Effects of Emulsion-Based Resonant Infrared Matrix Assisted Pulsed Laser Evaporation (RIR-MAPLE) on the Molecular Weight of Polymers

Abstract
:
1. Introduction
2. Results and Discussion
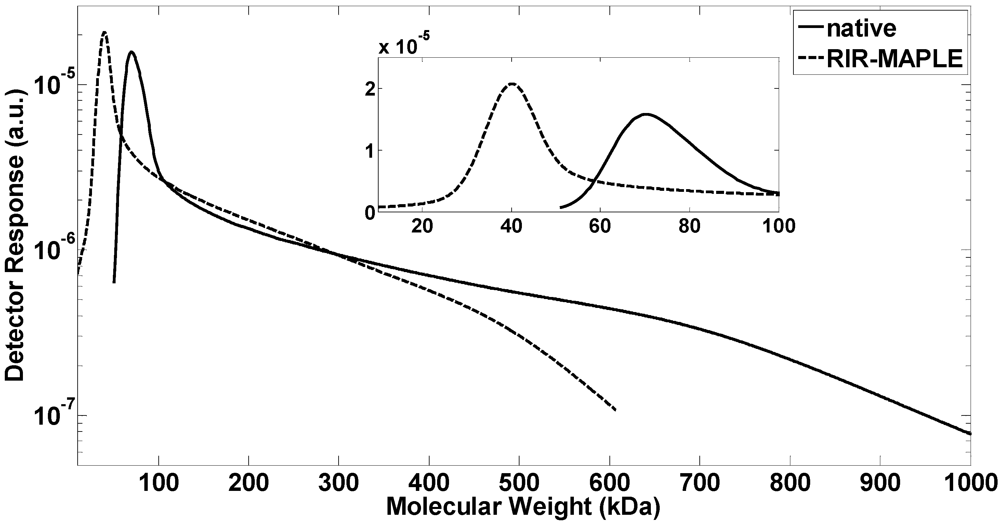
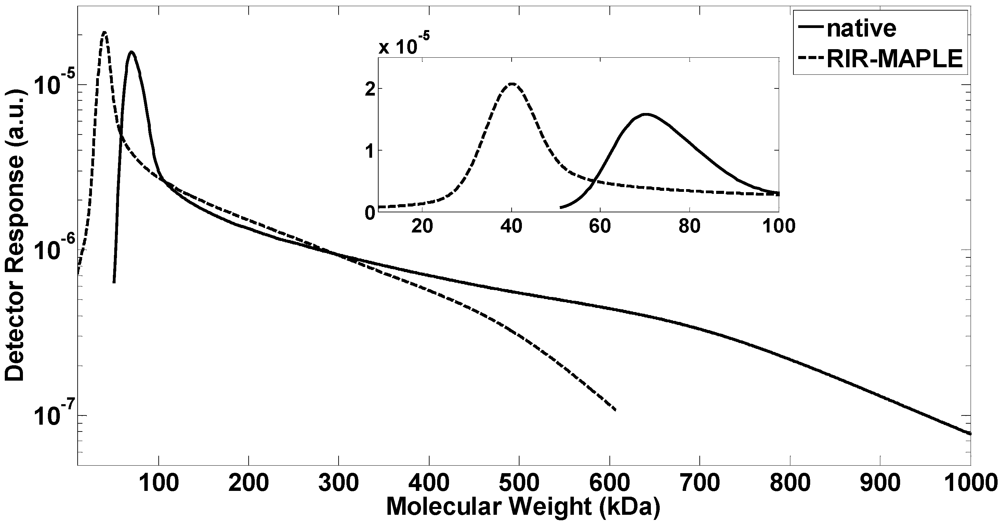
2.1. GPC Measurement of Polymer Molecular Weight

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
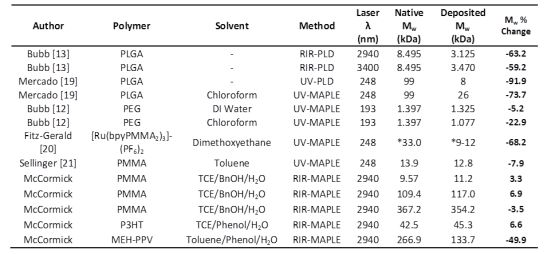
| Author | Polymer | Solvent | Method | Laser λ (nm) | Native Mw (kDa) | Deposited Mw (kDa) | Mw % Change |
|---|---|---|---|---|---|---|---|
| Bubb [13] | PLGA | - | RIR-PLD | 2940 | 8.495 | 3.125 | −63.2 |
| Bubb [13] | PLGA | - | RIR-PLD | 3400 | 8.495 | 3.470 | −59.2 |
| Mercado [19] | PLGA | - | UV-PLD | 248 | 99 | 8 | −91.9 |
| Mercado [19] | PLGA | Chloroform | UV-MAPLE | 248 | 99 | 26 | −73.7 |
| Bubb [12] | PEG | DI Water | UV-MAPLE | 193 | 1.397 | 1.325 | −5.2 |
| Bubb [12] | PEG | Chloroform | UV-MAPLE | 193 | 1.397 | 1.077 | −22.9 |
| Fitz-Gerald [20] | [Ru(bpyPMMA2)3]-(PF6)2 | Dimethoxyethane | UV-MAPLE | 248 | *33.0 | *9–12 | −68.2 |
| Sellinger [21] | PMMA | Toluene | UV-MAPLE | 248 | 13.9 | 12.8 | −7.9 |
| McCormick | PMMA | TCE/BnOH/H2O | RIR-MAPLE | 2940 | 9.57 | 11.2 | 3.3 |
| McCormick | PMMA | TCE/BnOH/H2O | RIR-MAPLE | 2940 | 109.4 | 117.0 | 6.9 |
| McCormick | PMMA | TCE/BnOH/H2O | RIR-MAPLE | 2940 | 367.2 | 354.2 | −3.5 |
| McCormick | P3HT | TCE/Phenol/H2O | RIR-MAPLE | 2940 | 42.5 | 45.3 | 6.6 |
