Tunable Crystal-to-Crystal Phase Transition in a Cadmium Halide Chain Polymer




Abstract
: The chain polymer (X = Cl, Br; py = pyridine) undergoes a fully reversible phase transition between a monoclinic low-temperature and an orthorhombic high-temperature phase. The transformation can be directly monitored in single crystals and can be confirmed for the bulk by powder diffraction. The transition temperature can be adjusted by tuning the composition of the mixed-halide phase: Transition temperatures between 175 K up to the decomposition of the material at ca. 350 K are accessible. Elemental analysis, ion chromatography and site occupancy refinements from single-crystal X-ray diffraction agree with respect to the stoichiometric composition of the samples.1. Introduction
Coordination polymers and inorganic-organic hybrid materials feature a plethora of topologies, and they often show intriguing properties. Over the last decade, our group has devoted systematic studies to halide-bridged polymers of divalent cations; the subject has recently been reviewed [1]. Several compounds in this class undergo single-crystal to single-crystal transformations: We have encountered solid state reactivity [2], thermal degradation [3], and phase transitions [4]. In the context of a charge density study [5], we envisaged the well-crystallizing chain polymer as a suitable candidate for high resolution X-ray diffraction experiments. We had previously determined its room temperature structure [6]; upon cooling, the compound undergoes a phase transition. In the present contribution, we wish to report the details about this rare example of a fully reversible single-crystal to single-crystal phase transformation. We communicate the twinned low-temperature structure of the material, and we describe the temperature dependence of the transformation in the isomorphous series as summarized in Scheme 1.
2. Results and Discussion
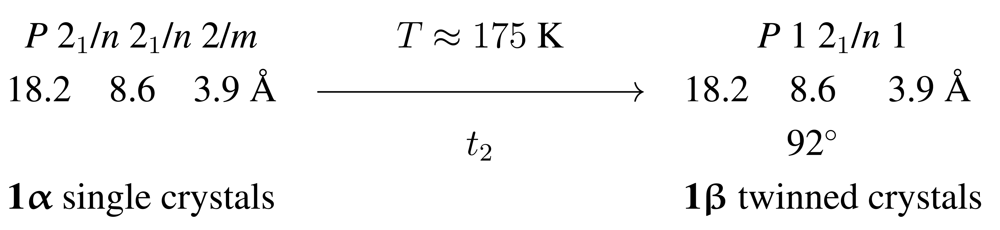
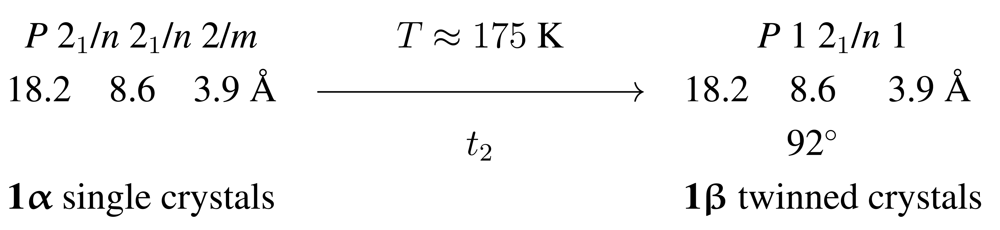
1 follows the general trend observed for Cd compounds with stoichiometric composition [CdBr2L2] (L = donor ligand) [1,6–10] and represents a halide-bridged chain polymer (Scheme 1). At room temperature the compound crystallizes in the orthorhombic space group Pnnm, with the Cd cations in Wyckoff position 2a. The associated site symmetry of the metal is 2/m; all atoms of the pyridine ligands are situated in the crystallographic mirror plane, for symmetry reasons perpendicular with respect to the Cd…Cd vector [6]. Upon cooling, a reversible crystal-to-crystal phase transition is observed. Scheme 2 illustrates the symmetry relationship between the high and low temperature structures. In agreement with the symmetry principle [11], single crystals of the room temperature phase 1α are converted into twins of the low temperature phase 1β. The latter crystallizes in a lattice-equivalent (translationengleich, hence t) subgroup of the former; the index of the subgroup P21/n in space group Pnnm is 2, thus resulting in the symbol t2 for the phase transition.
