Hybrid Block Copolymers Constituted by Peptides and Synthetic Polymers: An Overview of Synthetic Approaches, Supramolecular Behavior and Potential Applications


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
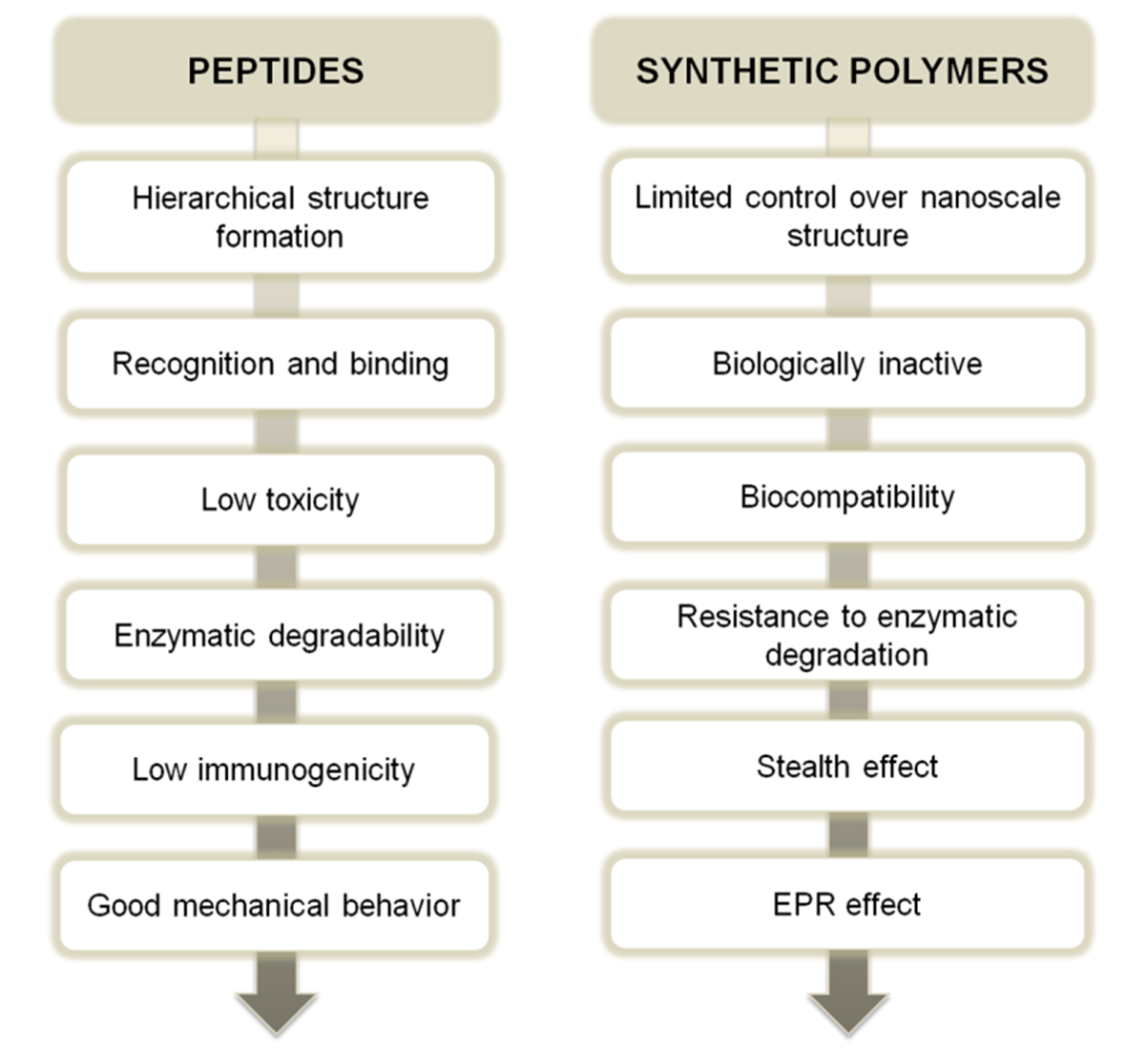
2. Synthetic Approaches for Peptide-Synthetic Polymer Hybrids

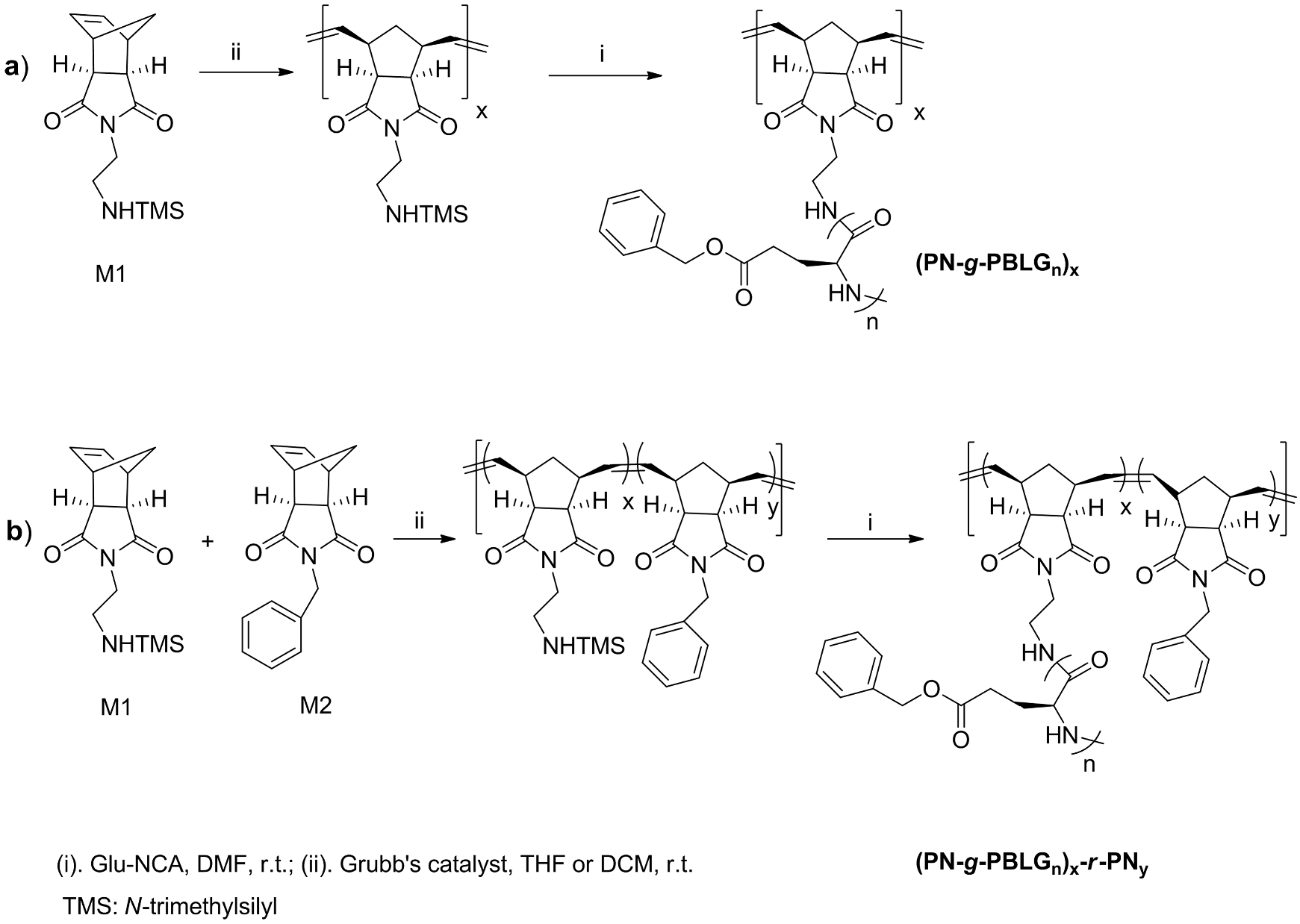
2.1. Liquid-Phase Peptide Synthesis. Preparation of Peptide-Synthetic Polymer Hybrids in Liquid-Phase
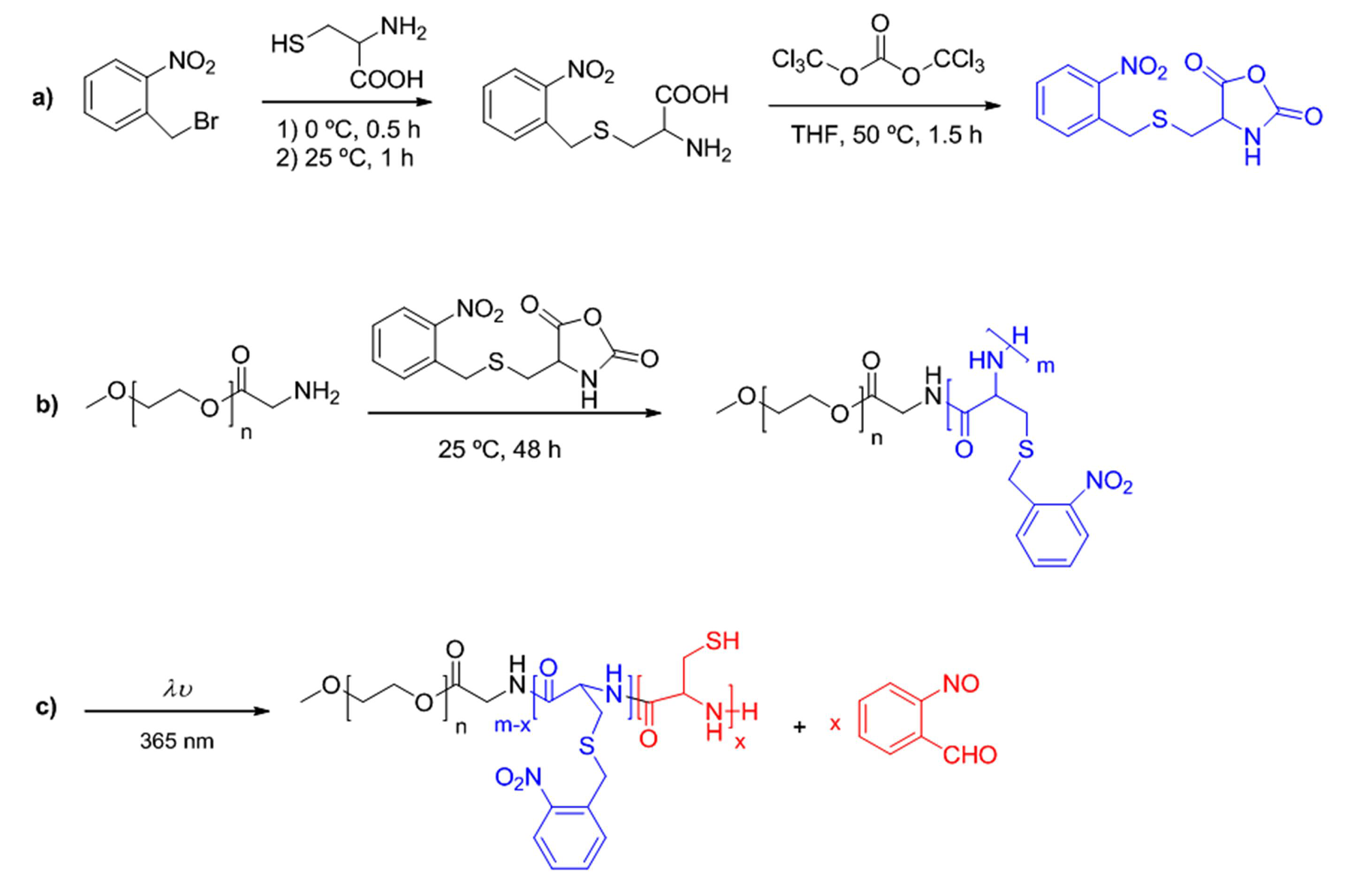
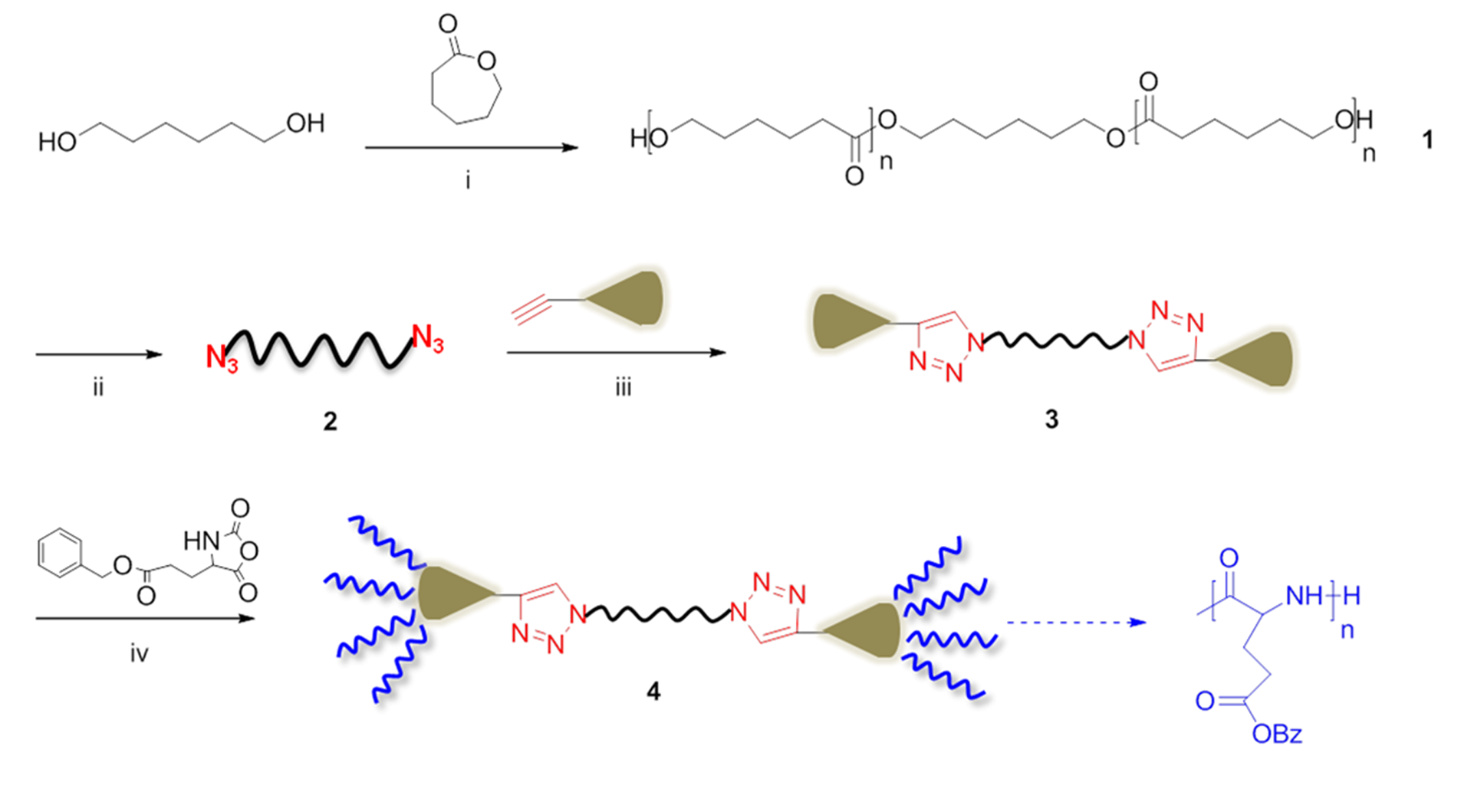
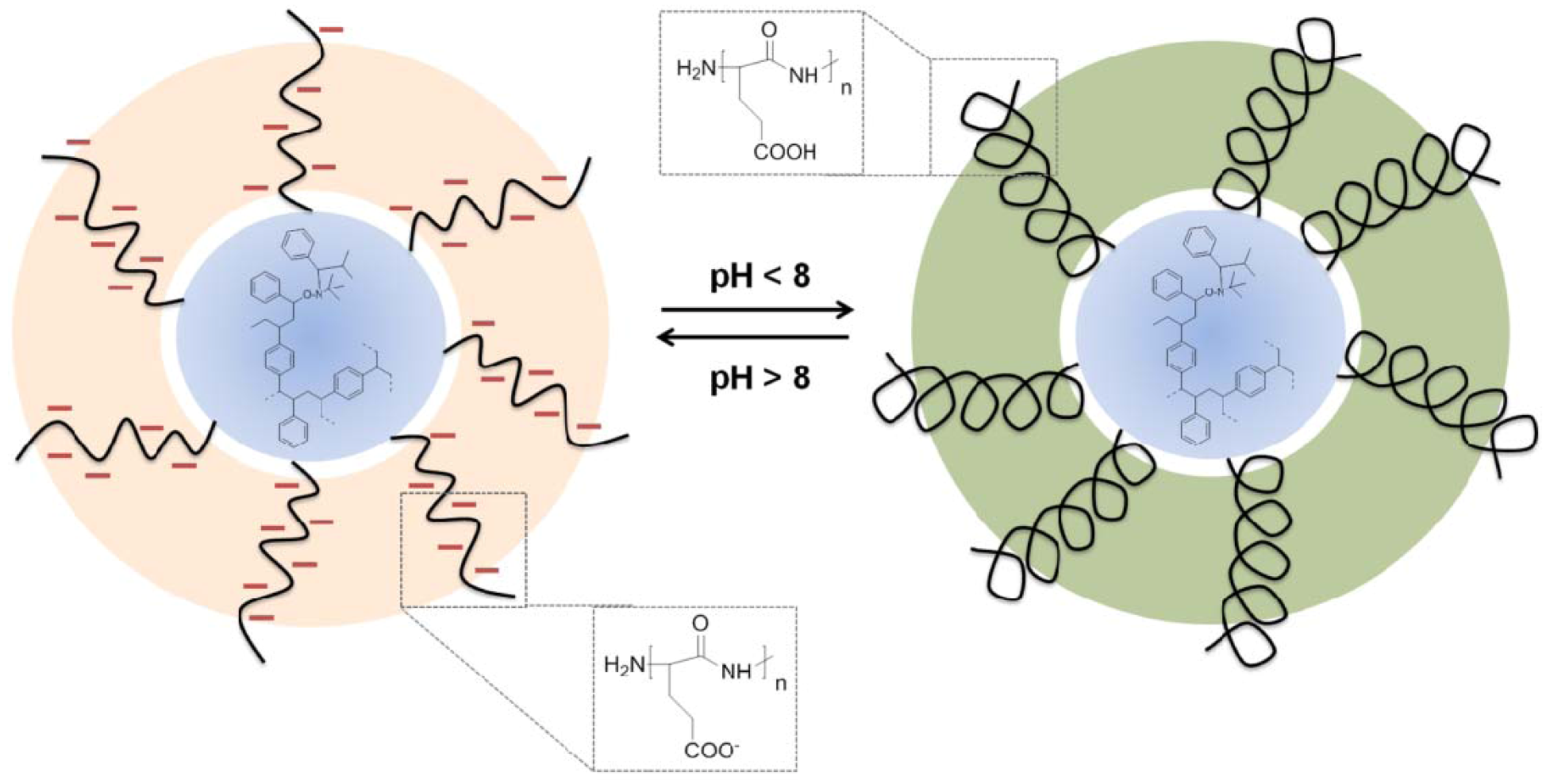
2.2. Solid-Phase Peptide Synthesis. Preparation of Peptide-Synthetic Polymer Hybrids in Liquid- or Solid-Phase
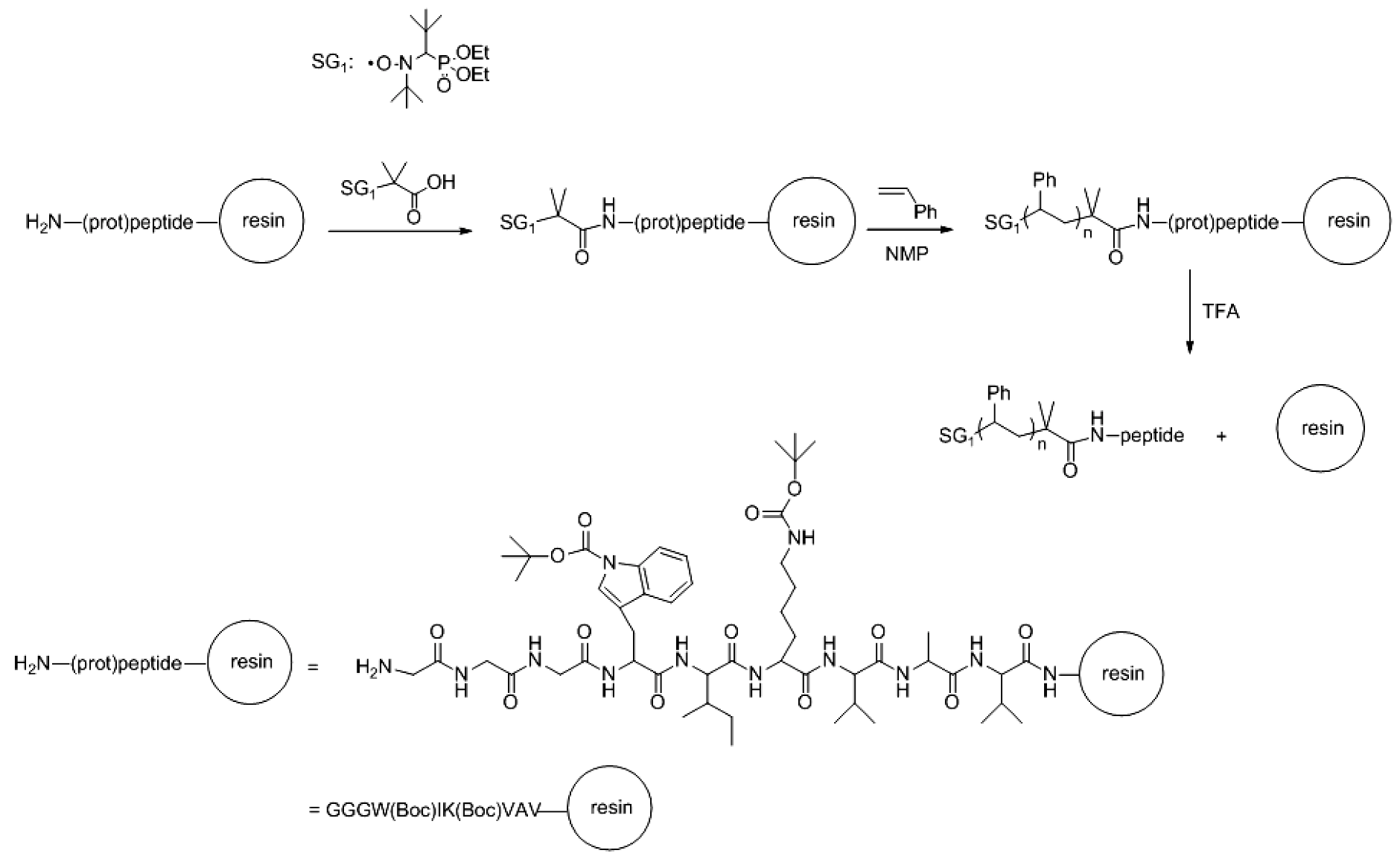
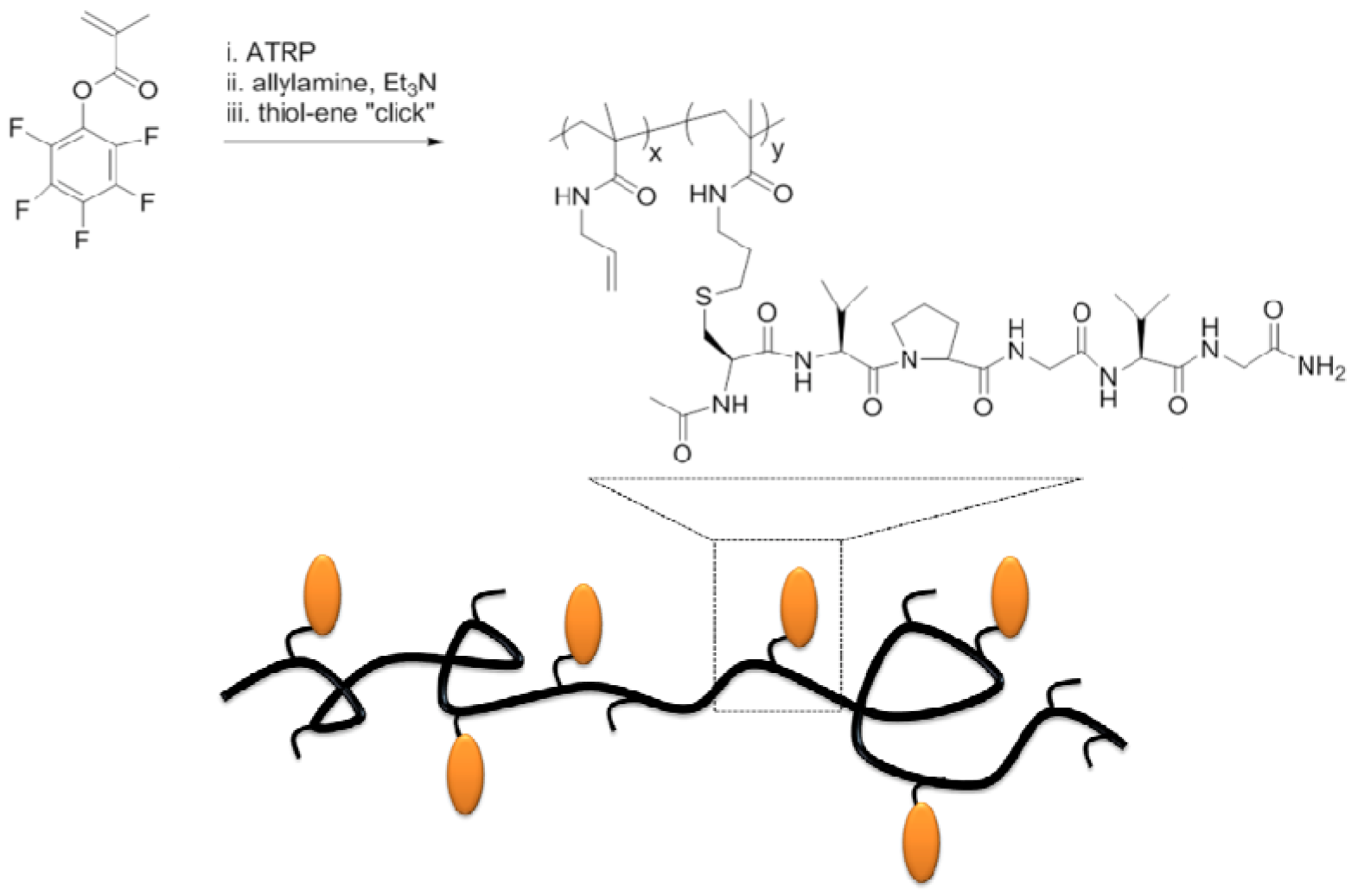
3. Supramolecular Organization of Peptide-Synthetic Polymer Hybrids
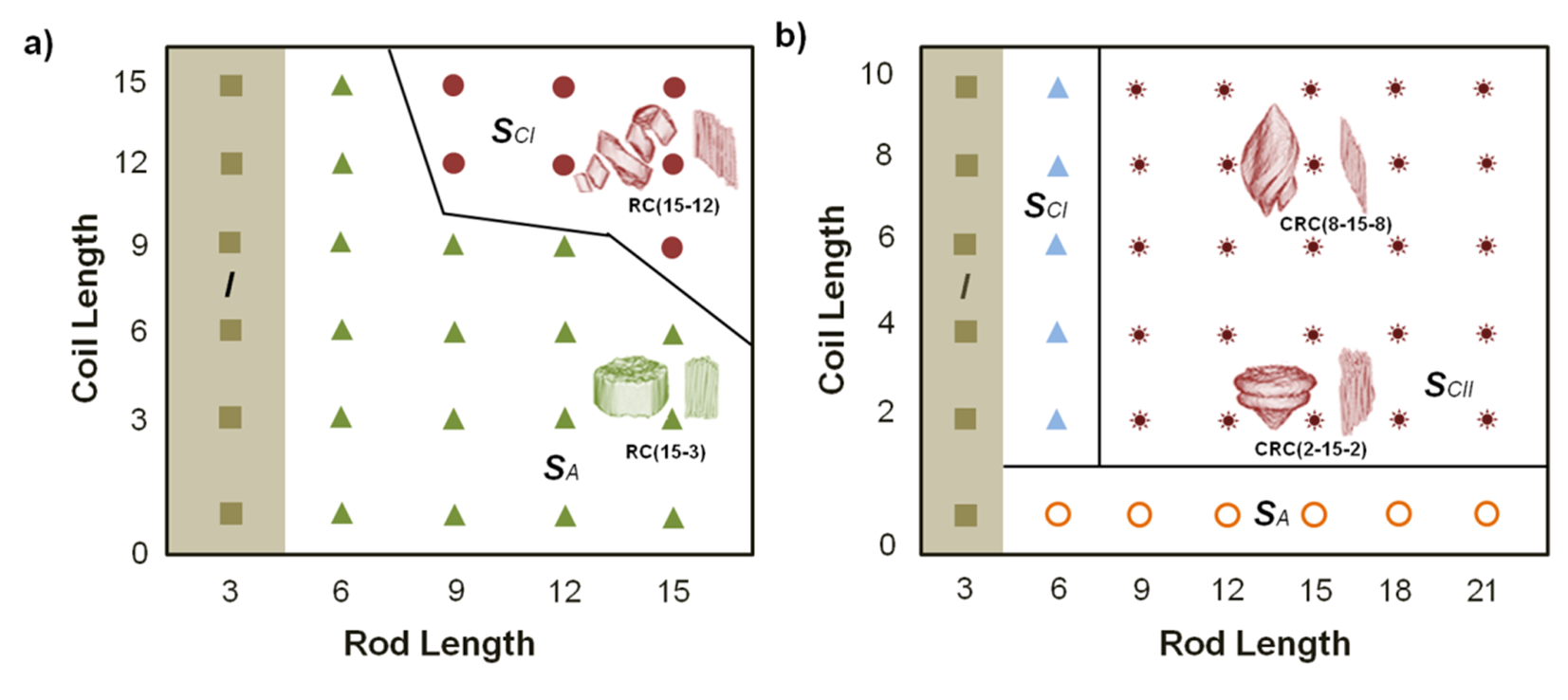
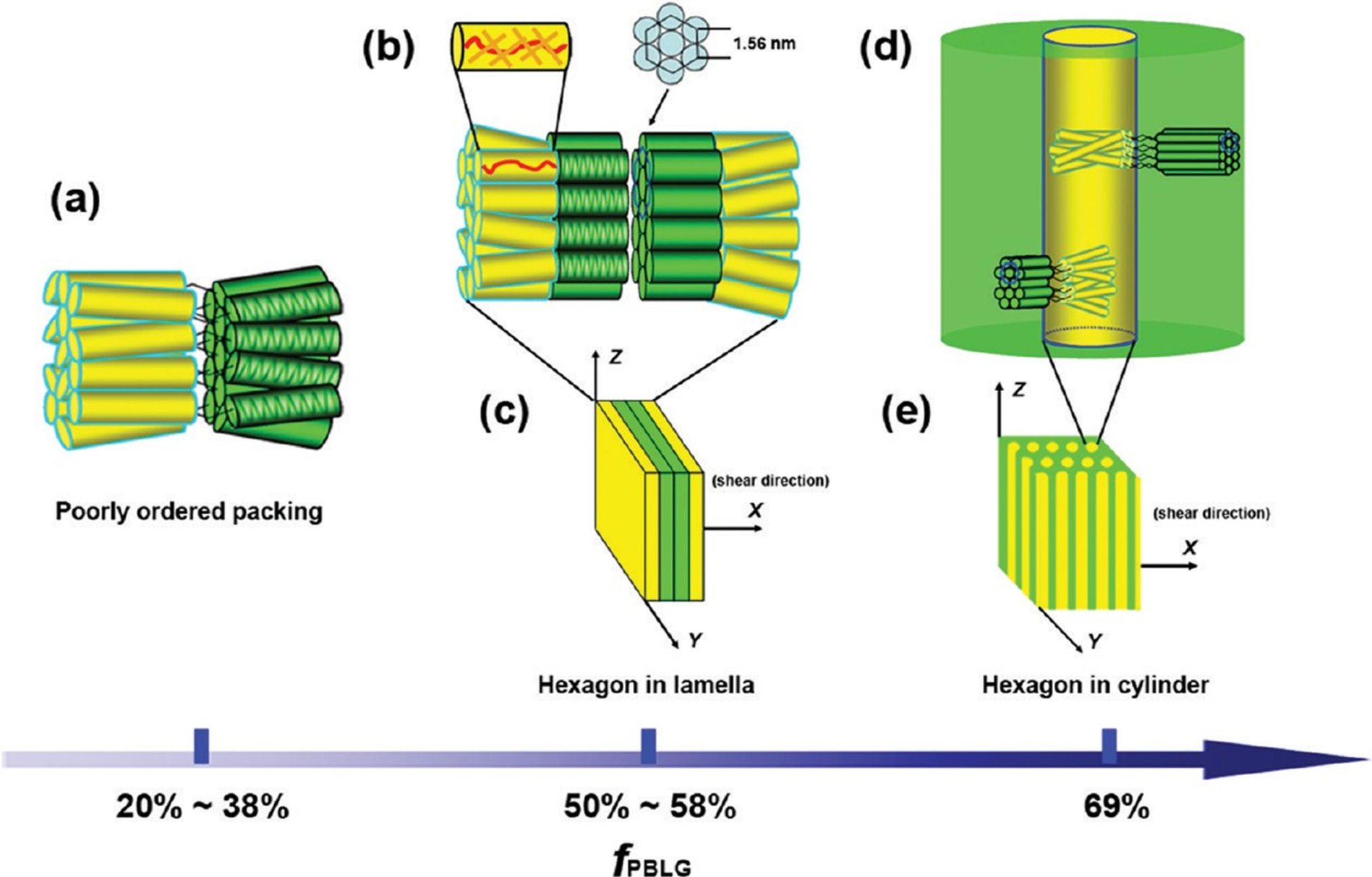



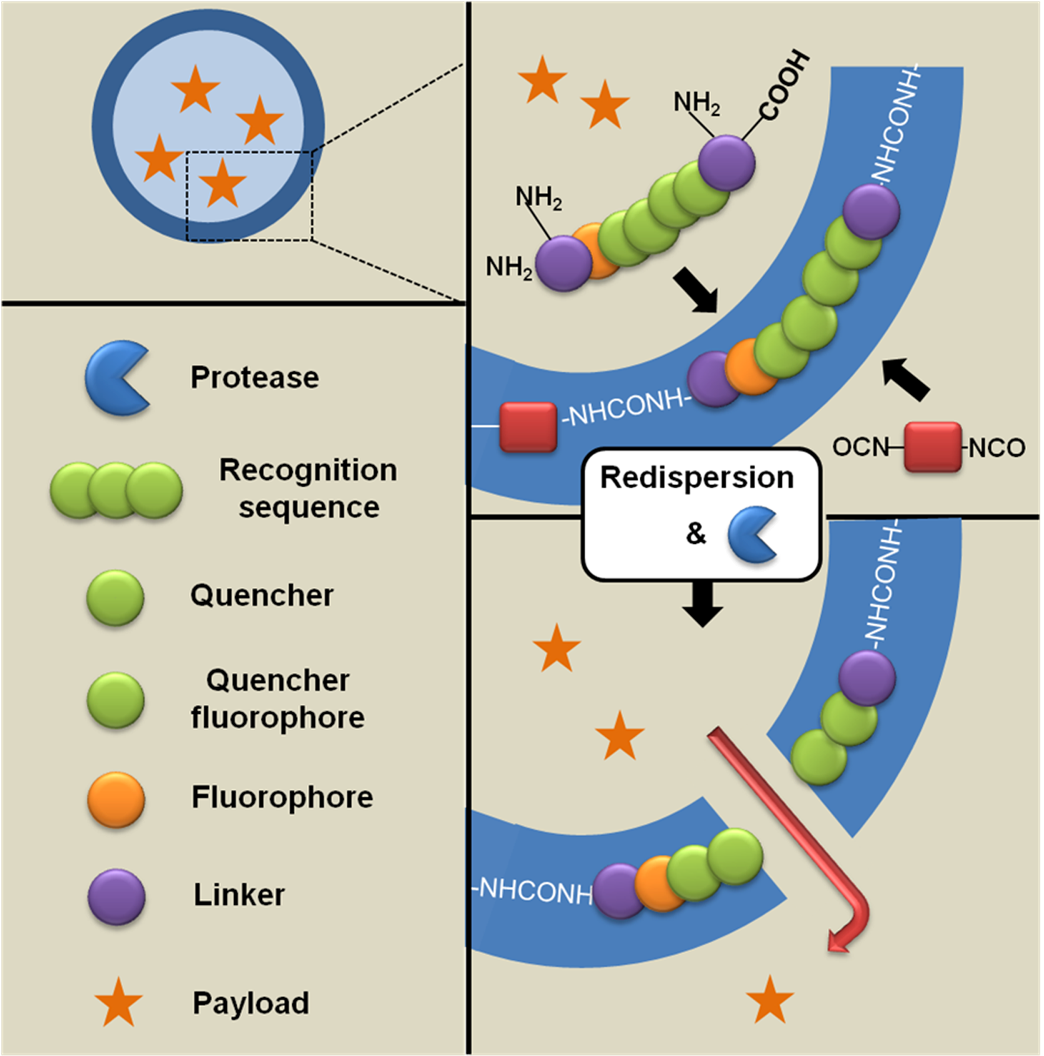
4. Potential Applications of Peptide-Synthetic Polymer Hybrids
4.1. Biological Evaluations (Cytotoxicity)
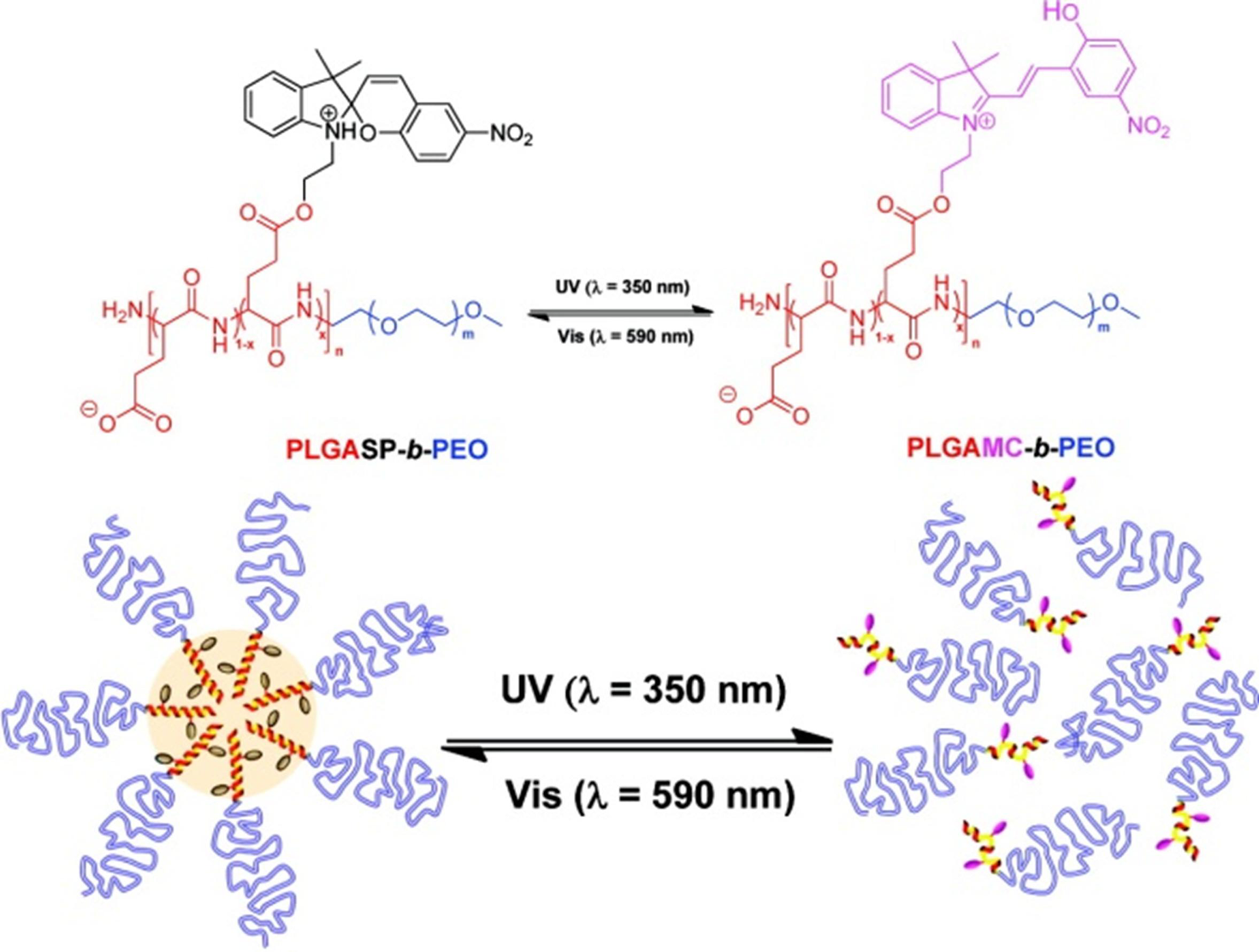
4.2. Self-Assembled Nanoparticles/Micelles for Drug/Enzyme/Gene Delivery
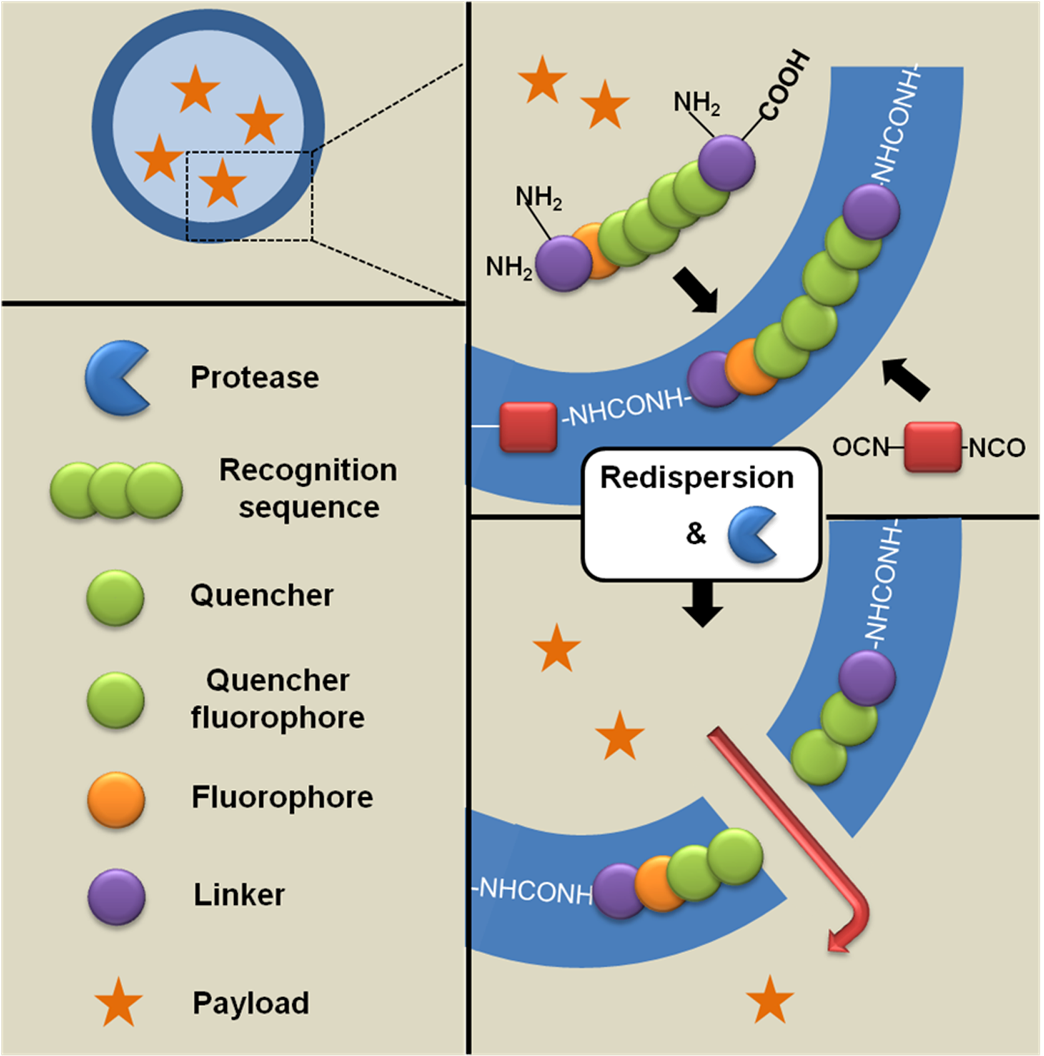
4.3. Crosslinked Hydrogels and Scaffolds for Tissue Engineering
5. Conclusions
Acknowledgments
References
- Schlaad, H.; Antonietti, M. Block copolymers with amino acid sequences: molecular chimeras of polypeptides and synthetic polymers. Eur. Phys. J. E 2003, 10, 17–23. [Google Scholar] [CrossRef]
- Klok, H.A.; Ayres, L. Peptide Hybrid Polymers; Klok, H.A., Schlaad, H., Eds.; Springer: Berlin, Germany, 2006. [Google Scholar]
- Börner, H.G. Strategies exploiting functions and self-assembly properties of bioconjugates for polymer and materials sciences. Prog. Polym. Sci. 2009, 34, 811–851. [Google Scholar] [CrossRef]
- Lutz, J.F.; Börner, H.G. Modern trends in polymer bioconjugates design. Prog. Polym. Sci. 2008, 33, 1–39. [Google Scholar] [CrossRef]
- Gallot, B. Comb-like and block liquid crystalline polymers for biological applications. Prog. Polym. Sci. 1996, 21, 1035–1088. [Google Scholar] [CrossRef]
- Langer, R.; Tirrell, D.A. Designing materials for biology and medicine. Nature 2004, 428, 487–491. [Google Scholar]
- Gauthier, M.A.; Klok, H.A. Peptide/protein-polymer conjugates: Synthetic strategies and design concepts. Chem. Commun. 2008, 23, 2591–2611. [Google Scholar] [CrossRef]
- Le Droumaguet, B.; Velonia, K. Click Chemistry: A powerful tool to create polymer-based macromolecular chimeras. Macromol. Rapid. Commun. 2008, 29, 1073–1089. [Google Scholar] [CrossRef]
- Klok, H.A. Peptide/Protein-synthetic polymer conjugates: Quo Vadis. Macromolecules 2009, 42, 7990–8000. [Google Scholar] [CrossRef]
- Marsden, H.R.; Kros, A. Polymer-peptide block copolymers—An overview and assessment of synthesis methods. Macromol. Biosci. 2009, 9, 939–951. [Google Scholar] [CrossRef]
- Deming, T.J. Polypeptide and polypeptide hybrid copolymer synthesis via NCA polymerization. In Peptide Hybrid Polymers; Klok, H.A., Schlaad, H., Eds.; Springer: Berlin, Germany, 2006. [Google Scholar]
