Ring Opening Metathesis Polymerization of Norbornene and Derivatives by the Triply Bonded Ditungsten Complex Na[W2(µ-Cl)3Cl4(THF)2]·(THF)3


 ,
, 
Abstract
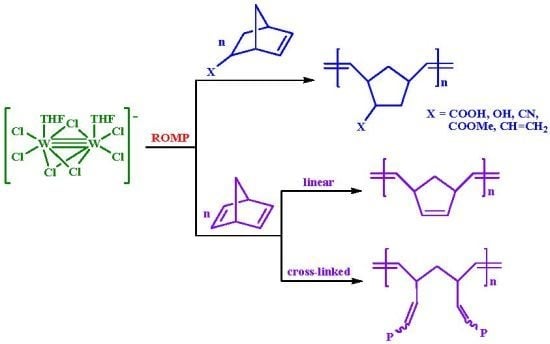
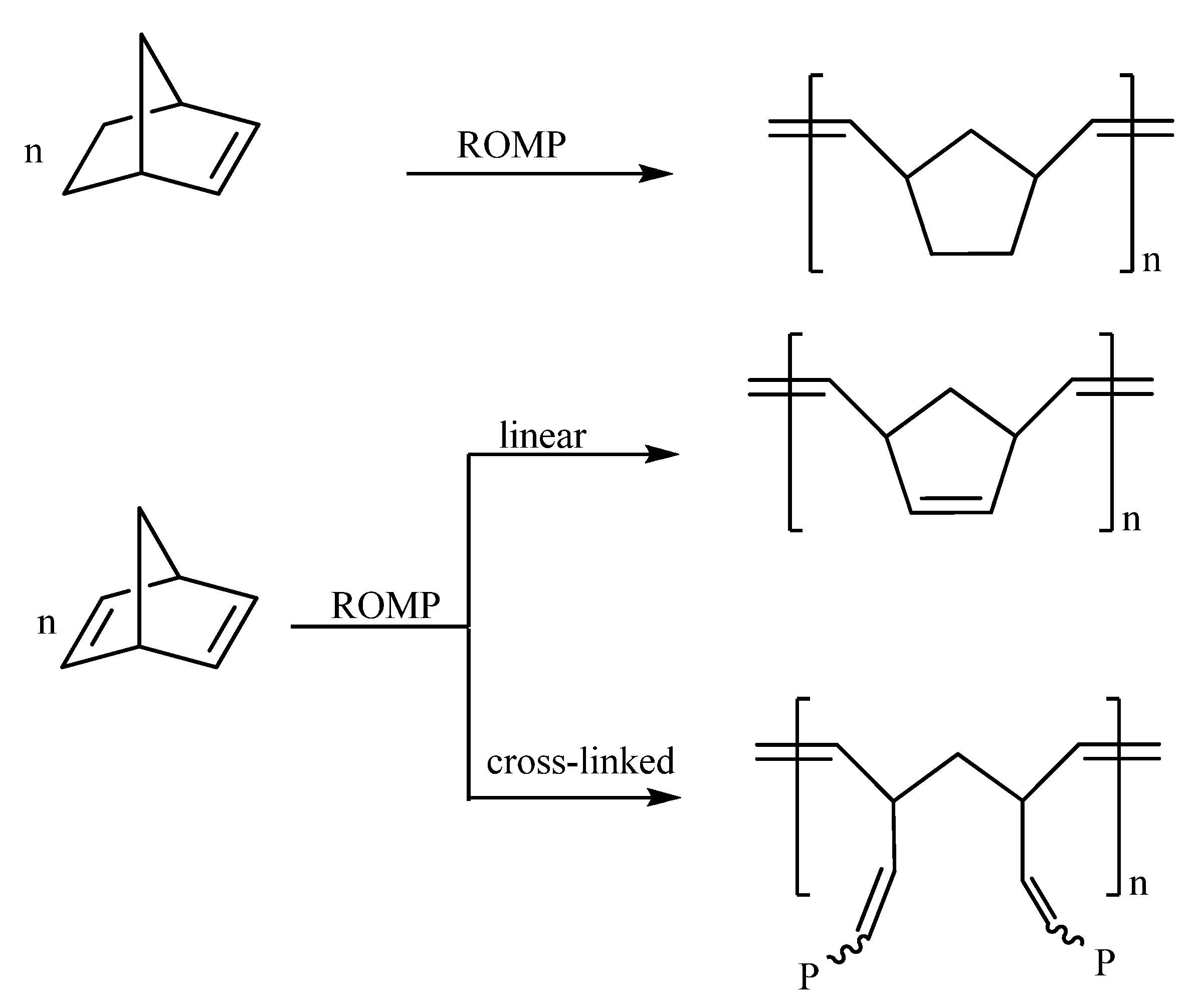
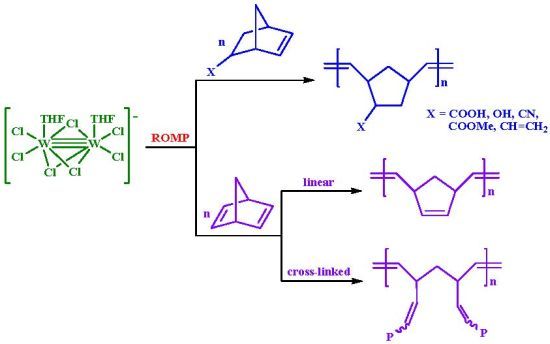
:
1. Introduction
- (i) ill-defined systems, including the classical high oxidation state halides (e.g., MoCl5, WCl6), oxohalides (e.g., WOCl4), alkoxohalides (e.g., [(RO)xWCl6−x], x =1–3) and oxoclusters (e.g., [W6O19]2−) exhibiting small to moderate activity; these become very reactive when activated by organometallic cocatalysts (e.g., SnMe4, AlEt3);
2. Experimental Section
2.1. General
2.2. Catalytic Reactions
2.3. Catalytic Reactions in NMR Tubes
2.4. Polymer Microstructure

{kind=link}
{kind=link}
{kind=link}
 | 1H NMR (CCl4, 300 MHz): 5.25 (s, 2H, H2,3 trans), 5.10 (s, 2H, H2,3 cis), 2.73 (broad, s, 2H, H1,4 cis), 2.37 (broad, s, 2H, H1,4 trans), 1.95–1.60 (broad, m, 3H, H5a,6a,7a), 1.50–1.20 (broad, m, 2H, H5b,6b), 1.20–0.85 ppm (broad, m, 1H, H7b); 13C NMR (CCl4, 75.4 MHz): 133.99 (s, C2,3 ccc), 133.21 (m, C2,3 ctt/ttt/ctc), 133.08 (s, C2,3 ttc), 43.67 (s, C1,4 tc), 43.44 (s, C1,4 tt), 42.88 (s, C7 cc), 42.25 (s, C7 ct/tc), 41.52 (s, C7 tt), 38.88 (s, C1,4 cc), 38.67 (s, C1,4 ct), 33.39 (s, C5,6 cc), 33.19 (s, C5,6 ct), 32.61 (s, C5,6 tc), 32.43 ppm (s, C5,6 tt). |