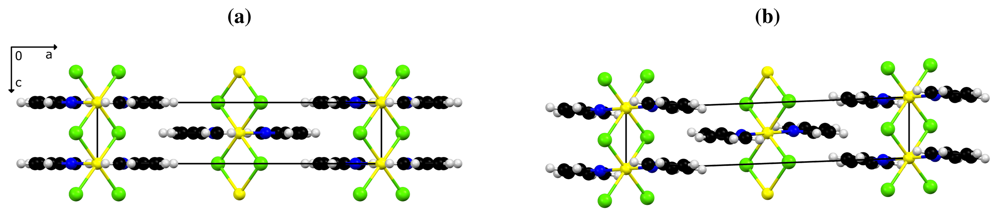
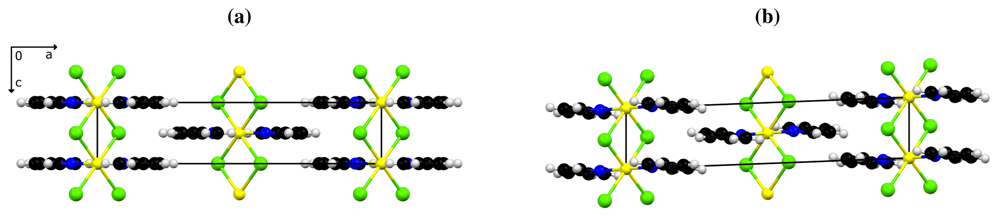
Despite the systematic twinning, a satisfactory structure model could be derived for 1β. The site symmetry of the cations is reduced to 1̲ and allows for tilting of the pyridine ring as shown in Figure 1.
The low temperature phase 1β is isomorphous to the crystal structure of the analogous chloro derivative 2 at room temperature [6]. This structural relationship, together with the chemical similarity and straightforward preparation of the compounds, induced us to investigate the effect of isomorphous replacement on the phase-transition temperature: As outlined above, this transformation occurs around 175 K for the bromo compound 1 and is either not detectable at all or associated with a considerably higher temperature in the chloro polymer 2. In the quest for a conveniently and predictably tunable property as a function of the chemical composition, we prepared the series of mixed-halide phases with 0 ≤ x ≤ 1. With the help of a combination of analytical techniques, we have been able to reliably address the stoichiometry in these mixed-halide derivative, to ensure their phase purity and to detect their phase-transition temperatures.
We will first focus on the Br:Cl ratio in the mixed halide compounds. Microanalysis represents an obvious and simple approach. For both end members of the series, i.e., 1 and 2, satisfactory analyses were available; the C content of the compounds covers the range between 27.9% and 35.2% and N should range from 6.5% to 8.2%. The experimental data indicated a clear trend in favor of Br-enriched solids with respect to the reaction stoichiometry.
