Li, T.; Rawlinson, D.; Emrick, T.; et al. Interfacial assembly of turnip yellow mosaic virus nanoparticles. Langmuir 2009, 25, 5168–5176. [Google Scholar] [CrossRef]
- He, J.; Niu, Z.; Tangirala, R.; Wang, J.-Y.; Wei, X.; Kaur, G.; Wang, Q.; Jutz, G.; Böker, A.; Lee, B.; et al. Self-assembly of tobacco mosaic virus at oil/water interfaces. Langmuir 2009, 25, 4979–4987. [Google Scholar] [CrossRef]
- Schulz, A.; Fioroni, M.; Linder, M.B.; Nessel, A.; Bocola, M.; Subkowski, T.; Schwaneberg, U.; Böker, A.; Rodríguez-Ropero, F. Exploring the mineralization of hydrophobins at a liquid interface. Soft Matter 2012, 8, 11343–11352. [Google Scholar] [CrossRef]
- Pai, S.S.; Przybycien, T.M.; Tilton, R.D. Protein PEGylation attenuates adsorption and aggregation on a negatively charged and moderately hydrophobic polymer surface. Langmuir 2010, 26, 18231–18238. [Google Scholar] [CrossRef]
- Hu, Y.; Samanta, D.; Parelkar, S.S.; Hong, S.W.; Wang, Q.; Russell, T.P.; Emrick, T. Ferritin-polymer conjugates: Grafting chemistry and integration into nanoscale assemblies. Adv. Funct. Mater. 2010, 20, 3603–3612. [Google Scholar] [CrossRef]
- Mougin, N.C.; van Rijn, P.; Park, H.; Müller, A.H.E.; Böker, A. Hybrid capsules via self-assembly of thermoresponsive and interfacially active bionanoparticle-polymer conjugates. Adv. Funct. Mater. 2011, 21, 2470–2476. [Google Scholar] [CrossRef]
- van Rijn, P.; Park, H.; Özlem Nazli, K.; Mougin, N.C.; Böker, A. Self-assembly process of soft ferritin-PNIPAAm conjugate bionanoparticles at polar-apolar interfaces. Langmuir 2013, 29, 276–284. [Google Scholar] [CrossRef]
- van Rijn, P.; Mougin, N.C.; Franke, D.; Park, H.; Böker, A. Pickering emulsion templated soft capsules by self-assembling cross-linkable ferritin-polymer conjugates. Chem. Commun. 2011, 47, 8376–8378. [Google Scholar]
- van Rijn, P.; Mougin, N.C.; Böker, A. Hierarchical structures via self-assembling protein-polymer hybrid building blocks. Polymer 2012, 53, 6045–6052. [Google Scholar] [CrossRef]
- Liu, Z.; Qiao, J.; Niu, Z.; Wang, Q. Natural supramolecular building blocks: From virus coat proteins to viral nanoparticles. Chem. Soc. Rev. 2012, 41, 6178–6194. [Google Scholar] [CrossRef]
- Lee, L.A.; Niu, Z.; Wang, Q. Viruses and virus-like protein assemblies—Chemically programmable nanoscale building blocks. Nano Res. 2010, 2, 349–364. [Google Scholar]
- de la Escosura, A.; Nolte, R.J.M.; Cornelissen, J.J.L.M. Viruses and protein cages as nanocontainers and nanoreactors. J. Mater. Chem. 2009, 19, 2274–2278. [Google Scholar] [CrossRef]
- Holder, P.G.; Finley, D.T.; Stephanopoulos, N.; Walton, R.; Clark, D.S.; Francis, M.B. Dramatic thermal stability of virus-polymer conjugates in hydrophobic solvents. Langmuir 2010, 26, 17383–17388. [Google Scholar]
- Manzenrieder, F.; Luxenhofer, R.; Retzlaff, M.; Jordan, R.; Finn, M.G. Stabilization of virus-like particles with poly(2-oxazoline)s. Angew. Chem. Int. Ed. 2011, 50, 2601–2605. [Google Scholar] [CrossRef]
- Pokorski, J.K.; Breitenkamp, K.; Liepold, L.O.; Qazi, S.; Finn, M.G. Functional virus-based polymer-protein nanoparticles by atom transfer radical polymerization. J. Am. Chem. Soc. 2011, 133, 9242–9245. [Google Scholar] [CrossRef]
- Takeuchi, T.; Hishiya, T. Molecular imprinting of proteins emerging as a tool for protein recognition. Org. Biomol. Chem. 2008, 6, 2459–2467. [Google Scholar] [CrossRef]
- Gao, J.; Tian, H.; Wang, Y.; Yang, Q.; Liu, D.; Mi, H. The design of protein-imprinted polymers as antibody substitutes for investigating protein-protein interactions. Biomaterials 2012, 33, 3344–3352. [Google Scholar] [CrossRef]
- Ying, X.; Cheng, G.; Li, X. The imprinting induce-fit model of specific rebinding of macromolecularly imprinted polymer microspheres. J. Appl. Polym. Sci. 2011, 122, 1847–1856. [Google Scholar] [CrossRef]
