Polymerization in Liquid Crystal Medium: Preparation of Polythiophene Derivatives Bearing a Bulky Pyrimidine Substituent


Abstract
:1. Introduction
2. Experimental Section
2.1. Materials and methods
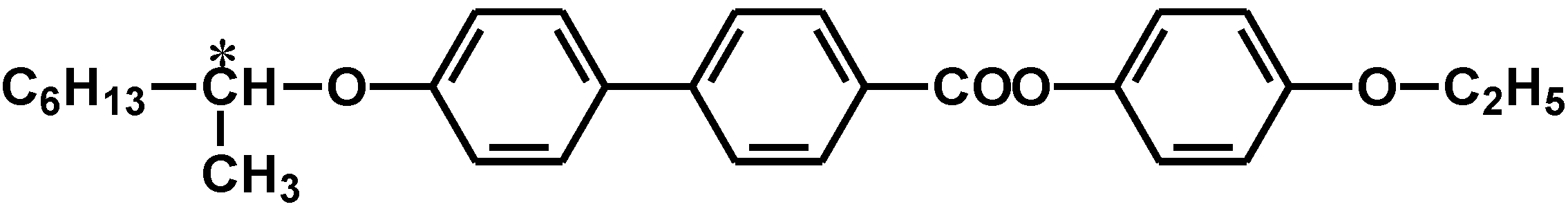
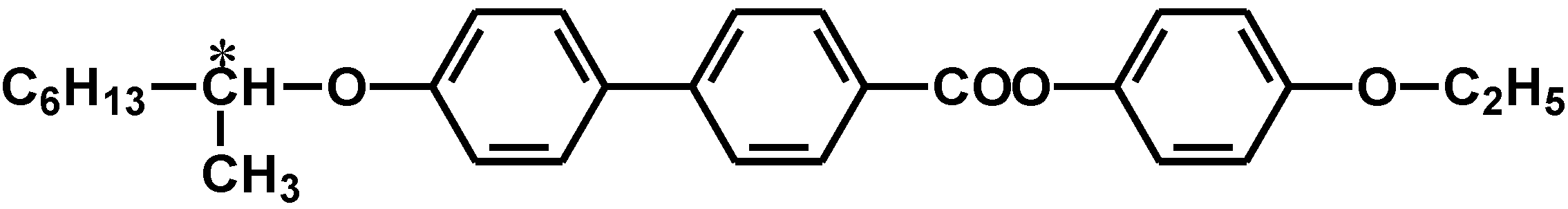
2.2. Synthesis of cholesteric liquid crystal medium
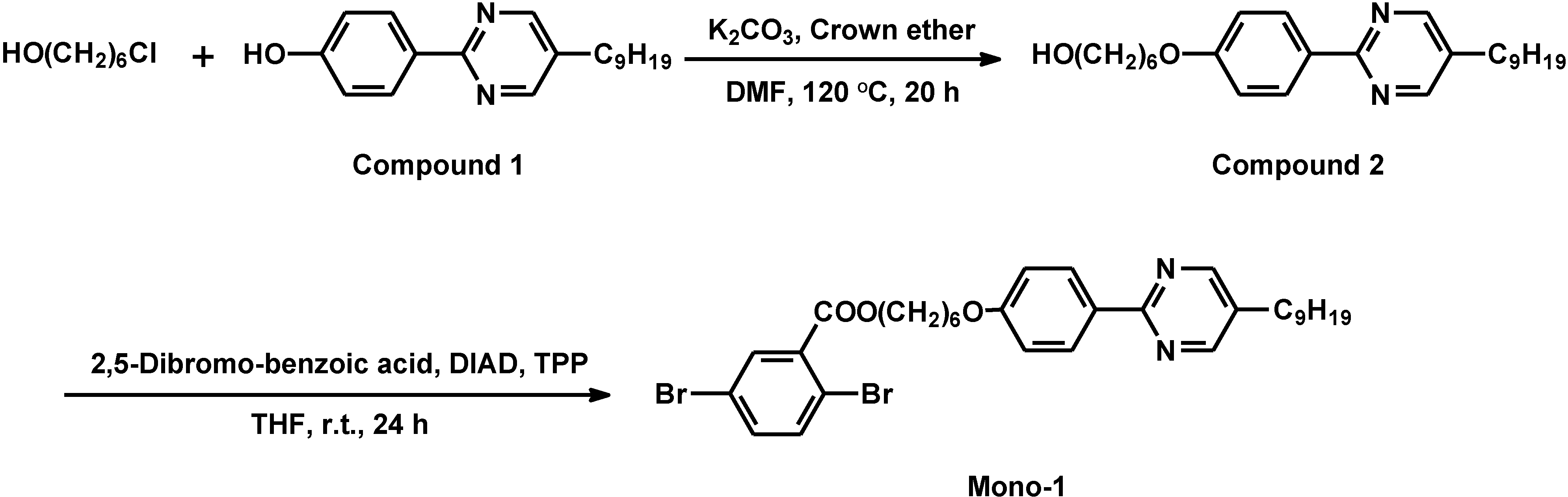
