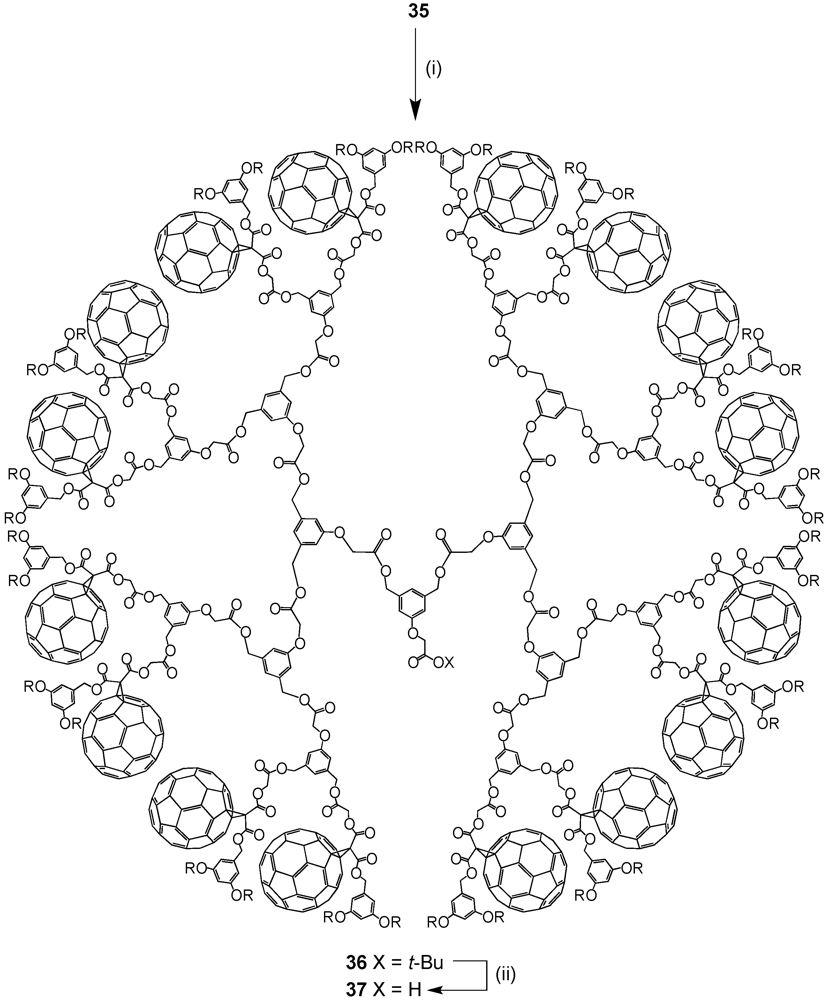
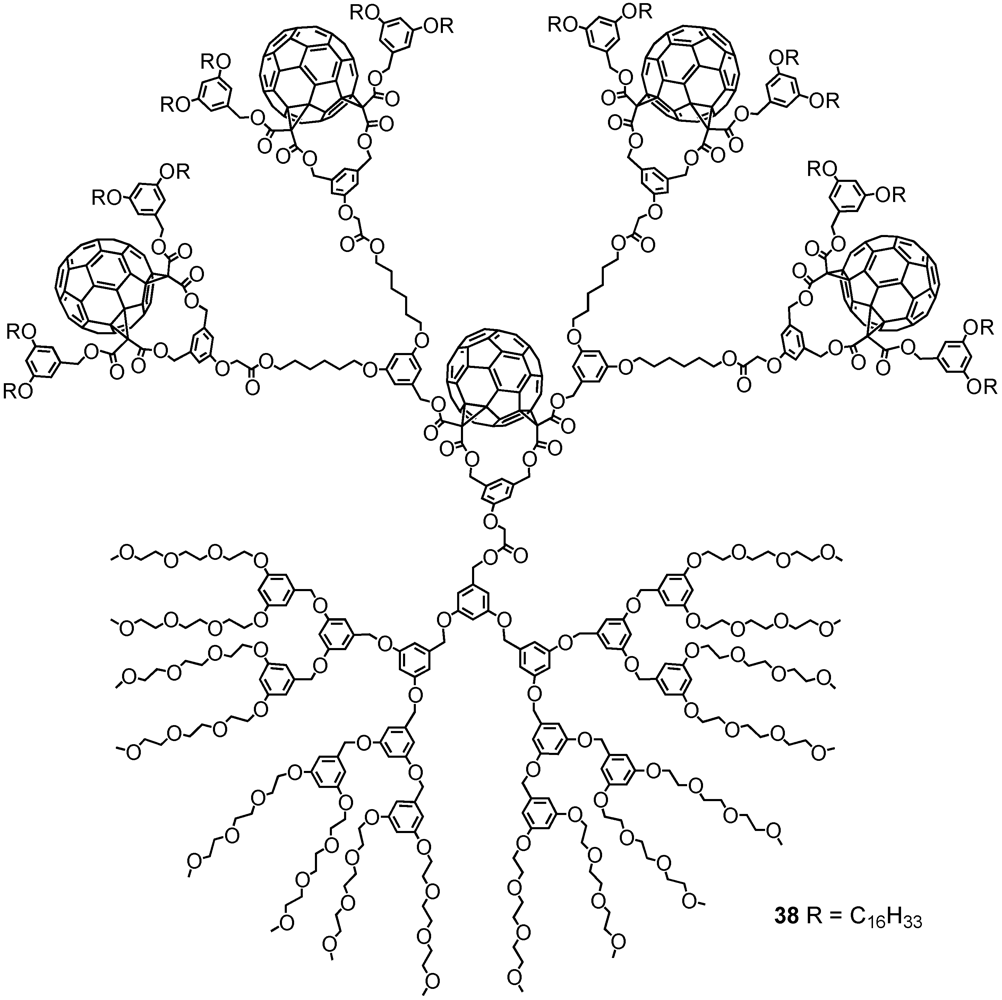
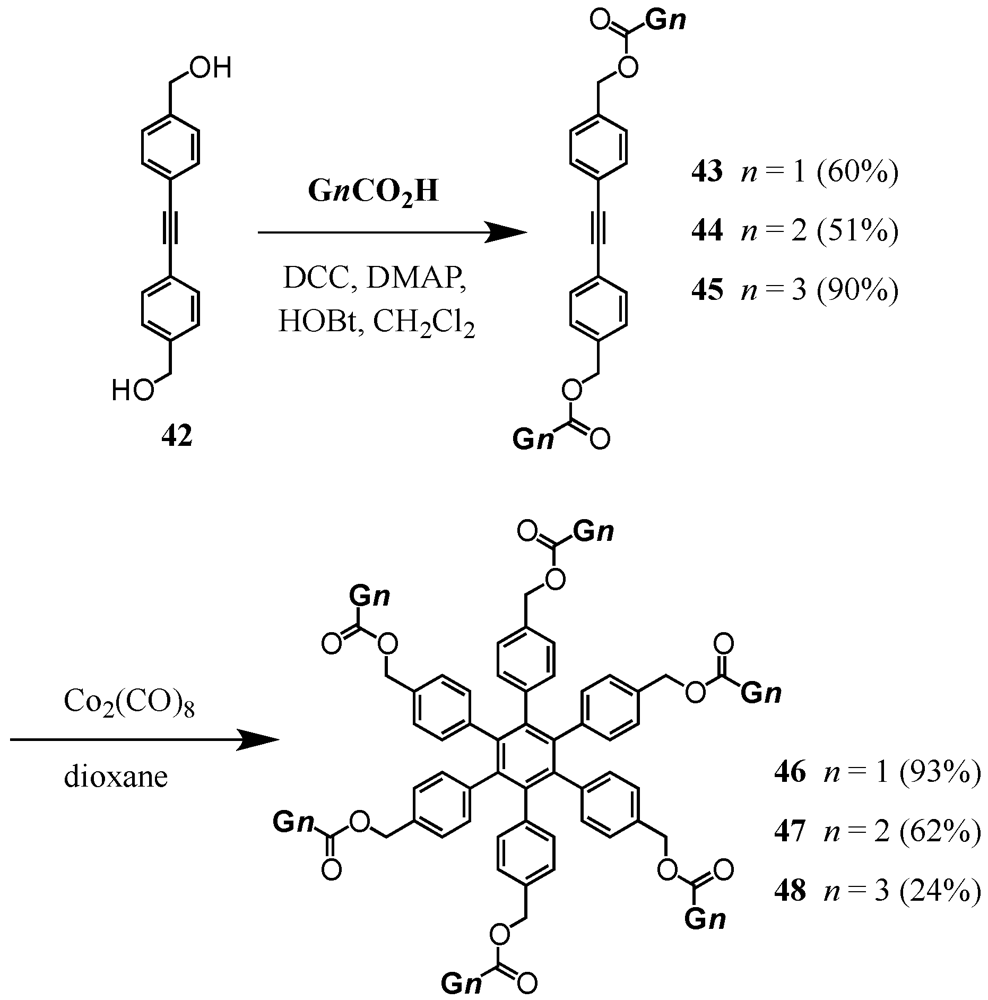
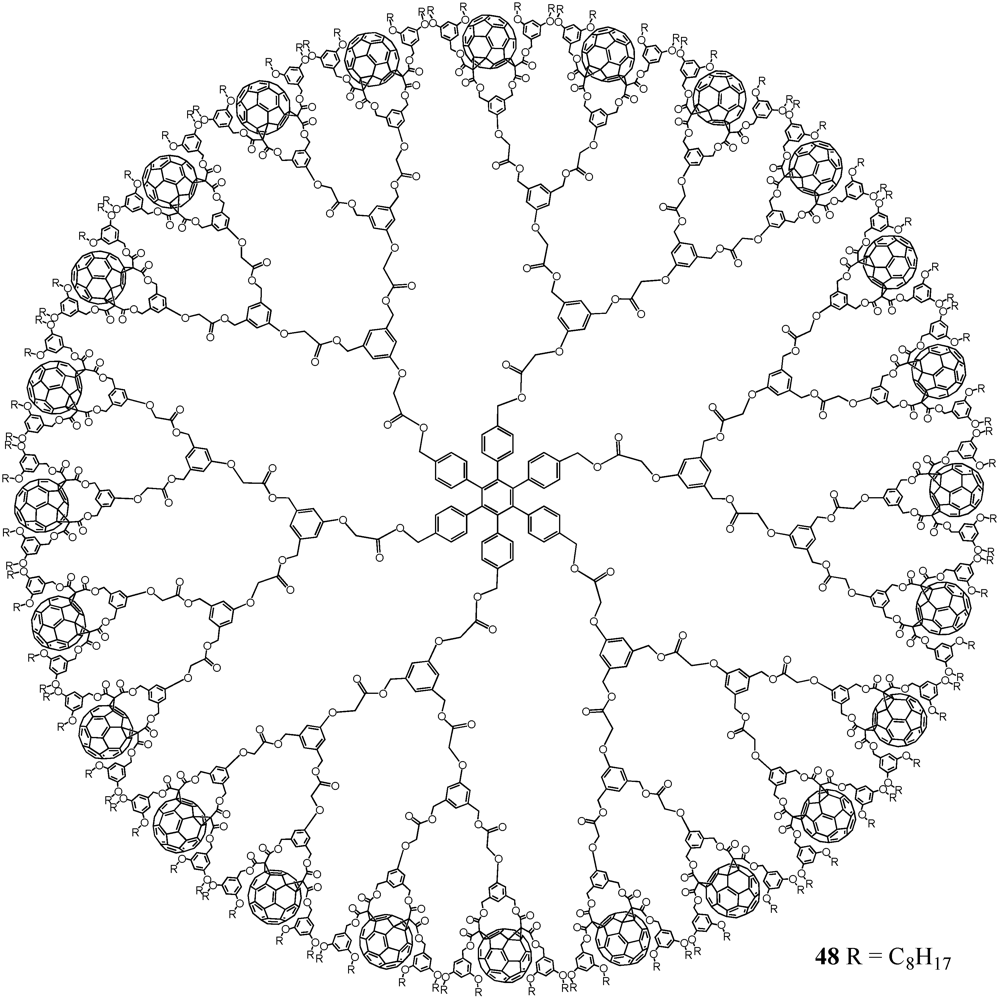
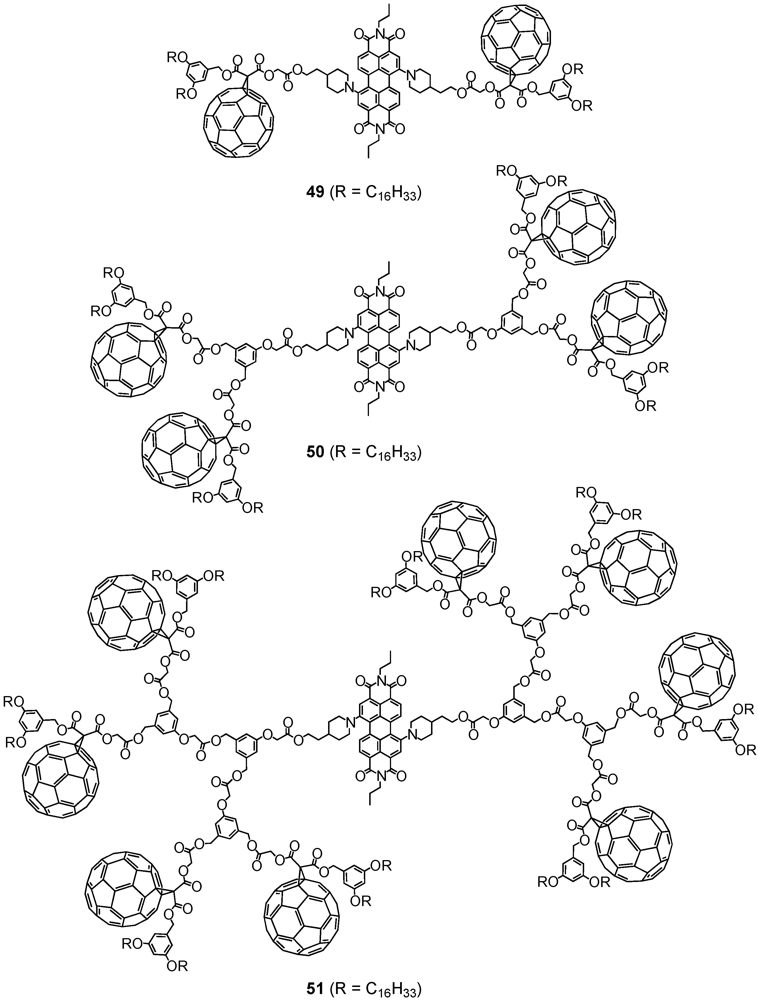
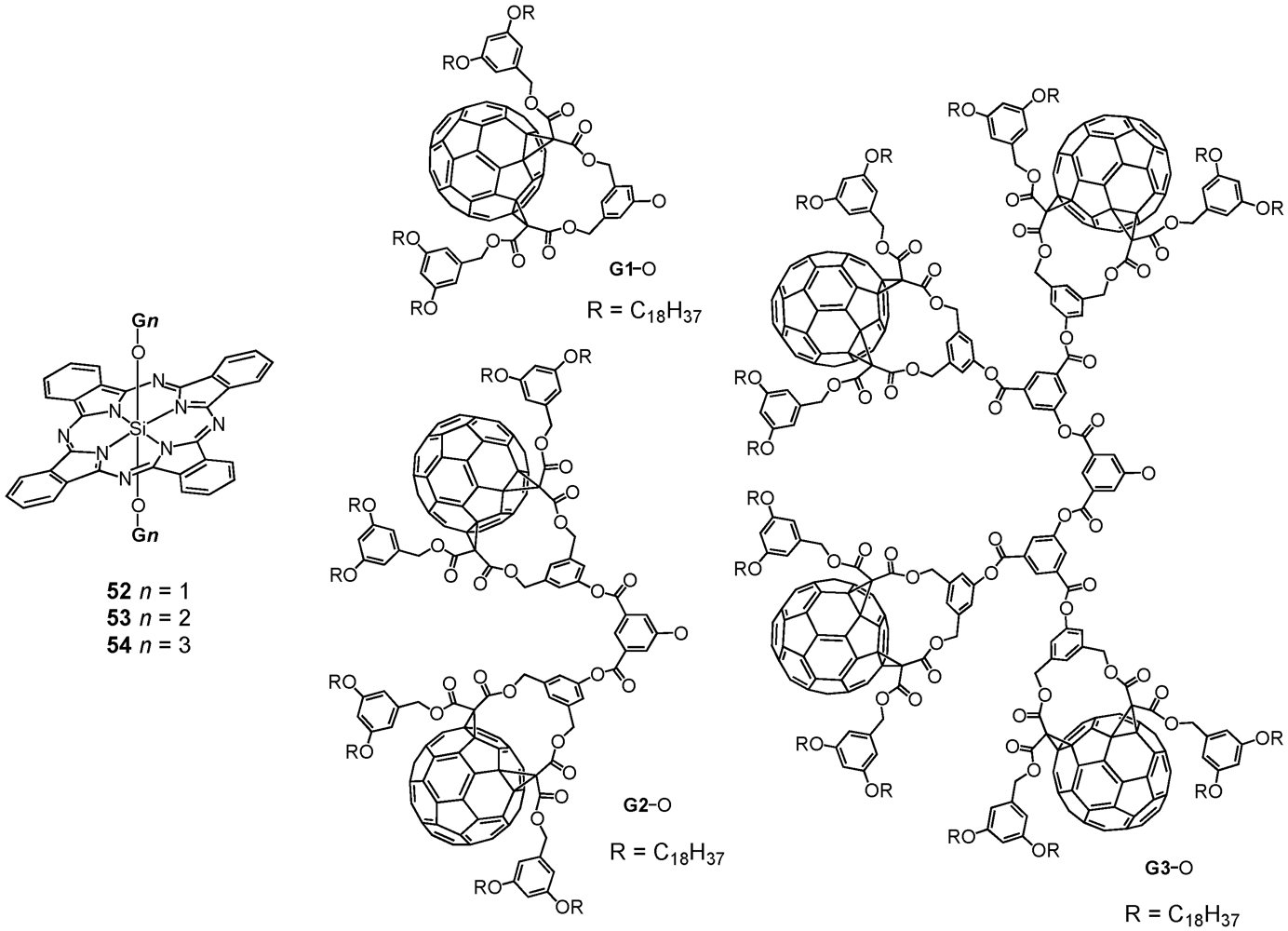
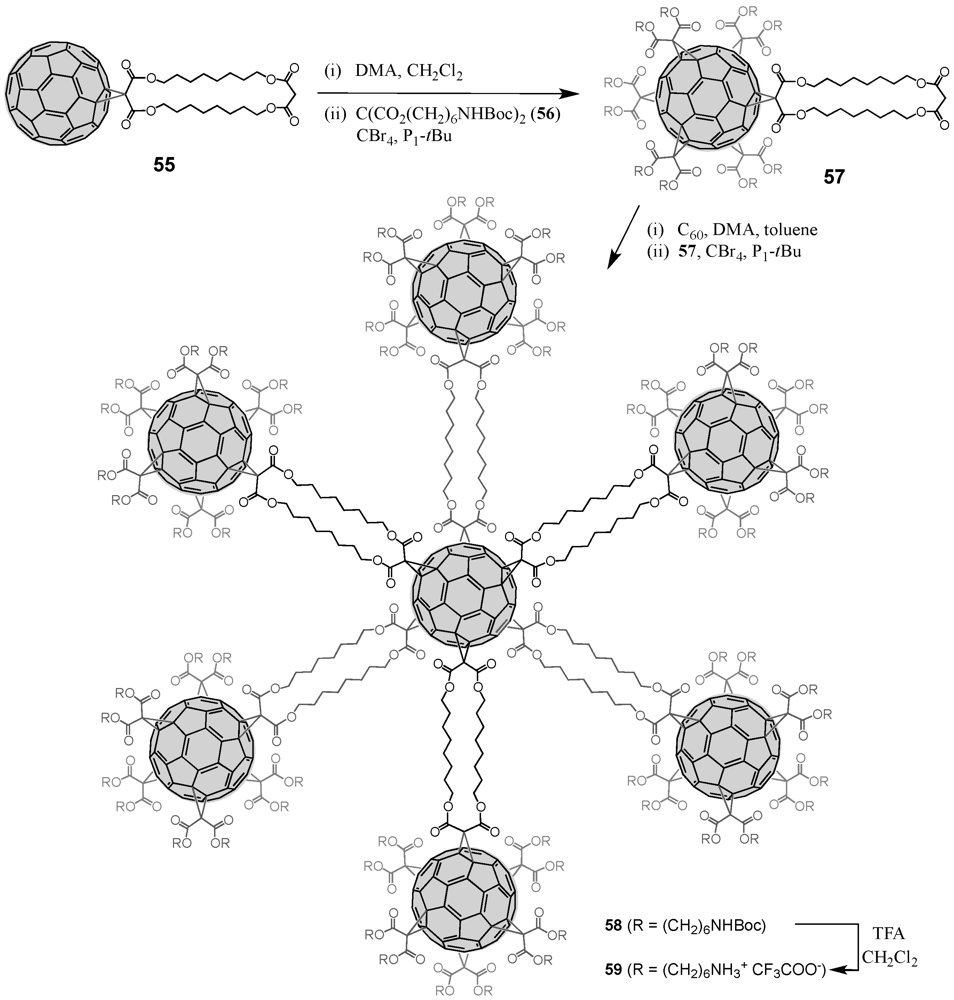
2. Divergent Synthesis
The general principle of a dendrimer synthesis according to the divergent method proceeds stepwise from a multi-functionalized core building block, to whose reactive coupling sites are attached new branching units in the form of dendritic branches via a reactive terminal functionality [
2,
3,
4]. Activation through for instance deprotection regenerates new reactive coupling sites for further branching units under creation of a new dendrimer generation with each branching unit. The iterative synthetic sequence progressively yields higher generations and permits the dendrimer to grow from the inside outwards. The divergent method makes attainable high-molecular nano-architectures, while at the same time, the exponential growth of the corresponding dendrimer structure may lead to structural defects since complete functionalization cannot always be ensured. Likewise, purification of structurally perfect from defective dendrimers often becomes tedious or almost impossible resulting from their very similar properties.