We envisaged ion chromatography as an alternative analytical method: Different anions in aqueous solutions can be quantitatively detected. For this purpose, the corresponding samples were decomposed by removal of the pyridine at elevated temperatures (cf. 4); the ratio between bromide and chloride ions in the residual mixed Cd halides showed very good agreement with the results from microanalyses.
The preparations were conducted as reactant diffusion experiments and yielded crystalline powders as well as crystals of sufficient size and quality for single-crystal X-ray diffraction. As the diffraction pattern represents the Fourier transform of the electron density and since the atomic scattering factors for the halides involved differ significantly, the halide occupancy could be obtained through refinement (Details about the single-crystal diffraction experiments are given in Section 4). Tables 1 and 2 summarize crystal data and convergence results (Supplementary crystallographic data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif).
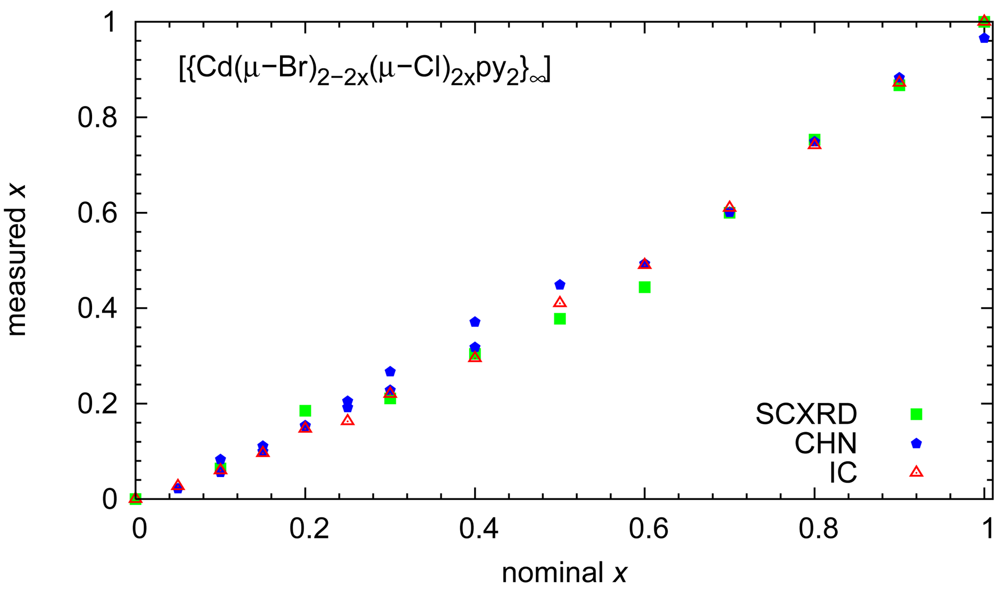
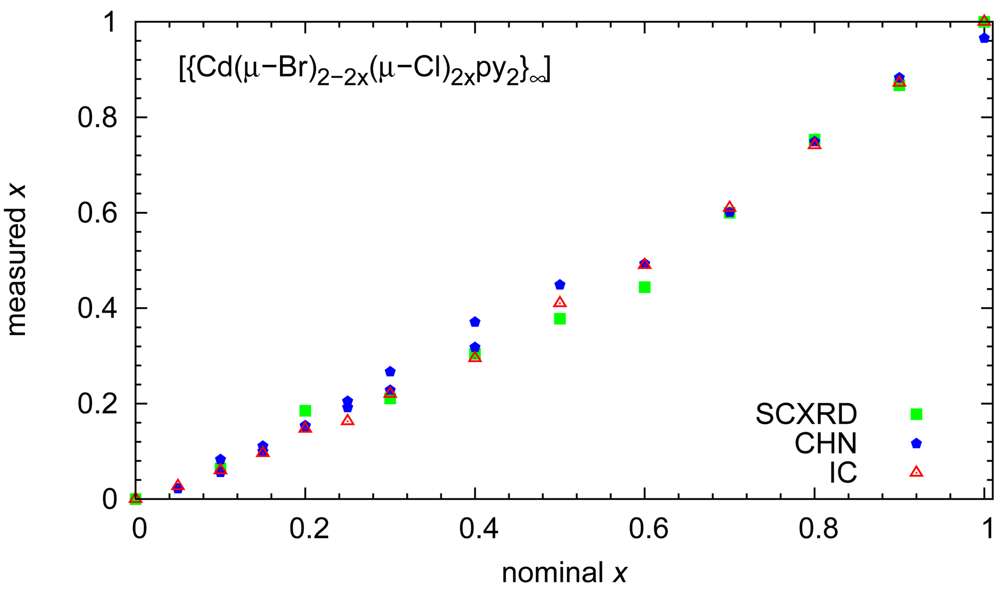
The results of our site occupancy refinements match the stoichiometries derived from the aforementioned chemical analyses. A synopsis of the chemical composition in the crystalline solids established by the different methods as a function of reactant stoichiometry is given in Figure 2.