| PNBE | |
 | 1H NMR (CCl4, 300 MHz): 5.26–5.37 (broad, 2H, H2,3), 2.76–3.10 (broad, s, 1H, H4), 2.26 (broad, s, 1H, H5), 1.80–2.10 (broad, m, 3H, H9), 1.10–1.60 (broad, m, 3H, H1,6,7); 13C NMR (CCl4, 75.4 MHz): 170.7 (s, C8), 140.9–127.4 (m, C2,3), 51.1 (s, C9), 48.6–34.8 ppm (s, C1,4,5,6,7). |
| PNBE-COOME | |
 | 1H NMR (CCl4, 300 MHz): 5.76 (broad, 1H, H8), 5.30 (broad, 2H, H2,3), 4.89–4.96 (broad, 2H, H9), 2.19–3.10 (broad, 3H, H1,4,5), 1.10–2.10 (broad, m, 2H, H6,7); 13C NMR (CCl4, 75.4 MHz): 141.7, 140.6 (s, C8), 135.6–130.3 (m, C2,3), 113.6, 113.1 (s, C9), 50.1, 47.9 (s, C5),45.6, 37.5 (s, C1), 45.6, 41.3 (s, C4), 42.8, 41.3, 39.5 (s, C7), 41.3 (s, C6). |
| PVNBE | |
 | 1H NMR (CDCl3, 300 MHz): 5.65 (s, 2H, H5,6 trans), 5.60 (s, 2H, H5,6 cis), 5.40 (m, 2H, H2,3 trans), 5.25 (m, 2H, H2,3 cis), 3.60 (m, 2H, H1,4 cis), 3.20 (m, 2H, H1,4 trans), 2.40 (m, 1H, H7), 1.27 ppm (m, 1H, H7); 13C NMR (CDCl3, 75.4MHz): 134.96 (s, C5,6 cc), 134.88 (s, C5,6 tt), 133.50 (s, C2,3 ct), 133.37 (s, C2,3 cc), 133.20 (s, C2,3 tt), 133.10 (s, C2,3 tc), 48.64 (s, C1,4 tc), 48.58 (s, C1,4 tt), 43.95 (s, C1,4 cc), 43.89 (s, C1,4 ct), 39.85 (s, C7 cc), 39.34 (s, C7 tc/ct), 38.80 ppm (s, C7 tt). |