- Bolisay, L.D.; Culver, J.N.; Kofinas, P. Molecularly imprinted polymers for tobacco mosaic virus recognition. Biomaterials 2006, 27, 4165–4168. [Google Scholar] [CrossRef]
- Verheyen, E.; Schillemans, J.P.; van Wijk, M.; Demeniex, M.-A.; Hennink, W.E.; van Nostrum, C.F. Challenges for the effective molecular imprinting of proteins. Biomaterials 2011, 32, 3008–3020. [Google Scholar] [CrossRef]
- Cumbo, A.; Lorber, B.; Corvini, P.F.-X.; Meier, W.; Shahgaldian, P. A synthetic nanomaterial for virus recognition produced by surface imprinting. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef]
- Zhang, X.; Smith, D.L.; Meriin, A.B.; Engemann, S.; Russel, D.E.; Roark, M.; Washington, S.L.; Maxwell, M.M.; Marsh, J.L.; Thompson, L.M.; et al. A potent small molecule inhibits polyglutamine aggregation in Huntington’s disease neurons and suppresses neurodegeneration in vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 892–897. [Google Scholar] [CrossRef]
- Karpuj, M.V.; Garren, H.; Slunt, H.; Price, D.L.; Gusella, J.; Becher, M.W.; Steinman, L. Transglutaminase aggregates huntingtin into nonamyloidogenic polymers, and its enzymatic activity increases in Huntington’s disease brain nuclei. Proc. Natl. Acad. Sci. USA 1999, 96, 7388–7393. [Google Scholar]
- Hexapeptide, P.; Zheng, J.; Liu, C.; Sawaya, M.R.; Vadla, B.; Khan, S.; Woods, R.J.; Eisenberg, D.; Goux, W.J.; Nowick, J.S. Macrocyclic β-sheet peptides that inhibit the aggregation of a tau-protein-derived hexapeptide. J. Am. Chem. Soc. 2011, 133, 3144–3157. [Google Scholar] [CrossRef]
- Marotta, N.P.; Cherwien, C.A.; Abeywardana, T.; Pratt, M.R. O-GlcNAc modification prevents peptide-dependent acceleration of α-synuclein aggregation. ChemBioChem 2012, 13, 2665–2670. [Google Scholar] [CrossRef]
- Rühs, P.A.; Adamcik, J.; Bolisetty, S.; Sánchez-Ferrer, A.; Mezzenga, R. A supramolecular bottle-brush approach to disassemble amyloid fibrils. Soft Matter 2011, 7, 3571–3579. [Google Scholar] [CrossRef]
- Skaat, H.; Chen, R.; Grinberg, I.; Margel, S. Engineered polymer nanoparticles containing hydrophobic dipeptide for inhibition of amyloid-β fibrillation. Biomacromolecules 2012, 13, 2662–2670. [Google Scholar] [CrossRef]
- le Droumaguet, B.; Nicolas, J.; Brambilla, D.; Mura, S.; Maksimenko, A.; de Kimpe, L.; Salvati, E.; Zona, C.; Airoldi, C.; Canovi, M.; et al. Versatile and efficient targeting using a single nanoparticulate platform: Application to cancer and Alzheimer’s disease. ACS Nano 2012, 6, 5866–5879. [Google Scholar] [CrossRef]
- Tan, H.; Jin, H.; Mei, H.; Zhu, L.; Wei, W.; Wang, Q.; Liang, F.; Zhang, C.; Li, J.; Qu, X.; et al. PEG-urokinase nanogels with enhanced stability and controllable bioactivity. Soft Matter 2012, 8, 2644–2650. [Google Scholar] [CrossRef]
- Muszanska, A.K.; Busscher, H.J.; Herrmann, A.; van der Mei, H.C.; Norde, W. Pluronic-lysozyme conjugates as anti-adhesive and antibacterial bifunctional polymers for surface coating. Biomaterials 2011, 32, 6333–6341. [Google Scholar]
- Browning, M.B.; Dempsey, D.; Guiza, V.; Becerra, S.; Rivera, J.; Russell, B.; Höök, M.; Clubb, F.; Miller, M.; Fossum, T.; et al. Multilayer vascular grafts based on collagen-mimetic proteins. Acta Biomater. 2012, 8, 1010–1021. [Google Scholar] [CrossRef]
- Fischlechner, M.; Donath, E. Viruses as building blocks for materials and devices. Angew. Chem. Int. Ed. 2007, 46, 3184–3193. [Google Scholar] [CrossRef]
- Tseng, R.J.; Tsai, C.; Ma, L.; Ouyang, J.; Ozkan, C.S.; Yang, Y. Digital memory device based on tobacco mosaic virus conjugated with nanoparticles. Nat. Nanotechnol. 2006, 1, 72–77. [Google Scholar]
- Evans, D.J. The bionanoscience of plant viruses: Templates and synthons for new materials. J. Mater. Chem. 2008, 18, 3746–3754. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).