| McCormick | MEH-PPV | Toluene/Phenol/H2O | RIR-MAPLE | 2940 | 266.9 | 133.7 | −49.9 |
| Author | Polymer | Solvent | Method | Laser λ (nm) | Native PDI | Deposited PDI |
|---|---|---|---|---|---|---|
| Bubb [13] | PLGA | - | RIR-PLD | 2940 | 1.22 | 3.05 |
| Bubb [13] | PLGA | - | RIR-PLD | 3400 | 1.22 | 2.63 |
| Mercado [19] | PLGA | - | UV-PLD | 248 | 1.73 | 2.70 |
| Mercado [19] | PLGA | Chloroform | UV-MAPLE | 248 | 1.73 | 2.00 |
| Bubb [12] | PEG | DI Water | UV-MAPLE | 193 | 1.02 | 1.06 |
| Bubb [12] | PEG | Chloroform | UV-MAPLE | 193 | 1.02 | 1.07 |
| Fitz-Gerald [20] | [Ru(bpyPMMA2)3]-(PF6)2 | Dimethoxyethane | UV-MAPLE | 248 | * | * |
| Sellinger [21] | PMMA | Toluene | UV-MAPLE | 248 | 1.71 | 1.53 |
| McCormick | PMMA | TCE/BnOH /H2O | RIR-MAPLE | 2940 | 1.12 | 1.04 |
| McCormick | PMMA | TCE/BnOH/H2O | RIR-MAPLE | 2940 | 1.15 | 1.19 |
| McCormick | PMMA | TCE/BnOH/H2O | RIR-MAPLE | 2940 | 1.45 | 1.59 |
| McCormick | P3HT | TCE/Phenol /H2O | RIR-MAPLE | 2940 | 2.20 | 2.04 |
| McCormick | MEH-PPV | Toluene/Phenol/H2O | RIR-MAPLE | 2940 | 2.29 | 2.18 |
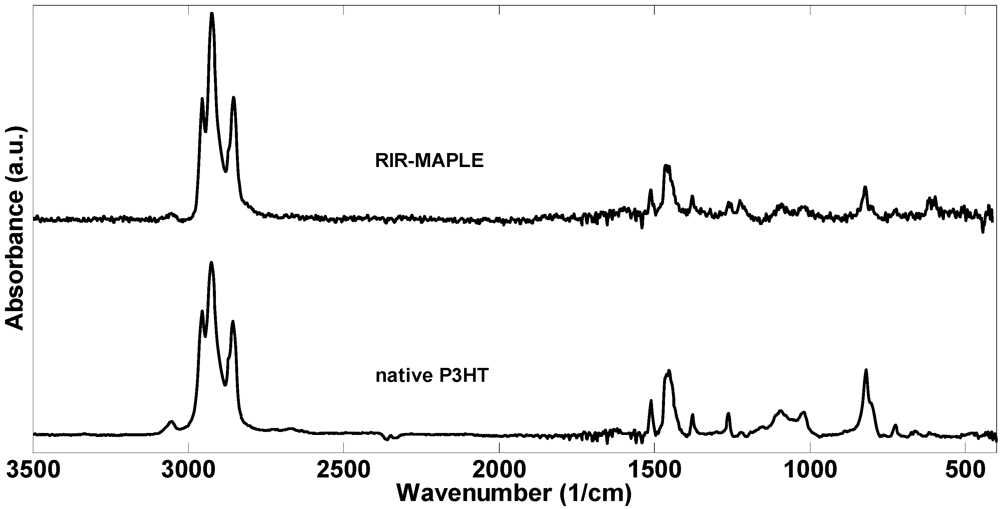
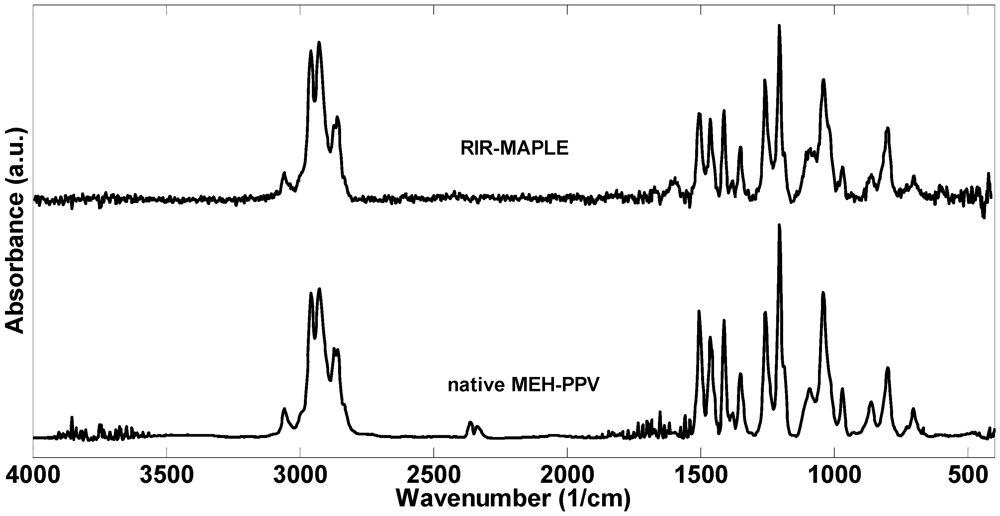
2.1. 1H NMR Spectroscopy and FTIR Spectroscopy of Polymers Deposited by RIR-MAPLE
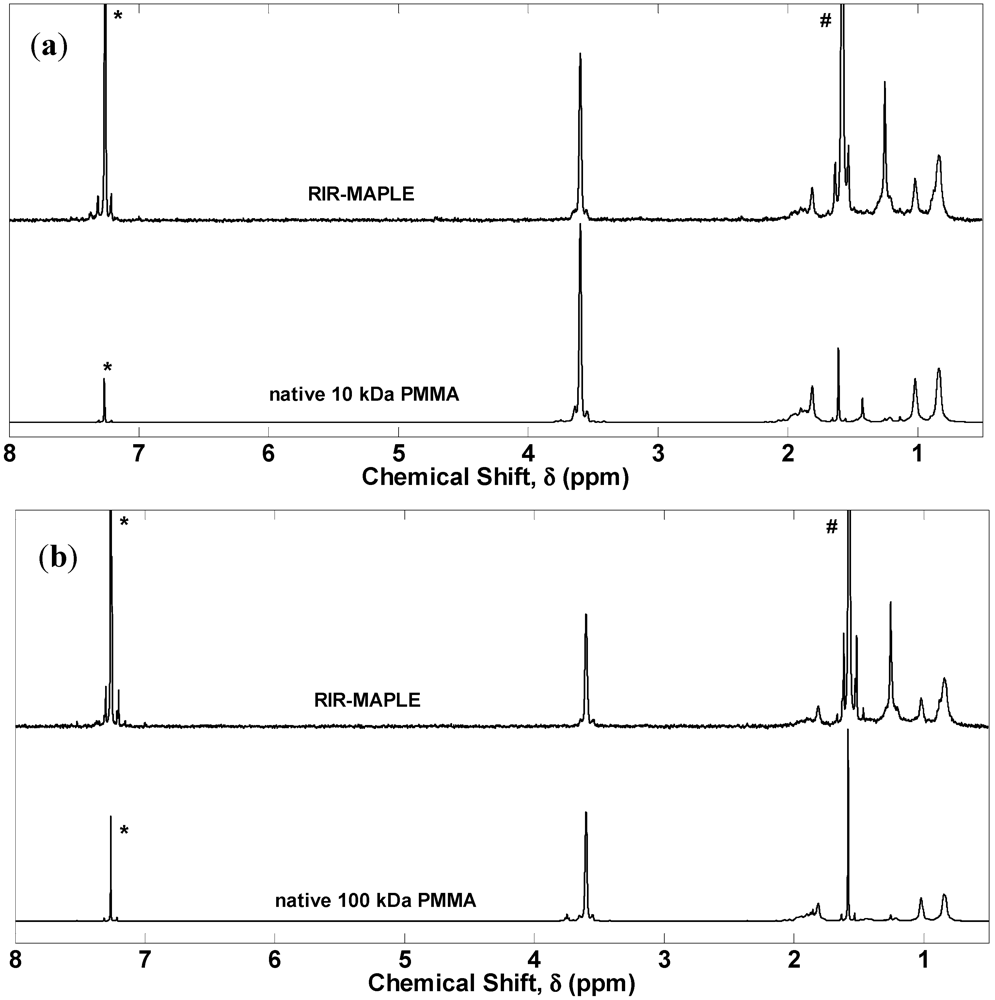
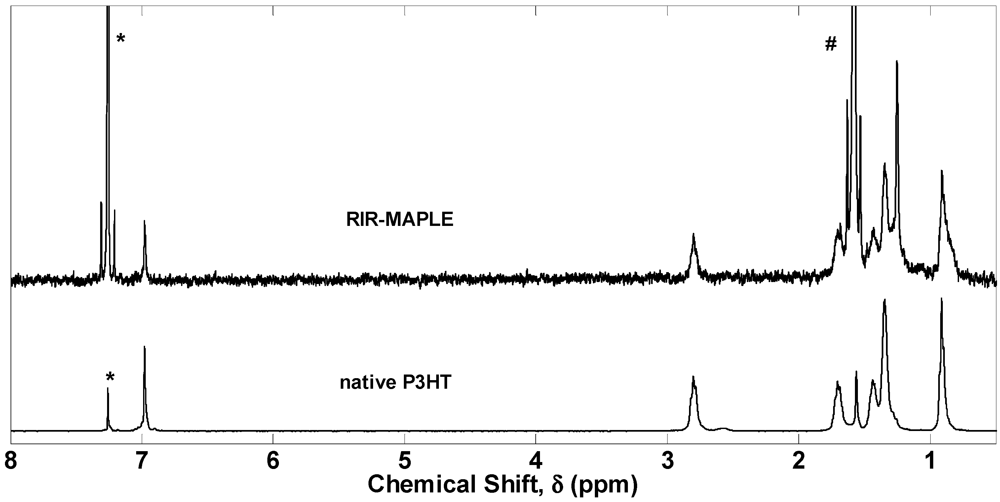
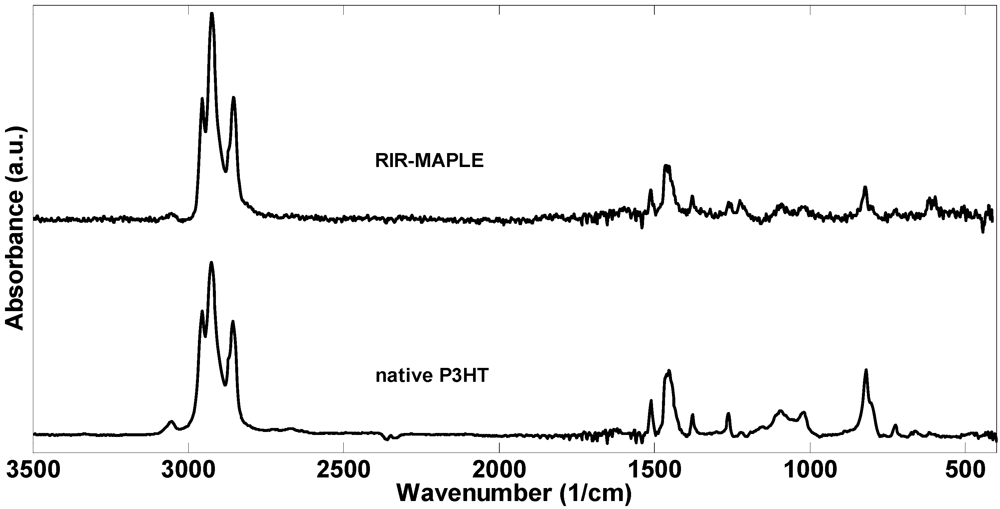