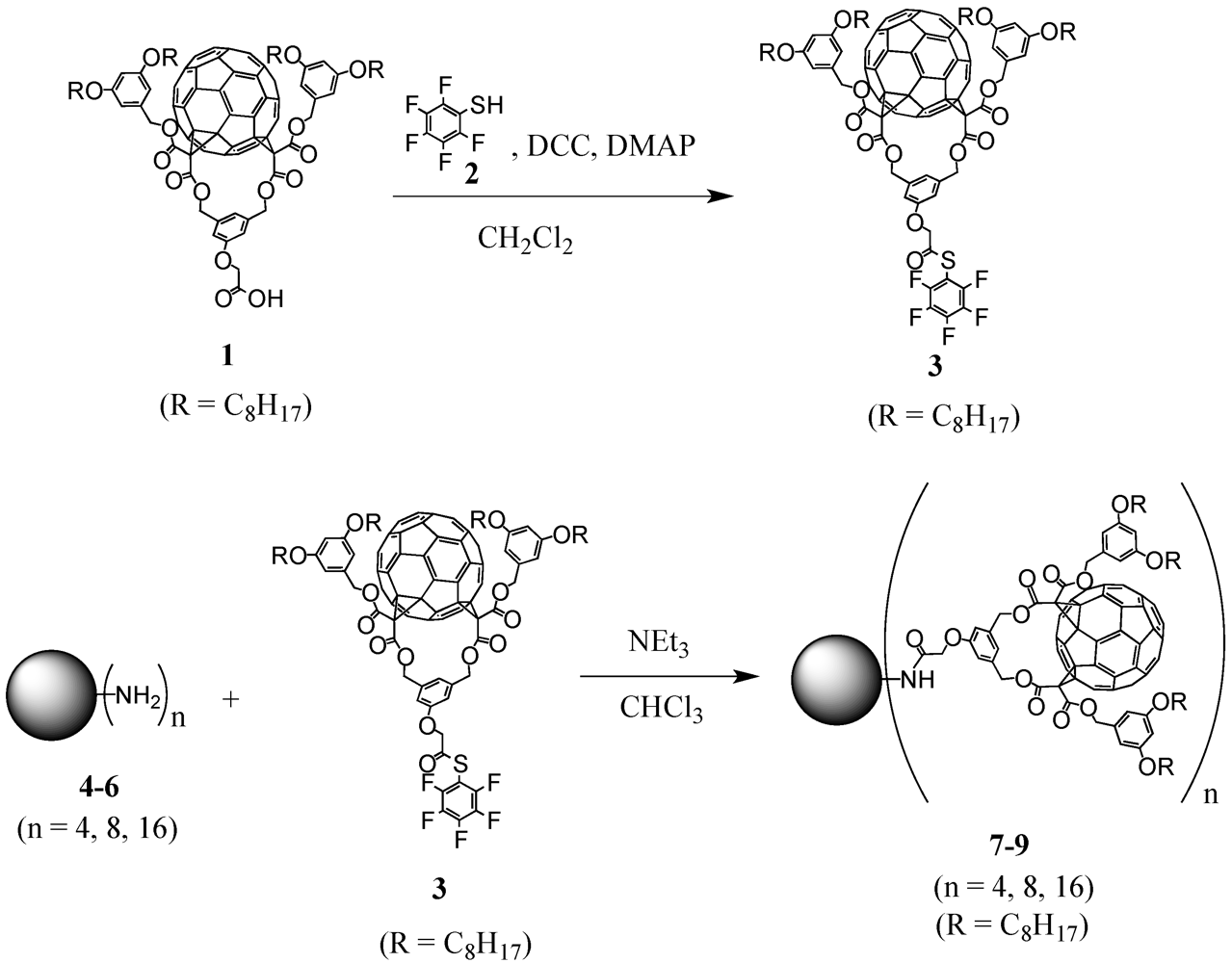
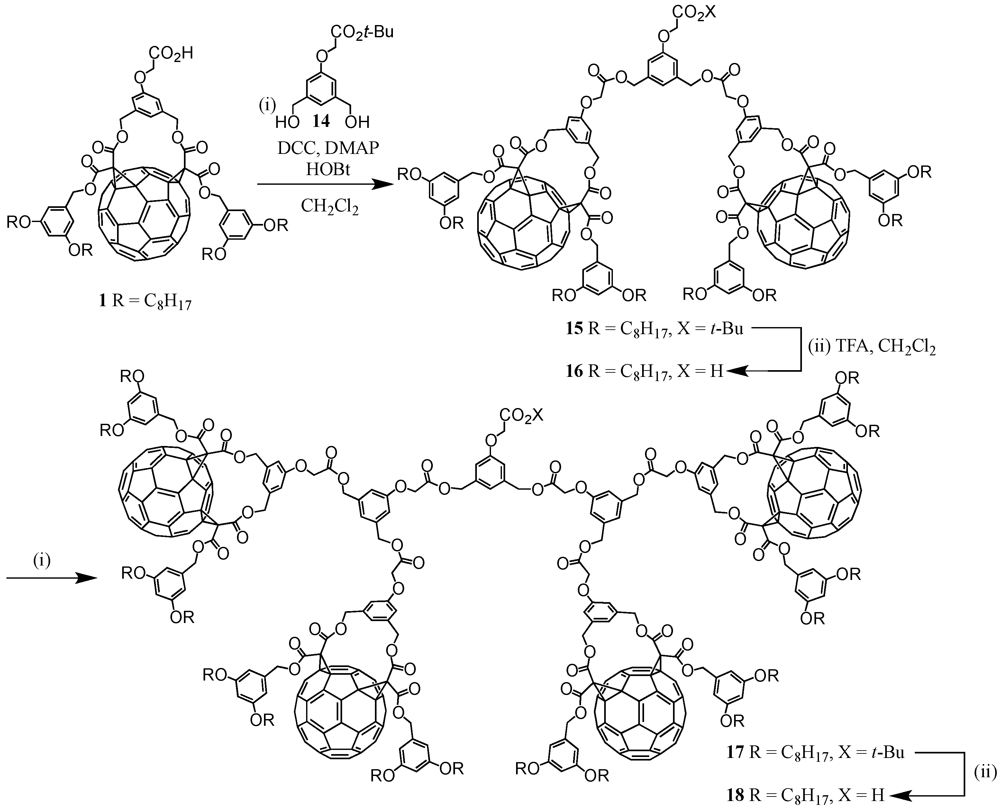
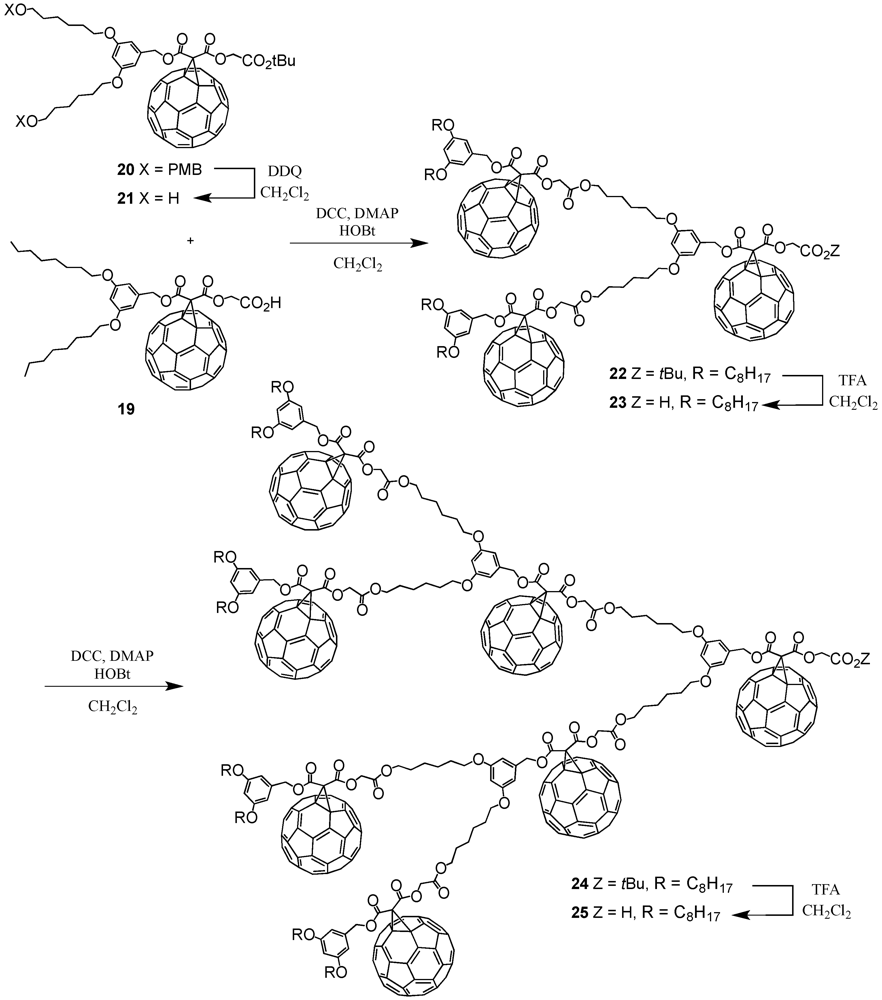
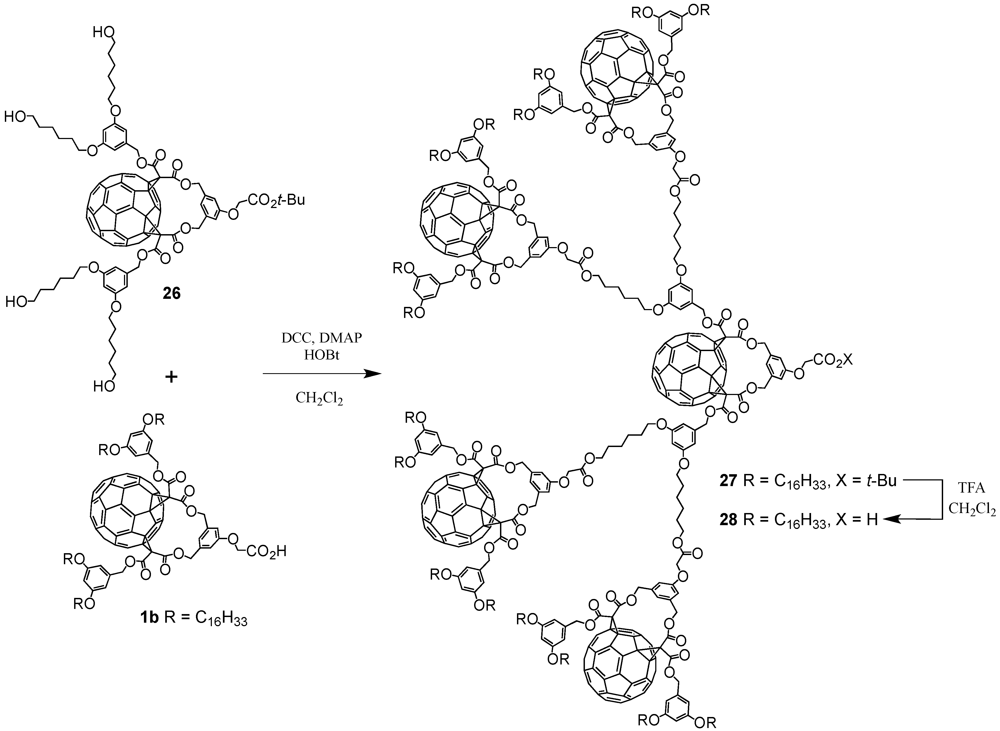
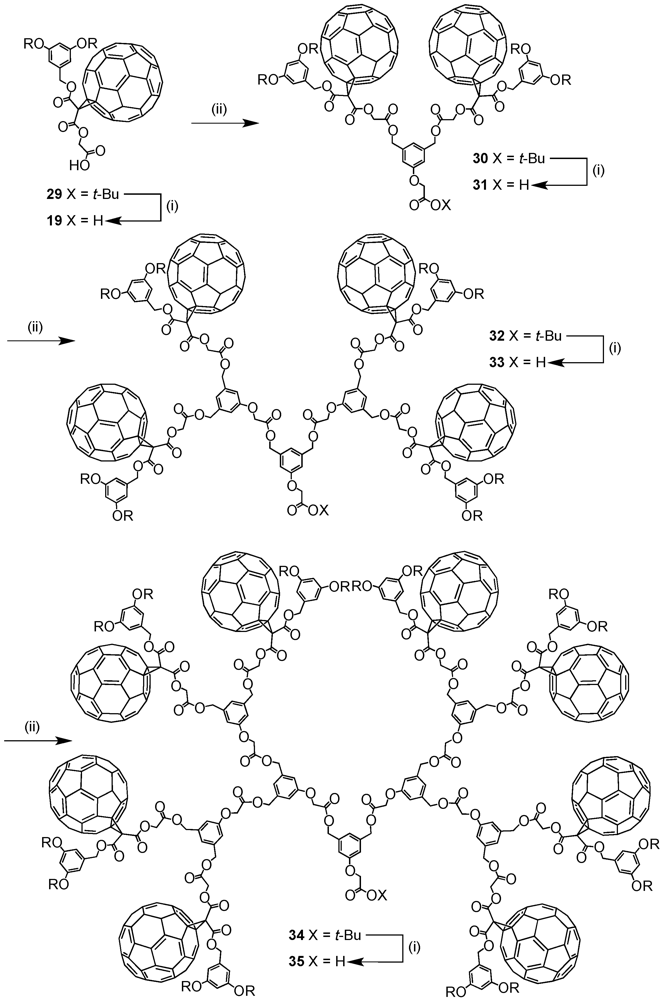
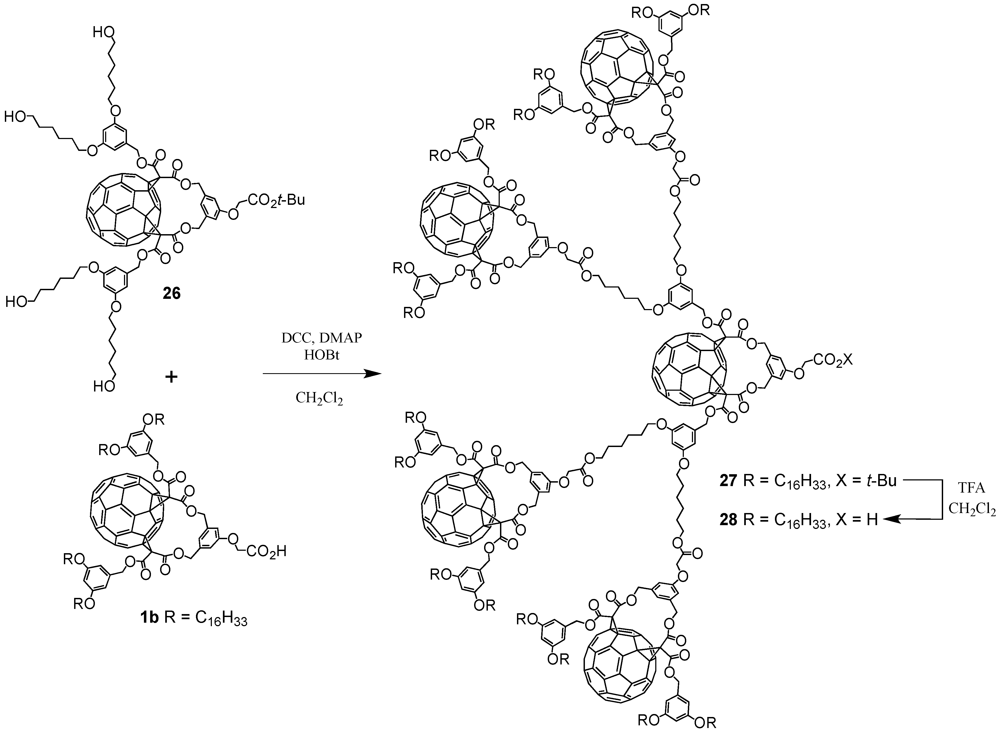
One of the key compounds in the preparation of fullerene-rich dendrimers has been
Cs-symmetrical fullerene bis-adduct
1 (
Scheme I). This functionalized fullerene derivative was obtained in ten steps according to a previously reported procedure [
13]. Briefly described, it involved the synthesis of an A
2B building block with two benzylic alcohol functions and a
t-butyl-protected carboxylic acid function starting from dimethyl 5-hydroxyisophthalate. In contrast, 5-(hydroxymethyl)benzene-1,3-diol was reacted with long alkyl chains followed by the reaction of the residual alcohol moiety with
Meldrums’ acid. Fusion of the two precursors gave a bismalonate which was reacted under classical Bingel conditions with C
60, I
2 and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) to afford carboxylic acid
1 after treatment with TFA. Importantly, the well-established strategy via 1,3-phenylenebis(methylene)-tethered bis-malonates produced regioselectively the
cis-2 addition pattern at C
60 [
14]. Acid
1 has then been engaged in the preparation of well-defined fullerene-rich nanostructures as obtained by the divergent approach.
Scheme I.
Synthesis of activated ester 1 and grafting to polypropylene imine (PPI) dendrimers 4-6 under the use of the divergent synthetic protocol.
Scheme I.
Synthesis of activated ester 1 and grafting to polypropylene imine (PPI) dendrimers 4-6 under the use of the divergent synthetic protocol.