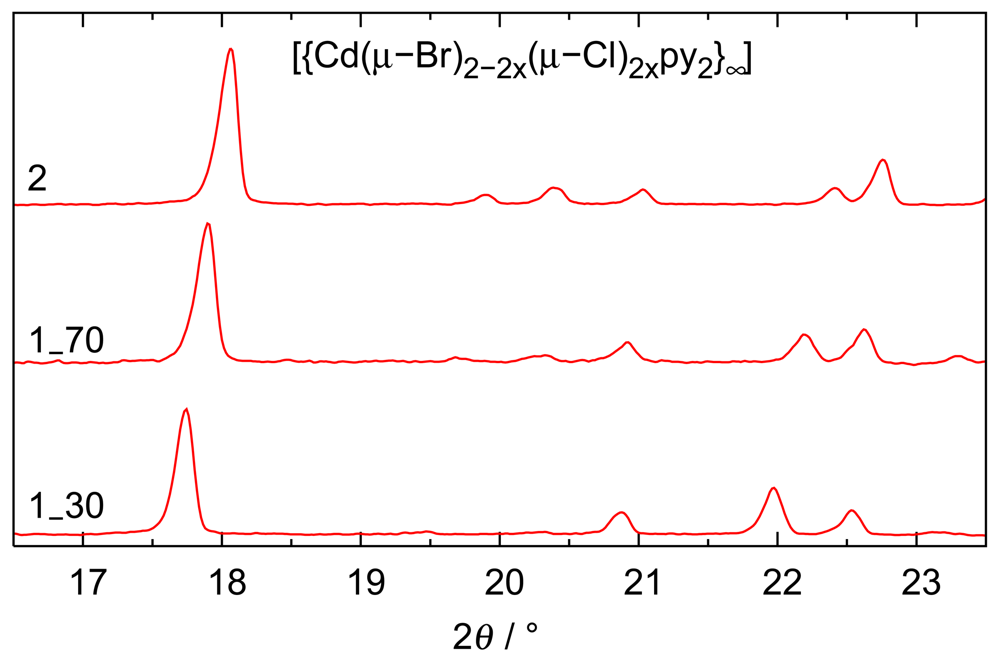
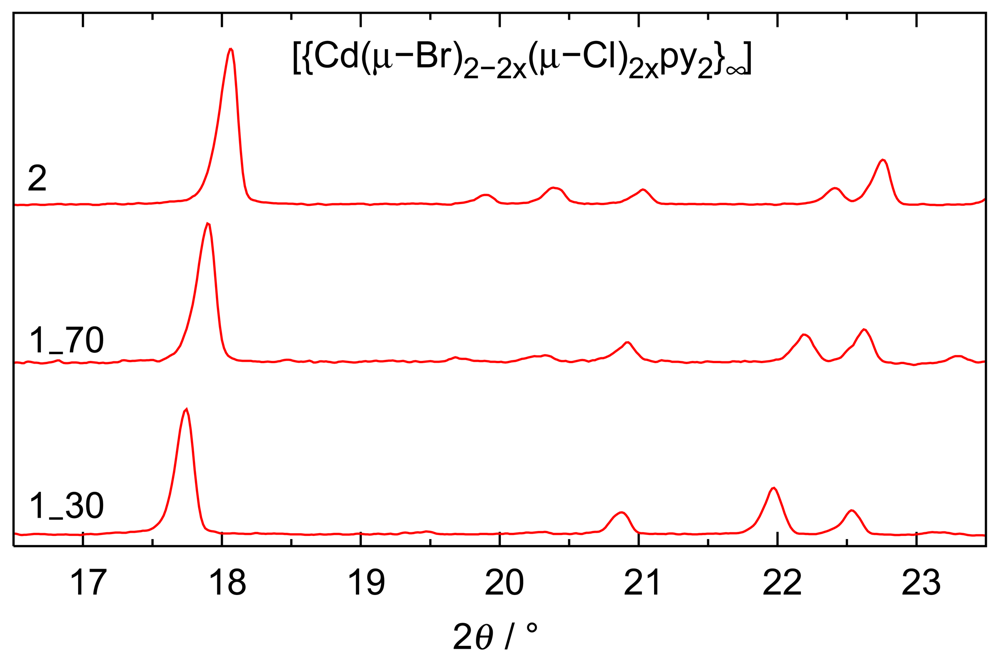
Beyond the comforting message that different analytical techniques agree with respect to the chemical composition, a second aspect is relevant: Electron density-based site occupancy refinements rely on single crystals and hence on a sample size of micrograms; microanalyses were performed in parallel on two samples, each with a mass of approximately 2 mg; finally, ion chromatography was based on even larger samples of ca. 15 mg of initial product. The agreement between these methods shows that the reaction products are homogeneous. This conclusion is corroborated by powder patterns routinely performed on all bulk preparations: the mixed halides show reflection widths comparable to the end members and the expected trend with respect to lattice parameters. Diffractograms for solids with different composition have been compiled in Figure 3.
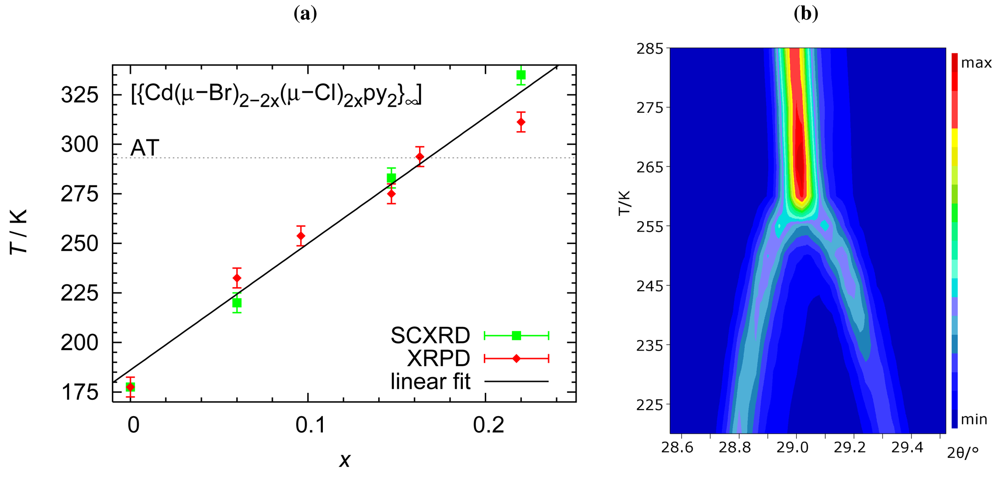
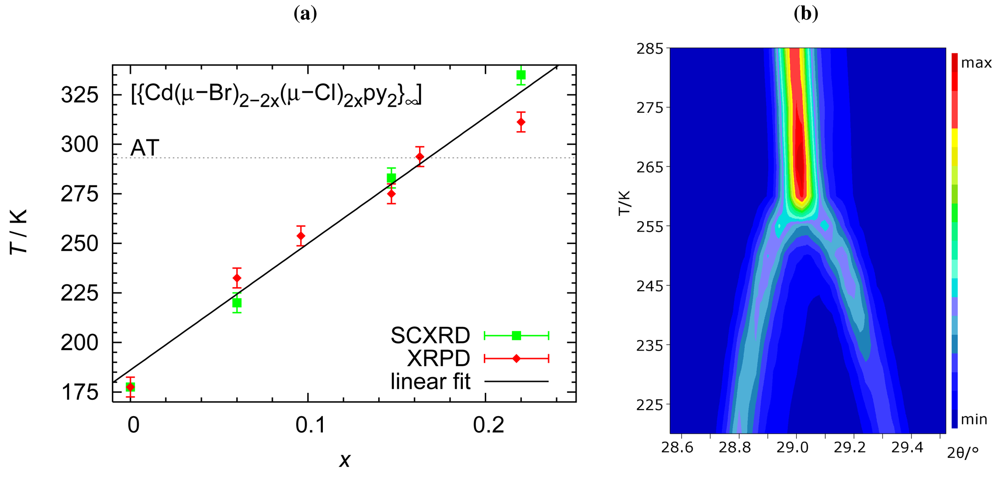
We will now address the target property, namely the temperature dependence of the phase transition. Variable temperature diffraction experiments were conducted both on single crystals and on powder. Both types of experiments not only agree with respect to the transition temperature but also consistently show the absence of hysteresis. Phase-transition temperatures established during sample heating and cooling do not differ significantly. Figure 4 summarizes the relationship between chemical composition and phase-transition temperature.