| PNBD |
3. Results and Discussion
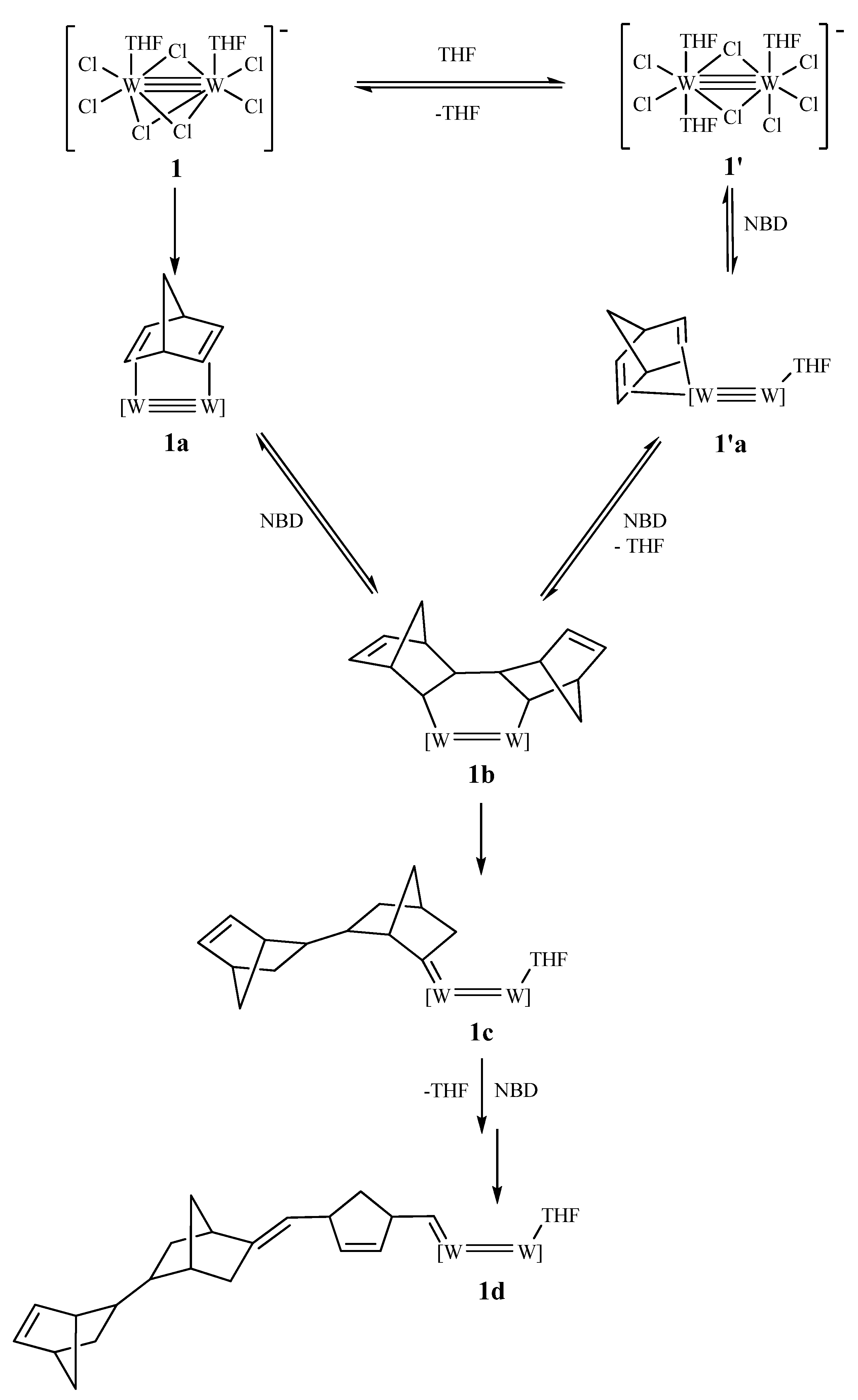
3.1. Catalyst and Polymerization Reactions
3.2. Polymerization of NBE and Derivatives
3.3. Polymerization of NBD
| Entry | Monomer | Solvent | t (h) | Yield (%) | Mw × 10−3 b | Mw/Mn | cis (%) |
|---|---|---|---|---|---|---|---|
| 1 | NBE | THF | 19 | 12 | 86.2 | 1.2 | 86 |
| 2 | CH2Cl2 | 1 | 96 | 529 | 1.2 | 86 | |
| 3 | dme | 48 | – c | – | – | – | |
| 4 | CH3CN | 48 | – c | – | – | – | |
| 5 | toluene | 24 | 37 | 296 | 2.9 | 86 | |
| 6 | Et2O | 20 | 94 | 422 | 1.4 | 86 | |
| 7 | CS2 | 24 | 93 | 1,174 | 1.4 | 80 | |
| 8 | NBE-COOH | CH2Cl2 | – | – c | – | – | – |
| 9 | NBE-OH | CH2Cl2 | – | – c | – | – | – |
| 10 | NBE-CN | CH2Cl2 | – | – c | – | – | – |
| 11 | NBE-COOMe | CH2Cl2 | 12 | > 99 | 685 | 1.15 | – |
| 12 | VNBE | CH2Cl2 | 8 | > 99 | 974 | 2.6 | – |
| 13 | NBD | – | 0.1 | >99 | – | – | – |
| 14 | THF | 4 | >99 | – | – | – | |
| 15 | toluene | 0.1 | >99 | – | – | – | |
| 16 | CH2Cl2 | 0.1 | >99 | – | – | – |
3.4. Mechanistic Considerations
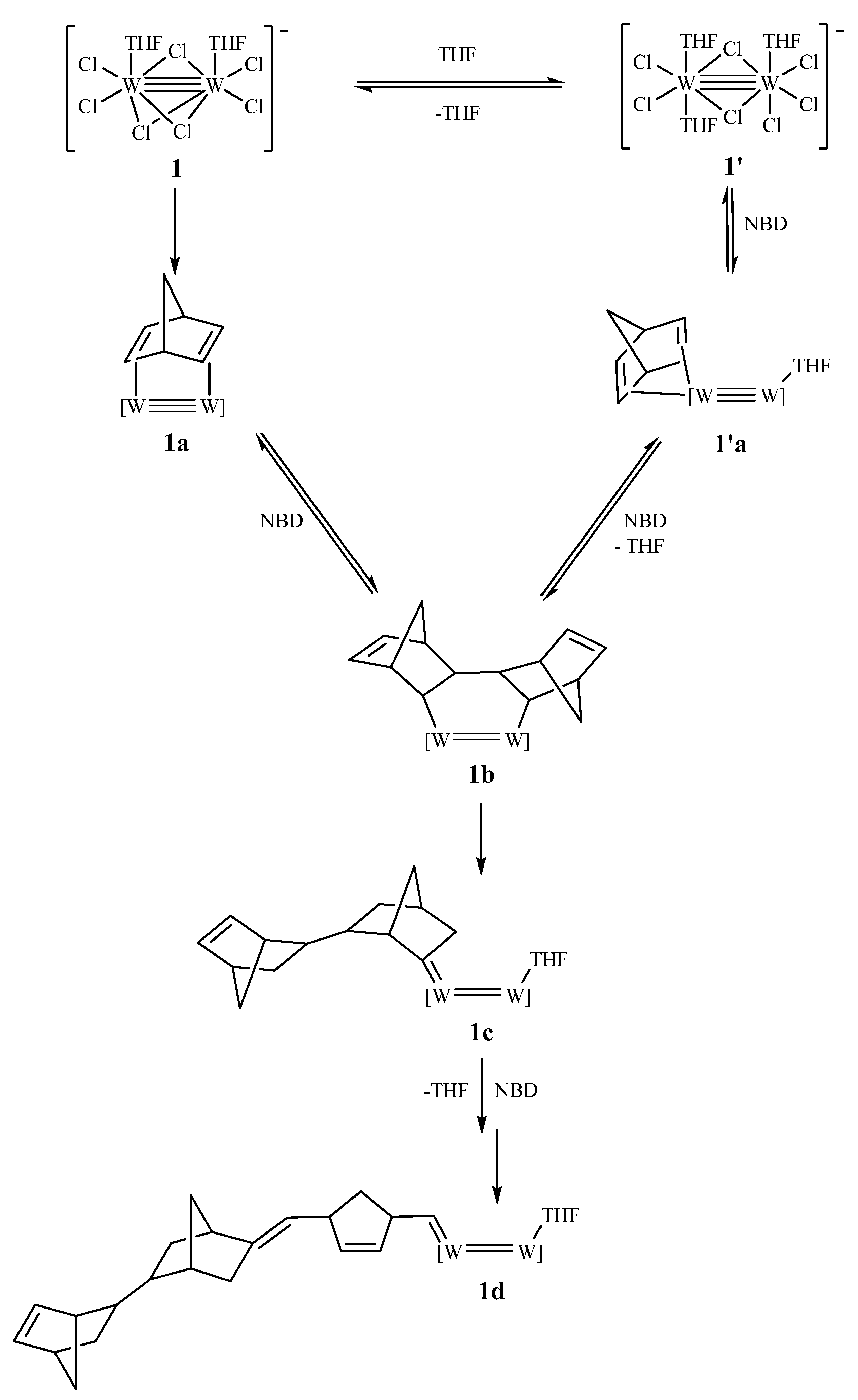
4. Conclusions