- König, H.M.; Kilbinger, A.F.M. Learning from nature: β-Sheet-mimicking copolymers get organized. Angew. Chem. Int. Ed. 2007, 46, 8334–8340. [Google Scholar] [CrossRef]
- van Hest, J.C.M. Biosynthetic-synthetic polymer conjugates. Polym. Rev. 2007, 47, 63–92. [Google Scholar] [CrossRef]
- Hadjichristidis, N.; Iatrou, H.; Pitsikalis, M.; Sakellariou, G. Synthesis of well-defined polypeptide-based materials via the ring-opening polymerization of α-amino acid N-carboxyanhydrides. Chem. Rev. 2009, 109, 5528–5578. [Google Scholar] [CrossRef]
- Habraken, G.J.M.; Heise, A.; Thornton, P.D. Block copolypeptides prepared by N-carboxyanhydride ring-opening polymerization. Macromol. Rapid Commun. 2011, 33, 272–286. [Google Scholar] [CrossRef]
- Yamashita, Y.; Iwaya, Y.; Ito, K. Block copolymerization, 9. Polymerization of the NCA of methyl D-glutamate by telechelic polystyrene having glycyl groups as active chain ends. Makromol. Chem. 1975, 176, 1207–1216. [Google Scholar] [CrossRef]
- Tang, H.; Li, Y.; Lahasky, S.H.; Sheiko, S.S.; Zhang, D. Core-shell molecular bottlebrushes with helical polypeptide backbone: Synthesis, characterization, and solution conformations. Macromolecules 2011, 44, 1491–1499. [Google Scholar] [CrossRef]
- Wang, J.; Lu, H.; Ren, Y.; Zhang, Y.; Morton, M.; Chen, J.; Lin, Y. Interrupted helical structure of grafted polypeptides in brush-like macromolecules. Macromolecules 2011, 44, 8699–8708. [Google Scholar] [CrossRef]
- Li, W.; Zhang, X.; Wang, J.; Qiao, X.; Liu, K.; Zhang, A. Peptidic molecular brushes with enhanced chirality. J. Polym. Sci. A Polym. Chem. 2012, 50, 4063–4072. [Google Scholar] [CrossRef]
- Jang, J.H.; Choi, Y.M.; Choi, Y.Y.; Joo, M.K.; Park, M.H.; Choi, B.G.; Kang, E.Y.; Jeong, B. pH/temperature sensitive chitosan-g-(PA-PEG) aqueous solutions as new thermogelling systems. J. Mater. Chem. 2011, 21, 5484–5491. [Google Scholar]
- Byrne, M.; Thornton, P.D.; Cryan, S.A.; Heise, A. Star polypeptides by NCA polymerisation from dendritic initiators: synthesis and enzyme controlled payload release. Polym. Chem. 2012, 3, 2825–2831. [Google Scholar] [CrossRef]
- Xia, J.; Chen, L.; Chen, J.; Tian, H.; Li, F.; Zhu, X.; Li, G.; Chen, X. Hydrophobic polyphenylalanine-grafted hyperbranched polyethyleneimine and its in vitro gene transfection. Macromol. Biosci. 2011, 11, 211–218. [Google Scholar] [CrossRef]
- Liu, G.; Dong, C.M. Photoresponsive Poly(S-(o-nitrobenzyl)-L-cysteine)-b-PEO from a L-cysteine N-carboxyanhydride monomer: Synthesis, self-assembly, and phototriggered drug release. Biomacromolecules 2012, 13, 1573–1583. [Google Scholar] [CrossRef]
- Wu, Y.; Ma, Q.; Song, X.R.; Zheng, Y.; Ren, W.; Zhang, J.K.; Ouyang, L.; Wu, F.B.; He, G. Biocompatible poly(ethylene glycol)-poly(γ-cholesterol-L-glutamate) copolymers: Synthesis, characterization, and in vitro studies. J. Polym. Sci. A Polym. Chem. 2012, 50, 4532–4537. [Google Scholar] [CrossRef]
- Peng, Y.L.; Huang, Y.; Chuang, H.J.; Kuo, C.Y.; Lin, C.C. Synthesis and characterization of biodegradable polylactides and polylactide-block-poly(Z-lysine) copolymers. Polymer 2010, 51, 4329–4335. [Google Scholar] [CrossRef]