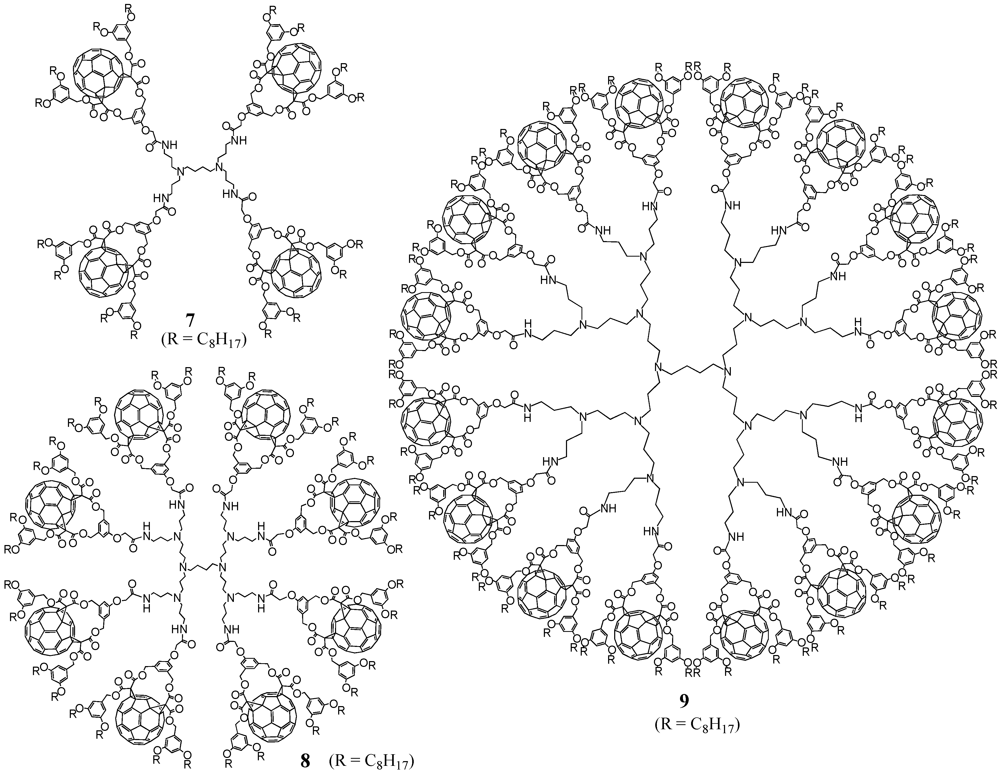
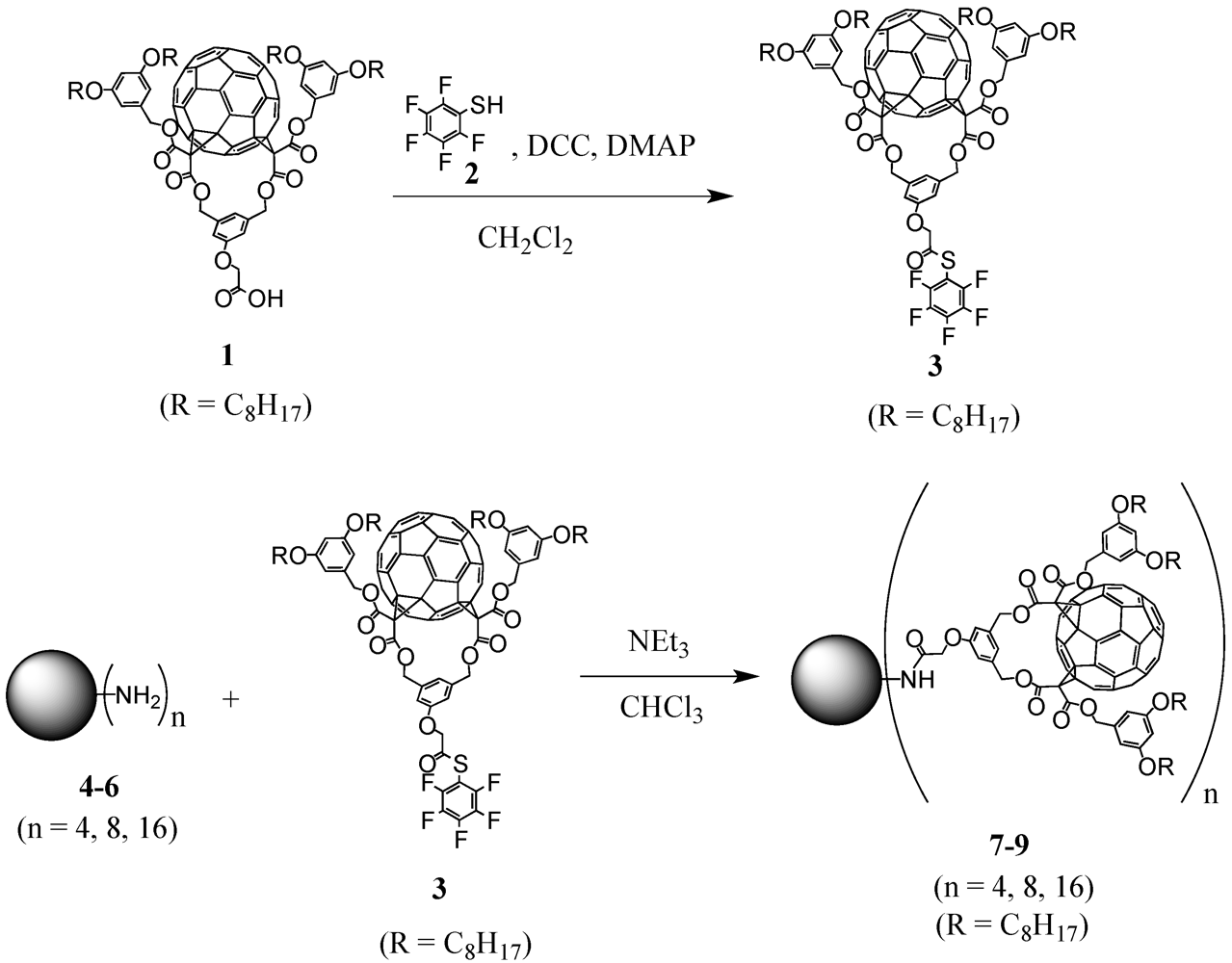
There are dendrimers available on the market like, e.g., polypropylene imine (POPAM or PPI) or polyamidoamine (PAMAM) dendrimers. In this first example of divergent preparation of fullerodendrimers, three generations with either 4, 8, or 16 surface amine groups have been used for decoration with a previously activated fullerene precursor (
Scheme I) [
15]. The choice of activation via pentafluorothiophenol ester
3 proved to be crucial for the functionalization. Owing to the nature of fullerene building block
1, reaction conditions for the activation may not be strongly acidic or basic to preserve the ester functions. In addition, the ultimate step to graft the modified precursor to the multiple peripheral amines requires extremely efficient and high-yielding reactions to ensure complete functionalization without forming defected dendrimer structures. The corresponding pentafluorothiophenol ester
3 met these criteria as has been illustrated at various examples in the literature [
16,
17,
18]. Accordingly, activated acid
3 was obtained in nearly quantitative yield upon reaction of carboxylic acid
1 with pentathiofluorothiophenol
2 in the presence of dicyclohexylcarbodiimide (DCC) and a catalytic amount of 4-dimethylaminopyridine (DMAP). Subsequent reaction of the resulting activated ester with PPI dendrimers
4-
6 of first to third generation using triethylamine as base provided the corresponding dendritic derivatives
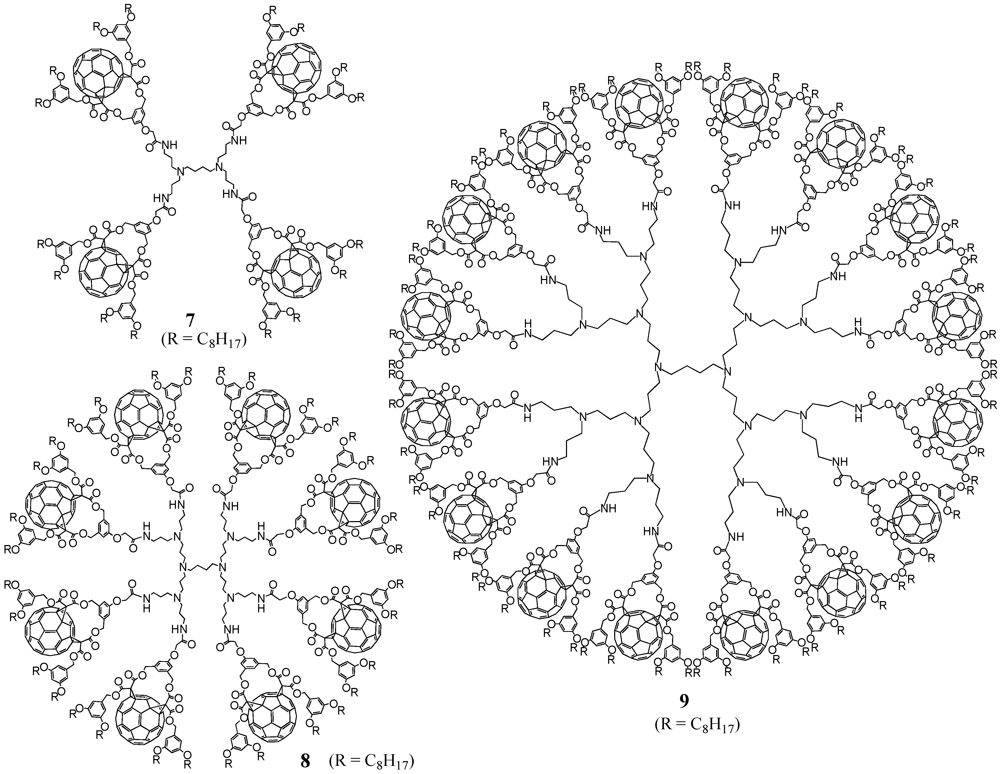
7-
9 in good yields (
Figure 1).
Figure 1.
Structures of fullerene-rich PPI-derived dendrimers 7-9.
Figure 1.
Structures of fullerene-rich PPI-derived dendrimers 7-9.
The fullerene-rich dendrimers are well soluble in a wide range of common organic solvents including CH2Cl2, CHCl3, THF or toluene, due to the four pendant alkyl chains per C60 unit and spectroscopic characterization was easily achieved with the 1H-NMR spectra of 7-9 to show the typical patterns of fullerene cis-2 bis-adducts and the expected additional signals arising from the PPI centrepart. Also MALDI-TOF mass spectrometry gave clear indication for the desired structures by depicting the expected molecular ion peaks. It is noteworthy that no peaks corresponding to defected dendrimers were observed in the mass spectra of 7-8, thus providing clear evidence for their monodispersity. On the contrary, the spectrum for 9 showing a high level of fragmentation prevented the observation of the expected molecular ion peak and its monodispersity could not be unambiguously demonstrated.
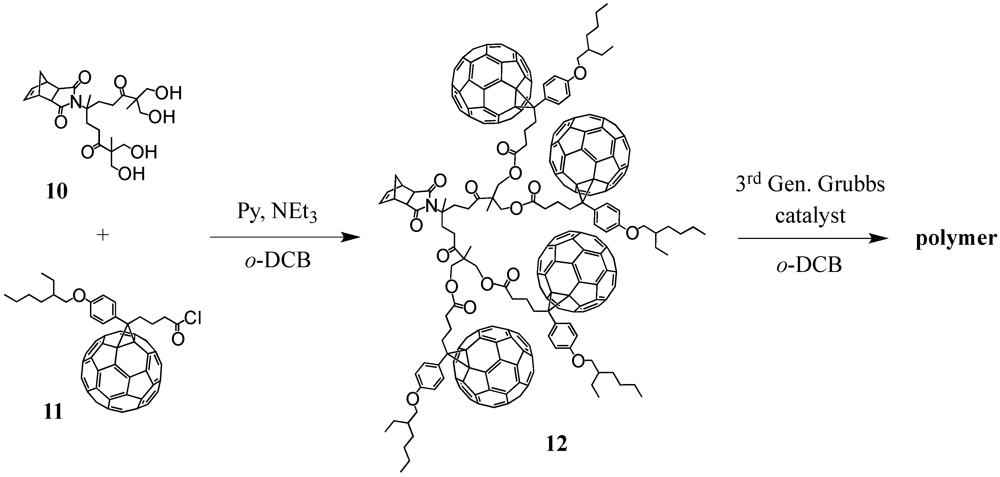
Very recently, Wudl
et al. presented a divergently grown dendronized norbornene derivative carrying four peripheral C
60 moieties [
19]. The synthetic protocol proceeded from a norbornene precursor bearing two terminal alcohol functions to which was reacted the anhydride of isopropylidene-2,2-bis(oxymethyl)propionic acid. After deprotection the four terminal hydroxyl groups of
10 became accessible for attachment of 4-(2-ethylhexyloxy)-[6,6]-phenyl C
61-butyric acid. Initial attempts to couple both building blocks via DMAP-catalyzed [4-(dimethylamino)-pyridinium
p-toluenesulfonate (DPTS)] esterification were not successful presumably due to steric hindrance between the acid functionality on the bulky fullerene cage and the peripheral alcohol groups in the dendron. On the contrary, conversion of the acid function into its acyl chloride
11 upon treatment with oxalyl chloride led to the target fullerodendron
12 (
Scheme II). Polymerization of the exo-norbornene monomer was then carried out using the fast initiating third generation Grubbs’ catalyst and the developed fullerene-rich linear polymer was claimed for possible applications in the field of polymer solar cells. Likewise, dendronized diblock copolymers have been reported [
20]. However, incorporation of C
60 units was attempted as the last step of the synthesis and did not lead to complete addition rather than a moderate coverage of approx. 50% of the peripheral long alkyl chains.
Scheme II.
Synthesis of fullerene-rich norbornene-centred compound 12.
Scheme II.
Synthesis of fullerene-rich norbornene-centred compound 12.
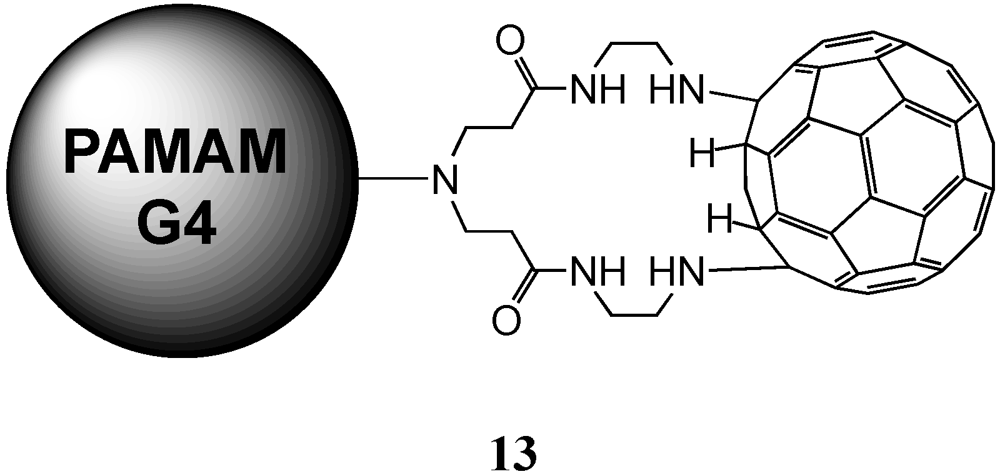
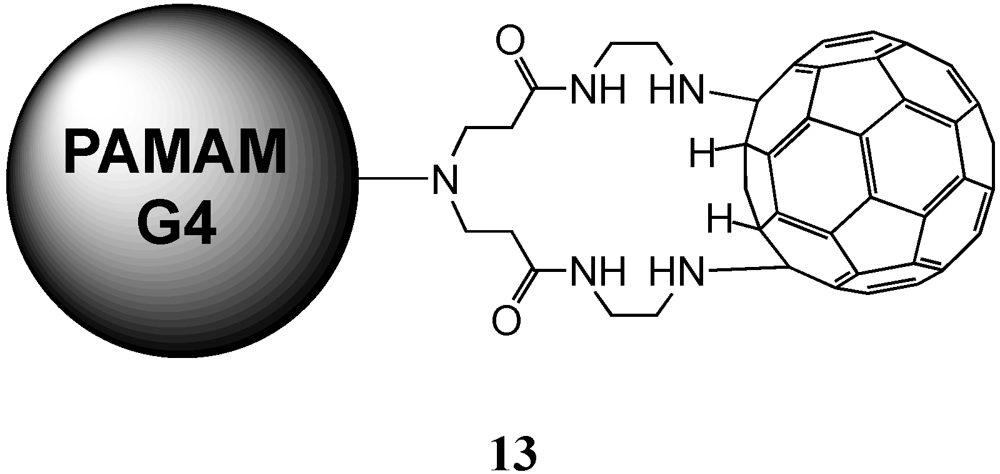
The group of Tomalia reported the preparation of a material containing a PAMAM dendrimer core coated with a shell of C
60 molecules based on the known reaction of amines with the electron-deficient fullerene moiety in the presence of a base [
21]. For this purpose, a large excess of fullerene has been dissolved in pyridine and it was slowly added a finely dispersed solution of G4 PAMAM dendrimer in pyridine. After stirring for one day and several purification steps, final material
13 was isolated in a 89% yield. As stated before, the reaction conditions as well as steric effects prevented complete derivatization hence obtaining a final product with a certain dispersity and a number of addends inferior to the total number of available primary amine groups,
i.e., 64. In addition, each fullerene could in principle react with more than one amine group as there were six independent pyracylene units per C
60 molecule. Indeed, the authors studied the number of bonded C
60 to the dendrimer surface in
13 by MALDI-TOF mass spectrometry and thermogravimetric analyses and it turned out that the results correspond to a C
60/dendrimer molar ratio of approximately 30:1. According to the authors, this number of almost exactly two terminal amine groups per fullerene suggests the bonding of two amine groups to most of the fullerenes (
Figure 2). This material was used to catalyse photooxidation of thioanisole by generation of singlet oxygen. The oxidation reactions was found to occur in both organic and aqueous solvents, with enhanced reactivity in aqueous solution, possibly due to a nanoreactor effect resulting from diffusion of hydrophobic reactant molecules into dendrimer cavities. Similarly, Godínez
et al. deposited multilayer films of PAMAM generation 0.0 dendrimers and C
60 on nanocrystalline TiO
2 electrodes to fabricate nanoassembled photoactive surfaces that absorbed in the visible region and offered a high molar extinction coefficient [
22].
Figure 2.
Proposed structure of 13 with covalently linked C60 at the surface of a polyamidoamine (PAMAM) G4-dendrimer.
Figure 2.
Proposed structure of 13 with covalently linked C60 at the surface of a polyamidoamine (PAMAM) G4-dendrimer.
4. Molecular Self-Assembly of Fullerene-Containing Dendrons
The approach via self-assembly of dendrons using non-covalent interactions is particularly well-suited for the preparation of fullerene-rich macromolecules. Upon preparation of the dendritic branches the different moieties self-organize thus creating the dendritic macromolecular structure. This way, the often-encountered tedious final synthetic coupling reactions with a given multifunctional core can be avoided, as can problems regarding side reactions that may occur with potentially reactive functional groups like C
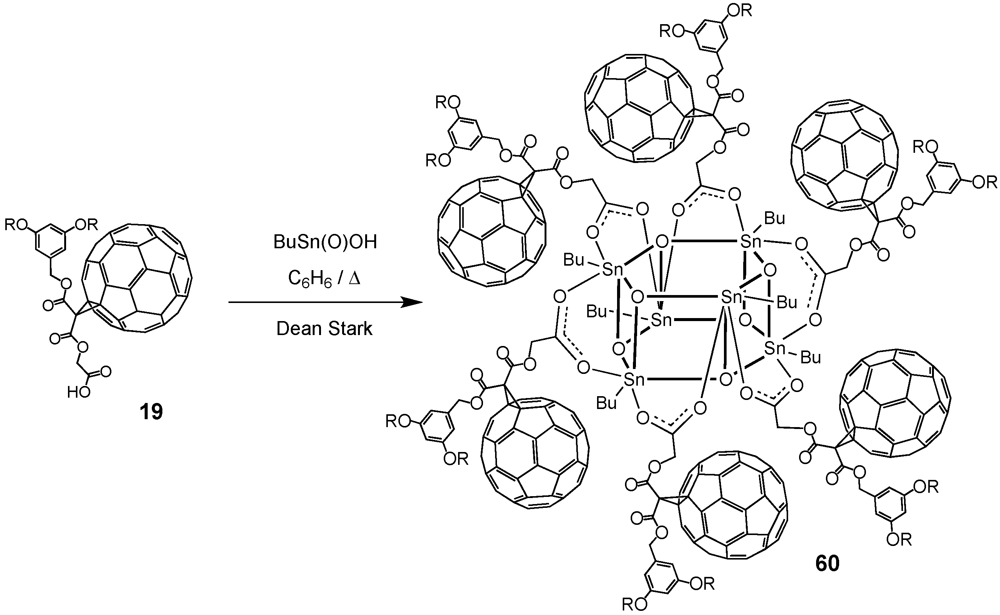
60. Nierengarten
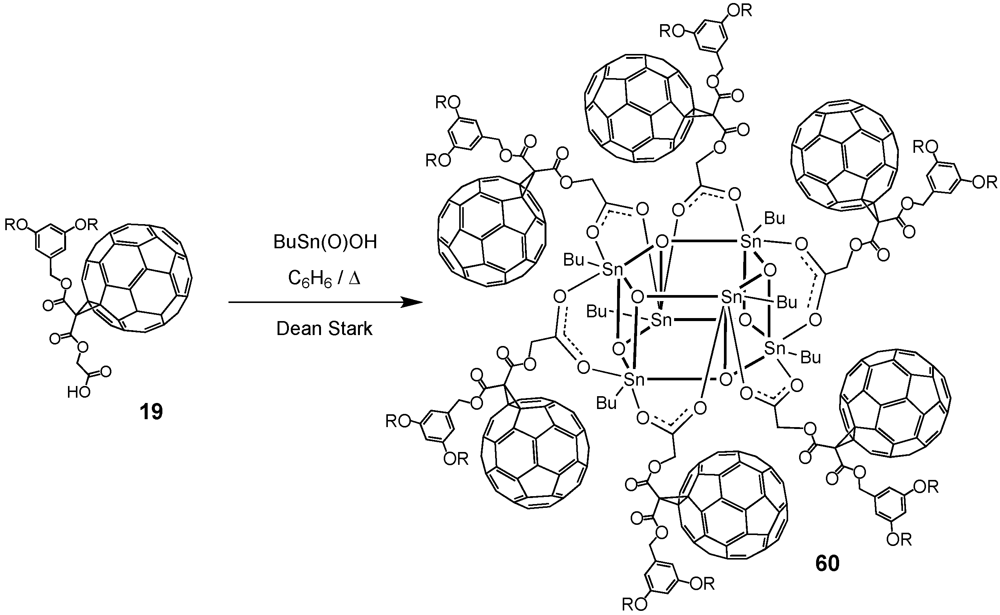
et al. exploited the self-assembly approach for the construction of organooxotin-derived clusters (
Scheme X) [
39]. Simple heating of an equimolar mixture of
19 and
nBuSn(O)OH in benzene to reflux for 12 h using a Dean-Stark trap afforded the hexameric organostannoxane derivative
60 in 99% yield. The six tin atoms were chemically equivalent as well as the six trivalent oxygen atoms. The Sn–O framework of the molecule could be described as a drum with top and bottom faces each being comprised of a six-membered (–Sn–O–)
3 tri-stannoxane ring. The drum faces were joined together by six Sn–O bonds containing tri-coordinated oxygen atoms. The sides of the drum were thus comprised of six four-membered (–Sn–O–)
2 distannoxane rings, each of which was spanned by a carboxylate group that formed a symmetrical bridge between two tin atoms.