Apparently, thermal analysis also represents a suitable analytical technique for monitoring the phase transitions. Decomposition of the samples, e.g., observed in preliminary studies of the pyridine removal prior to ion chromatography, could easily be detected both as DSC and TG signals. It must be noted, however, that we have not been able to obtain reproducible and significant DSC signals in the context of the reversible phase transitions: They are obviously associated with enthalpies too small to be reliably detected.
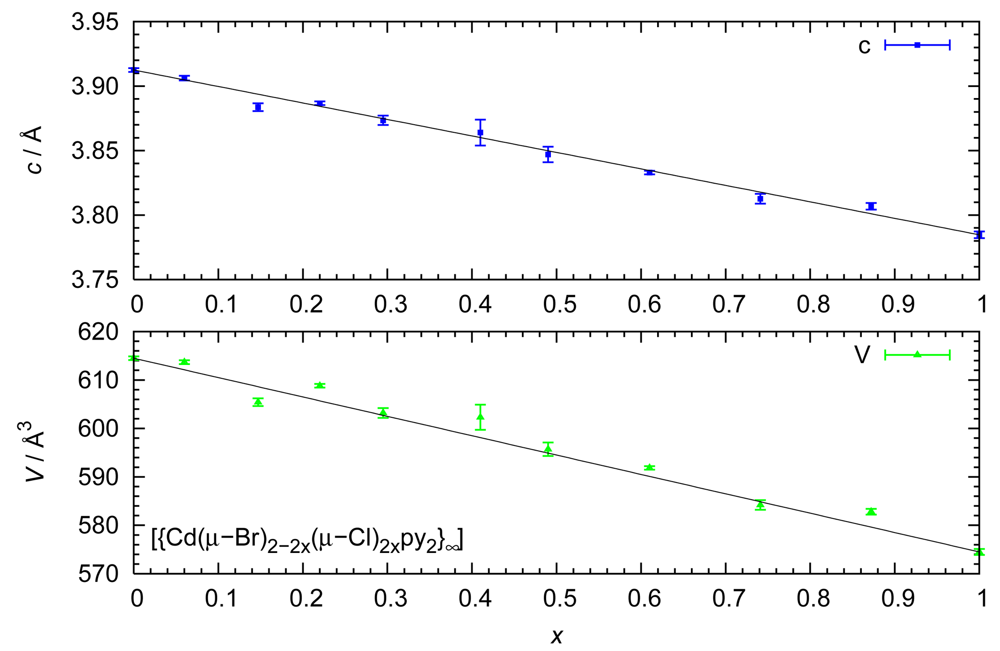
Although the relationship between transition temperature and chemical composition is the main focus of this contribution, it should be mentioned that other quantities also vary systematically as a function of the halide ratio. Especially the unit cell volume and the separation between adjacent Cd cations along a polymer strand show linear decrease for increasing percentage of the smaller chloride ions (Figure 5).
3. Conclusions
The crystal-to-crystal phase transition in and the isomorphism between its low-temperature phase and the crystal structure of the analogous chloro derivative induced us to address the transition temperature as a property tunable by chemical composition. Halide content has been proven a very sensitive criterion to adjust this property over a rather wide range between 175 K and the decomposition of the material around 350 K: An increase of the molar fraction of 0.05 for the chloro content in the mixed halides increases the transition temperature for ca. 30 K. We are well aware of the fact that cadmium halides cannot represent readily applicable materials; we are, however, confident that analogous relationships can be detected for less toxic compounds in the realm of coordination chemistry and that a similar combination of techniques will lead to fascinating insights into the structural response on subtle changes in chemical composition.