- Jacobs, J.; Pound-Lana, G.; Klumperman, B. Poly(N-vinylpyrrolidone-b-(gamma-benzyl-L-glutamate))—synthesis and self-assembly into pH-sensitive micelles. Polym. Chem. 2012, 3, 2551–2560. [Google Scholar] [CrossRef]
- Xiang, L.; Shen, L.J.; Long, F.; Yang, K.; Fan, J.B.; Li, Y.J.; Xiang, J.N.; Zhu, M.Q. A convenient method for the synthesis of the amphiphilic triblock copolymer poly(L-lactic acid)-block-poly(L-lysine)-block-poly(ethylene glycol) monomethyl ether. Macromol. Chem. Phys. 2011, 212, 563–573. [Google Scholar] [CrossRef]
- You, Y.; Chen, Y.; Hua, C.; Dong, C.M. Synthesis and thermoreversible gelation of dendron-like polypeptide/linear poly(ε-caprolactone)/dendron-like polypeptide triblock copolymers. J. Polym. Sci. A Polym. Chem. 2010, 48, 709–718. [Google Scholar] [CrossRef]
- Ray, J.G.; Naik, S.S.; Hoff, E.A.; Johnson, A.J.; Ly, J.T.; Easterling, C.P.; Patton, D.L.; Savin, D.A. Stimuli-responsive peptide-based ABA-triblock copolymers: Unique morphology transitions with pH. Macromol. Rapid Commun. 2012, 33, 819–826. [Google Scholar] [CrossRef]
- Wang, K.; Luo, G.F.; Liu, Y.; Li, C.; Cheng, S.X.; Zhuo, R.X.; Zhang, X.Z. Redox-sensitive shell cross-linked PEG-polypeptide hybrid micelles for controlled drug release. Polym. Chem. 2012, 3, 1084–1090. [Google Scholar] [CrossRef]
- Pintauer, T.; Matysjaszewski, K. Atom transfer radical addition and polymerization reactions catalyzed by ppm amounts of copper complexes. Chem. Soc. Rev. 2008, 6, 1087–1097. [Google Scholar] [CrossRef]
- Boyer, C.; Bulmus, V.; Davis, T.P.; Ladmiral, V.; Liu, J.; Perrier, S. Bioapplications of RAFT polymerization. Chem. Rev. 2009, 11, 5402–5436. [Google Scholar]
- Gregory, A.; Stenzel, M.H. Complex polymer architectures via RAFT polymerization: From fundamental process to extending the scope using click chemistry and nature’s building blocks. Prog. Polym. Sci. 2012, 37, 38–105. [Google Scholar] [CrossRef]
- Grubbs, R.B. Nitroxide-mediated radical polymerization: Limitations and versatility. Polym. Rev. 2011, 51, 104–137. [Google Scholar] [CrossRef]
- Habraken, G.J.M.; Peeters, M.; Thornton, P.D.; Koning, C.E.; Heise, A. Selective enzymatic degradation of self-assembled particles from amphiphilic block copolymers obtained by the combination of N-carboxyanhydride and nitroxide-mediated polymerization. Biomacromolecules 2011, 12, 3761–3769. [Google Scholar] [CrossRef]
- Knoop, R.J.I.; de Geus, M.; Habraken, G.J.M.; Koning, C.E.; Menzel, H.; Heise, A. Stimuli responsive peptide conjugated polymer nanoparticles. Macromolecules 2010, 43, 4126–4132. [Google Scholar] [CrossRef]
- Merrifield, R.B. Solid phase peptide synthesis. I. The synthesis of a tetrapeptide. J. Am. Chem. Soc. 1963, 85, 2149–2154. [Google Scholar] [CrossRef]
- Merrifield, R.B. Solid phase synthesis. Angew. Chem. Int. Ed. Engl. 1985, 10, 799–810. [Google Scholar] [CrossRef]
- White, P.D.; Chan, W.C. Basic principles. In Fmoc Solid Phase Peptide Synthesis: A Practical Approach; Chan, W.C., White, P.D., Eds.; Oxford University Press: New York, NY, USA, 2000. [Google Scholar]