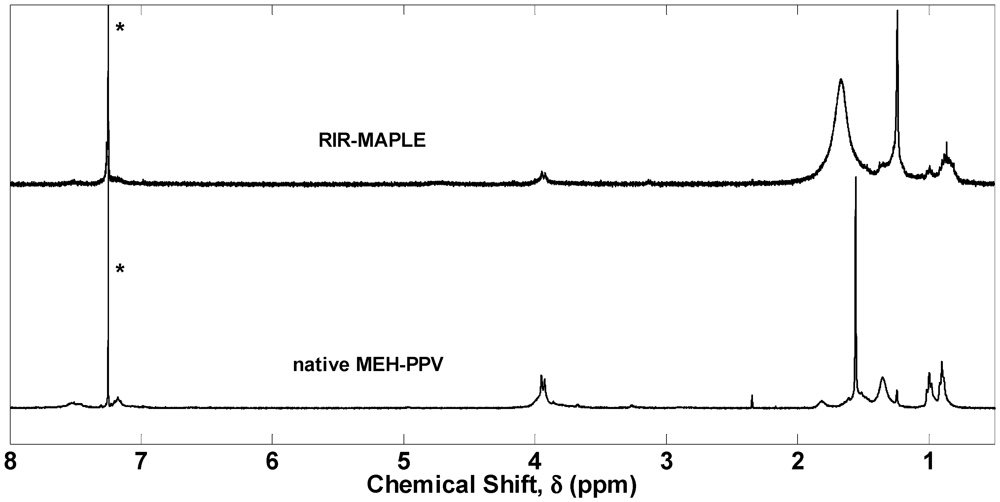
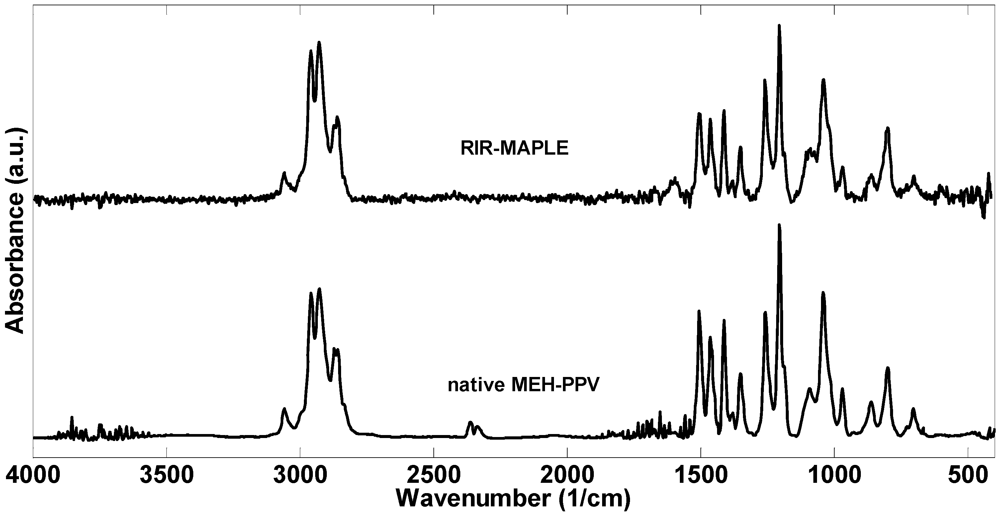
3. Experimental Section
4. Conclusions
Acknowledgments
References
- Chochos, C.L.; Choulis, S.A. How the structural deviations on the backbone of conjugated polymers influence their optoelectronic properties and photovoltaic performance. Prog. Polym. Sci. 2011, 36, 1326–1414. [Google Scholar] [CrossRef]
- Kline, R.J.; McGehee, M.D.; Kadnikova, E.N.; Liu, J.; Fréchet, J.M.J. Controlling the field-effect mobility of regioregular polythiophene by changing the molecular weight. Adv. Mater. 2003, 15, 1519–1522. [Google Scholar] [CrossRef]
- Zen, A.; Pflaum, J.; Hirschmann, S.; Zhuang, W.; Jaiser, F.; Asawapirom, U.; Rabe, J.; Scherf, U.; Neher, D. Effect of molecular weight and annealing of poly(3-hexylthiophene)s on the performance of organic field-effect transistors. Adv. Funct. Mater. 2004, 14, 757–764. [Google Scholar] [CrossRef]
- Schilinsky, P.; Asawapirom, U.; Scherf, U.; Biele, M.; Brabec, C.J. Influence of the molecular weight of poly(3-hexylthiophene) on the performance of bulk heterojunction solar cells. Chem. Mater. 2005, 17, 2175–2180. [Google Scholar]
- Hosoi, K.; Mori, T.; Mizutani, T.; Yamamoto, T.; Kitamura, N. Effects of molecular weight on polyfluorene-based polymeric light emitting diodes. Thin Solid Films 2003, 438–439, 201–205. [Google Scholar]