Scheme X.
Preparation of first generation organooxotin cluster 60.
Scheme X.
Preparation of first generation organooxotin cluster 60.
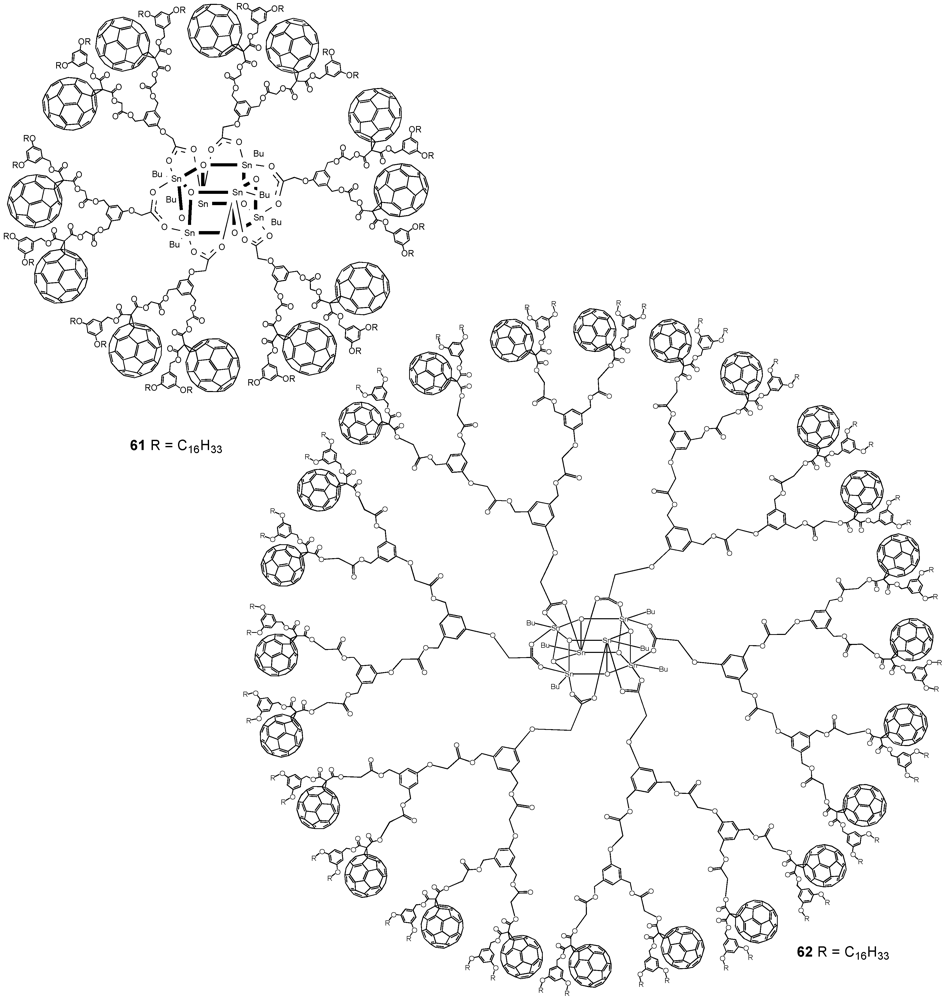
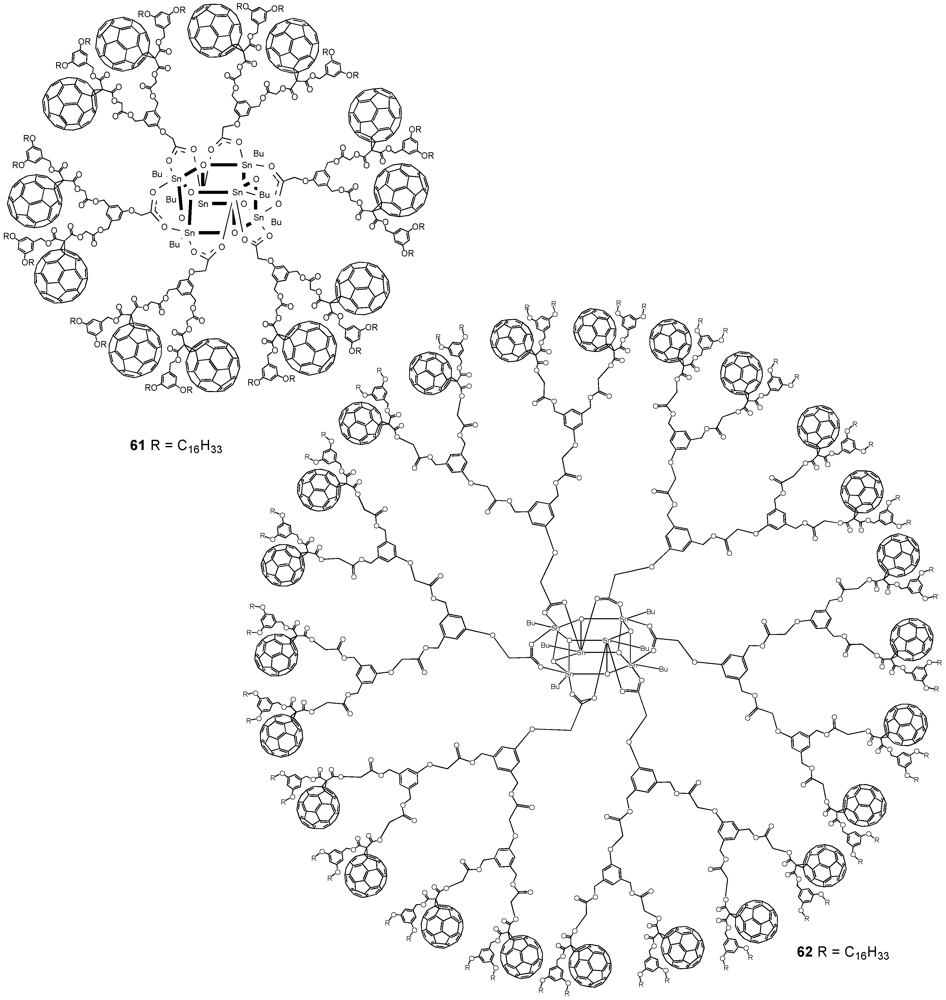
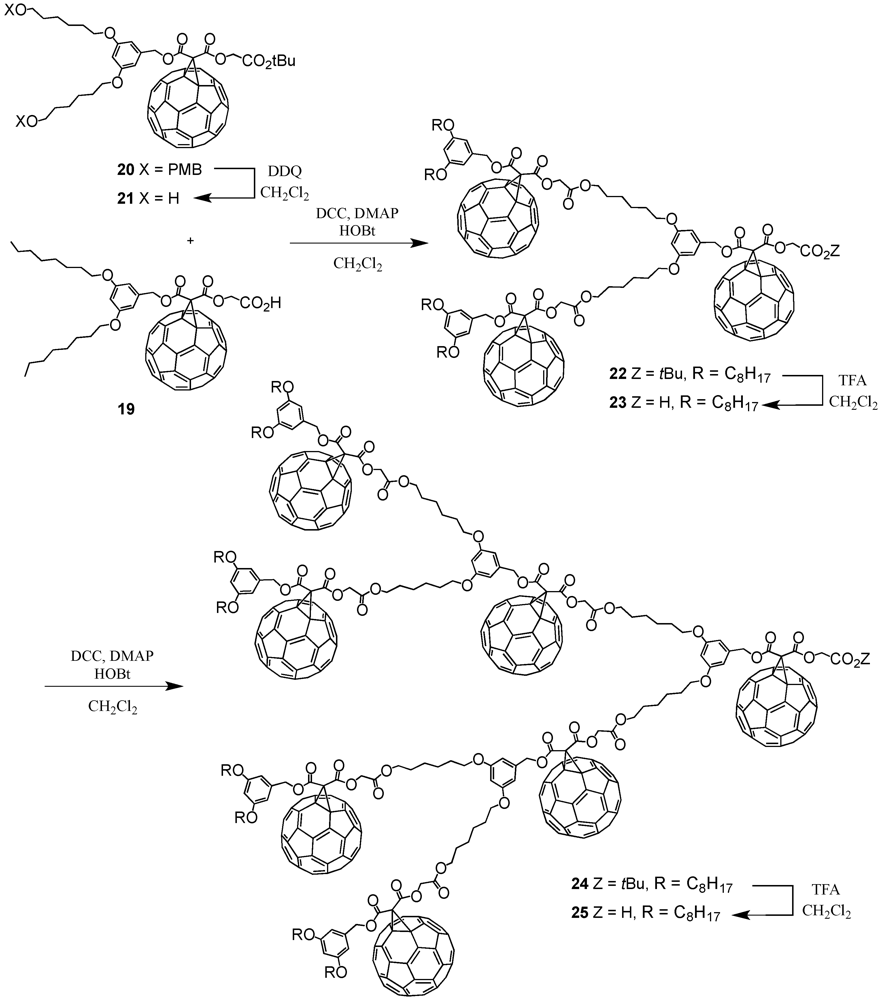
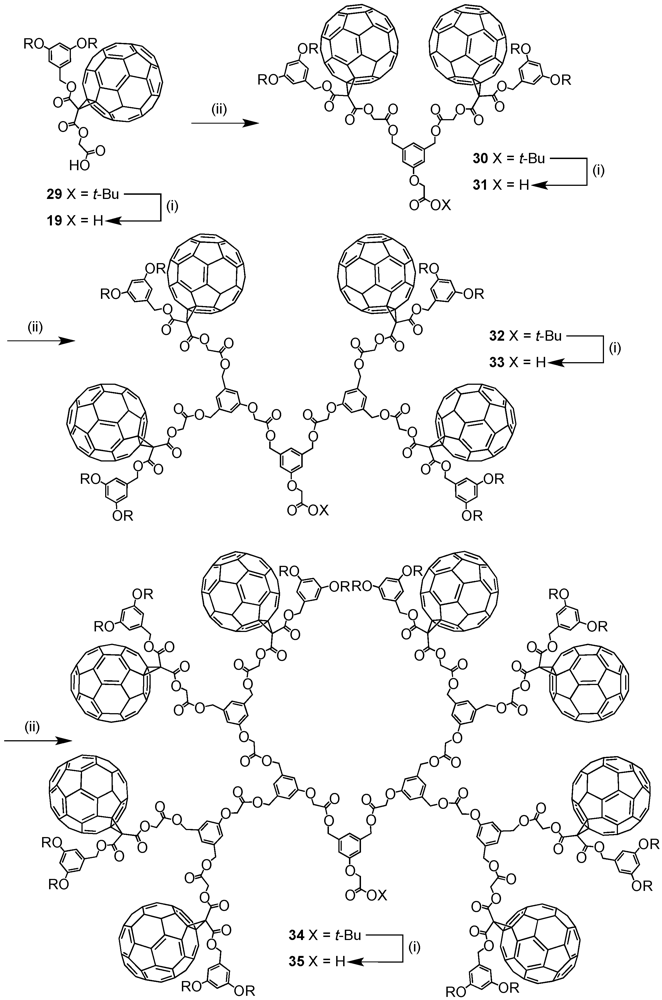
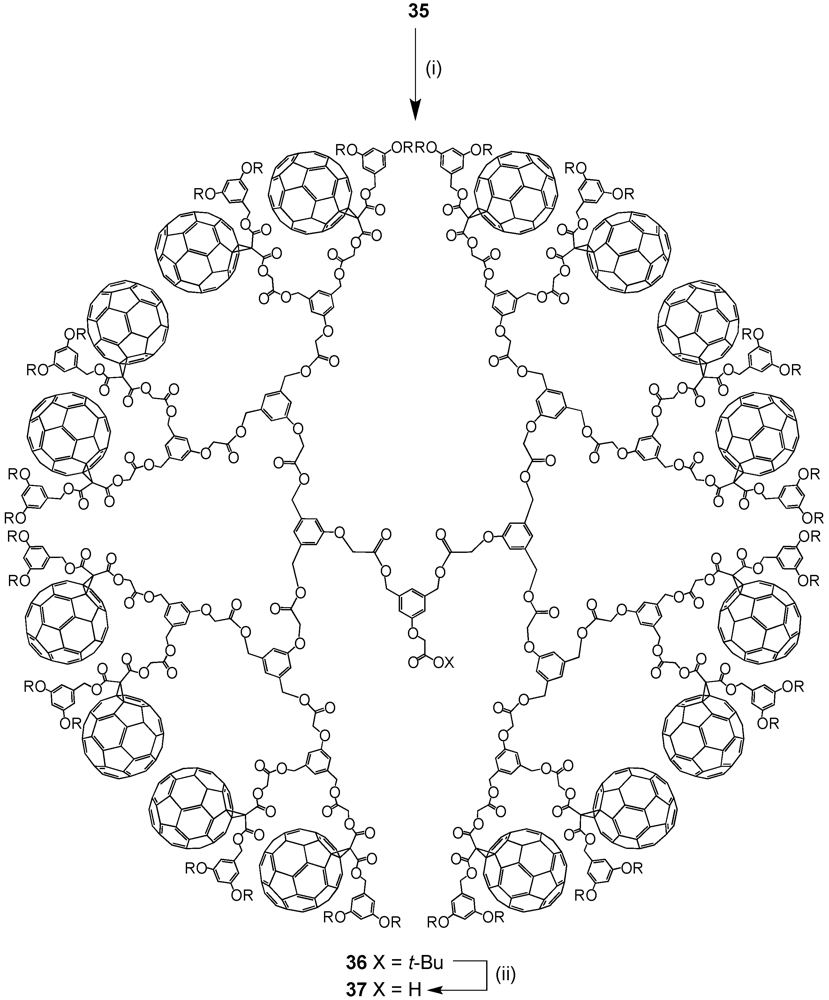
The optimized reaction conditions used for the preparation of
60 from carboxylic acid
19 were then applied to fullerodendrons
31 and
33. The corresponding organostannoxane derivatives
61-62 were thus obtained in almost quantitative yields (
Figure 8) [
40]. Though the size of the dendron increased significantly upon going to generation three, the self-assembly process was not severely affected by the increased steric demand of the starting carboxylic acids with the close to quantitative formation of the final ensemble after 12 h. The latter observations were in contrast with previous findings as described before for which slow reactions and often only moderate yields were obtained for such third generation derivatives when compared to the corresponding first and second generation. Apart of the proton and carbon NMR spectra displaying the characteristic signals of the starting dendritic carboxylic acids and the expected additional resonances ascribable to the
n-butyl chains, the respective
119Sn NMR spectra brought final structural proof. The equivalence of all peripheral fullerene subunits as expected for a six-fold symmetric assembly with a drum-shaped organostannoxane core was found as a single
119Sn resonance observed at
ca. −480 ppm.
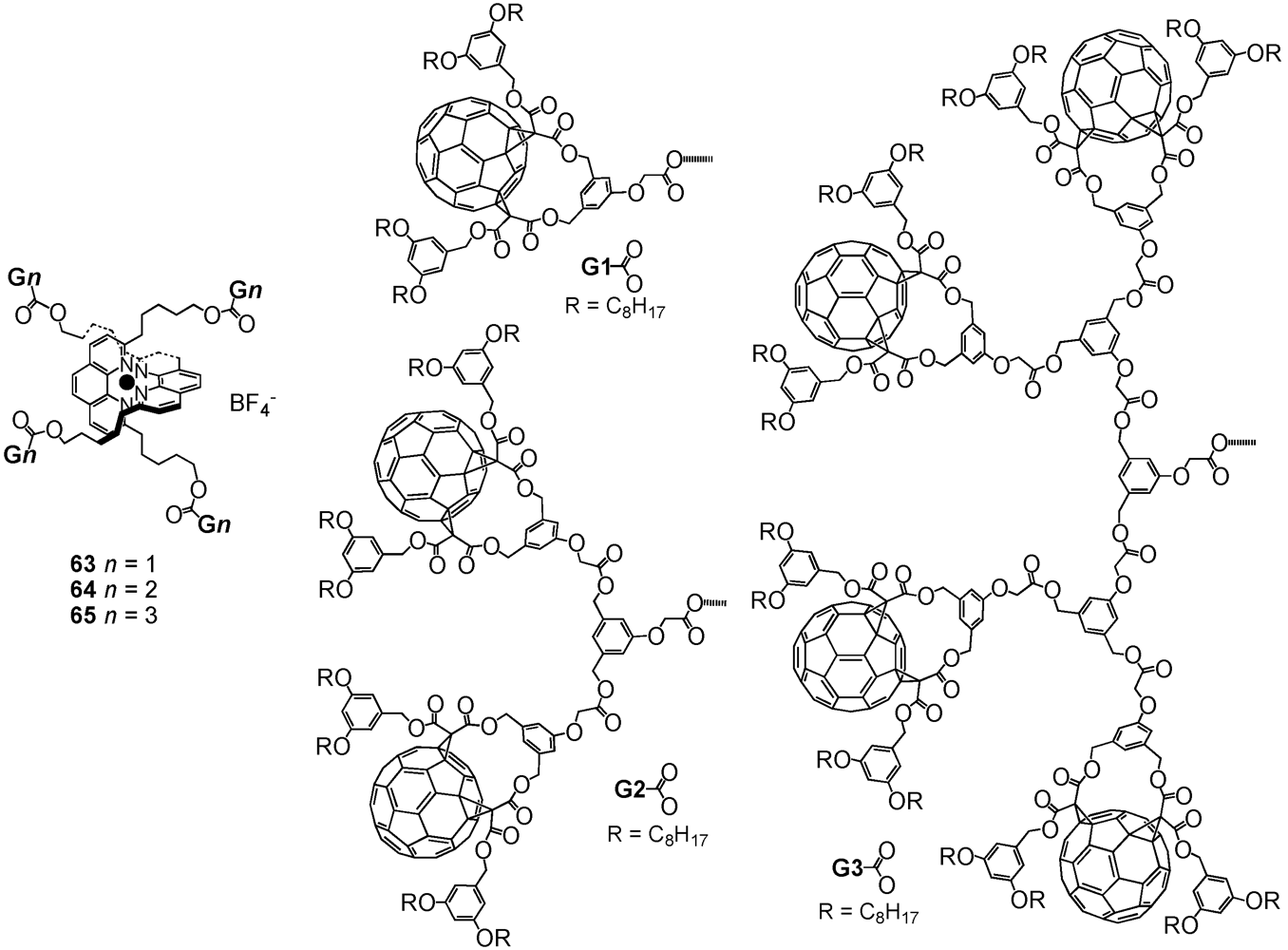
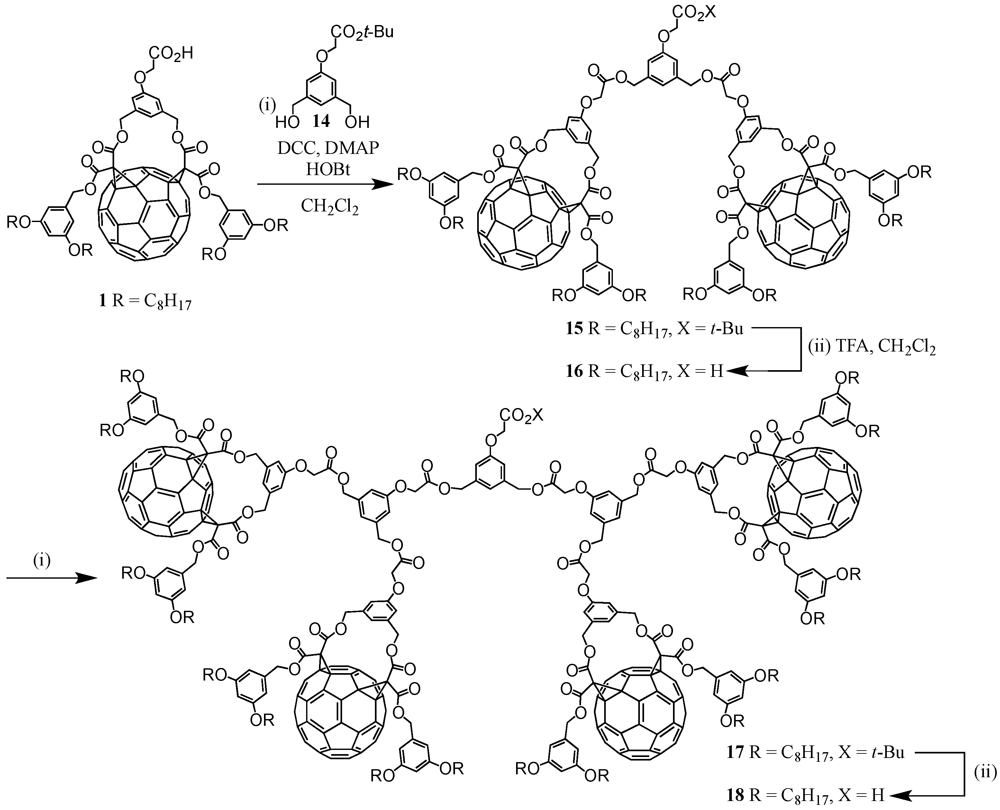
The use of metal coordination is perhaps the most widely developed method for the directed assembly of dendritic superstructures. In most cases, the metal center forms the core of the macromolecule, in which dendrons owing to their ligating groups at the focal point are able to be coordinated around a single, central metal ion. Nierengarten
et al. used this method for the construction of large bis(1,10-phenanthroline)copper(I)-derived metal complexes
63-
65 of first to third generation with up to 16 fullerene surface units (
Figure 9) [
41,
42]. The synthetic procedure started from a modified 1,10-phenanthroline building block containing at the 2,9 positions pentanol groups. To these hydroxyl functions were then attached dendritic fullerene precursors
1,
16 and
18 using classical DCC/DMAP/HOBt conditions. The dendritic ligands have thus been obtained in decreasing yields of 75, 48 and 24% yields. Treatment with Cu(MeCN)
4BF
4 in a 2:1 mixture of CH
2Cl
2/MeCN provided the target metal-based compounds
63-
65 in good yields. However, due to problems during the purification process and the ionic metal complex character of the dendrimers, the isolated yields ranging from 56 for
63 to 33% for
65 were considerably lower as some material remained adsorbed on the column stationary phase. Structural elucidation by the NMR technique was easily achieved and clearly indicated complex formation by significant shifts of approx. 0.6 ppm as observed for the methylene protons at 2,9 positions. From the electronic properties of dendrimers
63-
65, it could be deduced that the Cu(I) central core was strongly affected by the dendritic surrounding,
i.e., they were embedded in a
dendritic black box. Whereas for the lowest generation
63, oxidation of the central core was still possible though with lower amplitude, for the larger generation compounds
64 and
65, oxidation was prevented by the large fractal branches. Furthermore, due to the increasing number of peripheral fullerene subunits in
64 to
65, there was less and less light available for the core and the small portion of light energy able to excite the central Cu(I) complex was returned to the external fullerenes by energy transfer.