- Becker, H.; Lucas, H.W.; Maul, J.; Pillai, V.N.R.; Anzinger, H.; Mutter, M. Polyethyleneglycols onto crosslinked polystyrenes: A new class of hydrophilic polymeric supports for peptide synthesis. Makromol. Chem. Rapid Commun. 1982, 3, 217–223. [Google Scholar] [CrossRef]
- Burkoth, T.S.; Benzinger, T.L.S.; Jones, D.N.S.; Hallenga, K.; Meredith, S.C.; Lynn, D.G. C-terminal PEG blocks the irreversible step in β-amyloid(10-35) fibrillogenesis. J. Am. Chem. Soc. 1998, 120, 7655–7656. [Google Scholar]
- Rosler, A.; Klok, H.A.; Hamley, I.W.; Castelletto, V.; Mykhaylyk, O.O. Nanoscale structure of poly(ethylene glycol) Hybrid block copolymers containing amphiphilic β-strand peptide sequences. Biomacromolecules 2003, 4, 859–863. [Google Scholar] [CrossRef]
- Hamley, I.W.; Krysmann, M.J. Effect of PEG crystallization on the self-assembly of PEG/peptide copolymers containing amyloid peptide fragments. Langmuir 2008, 24, 8210–8214. [Google Scholar] [CrossRef]
- Eckhardt, D.; Groenewolt, M.; Krause, E.; Börner, H.G. Rational design of oligopeptide organizers for the formation of poly(ethylene oxide) nanofibers. Chem. Commun. 2005, 2814–2816. [Google Scholar]
- Castelletto, V.; McKendrick, J.E.; Hamley, I.W.; Olsson, U.; Cenker, C. PEGylated amyloid peptide nanocontainer delivery and release system. Langmuir 2010, 26, 11624–11627. [Google Scholar] [CrossRef]
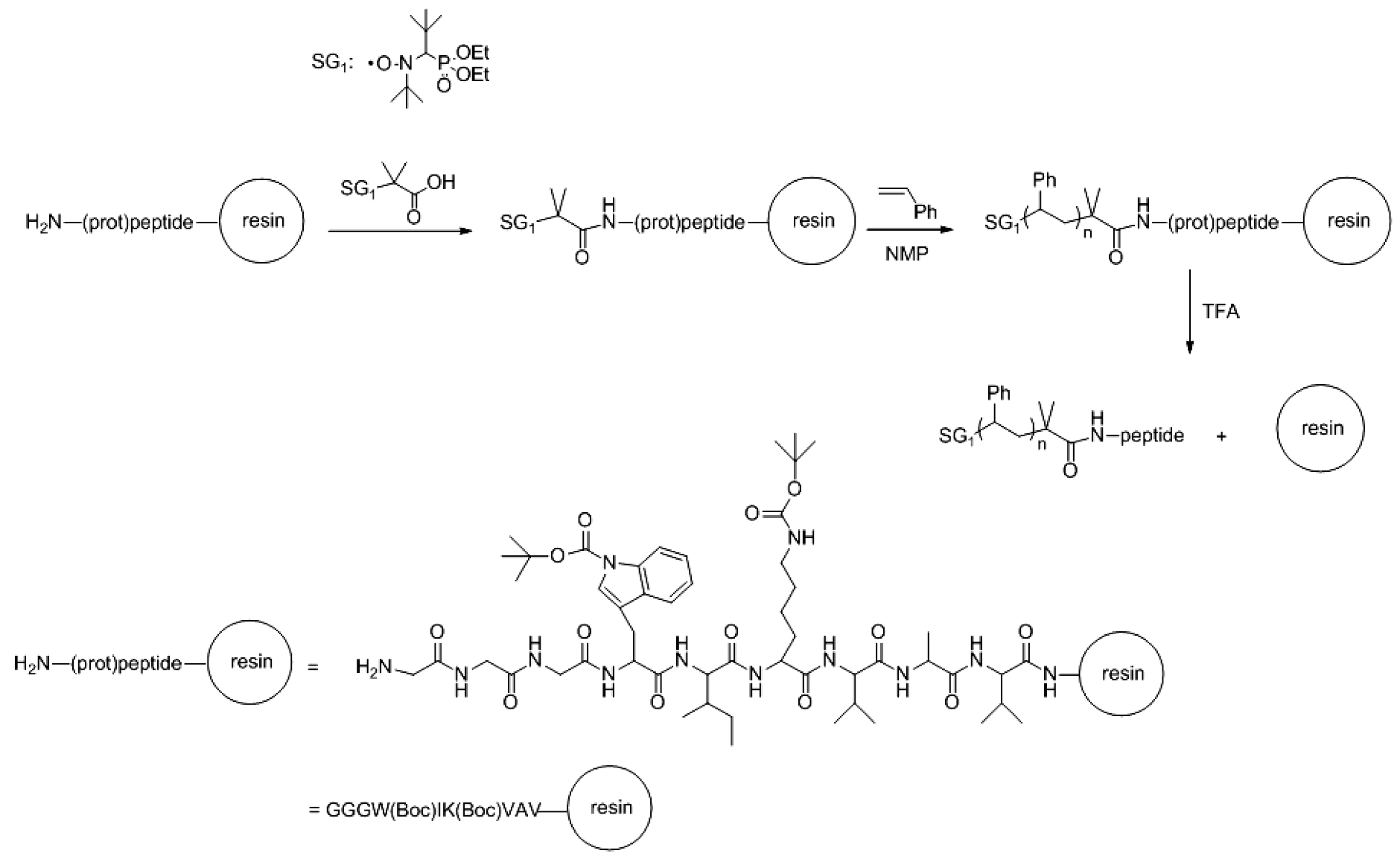
- Trimaille, T.; Mabrouk, K.; Monnier, V.; Charles, L.; Bertin, D.; Gigmes, D. SG1-Functionalized peptides as precursors for polymer-peptide conjugates: A straightforward approach. Macromolecules 2010, 43, 4864–4870. [Google Scholar] [CrossRef]
- de Graaf, A.J.; Mastrobattista, E.; Vermonden, T.; van Nostrum, C.F.; Rijkers, D.T.S.; Liskamp, R.M.J.; Hennink, W.E. Thermosensitive peptide-hybrid ABC block copolymers obtained by ATRP: Synthesis, self-assembly, and enzymatic degradation. Macromolecules 2012, 45, 842–851. [Google Scholar] [CrossRef]
- Paira, T.K.; Banerjee, S.; Mandal, T.K. Peptide-poly(ε-caprolactone) biohybrids by grafting-from ring-opening polymerization: synthesis, aggregation and crystalline properties. J. Polym. Sci. A Polym. Chem. 2012, 50, 2130–2141. [Google Scholar] [CrossRef]
- Paira, T.K.; Banerjee, S.; Raula, M.; Kotal, A.; Si, S.; Mandal, T.K. Peptide-polymer bioconjugates via atom transfer radical polymerization and their solution aggregation into hybrid micro/nanospheres for dye uptake. Macromolecules 2010, 43, 4050–4061. [Google Scholar] [CrossRef]
- Fuks, G.; Talom, R.M.; Gauffre, F. Biohybrid block copolymers: Towards functional micelles and vesicles. Chem. Soc. Rev. 2011, 40, 2475–2493. [Google Scholar] [CrossRef]
- Shi, H.; Liu, L.; Wang, X.; Li, J. Glycopolymer-peptide bioconjugates with antioxidant activity via RAFT polymerization. Polym. Chem. 2012, 3, 1182–1188. [Google Scholar] [CrossRef]
- Koga, T.; Iimura, M.; Higashi, N. Novel peptide-shelled dendrimer with dramatically changeable thermo-responsive character. Macromol. Biosci. 2012, 12, 1043–1047. [Google Scholar] [CrossRef]
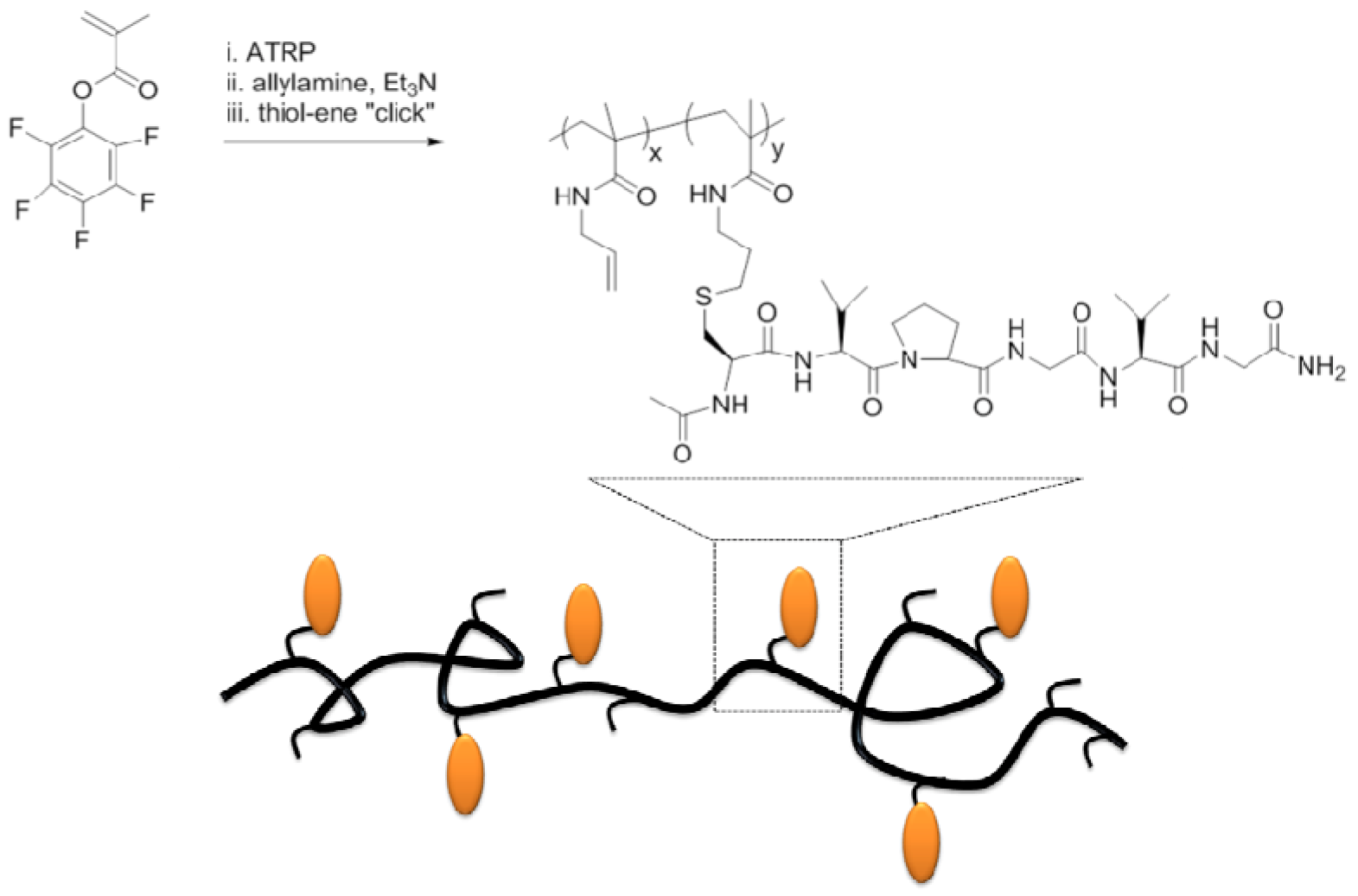
- Singha, N.K.; Gibson, M.I.; Koiry, B.P.; Dainal, M.; Klok, H.A. Side-chain peptide-synthetic polymer conjugates via tandem “Ester-Amide/Thiol-Ene” post-polymerization modification of poly(pentafluorophenyl methacrylate) obtained using ATRP. Biomacromolecules 2011, 12, 2908–2913. [Google Scholar] [CrossRef]