4. Experimental
4.1. General
Ion chromatography was measured at the Institute of Inorganic Chemistry, RWTH Aachen University, using a Metrohm 792 Basic IC. The chromatograph was calibrated with a standard solution of the anions F−, Cl−, , Br− and . Due to the pyridine contained in the compound the samples were prepared as follows.
A sample of approximately 15 mg of every product was placed into a vessel or flask, which was then evacuated to 10−3 mbar and heated in a sand bath at 220 °C. After 48 h the products were cooled to ambient temperature. 1H-NMR measurements were conducted on the prepared samples in D2O. The NMR spectra showed no signs of the pyridine 1H-resonances, meaning the product preparation was successful and yielded a mixture of CdBr2 and CdCl2 salts in the exact ratio as that of the unaltered product.
Aqueous solutions with a product concentration of 10–20 ppm were prepared using ultra-pure water. Finally the Br:Cl ratio was measured using the ion chromatograph. The ratio of Br and Cl in the compounds is the direct result of the ion chromatography graphs.
1H-NMR was measured at the Institute of Inorganic Chemistry, RWTH Aachen University. A Bruker Ultrashield™ plus 400 was used. TMS was added to all samples as standard.
CHN microanalysis was carried out at the Institute of Organic Chemistry, RWTH Aachen University, using a HERAEUS CHNO-Rapid.
Standard X-ray powder diffraction was done by using a Stoe Imageplate-detector IP-PSD with Cu-Kα1-radiation at the Institute of Inorganic Chemistry, RWTH Aachen University.
4.2. Synthesis
All chemicals were used as purchased without purification: CdBr2 4H2O (99.5%, Riedel-de Haën), CdCl2 (99%, Fluka), pyridine (py) (99%, Merck). The products were synthesized using diffusion experiments. The general method will be described with the synthesis of the pure bromine product 1.
4mL of aqueous CdBr2·4H2O (0.4 mmol, 1 eq.) of a 0.1 M standard solution of CdBr2·4H2O were given into a test tube and overlaid with an intermediate layer of 2 mL of pure H2O. A final layer of 6 mL acetone was then carefully added above the intermediate layer. Finally pyridine (0.063 g, 0.8 mmol, 2 eq.) was added to the test tube. The test tube was kept sealed for 48 h. The reaction mixture was decanted and the product was washed with acetone and left to dry in air. The experiment yielded colorless, odorless crystalline needles.
In order to synthesize the mixed halides a second 0.1 M standard solution of CdCl2 was prepared. Depending on the desired halide ratio different amounts of the standard solutions were combined in the initial 4mL layer, e.g., for a desired 1:1 ratio, 2mL of the CdBr2 4H2O standard solution (0.2 mmol, 0.5 eq) and 2 mL of the CdCl2 standard solution (0.2 mmol, 0.5 eq) were mixed in the test tube, followed by the pure H2O and acetone layers. In the end, pyridine (0.063 g, 0.8 mmol, 2 eq.) was added.
This procedure was repeated for different chlorine percentages varying between 0% and 100%. The isolated yields in crystalline product vary between 50% and 70%.
4.3. Single Crystal X-Ray Diffraction
The crystals were mounted on glass fibers. Geometry and intensity data were collected in ω-scan mode with an APEX CCD area detector [13] on a Bruker D8 goniometer equipped with an Incoatec microsource with multi-layer optics using Mo-Kα-radiation (λ = 0.71073 Å). An Oxford Cryostream 700 cooler was used for low-temperature measurements. The data were processed with the program SAINT+ [14].
To non-twinned data sets multi-scan absorption corrections were applied with the programs SADABS [15] or MULABS [16]. The diffraction patterns of twinned crystals in their low temperature phase were indexed with CELL NOW [17] with real space starting vectors ranging between 2 and 20 Å. Integrated intensities of each domain were corrected for absorption and merged with the help of TWINABS [18].
The structures of 1 in the high and low temperature phase were initially solved by the Patterson method [19]; this solution was transferred to the remaining isomorphous crystals. Full matrix least-squares refinement was performed on F2 [19] with anisotropic displacement parameters for all non-hydrogen atoms. Hydrogen atoms were placed in idealized positions (C–H = 0.95 Å) and included as riding with Uiso(H) = 1.2Ueq(non-H).