Figure 8.
Fullerene-rich tin drum shaped clusters 61-62.
Figure 8.
Fullerene-rich tin drum shaped clusters 61-62.
Figure 9.
Structures of G1 to G3 fullerene-rich Cu(II) phenanthroline-based metal complexes 63-65.
Figure 9.
Structures of G1 to G3 fullerene-rich Cu(II) phenanthroline-based metal complexes 63-65.
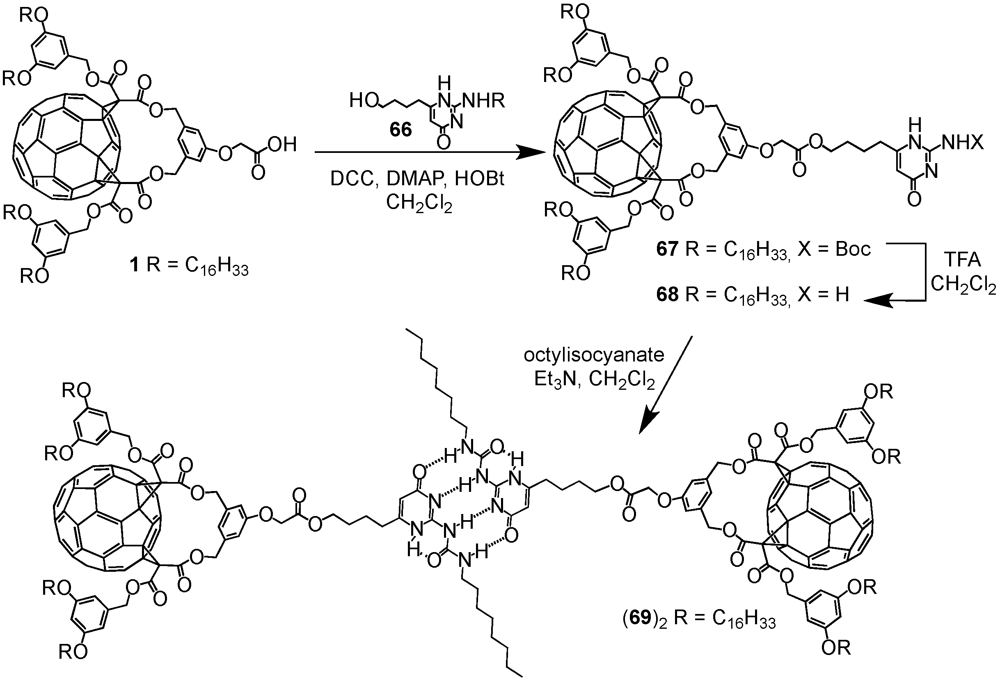
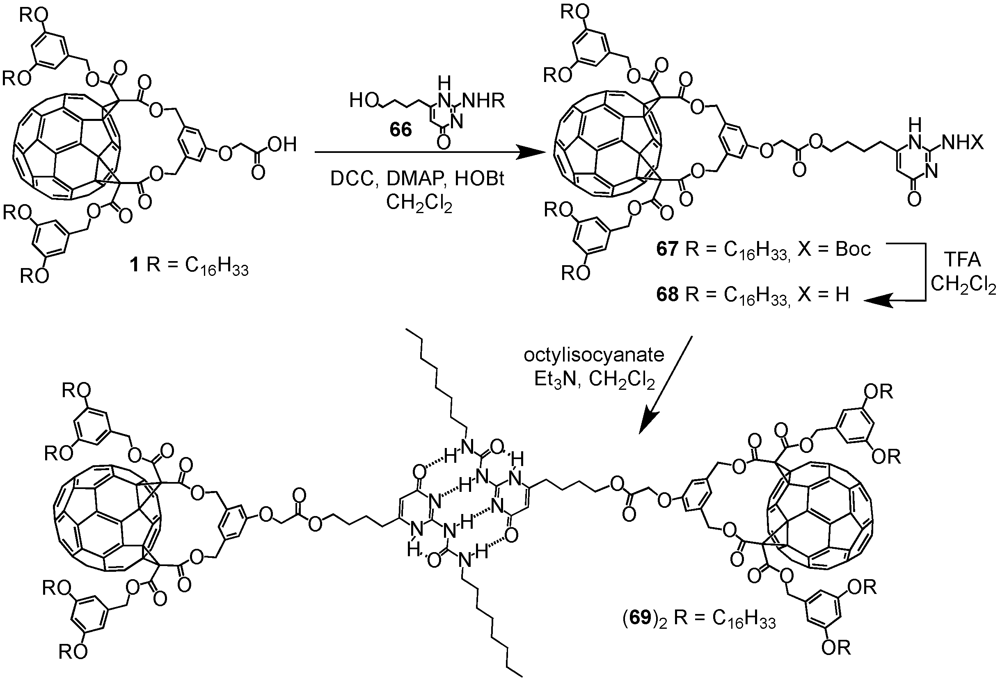
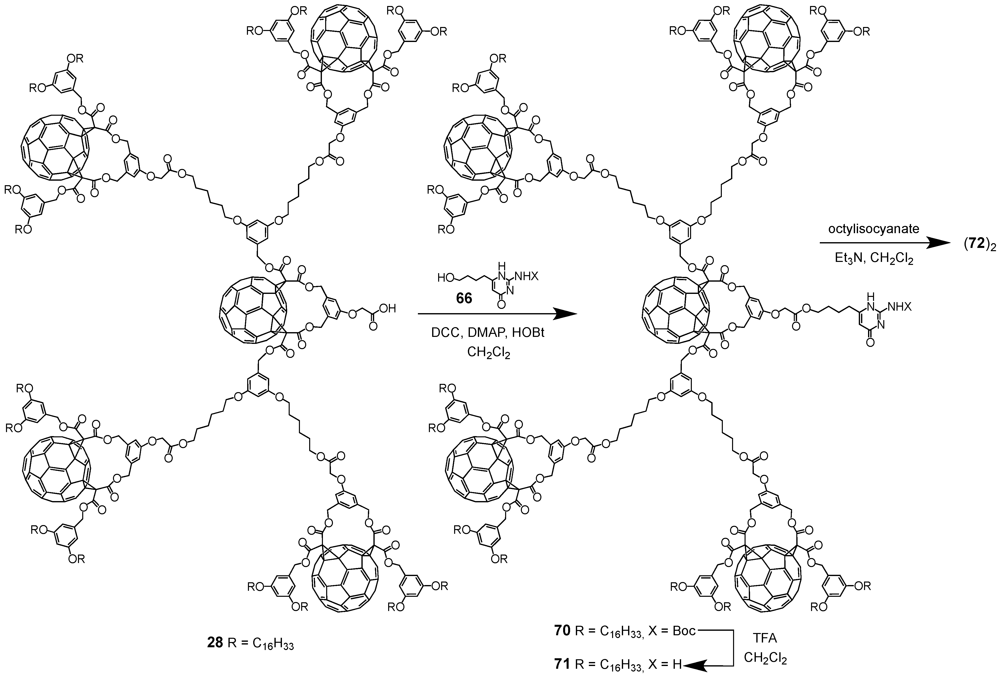
A key concept in supramolecular chemistry is the molecular self-assembly via hydrogen bonds. Such non-covalent interactions determine for instance the three-dimensional shape of proteins and nucleic bases. Presumably, the most important example constitutes the double helical structure of DNA that is largely due to hydrogen bonding between the base pairs upon linking one complementary strand to the other thus enabling replication. It is therefore not surprising that this concept has also been adopted in the preparation of fullerene-rich dendrimers. The group of Nierengarten
et al. has indeed demonstrated that the self-assembly of dendritic macromolecules using hydrogen-bonding interactions was particularly well-suited to construct such macromolecular ensembles. In this nexus, supramolecular dendrimer (
69)
2 has been obtained from the dimerization of a fullerene-functionalized dendron by a quadruple hydrogen bonding motif [
43]. The self-complementary arrays of four hydrogen bonds, originally developed by Meijer
et al., afforded remarkably stable dimers with high association constants in apolar organic solvents (
Ka 4 × 10
7 M
−1 in CHCl
3) [
44,
45].
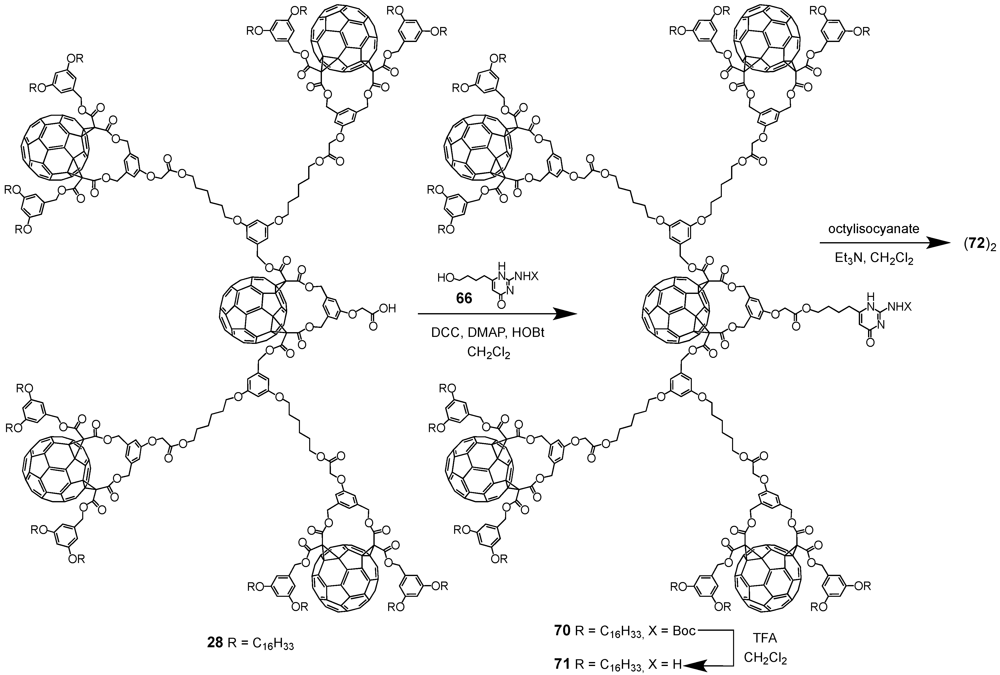
The required precursor namely a 2-ureido-4-[1H]pyrimidinone derivative bearing a residual Boc-protected amine function in position 2 and a 4-hydroxybutyl chain in position 6 has been obtained from diethyl 3-oxoheptanedioate following a three step procedure. To this end, dendrons
1 and
28 with either one or five C
60 units and a focal carboxylic acid function have been coupled using standard esterification conditions (DCC/DMAP/HOBt). The resulting esters
67 and
70 have been obtained in 58 or 70% yield, respectively. Initial attempts to purify the first generation compound by typical column chromatography resulted in rather low yields of 30% thanks to partial cleavage of the Boc protecting group of the amine function in position 7. On the contrary, gel permeation chromatography proved to be more efficient hence yielding target substrate
67 in 58% yield. Similar purification conditions have then also been applied for building block
70. The Boc-protecting groups of both dendrons were removed by treatment with an excess of TFA to afford amines
68 and
71 in good yields. Within the final step of the synthetic sequence, the dendritic amines were subjected to reaction with octylisocyanate in the presence of triethylamine to give the desired supramolecular fullerene dimers (
69)
2 and (
72)
2 in yields of 48 and 87% (
Scheme XI and
Scheme XII).
Scheme XI.
Preparation of supramolecular architecture (69)2 resulting from the dimerization via quadruple hydrogen bonding.
Scheme XI.
Preparation of supramolecular architecture (69)2 resulting from the dimerization via quadruple hydrogen bonding.
Spectroscopic characterisation via MALDI-TOF mass spectrometry indicated the formation of the proposed dimeric supramolecular structures. This technique, due to its mild ionization conditions and the concomitant marginal levels of fragmentations, was found to be a well-suited tool for characterizing such high-molecular-weight compounds. Even if the mass spectrum of (69)2 was dominated by the ion peak corresponding to the monomer 69, the molecular ion peak of dimer (69)2 was also clearly detected. Similarly, in the MALDI-TOF mass spectrum of fullerodendrimer (72)2 two peaks became apparent assignable to supramolecular dimer (72)2 and to the monomeric structure 72. These relatively low intensities as observed in the respective mass spectra could be easily rationalized by the rather weak non-covalent interactions. Nonetheless, the dimeric structures could be clearly detected and the absence of peaks corresponding to defected dendrons provided clear evidence for their monodispersity.
Definitive evidence for dimer structures of 69 and 72 could be deduced from the proton NMR measurements conducted in CDCl3. For both dendritic assemblies, signals corresponding to single compounds were detected. Apart from the characteristic features emanating from Cs symmetrical 1,3-phenylenebis(methylene)-tethered fullerene cis-2-bis-adducts, and the additional typical signals from the different subunits of the modified fullerene precursors, large downfield shifts were found for the protons of the hydrogen-bonding motif. In both cases, the urea NH protons were found at δ = 11.8 and 10.1 ppm and the intramolecularly chelated pyrimidinone NH at δ = 13.2 ppm. This observation is fully consistent with four donor–donor–acceptor–acceptor (DDAA) hydrogen bonds in the supramolecular fullerene–dimer system.
Scheme XII.
Preparation of supramolecular architecture (69)2 resulting from the dimerization via quadruple hydrogen bonding.
Scheme XII.
Preparation of supramolecular architecture (69)2 resulting from the dimerization via quadruple hydrogen bonding.
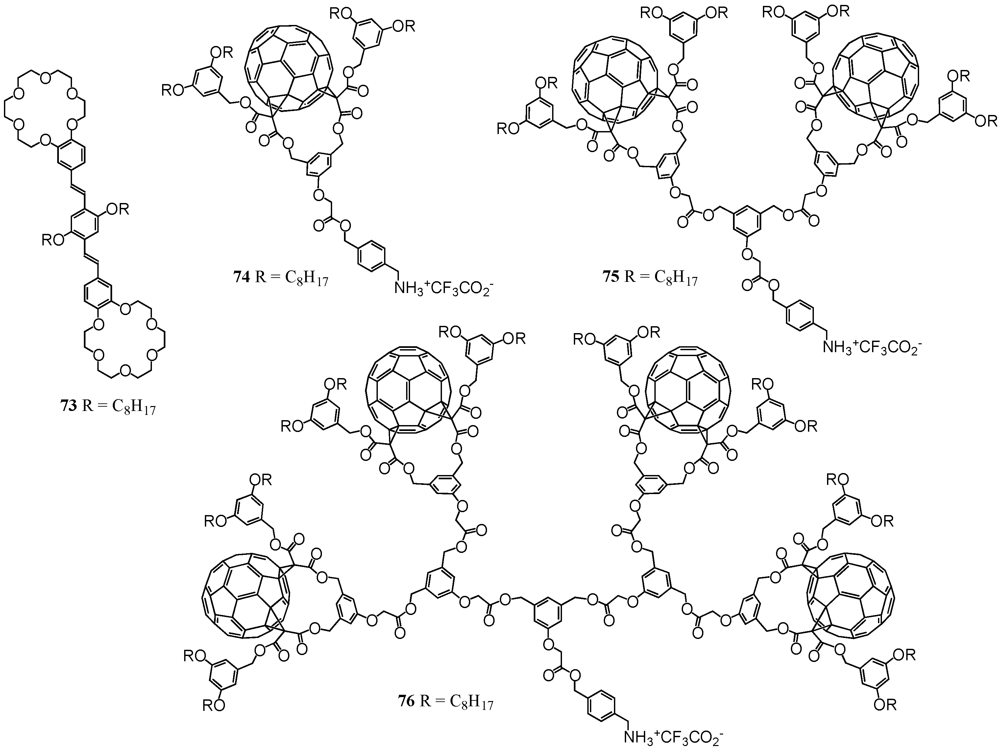
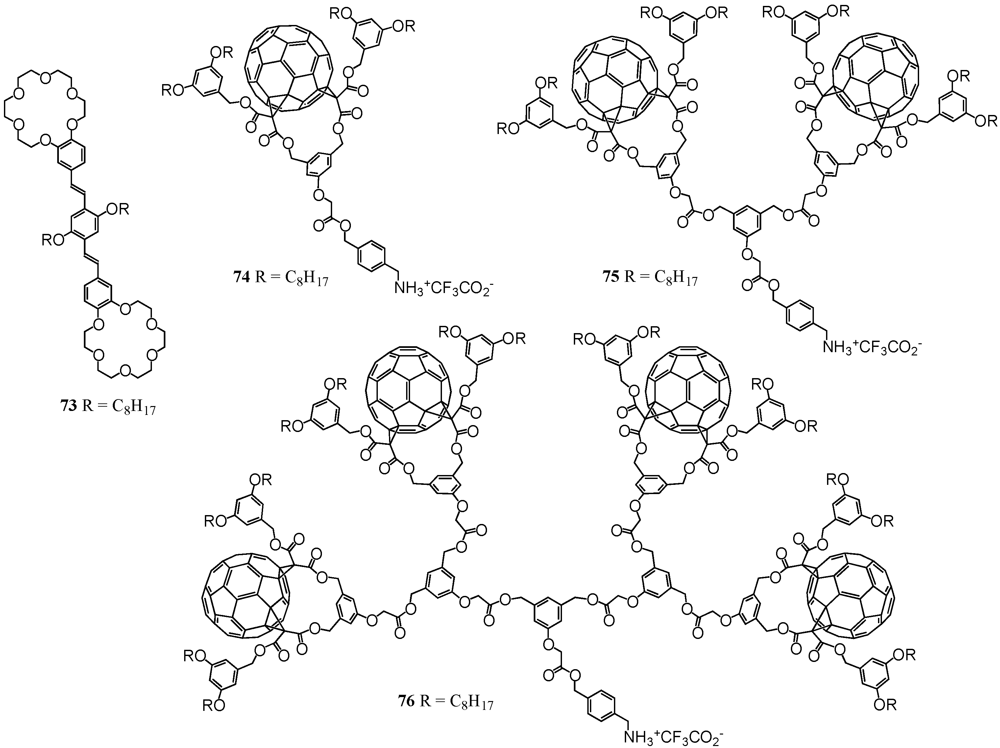
The ammonium–crown ether interaction is well-known in the field of molecular self-assembly. The diameter of the crown ether macrocycle largely determines the binding affinity under formation of host-guest complexes. One of the most prominent examples is 18-crown-6 that has a high affinity towards alkali ions such as potassium but is also famous to form quite stable complexes with protonated amines. Though the interactions are relatively weak for the latter case, they have been exploited for the association with fullerodendritic derivatives [
46]. Correspondingly, ditopic OPV compound
73 has been equipped with two 18-crown-6 units (
Figure 10). On the other hand, a Boc-protected amine has been coupled to fullerene dendron
1 using DCC/DMAP/HOBt. The final step then involved deprotection with TFA to readily afford the cationic ammonium trifluoroacetate salt
74 in 86% yield (
Figure 10).