- Tsurkan, M.V.; Chwalek, K.; Levental, K.R.; Freudenberg, U.; Werner, C. Modular starPEG-heparin gels with bifunctional peptide linkers. Macromol. Rapid Commun. 2010, 31, 1529–1533. [Google Scholar] [CrossRef]
- Börner, H.G.; Schlaad, H. Bioinspired functional block copolymers. Soft Matter 2007, 3, 394–408. [Google Scholar] [CrossRef]
- Hamley, I. Block copolymers in solution. In Fundamentals and Applications; Wiley: Chichester, UK, 2007. [Google Scholar]
- Yesodha, S.K.; Pillai, C.K.S.; Tsutsumi, N. Stable polymeric materials for nonlinear optics: A review based on azobenzene systems. Prog. Polym. Sci. 2004, 29, 45–74. [Google Scholar] [CrossRef]
- Cho, M.J.; Choi, D.H.; Sullivan, P.A.; Akelaitis, A.J.P.; Dalton, L.R. Recent progress in second-order nonlinear optical polymers and dendrimers. Prog. Polym. Sci. 2008, 33, 1013–1058. [Google Scholar] [CrossRef]
- Cornelissen, J.; Fischer, M.; Sommerdijk, N.; Nolte, R.J.M. Helical superstructures from charged poly(styrene)-poly(isocyanodipeptide) block copolymers. Science 1998, 280, 1427–1430. [Google Scholar]
- Klok, H.A.; Langenwalter, J.F.; Lecommandoux, S. Self-assembly of peptide-based diblock oligomers. Macromolecules 2000, 33, 7819–7826. [Google Scholar] [CrossRef]
- Hung, J.H.; Lin, Y.L.; Sheng, Y.J.; Tsao, H.K. Structure-photophysical property relationship of conjugated rod-coil block copolymers in solutions. Macromolecules 2012, 45, 2166–2170. [Google Scholar] [CrossRef]
- Antonietti, M.; Förster, S. Vesicles and liposomes: A self-assembly principle beyond lipids. Adv. Mater. 2003, 15, 1323–1333. [Google Scholar] [CrossRef]
- Chécot, F.; Lecommandoux, S.; Gnanou, Y.; Klok, H.A. Water-soluble stimuli-responsive vesicles from peptide-based diblock copolymers. Angew. Chem. Int. Ed. 2002, 41, 1339–1343. [Google Scholar] [CrossRef]
- Kukula, H.; Schlaad, A.; Antonietti, M.; Förster, S. The formation of polymer vesicles or “peptosomes” by polybutadiene-block-poly(L-glutamate)s in dilute aqueous solution. J. Am. Chem. Soc. 2002, 124, 1658–1663. [Google Scholar] [CrossRef]
- Holowka, E.P.; Sun, V.Z.; Kamei, D.T.; Deming, T.J. Polyarginine segments in block copolypeptides drive both vesicular assembly and intracellular delivery. Nat. Mater. 2007, 6, 52–57. [Google Scholar] [CrossRef]
- Lecommandoux, S.; Rodríguez-Hernández, J. Reversible inside-out micellization of pH-responsive and water-soluble vesicles based on polypeptide diblock copolymers. J. Am. Chem. Soc. 2005, 127, 2026–2027. [Google Scholar] [CrossRef]
- Zhou, Q.H.; Zheng, J.K.; Shen, Z.H.; Fan, X.H.; Chen, X.F.; Zhou, Q.F. Synthesis and hierarchical self-assembly of rod rod block copolymers via click chemistry between mesogen-jacketed liquid crystalline polymers and helical polypeptides. Macromolecules 2010, 43, 5637–5646. [Google Scholar]
- Hermes, F.; Otte, K.; Brandt, J.; Grawert, M.; Borner, H.G.; Schlaad, H. Polypeptide-based organogelators: Effects of secondary structure. Macromolecules 2011, 44, 7489–7492. [Google Scholar] [CrossRef]