- Pate, K.R.; Lantz, A.; Dhawan, T.; Vo-Dinh, R.; Stiff-Roberts, A.D. Resonant infrared matrix-assisted pulsed laser evaporation of inorganic nanoparticles and organic/inorganic hybrid nanocomposites. AIP Conf. Proc. 2010, 1278, 813–823. [Google Scholar]
- Pate, R.; Stiff-Roberts, A.D. The impact of laser-target absorption depth on the surface and internal morphology of matrix-assisted pulsed laser evaporated conjugated polymer thin films. Chem. Phys. Lett. 2009, 477, 406–410. [Google Scholar] [CrossRef]
- Pate, R.; McCormick, R.; Chen, L.; Zhou, W.; Stiff-Roberts, A. Rir-maple deposition of conjugated polymers for application to optoelectronic devices. Appl. Phys. A 2011. [Google Scholar] [CrossRef]
- Pate, R.; Lantz, K.R.; Stiff-Roberts, A.D. Tabletop resonant infrared matrix assisted pulsed laser evaporation of light emitting organic thin films. IEEE J. Quantum. Elect. 2008, 14, 1022–1030. [Google Scholar] [CrossRef]
- Pate, R.; Lantz, K.R.; Stiff-Roberts, A.D. Resonant infrared matrix-assisted pulsed laser evaporation of cdse colloidal quantum dot/poly[2-methoxy-5-(2′-ethylhexyloxy)-1,4-(1-cyano vinylene)phenylene] hybrid nanocomposite thin films. Thin Solid Films 2009, 517, 6798–6802. [Google Scholar] [CrossRef]
- Piqué, A.; McGill, R.A.; Chrisey, D.B.; Leonhardt, D.; Mslna, T.E.; Spargo, B.J.; Callahan, J.H.; Vachet, R.W.; Chung, R.; Bucaro, M.A. Growth of organic thin films by the matrix assisted pulsed laser evaporation (maple) technique. Thin Solid Films 1999, 355–356, 536–541. [Google Scholar]