- (a) Complex 1 is an efficient homogeneous and/or heterogeneous unicomponent initiator for the ROMP of NBE, providing high molecular weight polymers in high yields and high stereoselectivity (86% cis for PNBE). Strongly coordinating pendant groups (–COOH, –OH, –CN) deactivate 1, softer ones do not affect its reactivity (–COOMe), whereas less strained double bonds remain unaffected (–CH=CH2). NBD reacts fast and quantitatively giving insoluble polymers.
- (b) The presence of two labile cis-THF ligands in 1 seems necessary for catalytic activity, as this derives from the inactivity of 2 or the very small activity of 3 under the same conditions.
- (c) In situ monitoring of the reactions (1/NBD or NBE) by 1H NMR allows the observation of the active alkylidenes of the propagating chains, but the in-depth mechanistic aspects of these reactions remain a cloudy landscape.
- (d) The reactivity of 1 for the ROMP of NBE is higher than that of the classical mononuclear initiators and derivatives thereof, resembling more to the bi- or multicomponent ones (e.g., WCl6/organometallic co-catalysts), and to the well-defined high-valent carbenes. Its cis-specificity is more enhanced than that of the above mentioned systems, and is comparable to that observed with the low-valent carbenes (Katz) and the high-valent stereoselective design catalysts (Schrock, Basset).