- Kakwere, H.; Payne, R.J.; Jolliffe, K.A.; Perrier, S. Self-assembling macromolecular chimeras: Controlling fibrillization of a beta-sheet forming peptide by polymer. Soft Matter 2011, 7, 3754–3757. [Google Scholar] [CrossRef]
- Klok, H.A.; Langenwalter, J.F.; Lecommandoux, S. Self-assembly of peptide-based diblock oligomers. Macromolecules 2000, 33, 7819–7826. [Google Scholar] [CrossRef]
- Losik, M.; Kubowicz, S.; Smarsly, B.; Schlaad, H. Solid-state structure of polypeptide-based rod-coil block copolymers: Folding of helices. Eur. Phys. J. E. 2004, 15, 407–411. [Google Scholar] [CrossRef]
- Schlaad, H.; Smarsly, B.; Losik, M. The role of chain-length distribution in the formation of solid-state structures of polypeptide-based rod-coil block copolymers. Macromolecules 2004, 37, 2210–2214. [Google Scholar] [CrossRef]
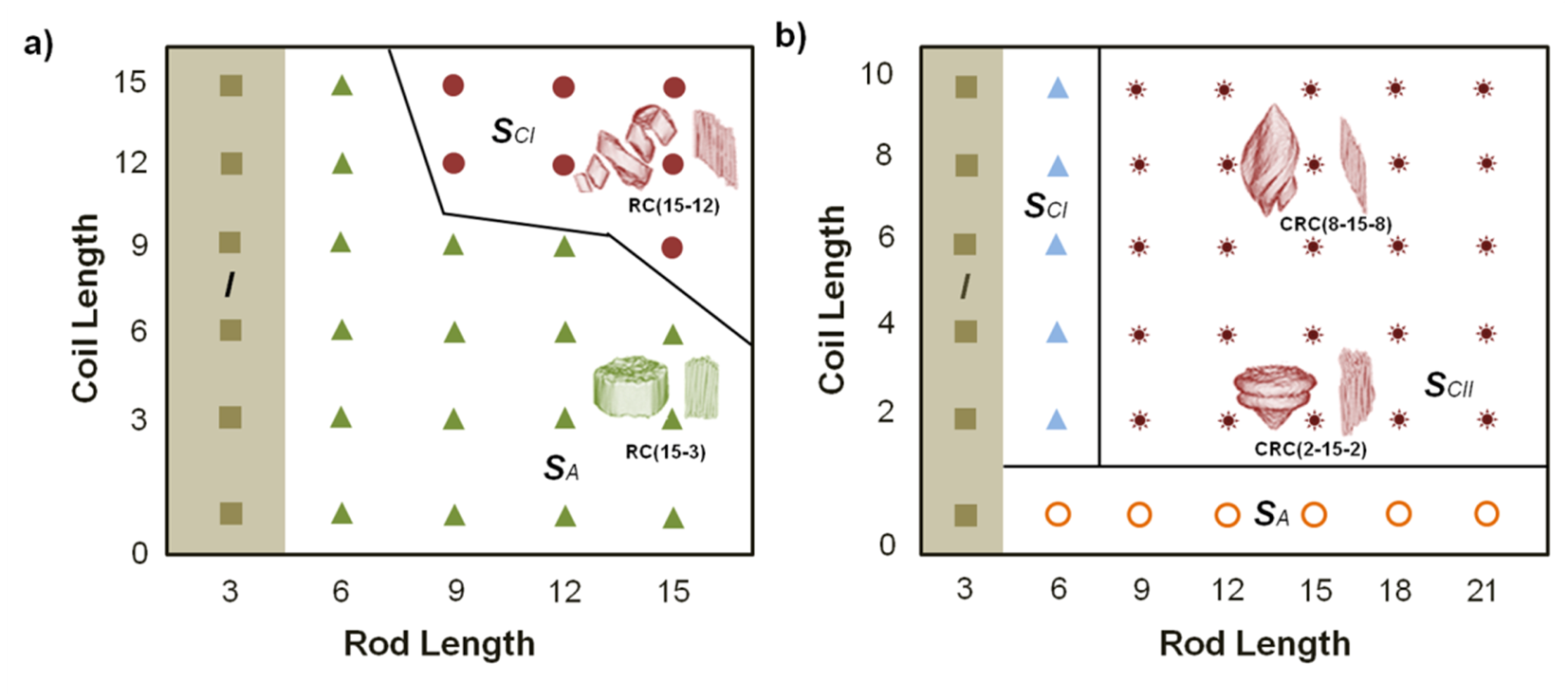
- Sanchez-Ferrer, A.; Mezzenga, R. Secondary structure-induced micro- and macrophase separation in rod-coil polypeptide diblock, triblock, and star-block copolymer. Macromolecules 2010, 43, 1093–1100. [Google Scholar] [CrossRef]
- Junnila, S.; Houbenov, N.; Hanski, S.; Iatrou, H.; Hirao, A.; Hadjichristidis, N.; Ikkala, O. Hierarchical smectic self-assembly of an ABC miktoarm star terpolymer with a helical polypeptide arm. Macromolecules 2010, 43, 9071–9076. [Google Scholar]
- Junnila, S.; Houbenov, N.; Karatzas, A.; Hadjichristidis, N.; Hirao, A.; Iatrou, H.; Ikkala, O. Side-chain-controlled self-assembly of polystyrene-polypeptide miktoarm star copolymers. Macromolecules 2012, 45, 2850–2856. [Google Scholar] [CrossRef]
- Nuhn, H.; Klok, H.A. Aqueous solution self-assembly of polystyrene-b-poly(L-lysine) diblock oligomers. Eur. Polym. J. 2011, 47, 782–791. [Google Scholar] [CrossRef]
- Itoh, T.; Hatanaka, T.; Ihara, E.; Inoue, K. Helix-coil transformation of poly(gamma-benzyl-L-glutamate) with polystyrene attached to the N or C terminus in trifluoroacetic acid-chloroform mixtures. Polym. J. 2012, 44, 189–194. [Google Scholar] [CrossRef]
- Gkikas, M.; Iatrou, H.; Thomaidis, N.S.; Alexandridis, P.; Hadjichristidis, N. Well-defined homopolypeptides, copolypeptides, and hybrids of poly(L-proline). Biomacromolecules 2011, 12, 2396–2406. [Google Scholar] [CrossRef]
- Rapoport, N. Physical stimuli-responsive polymeric micelles for anti-cancer drug delivery. Prog. Polym. Sci. 2007, 32, 962–990. [Google Scholar] [CrossRef]
- Kotharangannagari, V.K.; Sánchez-Ferrer, A.; Ruokolainen, J.; Mezzenga, R. Photoresponsive reversible aggregation and dissolution of rod-coil polypeptide diblock copolymers. Macromolecules 2011, 44, 4569–4573. [Google Scholar] [CrossRef]
- Oh, H.J.; Joo, M.K.; Sohn, Y.S.; Jeong, B. Secondary structure effect of polypeptide on reverse thermal gelation and degradation of L/DL-poly(alanine)-poloxamer-L/DL-poly(alanine) copolymers. Macromolecules 2008, 41, 8204–8209. [Google Scholar] [CrossRef]
- Kotharangannagari, V.K.; Sanchez-Ferrer, A.; Ruokolainen, J.; Mezzenga, R. Thermoreversible gel-sol behavior of rod-coil-rod peptide-based triblock copolymers. Macromolecules 2012, 45, 1982–1990. [Google Scholar] [CrossRef]