- Bubb, D.M.; Wu, P.K.; Horwitz, J.S.; Callahan, J.H.; Galicia, M.; Vertes, A.; McGill, R.A.; Houser, E.J.; Ringeisen, B.R.; Chrisey, D.B. The effect of the matrix on film properties in matrix-assisted pulsed laser evaporation. J. Appl. Phys. 2002, 91, 2055–2058. [Google Scholar]
- Bubb, D.M.; Toftmann, B.; Haglund, J.R.F.; Horwitz, J.S.; Papantonakis, M.R.; McGill, R.A.; Wu, P.W.; Chrisey, D.B. Resonant infrared pulsed laser deposition of thin biodegradable polymer films. Appl. Phys. A 2002, 74, 123–125. [Google Scholar]
- Bubb, D.M.; O’Malley, S.M.; Antonacci, C.; Simonson, D.; McGill, R.A. Matrix-assisted laser deposition of a sorbent oligomer using an infrared laser. J. Appl. Phys. 2004, 95, 2175–2177. [Google Scholar]
- Toftmann, B.; Papantonakis, M.R.; Auyeung, R.C.Y.; Kim, W.; O’Malley, S.M.; Bubb, D.M.; Horwitz, J.S.; Schou, J.; Johansen, P.M.; Haglund, R.F. Uv and rir matrix assisted pulsed laser deposition of organic meh-ppv films. Thin Solid Films 2004, 453–454, 177–181. [Google Scholar]
- Chrisey, D.B.; Piqué, A.; McGill, R.A.; Horwitz, J.S.; Ringeisen, B.R.; Bubb, D.M.; Wu, P.K. Laser deposition of polymer and biomaterial films. Chem. Rev. 2003, 103, 553–576. [Google Scholar] [CrossRef]
- Cristescu, R.; Mihaiescu, D.; Socol, G.; Stamatin, I.; Mihailescu, I.N.; Chrisey, D.B. Deposition of biopolymer thin films by matrix assisted pulsed laser evaporation. Appl. Phys. A 2004, 79, 1023–1026. [Google Scholar]