Figure 10.
Structures of ditopic OPV host 73 and ammonium-containing dendrons of first to third generation 74-76.
Figure 10.
Structures of ditopic OPV host 73 and ammonium-containing dendrons of first to third generation 74-76.
A mixture of compounds
73 and
74 self-assembled under formation of a stable 1:2 complex. UV/vis and fluorescence binding studies have been performed for the multicomponent photoactive devices in which the emission of the central ditopic receptor was dramatically quenched by the peripheral fullerene units. It turned out that the association constants were about one to two orders of magnitude higher than commonly observed for such systems, hence indicating additional recognition elements reinforcing the overall supramolecular structure. It was anticipated that favorable intramolecular fullerene–fullerene interactions were at the origin of this stabilization. Similarly, a two-center host–guest topography has been developed and it could be demonstrated that owing to the perfect complementarity of the two components, a bis-cationic substrate has been clicked on a ditopic crown ether derivative thus leading to a very stable non-covalent macrocyclic 1:1 complex [
47]. As this approach appeared to be modular and easily applicable to a wide range of functional groups for the preparation of new supramolecular architectures with tunable structural and electronic properties, it has also been applied for the preparation of even larger dendritic nanoscale architectures (
Figure 10) [
48]. Accordingly, dendritic branches
16 and
18 were involved in the coupling to the same Boc-protected alcohol under the same conditions as conducted for
1 to yield the two cationic species
75 and
76 in good yields. Complexation with ditopic receptor
73 resulted in even enhanced binding constants thus displaying that there were even more secondary weak intramolecular interactions such as π-π stacking and hydrophobic interactions prevalent. These results mirrored another example for a remarkable positive dendritic cooperative effect. The authors emphasized that the size of the dendritic building blocks did not constitute a severe limitation for the self-assembly of large dendritic architectures.
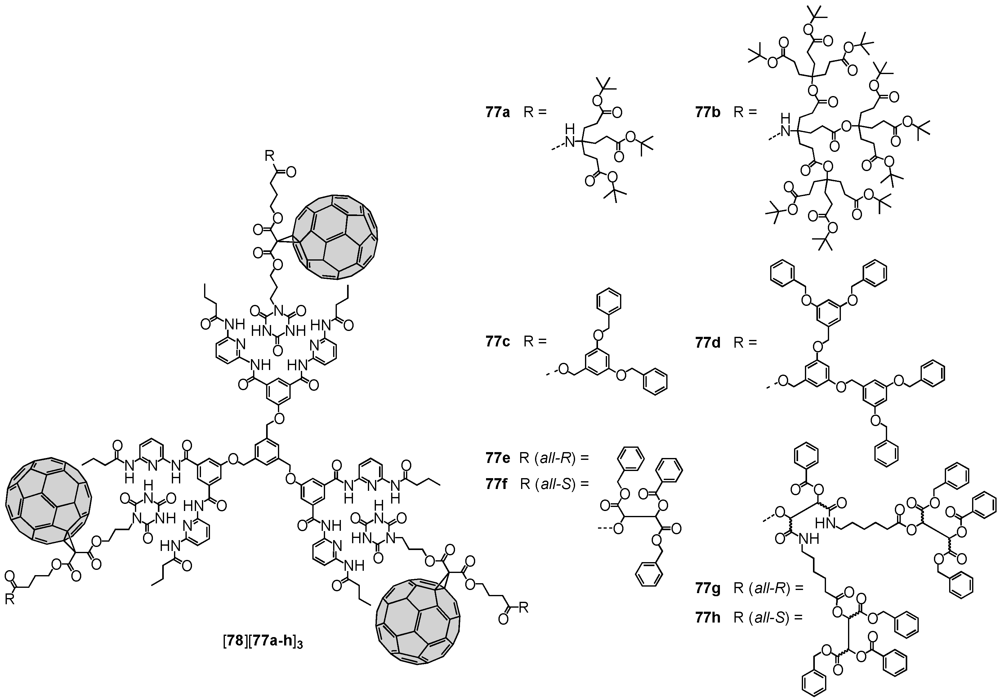
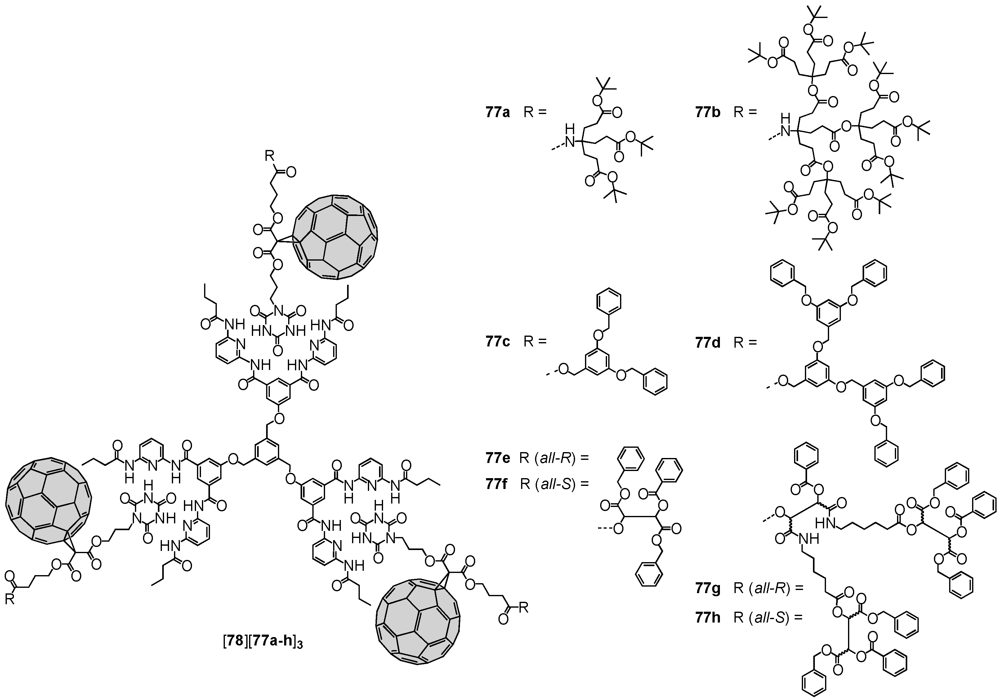
Hirsch
et al. used several (non-)dendritic mono-Bingel adducts for the self-assembly onto various Hamilton-receptor appended scaffolds via complementary hydrogen bonding [
49]. Consequently, an unsymmetric malonate has been functionalized under formation of ester bonds at one end with 3-bromo-1-propanol and on the other with
t-butyl 4-hydroxybutanoate. The latter was subjected to hydrolysis and the free carboxylic acid employed in esterification reactions with different dendrons. Nucleophilic mono-cyclopropanation then readily produced the fullerene-containing dendritic wedges. Initial attempts to form the Bingel-adduct prior to the modification by dendrons were not successful. Within the last step of the synthetic sequence, the cyanurate moiety was introduced through reaction of the residual bromide function with cyanuric acid to produce the desired dendronized fullerene derivatives
77a-h. Association studies with trivalent receptor
78 revealed a positive cooperativity, that originated from the preferably adopted planar
cis-cis conformation rather than the other two possible topologies,
i.e.,
trans-trans or
cis-trans (
Figure 11). The authors also encountered a size-dependent behavior,
i.e., very voluminous ligands disfavoured the introduction of the second and third ligand due to steric demand. This effect was found more pronounced for Newkome and Fréchet-type dendritic substituents in
77a-b and
77c-d, respectively. In continuation of research in this field, the same group reported on the self-assembly of the depsipeptide-derived fractal branches
77e-h onto different porphyrins to give photoactive supramolecular associates [
50,
51].
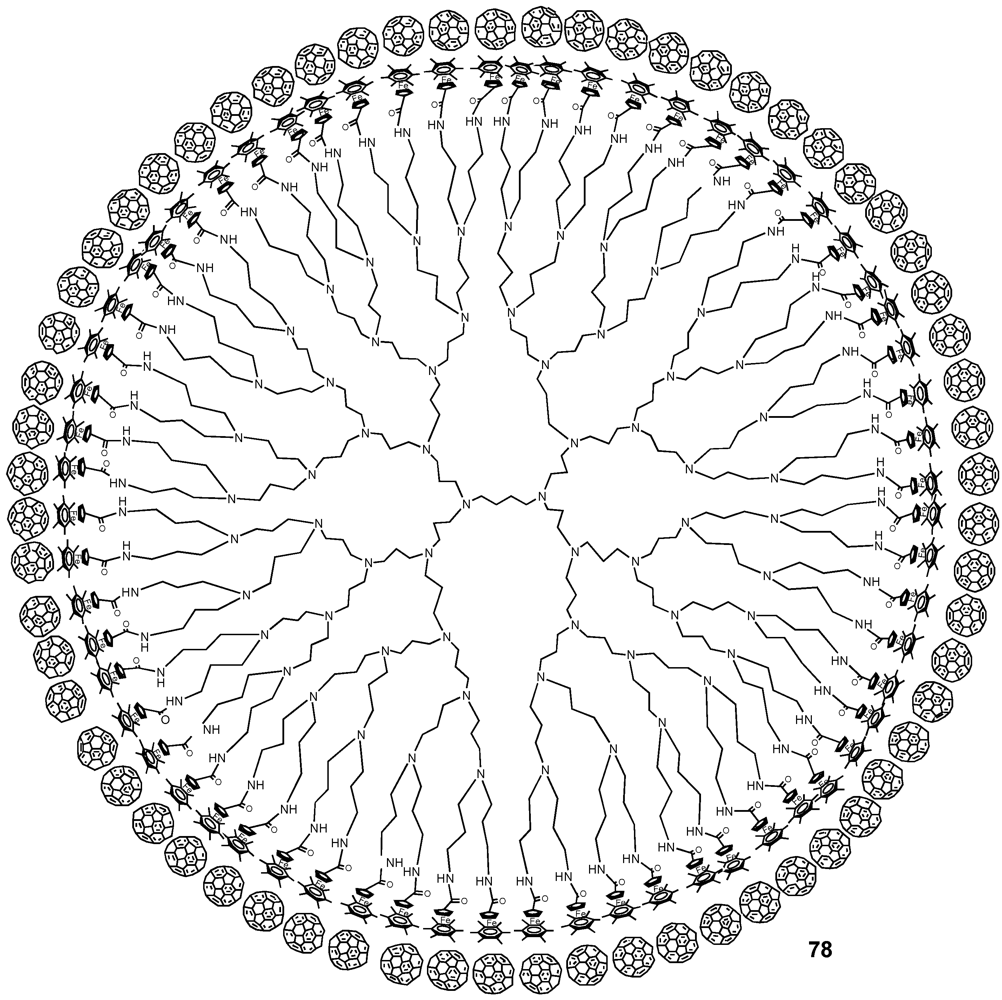
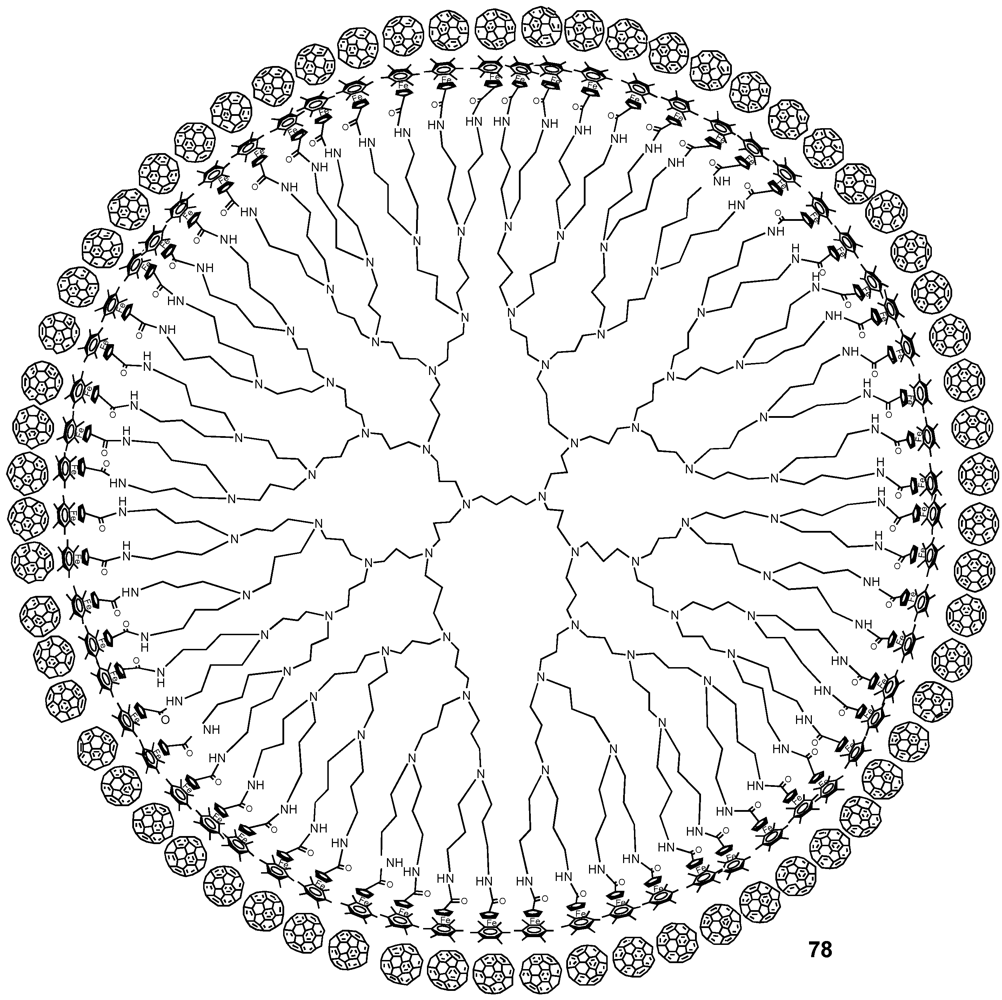
Figure 11.
Supramolecular assemblies of monodendronized fullerene derivatives 77a-h onto Hamilton-receptor functionalized host molecule 78.
Figure 11.
Supramolecular assemblies of monodendronized fullerene derivatives 77a-h onto Hamilton-receptor functionalized host molecule 78.
5. Molecular Self-Assembly of Fullerenes on Dendritic Scaffolds
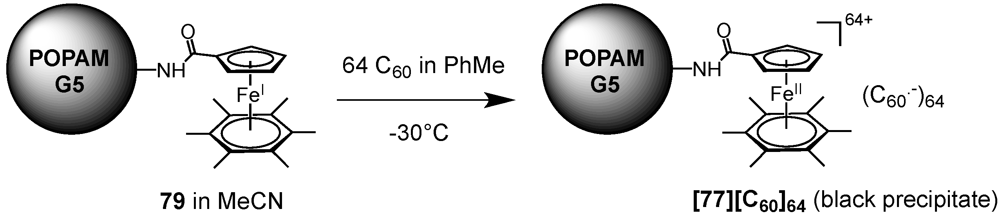
Similar to the work of Tomalia as described above for the divergent functionalization of dendrimer backbones, pristine C
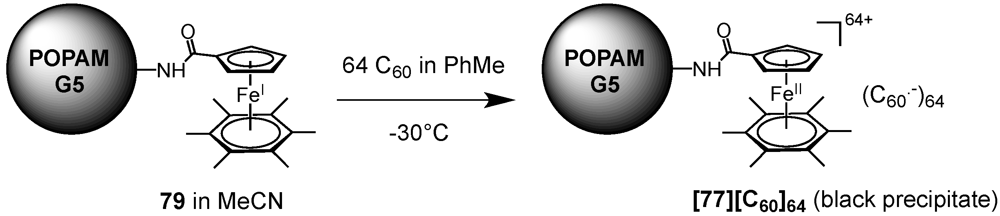
60 itself could also been assembled on dendritic templates by using purely electrostatic interactions. Astruc
et al. took advantage of the electrochemical properties of C
60 to assemble a fullerene-rich supramolecular dendritic structure [
52]. To an acetonitrile solution of one equivalent of dendrimer
79 consisting of a POPAM backbone and 64 peripheral ferrocene Fe(I) units was titrated at −30 °C a toluene solution containing pristine C
60. After the addition of 64 equivalents of fullerene meaning a 1:1 stoichiometry, the deep blue greenish color of the ferrocene-modified POPAM dendrimer
79 disappeared, while leaving a black precipitate
[79][C60]64 whose proposed structure is depicted in
Figure 12. This black-colored precipitate gave a clean quadrupole doublet for the Mössbauer spectrum whose parameters at 77 K were consistent with the presence of an Fe(II) sandwich complex. Its EPR spectrum recorded at 298 K shows the characteristic feature observed for a model compound obtained from the reaction of C
60 with the 19-electron complex [Fe(I)Cp(η
6-C
6Me
6)]. The authors concluded a probable C
60 reduction to its monoanion (
Scheme XIII), as designed for a process that is exergonic by 0.9 eV. The peripheral cationic Fe(II) units with their C
60- counteranion being very large, they were most likely located at the dendrimer periphery, presumably with rather tight ion pairs.
Figure 12.
Fifth-Generation polypropylene imine (POPAM) dendrimer decorated with 64 ferrocene Fe(II)/(C60·–) ion pairs.
Figure 12.
Fifth-Generation polypropylene imine (POPAM) dendrimer decorated with 64 ferrocene Fe(II)/(C60·–) ion pairs.
Scheme XIII.
Electrochemical process accompanying the formation of the ferrocene-C60 ion pairs.
Scheme XIII.
Electrochemical process accompanying the formation of the ferrocene-C60 ion pairs.
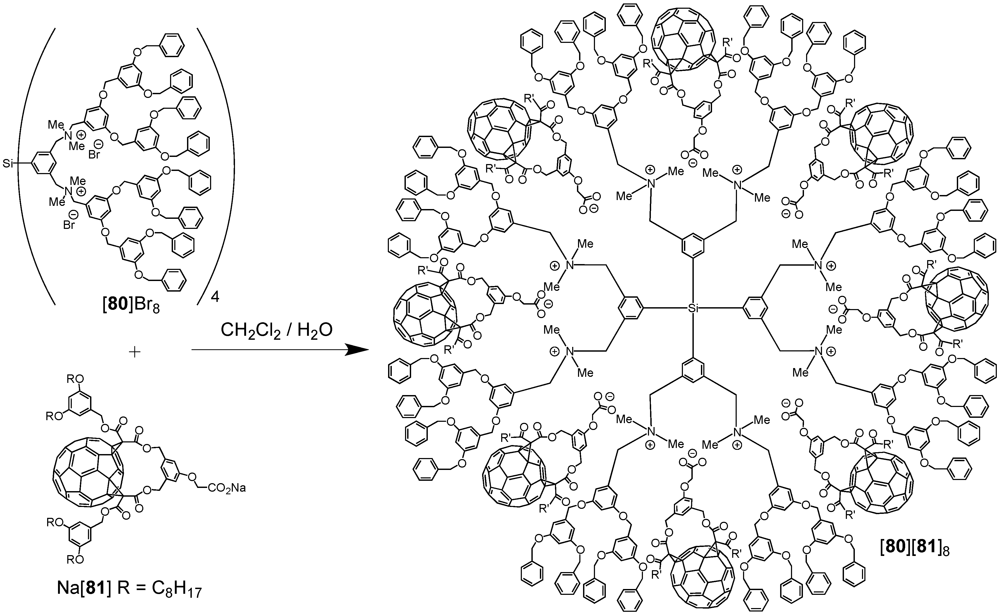
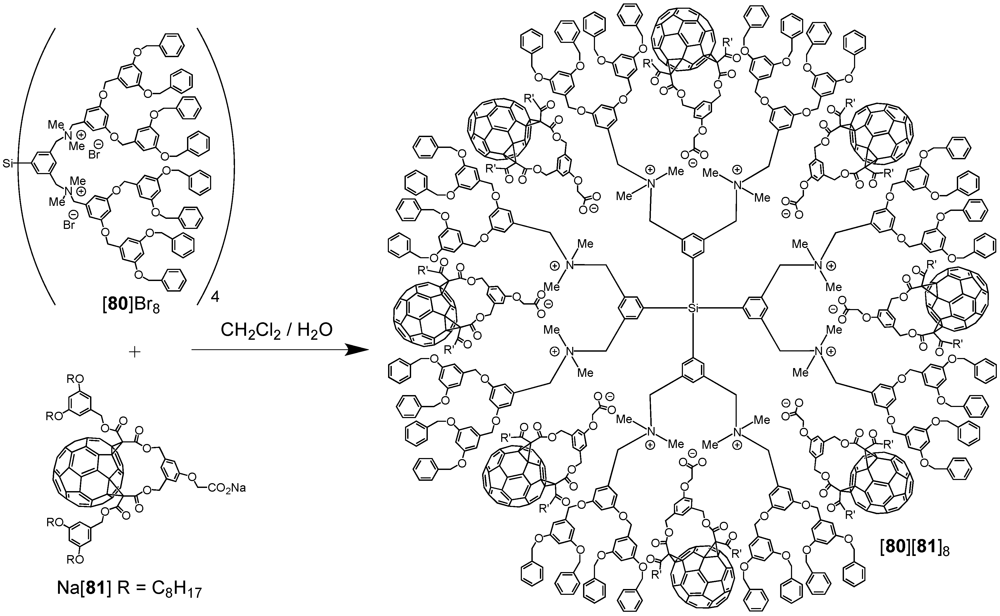
Another system based on electrostatic interactions has been reported by van Koten
et al. [
53]. A core-shell dendrimer with a cationic tetra[bis(benzylammonium)aryl] silane core has been used as a template for the assembly of fullerene-carboxylate derivatives via a straightforward anion exchange reaction of [
80]Br
8 with Na[
81] (
Scheme XIV). In contrast to dendrimer
80, the supramolecular fullerene-rich assembly [
80][
81]
8 was soluble in common organic solvents and its spectroscopic characterization could be easily achieved. In the
1H NMR spectrum of a solution of [
80][
81]
8 in CDCl
3, the specific signals of both [
80]
8+ and [
81]
- could clearly be observed. Furthermore, specific peak integrals showed that the octa-cationic dendritic moiety [
80]
8+ and the anions [
81]
− were present in a 1 to 8 molar ratio. In order to establish the molecular weight of the host-guest assembly, gel permeation chromatography coupled to a low angle laser light scattering (GPC/LALLS) instrument was performed using THF as eluent. The chromatogram displayed three peaks. The first one corresponded to the 1:8 host-guest octa-fullero-dendrimer assembly [
80][
81]
8. The two additional peaks with higher molecular weights were assigned to superstructures consisting of aggregated assemblies derived from [
80][
81]
8. Such behavior was commonly observed during the GPC analysis of poly-ionic macromolecules when an organic solvent was used as eluent. Importantly, no peaks corresponding to compounds with a molecular weight lower than [
80][
81]
8 were detected, which further substantiates the formation of a stoichiometric assembly between octa-cationic [
80]
8+ and eight [
81]
− anions.