- Chen, C.Y.; Wang, Z.H.; Li, Z.B. Thermoresponsive polypeptides from pegylated poly-L-glutamates. Biomacromolecules 2011, 12, 2859–2863. [Google Scholar] [CrossRef]
- Krishna, O.D.; Wiss, K.T.; Luo, T.Z.; Pochan, D.J.; Theato, P.; Kiick, K.L. Morphological transformations in a dually thermoresponsive coil-rod-coil bioconjugate. Soft Matter 2012, 8, 3832–3840. [Google Scholar]
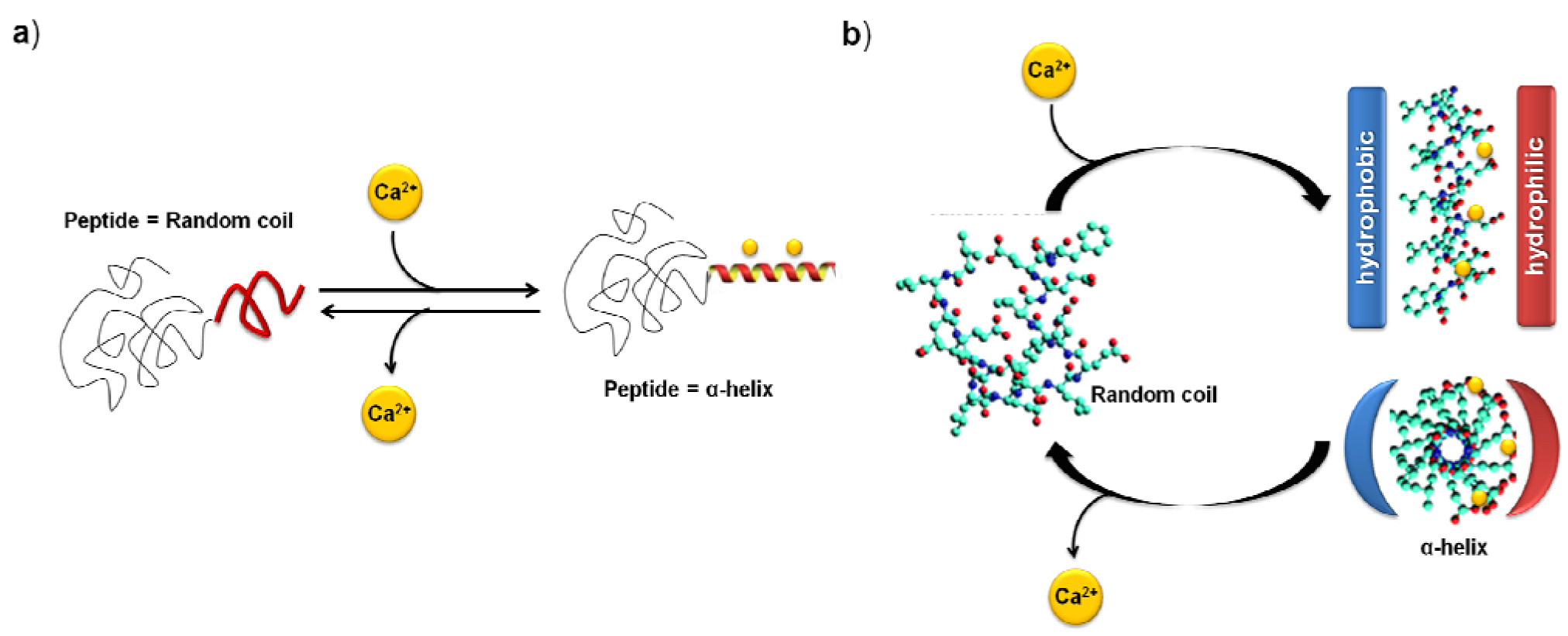
- Kühnle, R.I.; Gebauer, D.; Borner, H.G. Calcium ions as bioinspired triggers to reversibly control the coil-to-helix transition in peptide-polymer conjugates. Soft Matter 2011, 7, 9616–9619. [Google Scholar] [CrossRef]
- Tangbunsuk, S.; Whittell, G.R.; Ryadnov, M.G.; Vandermeulen, G.W.M.; Woolfson, D.N.; Manners, I. Metallopolymer-peptide hybrid materials: synthesis and self-assembly of functional, polyferrocenylsilane-tetrapeptide conjugates. Chem. Eur. J. 2012, 18, 2524–2535. [Google Scholar]
- Börner, H.G. Precision polymers—modern tools to understand and program macromolecular interactions. Macromol. Rapid Commun. 2011, 32, 115–126. [Google Scholar] [CrossRef]
- Rabotyagova, O.S.; Cebe, P.; Kaplan, D.L. Protein-based block copolymers. Biomacromolecules 2011, 12, 269–289. [Google Scholar] [CrossRef]
- Börner, H.G.; Kuhnle, H.; Hentschel, J. Making “smart polymers” smarter: Modern concepts to regulate functions in polymer science. J. Polym. Sci. A Polym. Chem. 2010, 48, 1–14. [Google Scholar]
- Oh, J.K. Polylactide (PLA-based) amphiphilic block copolymers: Synthesis, self-assembly, and biomedical applications. Soft Matter 2011, 7, 5096–5108. [Google Scholar] [CrossRef]
- Harada, A.; Kataoka, K. Supramolecular assemblies of block copolymers in aqueous media as nanocontainers relevant to biological applications. Prog. Polym. Sci. 2006, 31, 949–982. [Google Scholar] [CrossRef]
- Tyrrell, Z.L.; Shen, Y.Q.; Radosz, M. Fabrication of micellar nanoparticles for drug delivery through the self-assembly of block copolymers. Prog. Polym. Sci. 2010, 35, 1128–1143. [Google Scholar] [CrossRef]
- Andrieu, J.; Kotman, N.; Maier, M.; Mailander, V.; Strauss, W.S.L.; Weiss, C.K.; Landfester, K. Live monitoring of cargo release from peptide-based hybrid nanocapsules induced by enzyme cleavage. Macromol. Rapid Commun. 2012, 33, 248–253. [Google Scholar] [CrossRef]
- Maier, M.; Kotman, N.; Friedrichs, C.; Andrieu, J.; Wagner, M.; Graf, R.; Strauss, W.S.L.; Mailander, V.; Weiss, C.K.; Landfester, K. Highly site specific, protease cleavable, hydrophobic peptide-polymer nanoparticles. Macromolecules 2011, 44, 6258–6267. [Google Scholar]
- Apostolovic, B.; Deacon, S.P.E.; Duncan, R.; Klok, H.A. Hybrid polymer therapeutics incorporating bioresponsive, coiled coil peptide linkers. Biomacromolecules 2010, 11, 1187–1195. [Google Scholar] [CrossRef]
- Ramos, J.; Imaz, A.; Callejas-Fernandez, J.; Barbosa-Barros, L.; Estelrich, J.; Quesada-Perez, M.; Forcada, J. Soft nanoparticles (thermo-responsive nanogels and bicelles) with biotechnological applications: from synthesis to simulation through colloidal characterization. Soft Matter 2011, 7, 5067–5082. [Google Scholar]
- Kopecek, J.; Yang, J.Y. Smart self-assembled hybrid hydrogel biomaterials. Angew. Chem. Int. Ed. 2012, 51, 7396–7417. [Google Scholar] [CrossRef]