- Caricato, A.; Leggieri, G.; Martino, M.; Vantaggiato, A.; Valerini, D.; Cretì, A.; Lomascolo, M.; Manera, M.; Rella, R.; Anni, M. Dependence of the surface roughness of maple-deposited films on the solvent parameters. Appl. Phys. A 2010, 101, 759–764. [Google Scholar] [CrossRef]
- Mercado, A.L.; Allmond, C.E.; Hoekstra, J.G.; Fitz-Gerald, J.M. Pulsed laser deposition vs. Matrix assisted pulsed laser evaporation for growth of biodegradable polymer thin films. Appl. Phys. A 2005, 81, 591–599. [Google Scholar] [CrossRef]
- Fitz-Gerald, J.M.; Jennings, G.; Johnson, R.; Fraser, C.L. Matrix assisted pulsed laser deposition of light emitting polymer thin films. Appl. Phys. A 2005, 80, 1109–1112. [Google Scholar] [CrossRef]
- Sellinger, A.T.; Martin, A.H.; Fitz-Gerald, J.M. Effect of substrate temperature on poly(methyl methacrylate) nanocomposite thin films deposited by matrix assisted pulsed laser evaporation. Thin Solid Films 2008, 516, 6033–6040. [Google Scholar] [CrossRef]
- Wyatt, P.J. Light scattering and the absolute characterization of macromolecules. Anal. Chim. Acta 1993, 272, 1–40. [Google Scholar] [CrossRef]
- Abdou, M.S.A.; Holdcroft, S. Solid-state photochemistry of π-conjugated poly(3-alkylthiophenes). Can. J. Chem. 1995, 73, 1893–1901. [Google Scholar] [CrossRef]