Scheme XIV.
Anion exchange reaction of [80]Br8 with Na[81] leading to the fullerene-rich dendrimer [80][81]8.
Scheme XIV.
Anion exchange reaction of [80]Br8 with Na[81] leading to the fullerene-rich dendrimer [80][81]8.
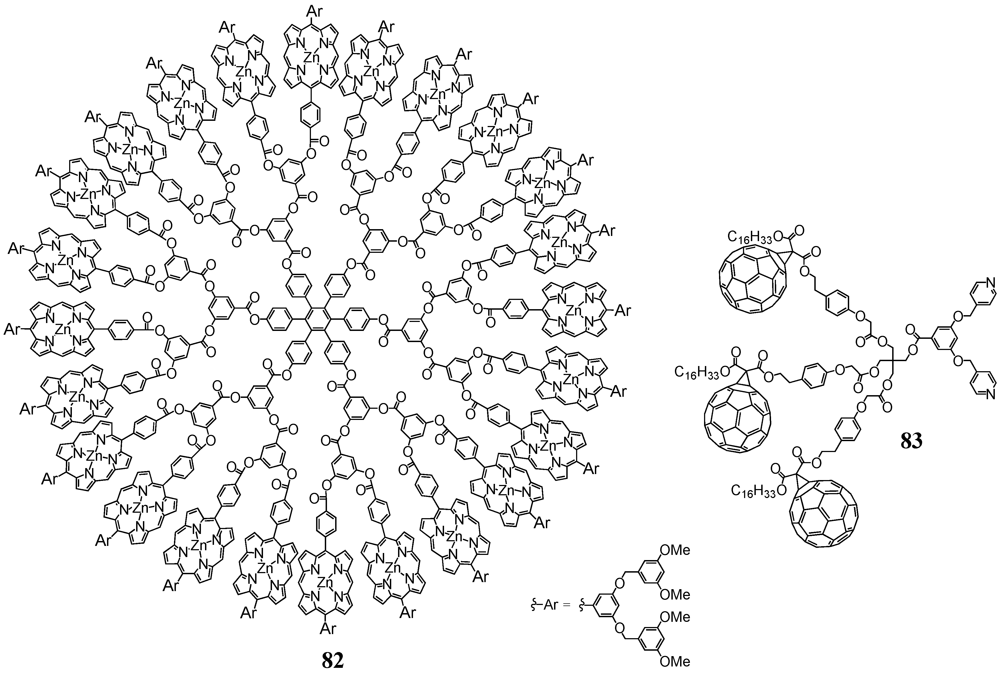
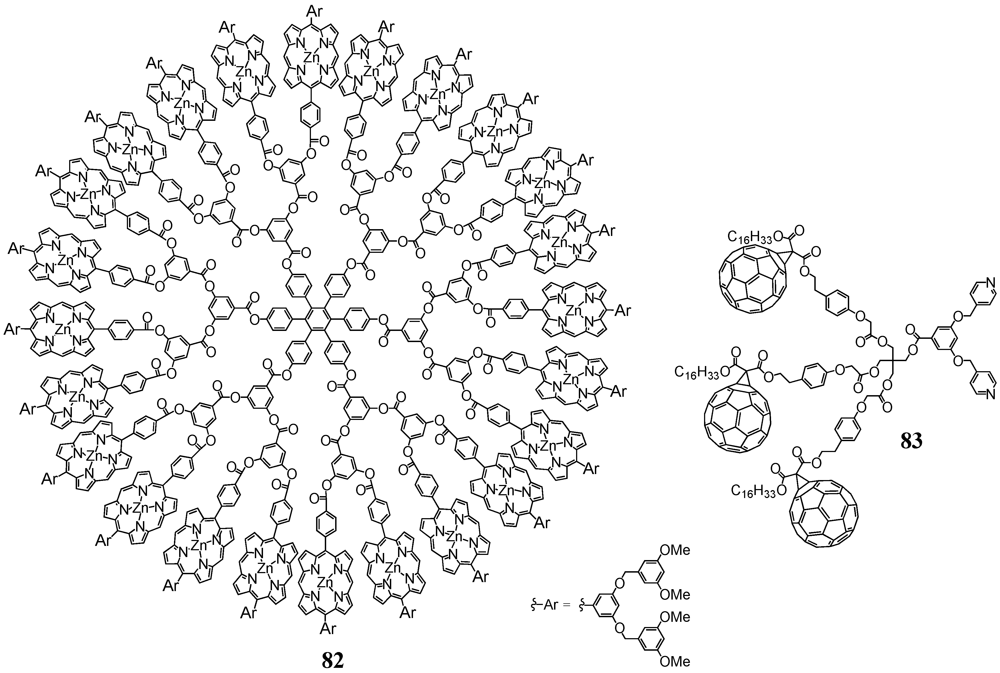
Aida
et al. contributed to the field by introducing the metal-assisted coordination of modified fullerene derivatives to a multi-porphyrin dendrimer [
54]. The synthetic protocol involved the preparation of a methanofullerene equipped with a long alkyl chain on one side and a spacered carboxylic acid on the other. This precursor then served for coupling to mono-, di- and trivalent bipyridine-terminated alcohols using DCC/DPTS as reagents for the esterification reactions to provide the three different ligands in moderate to good yields. The structure of the ligand
83 containing three C
60 moieties is depicted in
Figure 13. On the other hand, large dendritic architecture
82 was constructed for subsequent complexation. This dendrimer
82 consisted of a hexaphenylbenzene core unit, a polarylester fractal part, 24 zinc porphyrins and at the outside relatively small appended polyarylether dendrons to confer solubility to the system. All bipyridines have been demonstrated to strongly bind to the zinc porphyrins with an average binding affinity, as estimated by simply assuming a one-to-one coordination between the individual zinc porphyrin and pyridine units, of 1.2 × 10
6 M
−1. This value was more than two orders of magnitude greater than association constants reported for monodentate coordination between zinc porphyrins and pyridine derivatives and could be ascribed to the simultaneous coordination of two Zn centers of
82 by the two pyridine moieties of for instance
83. An in-depth study of the photophysical properties of this photactive device mirrored an almost quantitative intramolecular photoinduced electron transfer from the photoexcited porphyrins to the C
60 units as evidenced by means of steady-state emission spectroscopy and nanosecond flash photolysis measurements. Importantly, the ratio between the rate constants of charge separation and recombination for
[82][81]12 was found to be more than an order of magnitude greater than those reported for preceding porphyrin–fullerene supramolecular dyads and triads. The larger number of the C
60 units in large dendritic architecture
[82][81]12 could enhance the probability of electron transfer from the zinc porphyrin units and probably also the opportunity for this electron transfer through efficient energy migration along the densely packed Zn(II) porphyrin array.
Figure 13.
Polyporphyrin dendrimer 82 and bipyridine 83 for metal-ligand complexation studies.
Figure 13.
Polyporphyrin dendrimer 82 and bipyridine 83 for metal-ligand complexation studies.
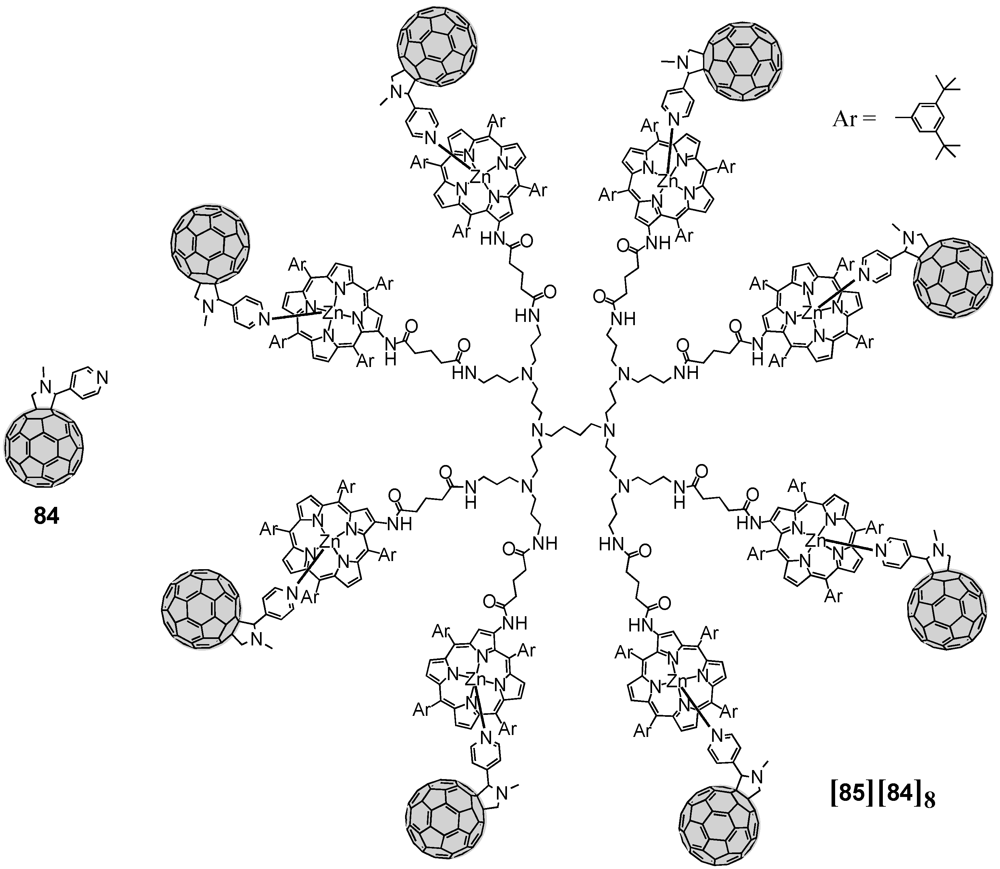
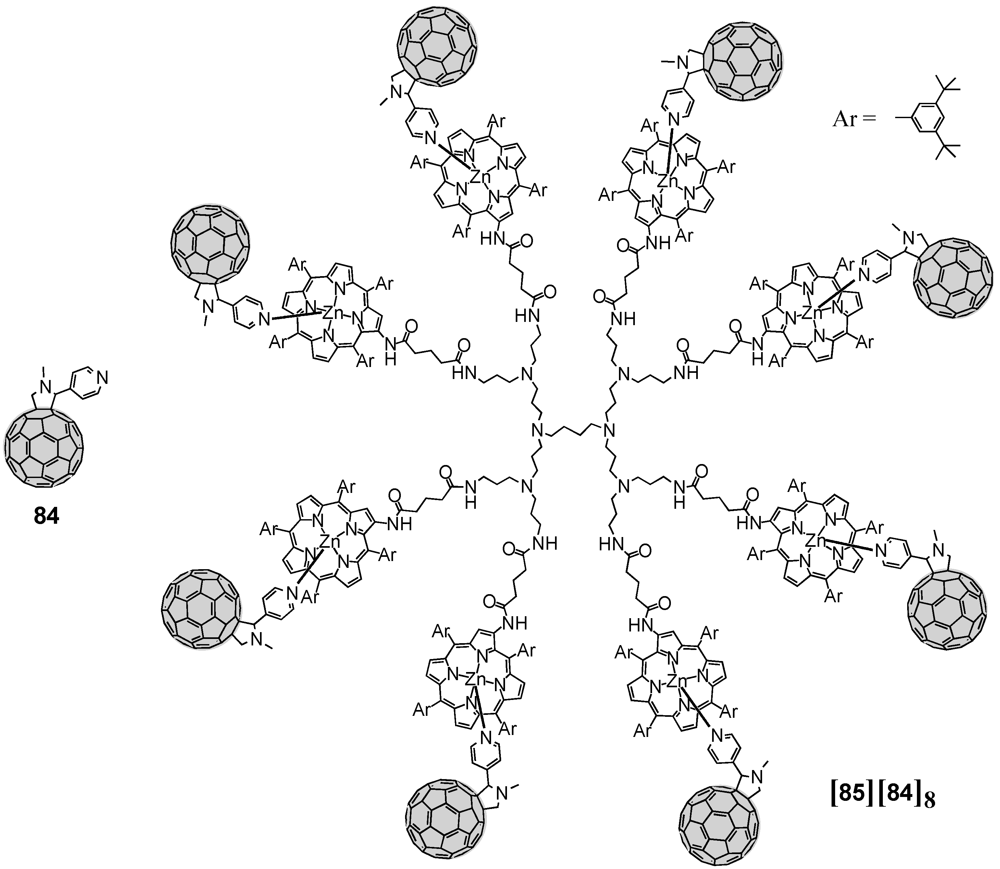
Very recently, large dendrimer architectures with up to 16 porphyrins (the second generation
85 is depicted in
Figure 14) discussed before were also engaged in the coordination of fullerene spheres equipped with one pyridine function [
55]. According to this, fulleropyrrolidine
84 bearing a pyridine has been obtained upon reaction of C
60 with
p-pyridine carboxaldehyde applying Prato conditions. Complexation of the pyridine subunit of fulleropyrrolidine
84 to the multiple zinc porphyrins of for instance
85 led to the supramolecular photoactive dendrimer assemblies. Multiple photosynthetic reaction centers combined with antenna complexes have thus been successfully constructed. The excited energy migration occurs efficiently between porphyrin units followed by charge separation in the supramolecular complex of the largest G3 dendrimer with pyridine-functionalized fulleropyrrolidine
84 as a result of the dendrimer effect. Measuring the charge-separated state of the supramolecular complex produced upon laser excitation had a particular long lifetime of 0.25 ms. Similarly, Ogawa
et al. exploited the same fulleropyrrolidine
84 for the complexation with zinc porphyrins [
56]. Upon the preparation of a π-conjugated porphyrin polymer, this molecular wire has been subjected to the coordination by
84. The electrical devices fabricated from the two electroactive units showed photo-responsive conduction with a tunneling mechanism at low temperatures and thermionic emission at high temperatures.
Figure 14.
Multiple metal-ligand complexation between polyporphyrin dendrimer 85 and fulleropyrrolidine 84.
Figure 14.
Multiple metal-ligand complexation between polyporphyrin dendrimer 85 and fulleropyrrolidine 84.
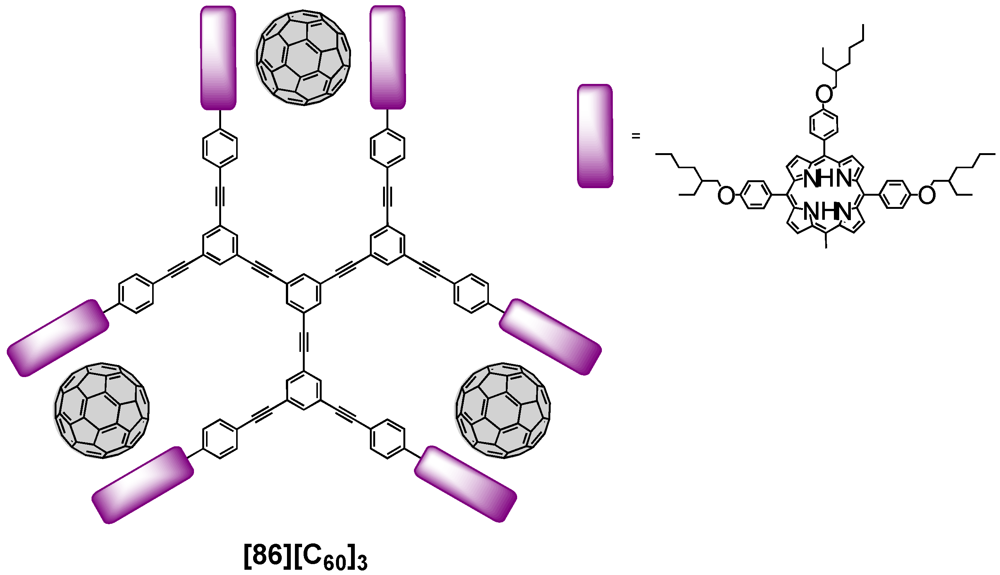
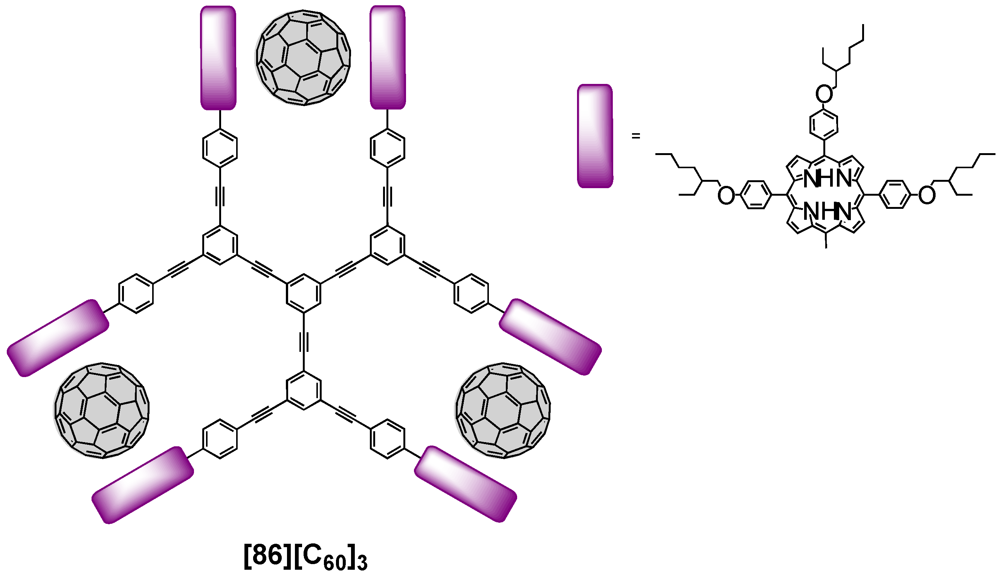
The supramolecular formation of host/guest complexes has also been described by Shinkai
et al. [
57]. They prepared a rigid star-shaped
D3-symmetric receptor that carried six peripheral zinc porphyrin moieties linked through phenylacetylene spacers (
Figure 15). This dendritic receptor had three rotational axes responsible for the spatial arrangement of the porphyrin-derived macrocycles. Addition of C
60 to a solution containing dendritic host
86 led to the formation of sandwich complexes in an allosteric manner resulting from the tweezering of one fullerene molecule by two zinc porphyrins. The complexation thereby strongly affected the molecular shape as the host-guest complexes suppressed rotational freedom upon formation. Binding studies furthermore evidenced considerably enhanced association constants owing to the binding in a positive allosteric manner.
Figure 15.
Rigid star-shaped receptor-fullerene complex [86][C60]3.
Figure 15.
Rigid star-shaped receptor-fullerene complex [86][C60]3.