Acknowledgments
References
- Ivin, K.J.; Mol, J.C. Olefin Metathesis and Metathesis Polymerization; Academic Press, Inc.: San Diego, CA, USA, 1997. [Google Scholar]
- Dragutan, V.; Streck, R. Catalytic Polymerization of Cycloolefins. Ionic, Ziegler-Natta and Ring-Opening Metathesis Polymerization; Elsevier Science B.V.: Amsterdam, The Netherlands, 2000. [Google Scholar]
- Grubbs, R.H. Handbook of Metathesis; Wiley-VCH: Weinheim, Germany, 2003. [Google Scholar]
- Frenzel, U.; Müller, B.K.M.; Nuyken, O. Handbook of Polymer Synthesis, 2nd; Kricheldorf, H.R., Nuyken, O., Swift, G., Eds.; Marcel Dekker: New York, NY, USA, 2005. [Google Scholar]
- Buchmeiser, M.R. Homogeneous metathesis polymerization by well-defined group VI and group VIII transition-metal alkylidenes: Fundamentals and applications in the preparation of advanced materials. Chem. Rev. 2000, 100, 1565–1604. [Google Scholar]
- Buchmeiser, M.R. Ring-opening metathesis polymerization. In Synthesis of Polymers: New Structures and Methods, 1st; Schlüter, A.D., Hawker, C.J., Sakamoto, J., Eds.; Wiley-VCH: Weinheim, Germany, 2012. [Google Scholar]
- Mol, J.C. Industrial applications of olefin metathesis. J. Mol. Catal. A Chem. 2004, 213, 39–45. [Google Scholar]
- Madan, R.; Srivastava, A.; Anand, R.C.; Varma, I.K. Polymers derived from bicylo[2.2.1]heptene and its derivatives. Prog. Polym. Sci. 1998, 23, 621–663. [Google Scholar] [CrossRef]
- Szymańska-Buzar, T. Structure and reactivity of tungsten(II) and molybdenum(II) compounds containing an M–M' bond. Coord. Chem. Rev. 2005, 249, 2195–2202. [Google Scholar]
- Yamaguchi, Y.; Fujita, A.; Suzuki, N.; Ito, T. Metathesis polymerization of norbornene and terminal acetylenes catalyzed by bis(acetonitrile) complexes of molybdenum and tungsten. J. Mol. Catal. A Chem. 2005, 240, 226–232. [Google Scholar]
- Kozmer, V.A.; Poletayeva, I.A.; Yufa, T.L. Cycloolefin polymerization initiated by transition metal-π-allylic complexes. J. Polym. Sci. A 1972, 10, 251–258. [Google Scholar]
- Katz, T.J. Olefin metatheses and related reactions initiated by carbene derivatives of metals in low-oxidation states. Angew. Chem. Int. Ed. 2005, 44, 3010–3019. [Google Scholar] [CrossRef]
- Schrock, R.R. Recent advances in high oxidation state Mo and W imido alkylidene chemistry. Chem. Rev. 2009, 109, 3211–3226. [Google Scholar]
- Adams, R.D.; Cotton, F.A. Catalysis by Di- and Polynuclear Metal Cluster Complexes; Wiley VCH: New York, NY, USA, 1999. [Google Scholar]
- Brumaghim, J.L.; Girolami, G.S. Ring-opening metathesis polymerization of norbornene by Cp*2Os2Br4 and related compounds. Organometallics 1999, 18, 1923–1938. [Google Scholar]
- Schrock, R.R.; Lopez, L.P.H.; Hafer, J.; Singh, R.; Sinha, A.; Müller, P. Olefin metathesis reactions initiated by d2 molybdenum or tungsten complexes. Organometallics 2005, 24, 5211–5213. [Google Scholar] [CrossRef]
- Barry, J.T.; Chisholm, M.H. Selective hydrogenations of dienes and olefins by [W2(OCH2But)6(py)2]. J. Chem. Soc. Chem. Commun. 1995, 1599–1600. [Google Scholar]
- Chisholm, M.H. Ditungsten hexaalkoxides: Templates for organometallic chemistry and catalysis. J. Chem. Soc. Dalton Trans. 1996, 1781–1791. [Google Scholar]
- Schrock, R.R. Synthesis of stereoregular ROMP polymers using molybdenum and tungsten imido alkylidene initiators. Dalton Trans. 2011, 40, 7484–7495. [Google Scholar] [CrossRef]
- van der Schaaf, P.A.; Abbenhuis, R.A.T.M.; van der Noort, W.P.A.; de Graaf, R.; Grove, D.M.; Smeets, W.J.J.; Spekn, A.L.; van Koten, G. Tungsten(VI) phenylimido alkylidene complexes containing a monoanionic O,N-chelating ligand and their isolated precursor complexes: X-ray structures of W(CH2SiMe3)3(=NPh)[OCPh2(2-py)] and W(=CHSiMe3)(CH2SiMe3)(=NPh)[OCPh2(2-py)]. Organometallics 1994, 13, 1433–1444. [Google Scholar]
- Basset, J.-M.; Leconte, M.; Lefebvre, F.; Hamilton, J.G.; Rooney, J.J. Stereoselectivity in cyclic and acyclic metathesis reactions. Macromol. Chem. Phys. 1997, 198, 3499–3506. [Google Scholar]
- Mutch, A.; Leconte, M.; Lefebvre, F.; Basset , J.-M. Effect of alcohols and epoxides on the rate of ROMP of norbornene by a ruthenium trichloride catalyst. J. Mol. Catal. A Chem. 1998, 133, 191–199. [Google Scholar] [CrossRef]