Twin formation is due to a phase transition rather than to intergrowth; not surprisingly, all twinned crystals studied in the context of this work exhibit domains of comparable volume. Refinement of the twinned crystals was therefore conducted on reflection files containing the intensities from both domains, and their volume ratio was included in the refinement as a variable. The partially occupied halide sites were constrained to share the same positional coordinates and anisotropic displacement parameters, and the sum of their occupancies was constrained to unity. More demanding models according to which the mutually exclusive halide sites do not precisely coincide have been tested but discarded: Despite their inherent complexity, they did not improve quality criteria such as residual electron density, standard uncertainties in derived geometric parameters or reliability factors.
In comparison to the other non-twinned samples, agreement factors of 1_50 and 1_60 are less satisfactory (see Table 2). We can only speculate about the reasons: Powder diffraction of the bulk material does not indicate line broadening or any change in the structure type. The chlorine content of approximately 50% in these samples might cause local distortions which lead to lower quality and higher mosaicity in the diffraction patterns of larger single crystals. This speculation is consistent with the fact that higher values for Rσ (σ(I)/I) in the single-crystal intensity data for 1_50 and 1_60 have been encountered.
4.4. Temperature-Dependent Powder X-Ray Diffraction
The temperature-dependent X-ray studies were performed to precisely determine the temperature of the phase transformation. The measurements were done using a Huber G645 Guinier powder diffractometer (transmission geometry, Cu-Kα1-radiation (λ = 1.54059 Å)) with a position-sensitive detector (STOE, Darmstadt) registering simultaneously a 2θ-range of 14°. The flat sample was cooled in an evacuated chamber via a closed-cycle refrigerator, allowing precise temperature control between 10 K and 350 K. The temperature was kept constant during the measurement. Scans were performed by cooling and heating in 5 K-steps to prove the nonexistence of hysteresis effects.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | 1 | 1_10 | 1_20 | 1_30 | 1_40 |
|---|---|---|---|---|---|
| Refined Cl ratio | - | 0.064 | 0.185 | 0.211 | 0.304 |
| Twin domain fraction | 0.504 | 0.489 | 0.418 | 0.412 | 0.464 |
| a/Å | 18.239(3) | 18.221(4) | 18.090(7) | 18.132(3) | 18.044(8) |
| b/Å | 8.6180(15) | 8.6318(18) | 8.629(3) | 8.6510(15) | 8.645(4) |
| c/Å | 3.9125(7) | 3.9062(9) | 3.8837(15) | 3.8867(7) | 3.8735(18) |
| β/° | 92.220(6) | 92.569(5) | 92.926(9) | 93.093(4) | 93.279(8) |
| V/Å3 | 614.51(19) | 613.7(2) | 605.4(4) | 608.79(19) | 603.2(5) |
| Variables refined | 71 | 72 | 72 | 72 | 72 |
| R1[I > 2σ(I)] | 0.0693 | 0.0595 | 0.0658 | 0.0406 | 0.0649 |
| wR2 (all reflections) | 0.2455 | 0.1722 | 0.1611 | 0.1284 | 0.1569 |
| CCDC number | 828355 | 828356 | 828357 | 828358 | 828359 |
| Parameter | 1_50 | 1_60 | 1_70 | 1_80 | 1_90 | 2 |
|---|---|---|---|---|---|---|
| Refined Cl ratio | 0.378 | 0.444 | 0.600 | 0.753 | 0.867 | - |
| a/Å | 18.03(2) | 17.956(12) | 17.901(3) | 17.790(9) | 17.767(6) | 17.680(6) |
| b/Å | 8.658(11) | 8.640(6) | 8.6410(15) | 8.627(4) | 8.630(3) | 8.597(3) |
| c/Å | 3.864(5) | 3.847(3) | 3.8330(7) | 3.8127(19) | 3.8068(13) | 3.7847(13) |
| β/° | 93.31(2) | 93.379(10) | 93.437(3) | 93.284(8) | 93.133(6) | 93.013(6) |
| V/Å3 | 602.3(13) | 595.7(7) | 591.83(18) | 584.2(5) | 582.8(3) | 574.5(3) |
| Variables refined | 71 | 71 | 71 | 71 | 71 | 70 |
| R1[I > 2σ(I)] | 0.0654 | 0.0669 | 0.0258 | 0.0336 | 0.0531 | 0.0327 |
| wR2 (all reflections) | 0.1564 | 0.1720 | 0.0610 | 0.0817 | 0.1076 | 0.0643 |
| CCDC number | 828360 | 828361 | 828362 | 828363 | 828364 | 828365 |
Acknowledgments
The authors thank Carina Merkens and Fangfang Pan for help with the single-crystal data collections. A DAAD scholarship for Amr Ramadan is gratefully acknowledged. We thank our colleagues of Griinenthal GmbH for their efforts with thermal analyses.