- Kang, E.Y.; Moon, H.J.; Joo, M.K.; Jeong, B. Thermogelling chitosan-g-(PAF-PEG) aqueous solution as an injectable scaffold. Biomacromolecules 2012, 13, 1750–1757. [Google Scholar] [CrossRef]
- Kang, E.Y.; Yeon, B.; Moon, H.J.; Jeong, B. PEG-l-PAF and PEG-d-PAF: Comparative study on thermogellation and biodegradation. Macromolecules 2012, 45, 2007–2013. [Google Scholar] [CrossRef]
- Choi, B.G.; Park, M.H.; Cho, S.H.; Joo, M.K.; Oh, H.J.; Kim, E.H.; Kwideok Park, K.; Han, D.K.; Jeong, B. In situ thermal gelling polypeptide for chondrocytes 3D culture. Biomaterials 2010, 31, 9266–9272. [Google Scholar] [CrossRef]
- Min Hee Park, M.H.; Bo Gyu Choi, B.G.; Jeong, B. Complexation-induced biomimetic long range fibrous orientation in a rigid-flexible block copolymer thermogel. Adv. Funct. Mater. 2012, 22, 5118–5125. [Google Scholar] [CrossRef]
- Yun, E.J.; Yon, B.; Joo, M.K.; Jeong, B. Cell Therapy for skin wound using fibroblast encapsulated poly(ethylene glycol)-poly(L-alanine) thermogel. Biomacromolecules 2012, 13, 1106–1111. [Google Scholar] [CrossRef]
- Hamley, I.W.; Cheng, G.; Castelletto, V. A thermoresponsive hydrogel based on telechelic PEG end-capped with hydrophobic dipeptides. Macromol. Biosci. 2011, 11, 1068–1078. [Google Scholar] [CrossRef]
- Grieshaber, S.E.; Nie, T.; Yan, C.Q.; Zhong, S.; Teller, S.S.; Clifton, R.J.; Pochan, D.J.; Kiick, K.L.; Jia, X.Q. Assembly properties of an alanine-rich, lysine-containing peptide and the formation of peptide/polymer hybrid hydrogels. Macromol. Chem. Phys. 2011, 212, 229–239. [Google Scholar] [CrossRef]
- Grieshaber, S.E.; Farran, A.J.E.; Bai, S.; Kiick, K.L.; Jia, X.Q. Tuning the properties of elastin mimetic hybrid copolymers via a modular polymerization method. Biomacromolecules 2012, 13, 1774–1786. [Google Scholar] [CrossRef]
- Chawla, K.; Yu, T.B.; Liao, S.W.; Guan, Z.B. Biodegradable and biocompatible synthetic saccharide-peptide hydrogels for three-dimensional stem cell culture. Biomacromolecules 2011, 12, 560–567. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Morell, M.; Puiggalí, J. Hybrid Block Copolymers Constituted by Peptides and Synthetic Polymers: An Overview of Synthetic Approaches, Supramolecular Behavior and Potential Applications . Polymers 2013, 5, 188-224. https://doi.org/10.3390/polym5010188
Morell M, Puiggalí J. Hybrid Block Copolymers Constituted by Peptides and Synthetic Polymers: An Overview of Synthetic Approaches, Supramolecular Behavior and Potential Applications . Polymers. 2013; 5(1):188-224. https://doi.org/10.3390/polym5010188
Chicago/Turabian StyleMorell, Mireia, and Jordi Puiggalí. 2013. "Hybrid Block Copolymers Constituted by Peptides and Synthetic Polymers: An Overview of Synthetic Approaches, Supramolecular Behavior and Potential Applications " Polymers 5, no. 1: 188-224. https://doi.org/10.3390/polym5010188
APA StyleMorell, M., & Puiggalí, J. (2013). Hybrid Block Copolymers Constituted by Peptides and Synthetic Polymers: An Overview of Synthetic Approaches, Supramolecular Behavior and Potential Applications . Polymers, 5(1), 188-224. https://doi.org/10.3390/polym5010188