- Olsson, O. Über die reduktion der wolframsäure und die tieferen oxydationsstufen des wolframs. I. Ber. Deutsch. Chem. Ges. 1913, 46, 566–582. [Google Scholar] [CrossRef]
- Saragas, N.; Floros, G.; Paraskevopoulou, P.; Psaroudakis, N.; Koinis, S.; Pitsikalis, M.; Mertis, K. Polymerization of terminal alkynes with a triply bonded ditungsten halo-complex. J. Mol. Catal. A Chem. 2009, 303, 124–131. [Google Scholar]
- Chisholm, M.H.; Eichhorn, B.W.; Folting, K.; Huffman, J.C.; Ontiveros, C.D.; Streib, W.E.; van der Sluys, W.G. Preparation and characterization of NaW2Cl7(THF)5. A synthetically useful precursor for X3W≡WX3 compounds where X = CH2-t-Bu, NMe2, and O-t-Bu. Inorg. Chem. 1987, 26, 3182–3186. [Google Scholar] [CrossRef]
- Paraskevopoulou, P.; Petalidou, E.; Panas, A.; Ioannou, M.; Koinis, S.; Psaroudakis, N.; Leventis, N.; Stavropoulos, P.; Mertis, K. Redox reactivity and comprehensive synthetic chemistry of the perchloroditungstate [W2(µ-Cl)3Cl6]n− (n = 3, 2, 1) anions in organic media. Polyhedron 2008, 27, 2859–2866. [Google Scholar] [CrossRef]
- Bergs, D.J.; Chisholm, M.H.; Folting, K.; Huffman, J.C.; Stahl, K.A. Preparation and characterization of the heptachlorobis(tetrahydrofuran)ditungstate and octachloro(tetrahydrofuran)ditungstate anions: [Ph4P][W2Cl7(THF)2] and [Ph4P][W2Cl8(THF)]. Inorg. Chem. 1988, 27, 2950–1954. [Google Scholar] [CrossRef]
- Düz, B.; Elbistan, C.K.; Ece, A.; Sevin, F. Application of carbon arc-generated Mo- and W-based catalyst systems to the ROMP of norbornene. Appl. Organometal. Chem. 2009, 23, 359–364. [Google Scholar]
- Bell, B.; Hamilton, J.G.; Law, E.E.; Rooney, J.J. A one-pot synthesis of comb polymers and hydrogels by the ring-opening metathesis polymerization reaction. Macromol. Rapid Commun. 1994, 15, 543–550. [Google Scholar]
- Vogel, N.; Théato, P. Controlled synthesis of reactive polymeric architectures using 5-norbornene-2-carboxylic acid pentafluorophenyl ester. Macromol. Symp. 2007, 249–250, 383–391. [Google Scholar]
- Gorski, M.; Szymańska-Buzar, T. Tungsten(II)-initiated ring-opening metathesis polymerization and other C–C bond forming reactions of 5-vinyl-2-norbornene. J. Mol. Cat. A Chem. 2006, 257, 41–47. [Google Scholar]
- Bell, B.; Hamilton, J.G.; Mackey, O.N.D.; Rooney, J.J. Microstructure of ring-opened polymers and copolymers of norbomadiene. J. Mol. Catal. 1992, 77, 61–73. [Google Scholar] [CrossRef]
- Cotton, F.A.; Walton, R.A. Multiple Bonds between Metal Atoms, 3rd ed; Springer: New York, NY, USA, 2005. [Google Scholar]
- Bencze, L.; Kraut-Vass, A.; Prókai, L. Mechanism of initiation of the metathesis of norbornene using W(CO)3Cl2(AsPh3)2 as catalyst. J. Chem. Soc. Chem. Comm. 1985, 911–912. [Google Scholar]
- Czeluśniak, I.; Szymańska-Buzar, T. Ring-opening metathesis polymerization of norbornene and norbornadiene by tungsten(II) and molybdenum(II) complexes. J. Mol. Catal. A Chem. 2002, 190, 131–143. [Google Scholar]
- Bencze, L.; Szalai, G.; Overton, T.L. Investigation of the catalytic properties of the thermally activated dichloro-tetracarbonyl-tungsten in olefin metathesis reaction. Inorg. Chim. Acta 1997, 254, 5–7. [Google Scholar] [CrossRef]
- Bencze, L.; Biró, N.; Szabó-Ravasz, B.; Mihichuk, L. Chemical transformations of cis-W(CO)4(C5H5N)2 in the ring-opening metathesis polymerization of norbornene. Can. J. Chem. 2004, 82, 499–503. [Google Scholar] [CrossRef]
- Farona, M.F.; Tucker, R.L. Studies on the mechanism of olefin metathesis promoted by a heterogeneous catalyst. J. Mol. Catal. 1980, 8, 85–90. [Google Scholar]
- Szymańska-Buzar, T.; Głowiak, T.; Czeluśniak, I. The initiation of ring-opening metathesis polymerisation of norbornadiene by seven-coordinate molybdenum(II) compounds. X-ray crystal structure of [Mo(µ-Cl)(SnCl3)(CO)3(η4-NBD)]. J. Organomet. Chem. 2001, 640, 72–78. [Google Scholar]
- Szymańska-Buzar, T.; Głowiak, T.; Czeluśniak, I. Reactivity of [Mo(µ-Cl)(SnCl3)(CO)3(NCMe)2] towards norbornadiene. X-ray crystal structure of [Mo(µ-Cl)(SnCl3)(CO)2(η4-C7H8)(NCMe)]. Polyhedron 2002, 21, 2505–2513. [Google Scholar] [CrossRef]
- Veige, A.S.; Wolczanski, P.T.; Lobkovsky, E.B. Dehydrogenation of [{(silox)3Nb}2(η-1,2;η-5,6-C8H8)] (silox = tBu3SiO) to [{(silox)3Nb}2(η-1,2;η-5,6-C8H6)] and its subsequent alkene-to-alkylidene rearrangement. Angew. Chem. Int. Ed. 2001, 40, 3629–3632. [Google Scholar] [CrossRef]