References
- Englert, U. Halide-bridged polymers of divalent metals with donor ligands—structures and properties. Coord. Chem. Rev. 2010, 254, 537–554. [Google Scholar]
- Hu, C.; Englert, U. Single-crystal to single-crystal transformation from chain polymer to two dimensional network at very low temperatures. Angew. Chem. Int. Ed. 2005, 44, 2281–2283. [Google Scholar]
- Merkens, C.; Kalf, I.; Englert, U. A new chain polymer via thermolysis: [Hg(μ-Br)2(3,5-Br2py)]∞. Z. Anorg. Allg. Chem. 2010, 636, 681–684. [Google Scholar]
- Hu, C.; Englert, U. Space filling versus symmetry: Two consecutive crystal-to-crystal phase transitions in a 2D network. Angew Chem. Int. Ed. 2006, 45, 3457–3459. [Google Scholar]
- Wang, R.; Lehmann, C.W.; Englert, U. Weak interactions in chain polymers [M(μ-X)2L2]∞ (M = Zn, Cd; X = Cl, Br; L = substituted pyridine)—an electron density study. Acta Crystallogr. 2009, B65, 600–611. [Google Scholar]
- Hu, C.; Li, Q.; Englert, U. Structural trends in one and two dimensional coordination polymers of cadmium(II) with halide bridges and pyridine type ligands. CrystEngComm 2003, 5, 519–529. [Google Scholar]
- Hu, C.; Englert, U. Coordination polymers based on cadmium(II) pyridine complexes: Synthesis, range of existence, and structure. CrystEngComm 2002, 4, 20–25. [Google Scholar]
- Englert, U.; Schiffers, S. catena-Poly[[aqua(pyridine N-oxide-κO)cadmium(II)]-di-μ-chloro]. Acta Crystallogr. 2006, E62, m295–m296. [Google Scholar]
- Cao, L.; Li, Q.; Englert, U. catena-Bis(μ-chloro)-(κ-O, κ-N-{2-thenoylhydrazine})-cadmium(II), a novel cadmium coordination polymer. J. Chem. Cryst. 2008, 38, 833–836. [Google Scholar]
- Turgunov, K.; Englert, U. catena-Poly[[bis[quinazolin-4(3H)-one-κN1]cadmium(II)]-di-μ-chlorido]. Acta Crystallogr. 2010, E66, m1457. [Google Scholar]
- Bärnighausen, H. Group-subgroup relations between space groups: A useful tool in crystal chemistry. MATCH Comm. Math. Chem. 1980, 9, 139–175. [Google Scholar]
- Macrae, C.F.; Bruno, I.J.; Chrisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; de Streek, J.; Wood, P.A. Mercury CSD 2.0—new features for the visualization and investigation of crystal structures. J. Appl. Cryst. 2008, 41, 466–470. [Google Scholar]
- SMART (Version 5.624). Program for Bruker CCD X-Ray Diffractometer Control; Bruker AXS Inc.: Madison, WI, USA, 2000. [Google Scholar]
- SAINT+ (Version 6.02). Program for Reduction of Data Collected on Bruker CCD Area Detector Diffractometer; Bruker AXS Inc.: Madison, WI, USA, 1999. [Google Scholar]
- Sheldrick, G.M.; SADABS (Version 2.03). Program for Empirical Absorption Correction of Area Detector Data; University of Göttingen: Göttingen, Germany, 1996. [Google Scholar]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. 2009, D65, 148–155. [Google Scholar]
- Sheldrick, G.M. CELL NOW. Program for Unit Cell Determination; Bruker-AXS Inc.: Madison, WI, USA, 2004. [Google Scholar]
- Sheldrick, G.M. TWINABS; University of Gottingen: Gottingen, Germany, 2006. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, A64, 112–122. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/.)
Share and Cite
Lamberts, K.; Kalf, I.; Ramadan, A.; Müller, P.; Dronskowski, R.; Englert, U. Tunable Crystal-to-Crystal Phase Transition in a Cadmium Halide Chain Polymer. Polymers 2011, 3, 1151-1161. https://doi.org/10.3390/polym3031151
Lamberts K, Kalf I, Ramadan A, Müller P, Dronskowski R, Englert U. Tunable Crystal-to-Crystal Phase Transition in a Cadmium Halide Chain Polymer. Polymers. 2011; 3(3):1151-1161. https://doi.org/10.3390/polym3031151
Chicago/Turabian StyleLamberts, Kevin, Irmgard Kalf, Amr Ramadan, Paul Müller, Richard Dronskowski, and Ulli Englert. 2011. "Tunable Crystal-to-Crystal Phase Transition in a Cadmium Halide Chain Polymer" Polymers 3, no. 3: 1151-1161. https://doi.org/10.3390/polym3031151
APA StyleLamberts, K., Kalf, I., Ramadan, A., Müller, P., Dronskowski, R., & Englert, U. (2011). Tunable Crystal-to-Crystal Phase Transition in a Cadmium Halide Chain Polymer. Polymers, 3(3), 1151-1161. https://doi.org/10.3390/polym3031151