- Mirau, P.A. A Practical Guide to Understanding the Nmr of Polymers; Wiley-Interscience: Hoboken, NJ, USA, 2005. [Google Scholar]
- Chen, T.-A.; Wu, X.; Rieke, R.D. Regiocontrolled synthesis of poly(3-alkylthiophenes) mediated by rieke zinc: Their characterization and solid-state properties. J. Am. Chem. Soc. 1995, 117, 233–244. [Google Scholar] [CrossRef]
- Rahman, M.H.; Chen, H.-L.; Chen, S.-A.; Chu, P.P.J. 1H nmr spectroscopic study of the solution structure of a conjugated polymer. J. Chin. Chem. Soc. 2010, 57, 490–495. [Google Scholar]
- Ram, M.K.; Sarkar, N.; Bertoncello, P.; Sarkar, A.; Narizzano, R.; Nicolini, C. Fabrication and characterization of poly[(2-methoxy-5-(2'-ethyl-hexyloxy)phenylene vinylene] (meh-ppv) langmuir-schaefer films and their application as photoelectrochemical cells. Synth. Metals 2001, 122, 369–378. [Google Scholar] [CrossRef]
- Collison, C.J.; Rothberg, L.J.; Treemaneekarn, V.; Li, Y. Conformational effects on the photophysics of conjugated polymers: A two species model for meh−ppv spectroscopy and dynamics. Macromolecules 2001, 34, 2346–2352. [Google Scholar] [CrossRef]
- Verma, D.; Ranga Rao, A.; Dutta, V. Surfactant-free cdte nanoparticles mixed meh-ppv hybrid solar cell deposited by spin coating technique. Solar Energy Mater. Solar Cells 2009, 93, 1482–1487. [Google Scholar] [CrossRef]
- Jørgensen, M.; Norrman, K.; Krebs, F.C. Stability/degradation of polymer solar cells. Solar Energy Mater. Solar Cells 2008, 92, 686–714. [Google Scholar] [CrossRef]