- Handzlik, J.; Stosur, M.; Kochel, A.; Szymańska-Buzar, T. Norbornadiene complexes of molybdenum(II) and their transformation to a catalyst for ring-opening metathesis polymerization: DFT calculations—X-ray crystal structure of a new norbornadiene complex [MoCl(GeCl3)(CO)3(η4-nbd)]. Inorg. Chim. Acta 2008, 361, 502–512. [Google Scholar]
- East, A.L.L.; Berner, G.M.; Morcom, A.D.; Mihichuk, L.M. Computational study of tungsten(II)-catalyzed rearrangements of norbornadiene. J. Chem. Theory Comput. 2008, 4, 1274–1282. [Google Scholar] [CrossRef]
- Jayaraman, A.; Berner, G.M.; Mihichuk, L.M.; East, A.L.L. Tungsten(II)-catalyzed rearrangements of norbornadiene: effects of alternative complexation stages. J. Mol. Catal. A Chem. 2011, 351, 143–153. [Google Scholar]
- Malinowska, A.; Czeluśniak, I.; Górski, M.; Szymańska-Buzar, T. A novel catalytic route to 2-bicyclo[2.2.1]hept-2-ylidenebicyclo[2.2.1]-heptane involving C–H bond activation of bicyclo[2.2.1]hept-2-ene. J. Mol. Catal. A Chem. 2005, 226, 259–262. [Google Scholar] [CrossRef]
- Górski, M.; Kochel, A.; Szymańska-Buzar, T. Photochemical reaction of W(CO)6 with GeCl4 as a source of germyl and germylene compounds acting as initiators for ring-opening metathesis polymerization of norbornene. J. Organomet. Chem. 2006, 691, 3708–3714. [Google Scholar]
- Malinowska, A.; Kochel, A.; Szymańska-Buzar, T. An anionic binuclear complex of tungsten(II), [(µ-Cl)3{W(SnCl3)(CO)3}2]−, and its reactivity towards norbornene. J. Organomet. Chem. 2007, 692, 3994–3999. [Google Scholar] [CrossRef]
- Kress, J.; Osborn, J.A.; Greene, R.M.E.; Ivin, K.J.; Rooney, J.J. The detection of “living” propagating tungsten-carbene complexes in the ring-opening polymerization of bicycloalkenes. J. Chem. Soc. Chem. Commun. 1985, 874–876. [Google Scholar]
- Percec, V.; Künzler, J. Monitoring the WCI6/(CH3)4Sn initiated polymerization of substituted acetylenes by 1H-NMR spectroscopy. Polym. Bull. 1991, 25, 483–490. [Google Scholar]
- Dzik, W.I.; Zhang, X.P.; de Bruin, B. Redox noninnocence of carbene ligands: Carbene radicals in (catalytic) C–C bond formation. Inorg. Chem. 2011, 50, 9896–9903. [Google Scholar]
- Kerby, M.C.; Eichhorn, B.W.; Doviken, L.; Vollhardt, K.P.C. Activated molybdenum–molybdenum quadruple bonds. 2. First example of alkyne additions to metal–metal quadruple bonds. Inorg. Chem. 1991, 30, 156–158. [Google Scholar]
- Byrnes, M.J.; Chisholm, M.H.; Gallucci, J.; Wilson, P.J. lkyne adducts of ditungsten tetrapivalate: [W2(κ2-O2CtBu)4(µ-RCCR')2], where R = R' = Me, Et, Ph and R = Me, R' = Ph. Organometallics 2002, 21, 2240–2247. [Google Scholar] [CrossRef]
- Bott, S.G.; Clark, D.L.; Green, M.L.H.; Mountford, P. Chloride-supported tungsten alkyne complexes. Synthesis, electronic and molecular structures of [W2Cl4(µ-Cl)2(µ-C2R2)(thf)2](R = H or Me) and [WCl4(η-C2Me2)(thf)](thf = tetrahydrofuran). J. Chem. Soc. Dalton Trans. 1991, 471–481. [Google Scholar]
- Chisholm, M.H.; Huffman, J.C.; Hampden-Smith, M.J. Metal alkoxides. Models for metal oxides. 15. Carbon–carbon and carbon–hydrogen bond activation in the reactions between ethylene and ditungsten hexaalkoxides: W2(OCH2-t-Bu)6(η2-C2H4)2, W2(OR)6(CH2)4(η2-C2H4), and W2(OR)6(µ-CCH2CH2CH2) (where R = CH2-t-Bu, i-Pr, c-C5H9, and c-C6H11). Preparations, properties, structures, and reaction mechanisms. J. Am. Chem. Soc. 1989, 111, 5284–5299. [Google Scholar] [CrossRef]
- Chisholm, M.H.; Streib, W.E.; Tiedtke, D.B.; Wu, D.-D. Organometallic chemistry of [W2(OCH2tBu)8] (M=M): substrate uptake and activation at a tungsten–tungsten double bond. Chem. Eur. J. 1998, 4, 1470–1479. [Google Scholar] [CrossRef]
- Lopez, L.P.H.; Schrock, R.R.; Müller, P. Dimers that contain unbridged W(IV)/W(IV) double bonds. Organometallics 2006, 25, 1978–1986. [Google Scholar]
- Barry, J.T.; Bollinger, J.C.; Chisholm, M.H.; Glasgow, K.C.; Huffman, J.C.; Lucas, E.A.; Lubkovsky, E.B.; Streib, W.E. Preparation and characterization of 1,3-butadiene and isoprene complexes, W2(OCH2tBu)6(diene)(py), and studies of the selective hydrogenation of 1,3-dienes. Organometallics 1999, 18, 2300–2308. [Google Scholar]
- Chisholm, M.H.; Rankel, L.A.; Bailey, W.I., Jr.; Cotton, F.A.; Murillo, C.A. µ-Allene-bis(cyclopentadieny1)tetracarbonyl-dimolybdenum; a bridging allene ligand. J. Am. Chem. Soc. 1977, 99, 1261–1262. [Google Scholar]