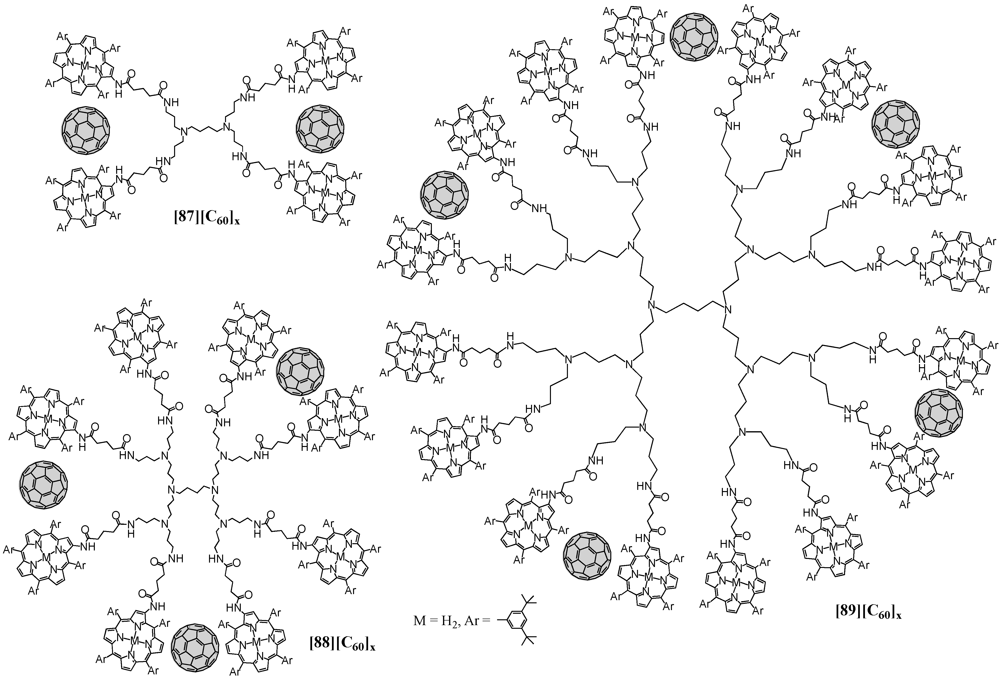
Similarly, a less rigid system has been introduced aiming at the first organic photovoltaic system using supramolecular complexes of porphyrin dendrimers with fullerenes [
58,
59,
60,
61]. Accordingly, terminal amines of generation one to three POPAM dendrimers have been modified through grafting of porphyrin derivatives endowed with an activated ester. Porphyrin dendrimers
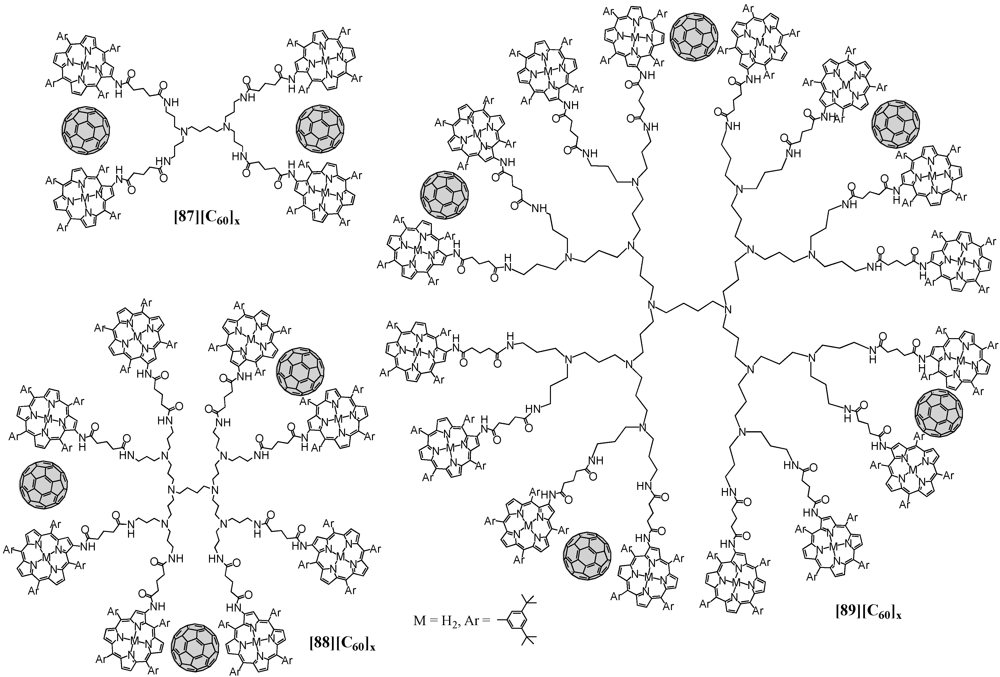
87-
89 were then employed in the formation of supramolecular assemblies with C
60 and using an acetonitrile/toluene mixed solvent system led to the clusterization of the nanoscale ensembles (
Figure 16). They have then been deposited onto nanostructured SnO
2 electrodes to show an efficient photoresponse in the visible and near-infrared regions, as well as a high photoenergy conversion efficiency due to the effective electron transfer from the excited porphyrin to fullerene within the dendritic matrix.
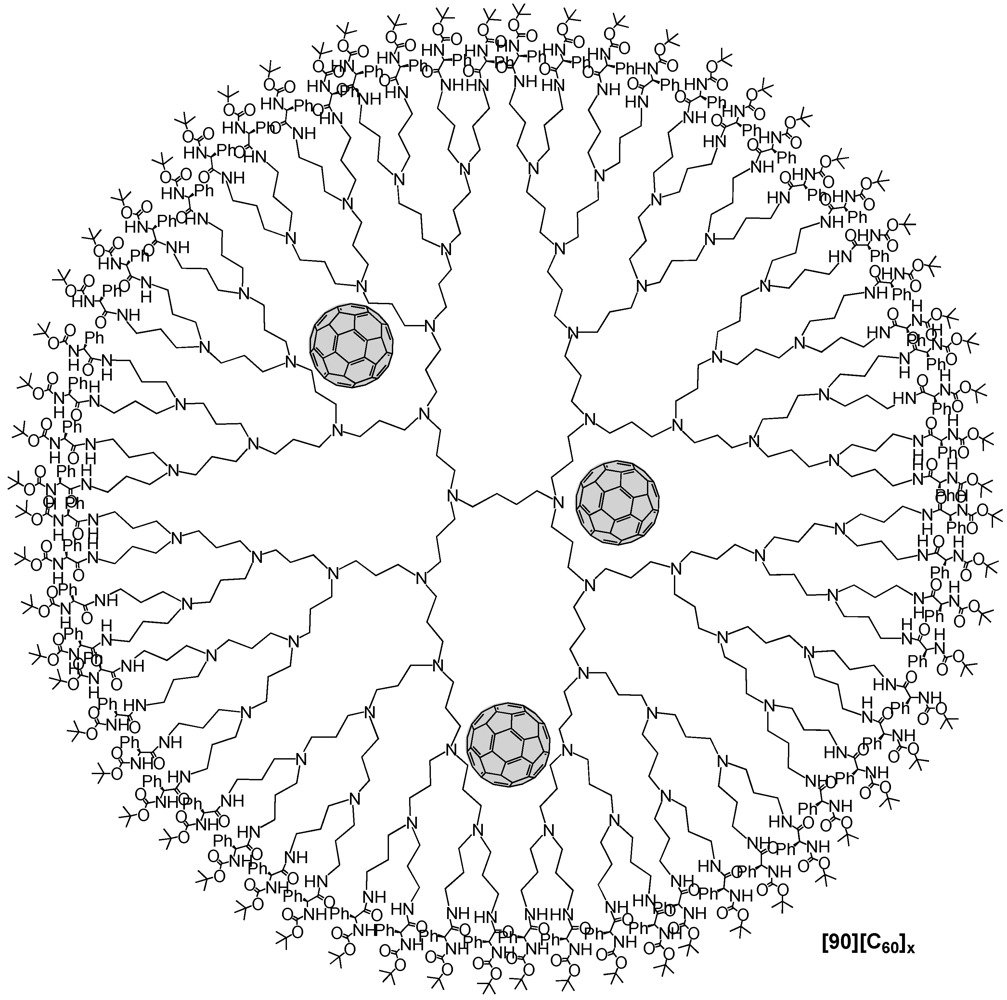
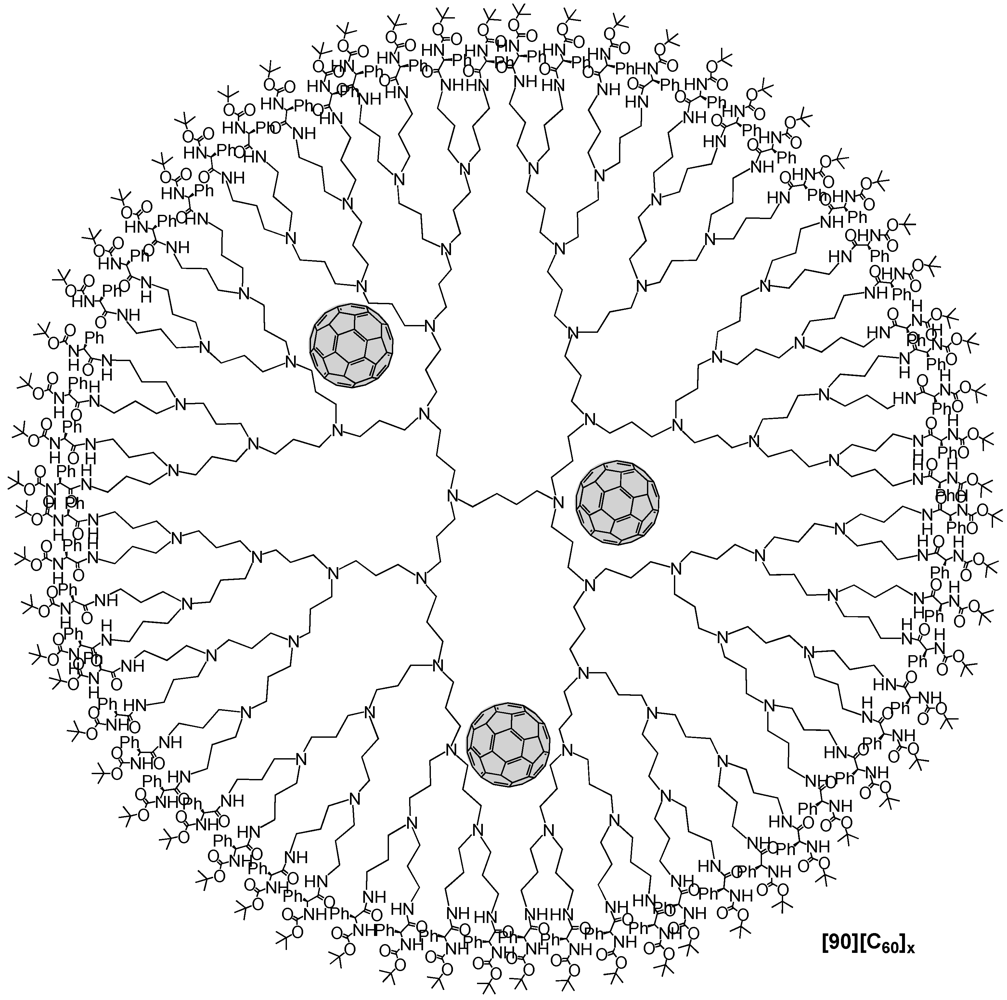
Márquez
et al. presented the inclusion of C
60 molecules within the internal cavities of a fifth generation POPAM dendrimer with 64 terminal amine groups [
62]. Following the procedure by Meijer, the product was obtained by adding C
60 to a solution of a POPAM fifth dendrimer in CH
2Cl
2 and the presence of triethyamine. The resulting mixture was stirred for one day upon which was added
N-t-Boc-L-phenylalanine
N-hydroxysuccinimide ester as bulky substituent in order to build a protecting shell at the dendrimer surface. After additional stirring for 24 h, the crude has been purified and characterized by MALDI-TOF mass spectrometry and UV-vis spectroscopy. It turned out that the final complex structure [
90][C
60]
x contained an average number of approx. 4 fullerenes that were encapsulated within the dendrimer voids (
Figure 17). The dense external shell created with the
N-
t-Boc-protected amino acid was thus successfully able to hermetically lock the internal space preventing the trapped fullerene molecules to escape. Interestingly, the fluorescence spectrum revealed that the confinement into a dendritic structure had a noticeable influence on the emission spectra of the guest molecule since under these conditions the emission band of C
60 experienced a strong bathochromic shift of
ca. 132 nm with respect to the emission of C
60 in solution (λ
em = 675 nm after
λexc = 285 nm). This impressive red shift turned out to be solvent-dependent due to favorable electronic communication between host and guest molecules that could be stronger due to the different solvent polarities that significantly affected the macromolecular dendrimer host structure.
Another approach towards supramolecular structures has been introduced by Kono
et al. [
63]. Decoration of the surface of G4-PAMAM dendrimer by
β-cyclodextrins (
β-CD) through reaction with the corresponding monotosylated
β-CD followed by the coupling of monomethoxy polyethylene glycol PEG 4-nitrophenyl carbonate gave the target dendrimer scaffold.
1H NMR studies revealed the average number of bound
β-CDs to be 29. It is a well-known phenomenon that cyclodextrins form non-covalent associates with fullerenes. For instance, in the case of
β-CD the ball-shaped C
60 is encapsulated within two cyclic oligosaccharides. Addition of pristine fullerene led indeed to the proposed host-guest conjugates with concentration reaching values of 2.8
μM aqueous fullerene solutions.
Figure 16.
Noncovalent ensembles of C60 with first to third generation POPAM dendrimers 87-89 decorated at the surface by multiple porphyrin macrocycles.
Figure 16.
Noncovalent ensembles of C60 with first to third generation POPAM dendrimers 87-89 decorated at the surface by multiple porphyrin macrocycles.
Figure 17.
Schematic representation of encapsulated fullerene moieties within the backbone of a POPAM G5 dendrimer according to Márquez
et al [
62].
Figure 17.
Schematic representation of encapsulated fullerene moieties within the backbone of a POPAM G5 dendrimer according to Márquez
et al [
62].
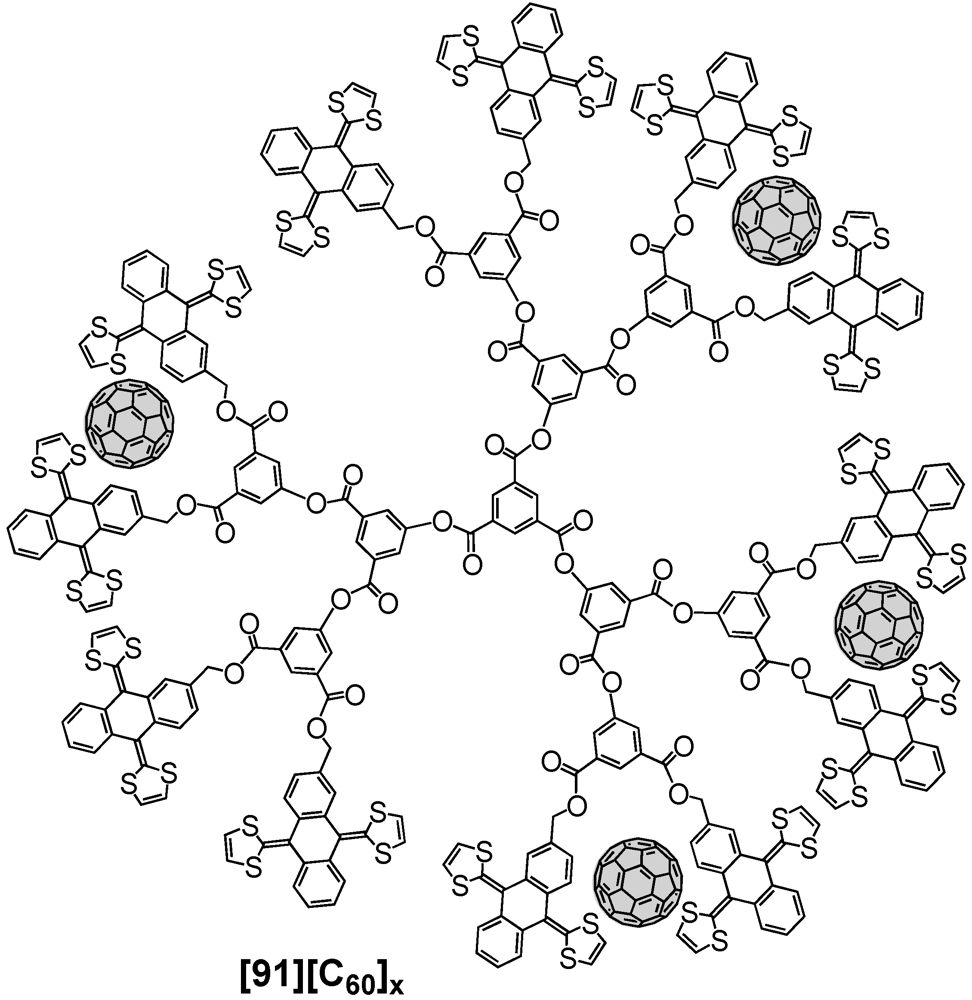
Martín
et al. also reported the tweezering of fullerene by extended tetrathiafulvalene (ex-TTF) units [
64]. The dendrimer skeleton has been prepared employing the general concept of repetitive synthetic sequences to provide the polyester backbone with either 6, 12 (
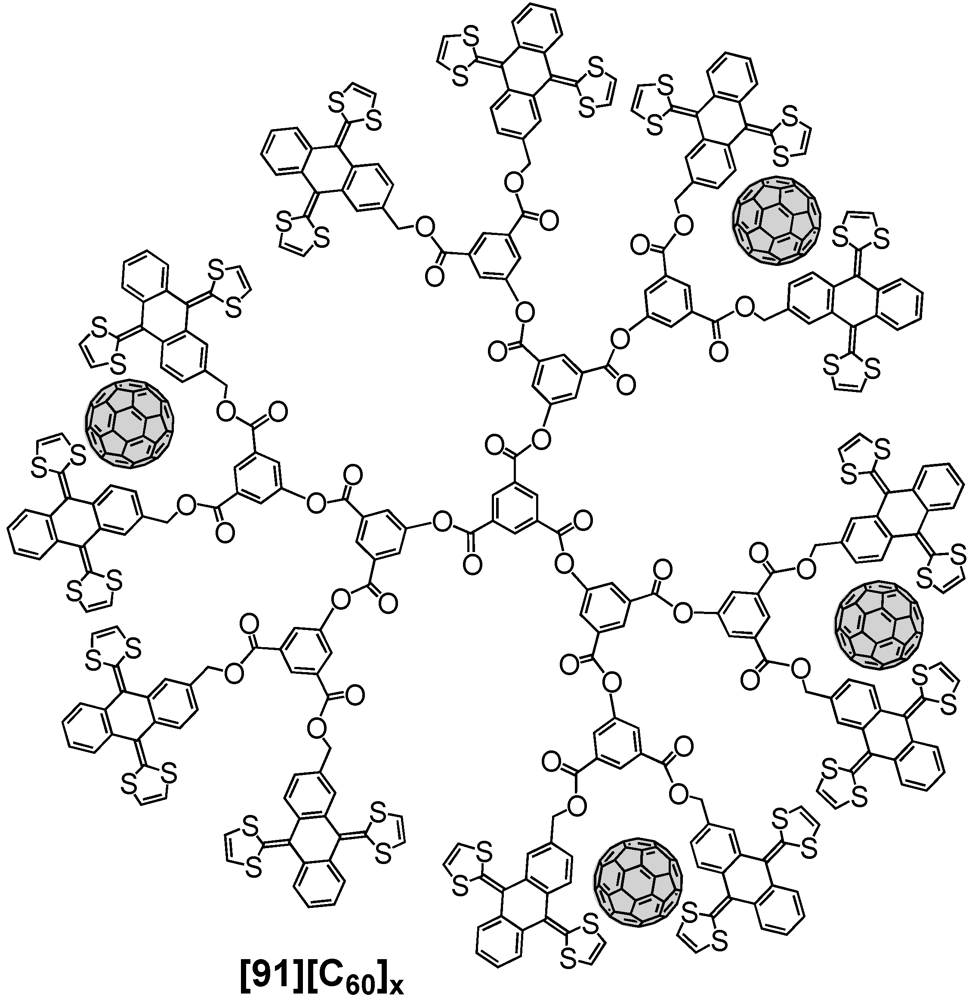
91) or 24 ex-TTF moieties at the dendrimer surface. In agreement with previous findings of the same group, tweezering of fullerene was envisaged and indeed, it was found that regardless of the dendrimer size C
60 could be accommodated. A schematic representation of a possible supramolecular associate of G3 ex-TTF dendrimer 90 and several C
60 units is depicted in
Figure 18. Initial UV/vis titration studies with the first generation species evidenced a positive cooperative binding of three equivalents of C
60 by the present six ex-TTF units. Similar experiments have then been conducted with the next higher generation structures, which exhibited a similar degree of cooperativity in the binding events. The authors explained the cooperative effect most probably to arise from the need to disassemble the dendrimers prior to complexation of C
60.
Figure 18.
Supramolecular complexation of C60 units at the surface of an ex-TFF appended third generation dendrimer 91.
Figure 18.
Supramolecular complexation of C60 units at the surface of an ex-TFF appended third generation dendrimer 91.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}