- Brandrup, J.; Immergut, E.H.; Grulke, E.A. Polymer Handbook, 4th ed; Wiley: New York, NY, USA, 1999. [Google Scholar]
- Heffner, G.W.; Pearson, D.S. Molecular characterization of poly(3-hexylthiophene). Macromolecules 1991, 24, 6295–6299. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
McCormick, R.D.; Lenhardt, J.; Stiff-Roberts, A.D. Effects of Emulsion-Based Resonant Infrared Matrix Assisted Pulsed Laser Evaporation (RIR-MAPLE) on the Molecular Weight of Polymers. Polymers 2012, 4, 341-354. https://doi.org/10.3390/polym4010341
McCormick RD, Lenhardt J, Stiff-Roberts AD. Effects of Emulsion-Based Resonant Infrared Matrix Assisted Pulsed Laser Evaporation (RIR-MAPLE) on the Molecular Weight of Polymers. Polymers. 2012; 4(1):341-354. https://doi.org/10.3390/polym4010341
Chicago/Turabian StyleMcCormick, Ryan D., Jeremy Lenhardt, and Adrienne D. Stiff-Roberts. 2012. "Effects of Emulsion-Based Resonant Infrared Matrix Assisted Pulsed Laser Evaporation (RIR-MAPLE) on the Molecular Weight of Polymers" Polymers 4, no. 1: 341-354. https://doi.org/10.3390/polym4010341
APA StyleMcCormick, R. D., Lenhardt, J., & Stiff-Roberts, A. D. (2012). Effects of Emulsion-Based Resonant Infrared Matrix Assisted Pulsed Laser Evaporation (RIR-MAPLE) on the Molecular Weight of Polymers. Polymers, 4(1), 341-354. https://doi.org/10.3390/polym4010341