- Bailey, W.I., Jr.; Chisholm, M.H.; Cotton, F.A.; Murillo, C.A.; Rankel, L.A. Reactions of metal-to-metal multiple bonds. 1. µ-Allene-bis(cyclopentadienyl)tetracarbonyldimolybdenum and -ditungsten compounds. Preparation, properties, and structural characterization. J. Am. Chem. Soc. 1978, 100, 802–807. [Google Scholar]
- Cayton, R.H.; Chisholm, M.H.; Hampden-Smith, M.J. An allene adduct of ditungsten hexa-tert-butoxide: prediction of a stabilized µ-η3-CH2CCH2 moiety. J. Am. Chem. Soc. 1988, 110, 4438–4440. [Google Scholar] [CrossRef]
- Crabtree, R.H. The Organometallic Chemistry of the Transition Metals, 4th ed; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2005. [Google Scholar]
- Kolesnichenko, V.; Swenson, D.C.; Messerle, L. Facile reduction of tungsten halides with nonconventional, mild reductants. I. Tungsten tetrachloride: several convenient solid-state syntheses, a solution synthesis of highly reactive (WCl4)x, and the molecular structure of polymeric tungsten tetrachloride. Inorg. Chem. 1998, 37, 3257–3262. [Google Scholar]
- Budzichowski, T.A.; Chisholm, M.H.; Folting, K.; Huffman, J.C.; Streib, W.E. Dimetal hepta- and octaalkoxide anions of molybdenum and tungsten, M2(OR)7− and M2(OR)82− (M≡M). Preparation, structures, oxidation, and a study of the thermal decomposition of M2(OR)7− to give W2(H)(O)(OR)6− where R = tBu and iPr. J. Am. Chem. Soc. 1995, 117, 7428–7440. [Google Scholar]
- Aime, S.; Arce, A.J.; Chiantore, O.; Gobetto, R.; Russo, A.; de Sanctis, Y. Ring-opening polymerisation of norbornene by (µ-H)2Os3(CO)10 complex. J. Organomet. Chem. 2001, 622, 43–46. [Google Scholar]
- Levisalles, J.; Rose-Munch, F.; Rudler, H.; Daran, J.-C.; Dromzée, Y.; Jeannin, Y. Dinuclear carbene complexes of tungsten as catalysts for metathetical polymerisation of alkenes and alkynes; X-ray crystal structure of the insertion product of but-2-yne into a µ-alkylidene carbon–tungsten bond. J. Chem. Soc. Chem. Commun. 1981, 152–154. [Google Scholar]
- Knox, S.A.R. 25 years with ruthenium. J. Organomet. Chem. 1990, 400, 255–272. [Google Scholar]
- Abraham, R.J.; Barlow, A.P.; Rowan, A.E. Substituent chemical shifts in NMR. Part 4—1H SCS in some 2-substituted norbornanes and bornanes. Magn. Res. Chem. 1989, 27, 1074–1084. [Google Scholar] [CrossRef]
- Dragutan, V.; Dragutan, I.; Dimonie, M. A Selective Route for Synthesis of Linear Polydicyclopentadiene. In Green Metathesis Chemistry: Great Challenges in Synthesis, Catalysis And Nanotechnology; Dragutan, V., Demonceau, A., Dragutan, I., Finkelshtein, E.S., Eds.; Springer: Dordrecht, The Netherlands, 2009. [Google Scholar]
- Bokaris, E.P.; Kosmas, M.M. All cis-poly(NBE) derived by the ROMP catalysts based on WCl6. J. Mol. Catal. A Chem. 2003, 192, 263–273. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Floros, G.; Saragas, N.; Paraskevopoulou, P.; Psaroudakis, N.; Koinis, S.; Pitsikalis, M.; Hadjichristidis, N.; Mertis, K. Ring Opening Metathesis Polymerization of Norbornene and Derivatives by the Triply Bonded Ditungsten Complex Na[W2(µ-Cl)3Cl4(THF)2]·(THF)3. Polymers 2012, 4, 1657-1673. https://doi.org/10.3390/polym4041657
Floros G, Saragas N, Paraskevopoulou P, Psaroudakis N, Koinis S, Pitsikalis M, Hadjichristidis N, Mertis K. Ring Opening Metathesis Polymerization of Norbornene and Derivatives by the Triply Bonded Ditungsten Complex Na[W2(µ-Cl)3Cl4(THF)2]·(THF)3. Polymers. 2012; 4(4):1657-1673. https://doi.org/10.3390/polym4041657
Chicago/Turabian StyleFloros, Georgios, Nikolaos Saragas, Patrina Paraskevopoulou, Nikolaos Psaroudakis, Spyros Koinis, Marinos Pitsikalis, Nikos Hadjichristidis, and Konstantinos Mertis. 2012. "Ring Opening Metathesis Polymerization of Norbornene and Derivatives by the Triply Bonded Ditungsten Complex Na[W2(µ-Cl)3Cl4(THF)2]·(THF)3" Polymers 4, no. 4: 1657-1673. https://doi.org/10.3390/polym4041657
APA StyleFloros, G., Saragas, N., Paraskevopoulou, P., Psaroudakis, N., Koinis, S., Pitsikalis, M., Hadjichristidis, N., & Mertis, K. (2012). Ring Opening Metathesis Polymerization of Norbornene and Derivatives by the Triply Bonded Ditungsten Complex Na[W2(µ-Cl)3Cl4(THF)2]·(THF)3. Polymers, 4(4), 1657-1673. https://doi.org/10.3390/polym4041657