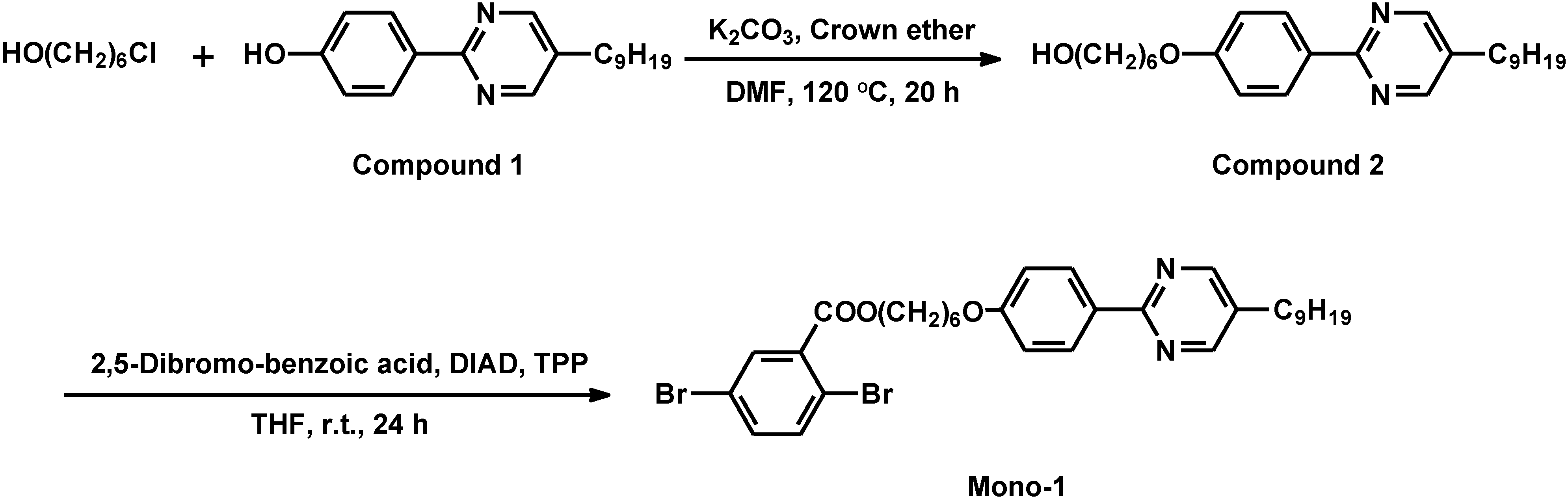
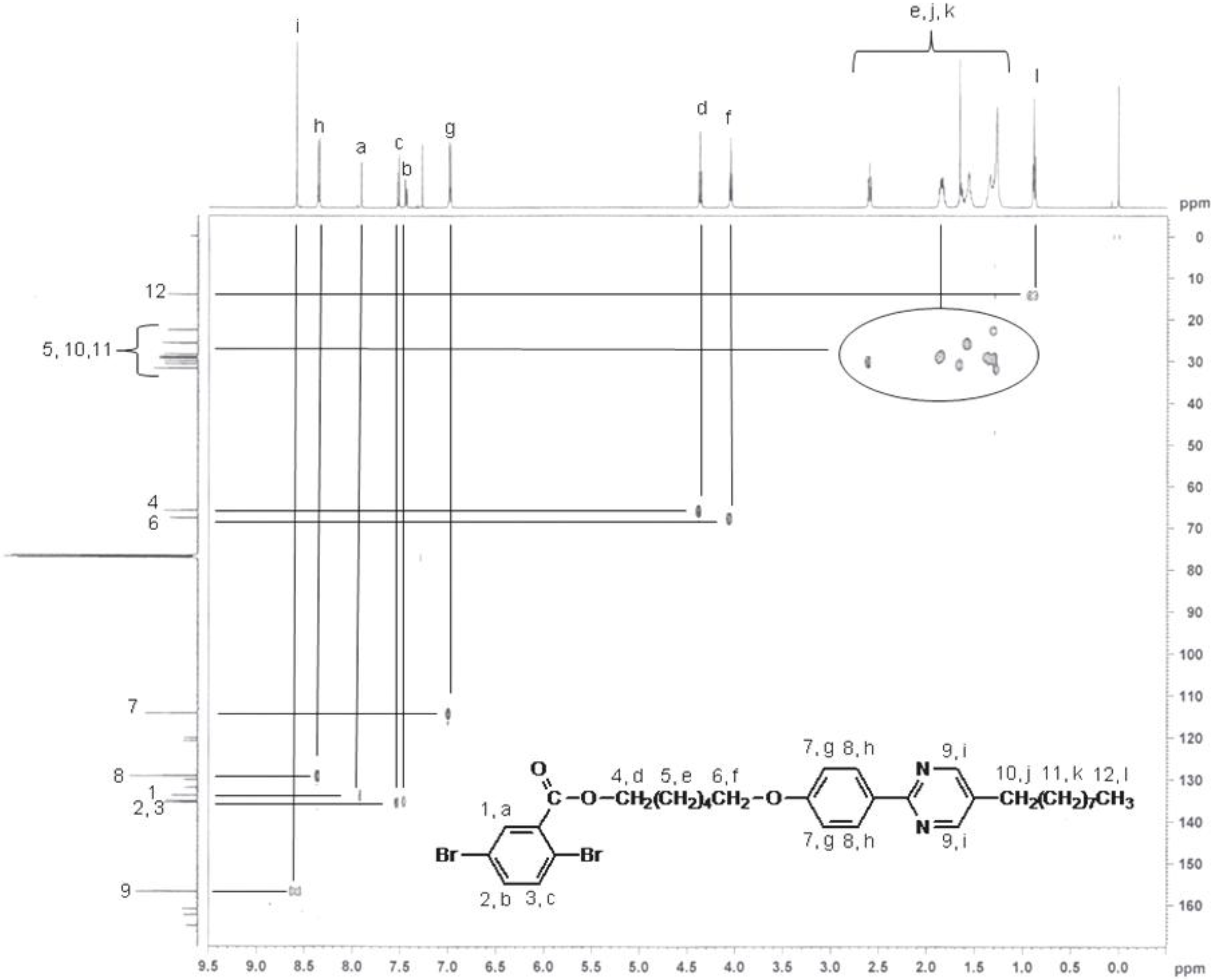
2.3. Synthesis of monomer
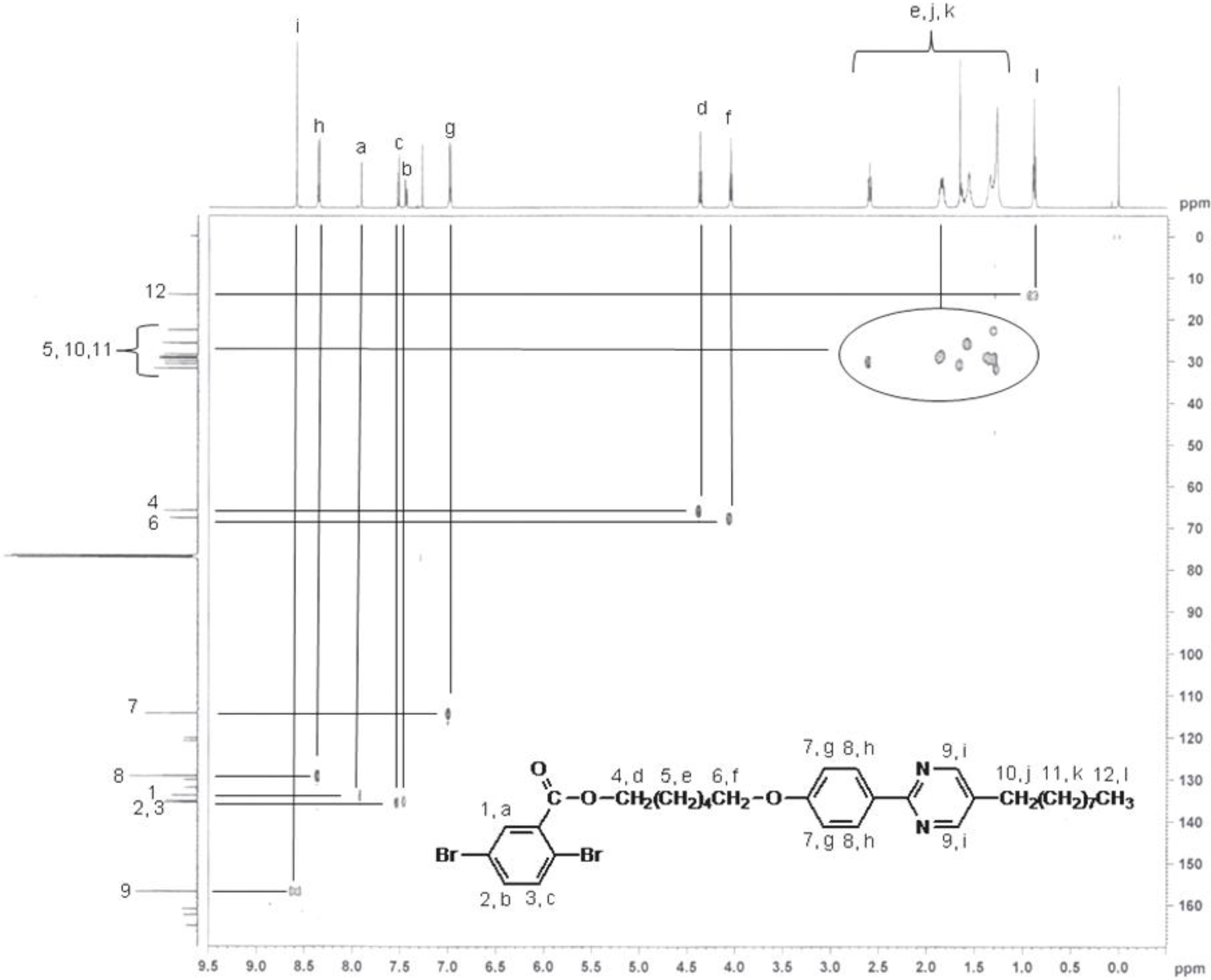
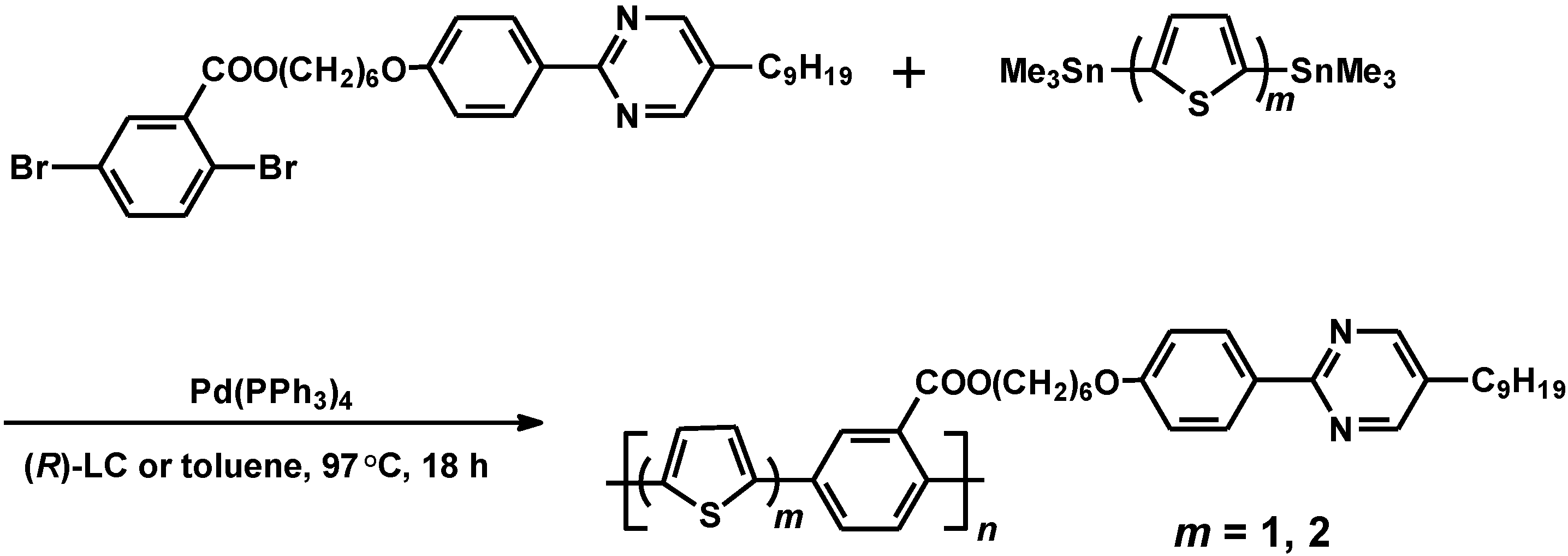
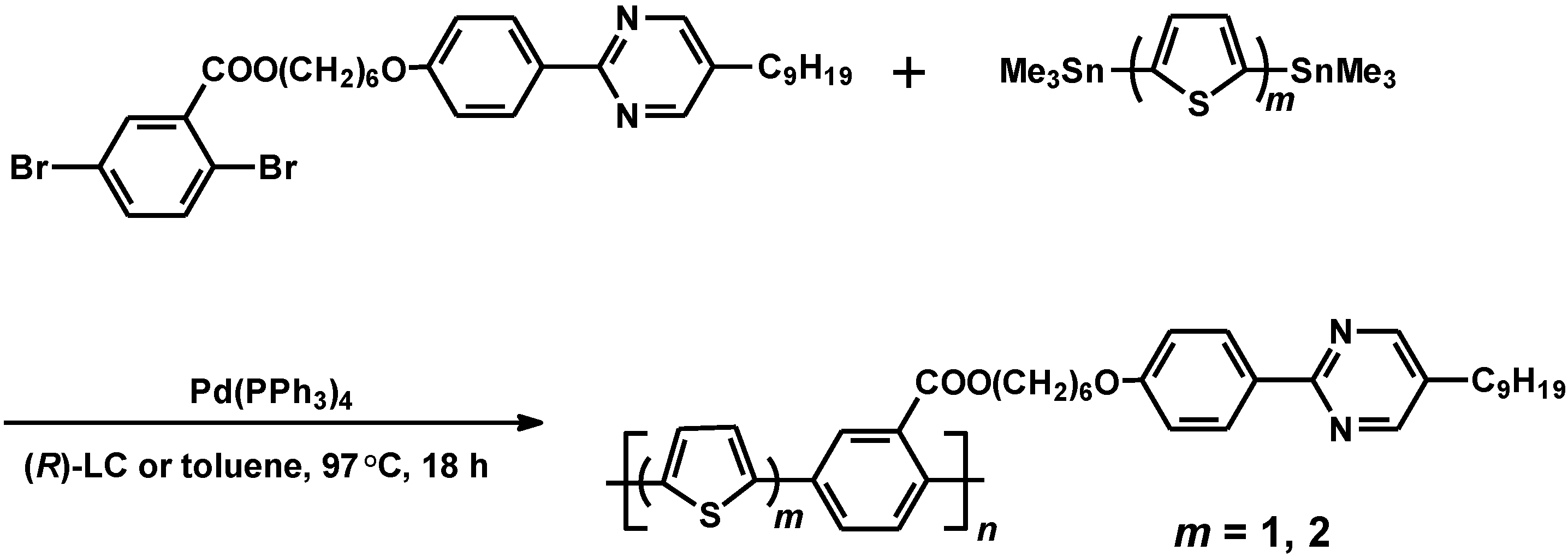
2.4. Polymerization
| Reaction medium | m = 1 | m = 2 |
| Toluene | P1T | P2T |
| (R)-LC | P1LC | P2LC |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | Mn[a] | Mw[a] | Mw/Mn[a] | Yield [%] |
|---|---|---|---|---|
| P1T | 4700 | 6900 | 1.5 | 90 |
| P2T | 3800 | 5700 | 1.5 | 84 |
| P1LC | 3700 | 5700 | 1.6 | 46 |
| P2LC | 3400 | 5900 | 1.7 | 66 |
3. Results and Discussion
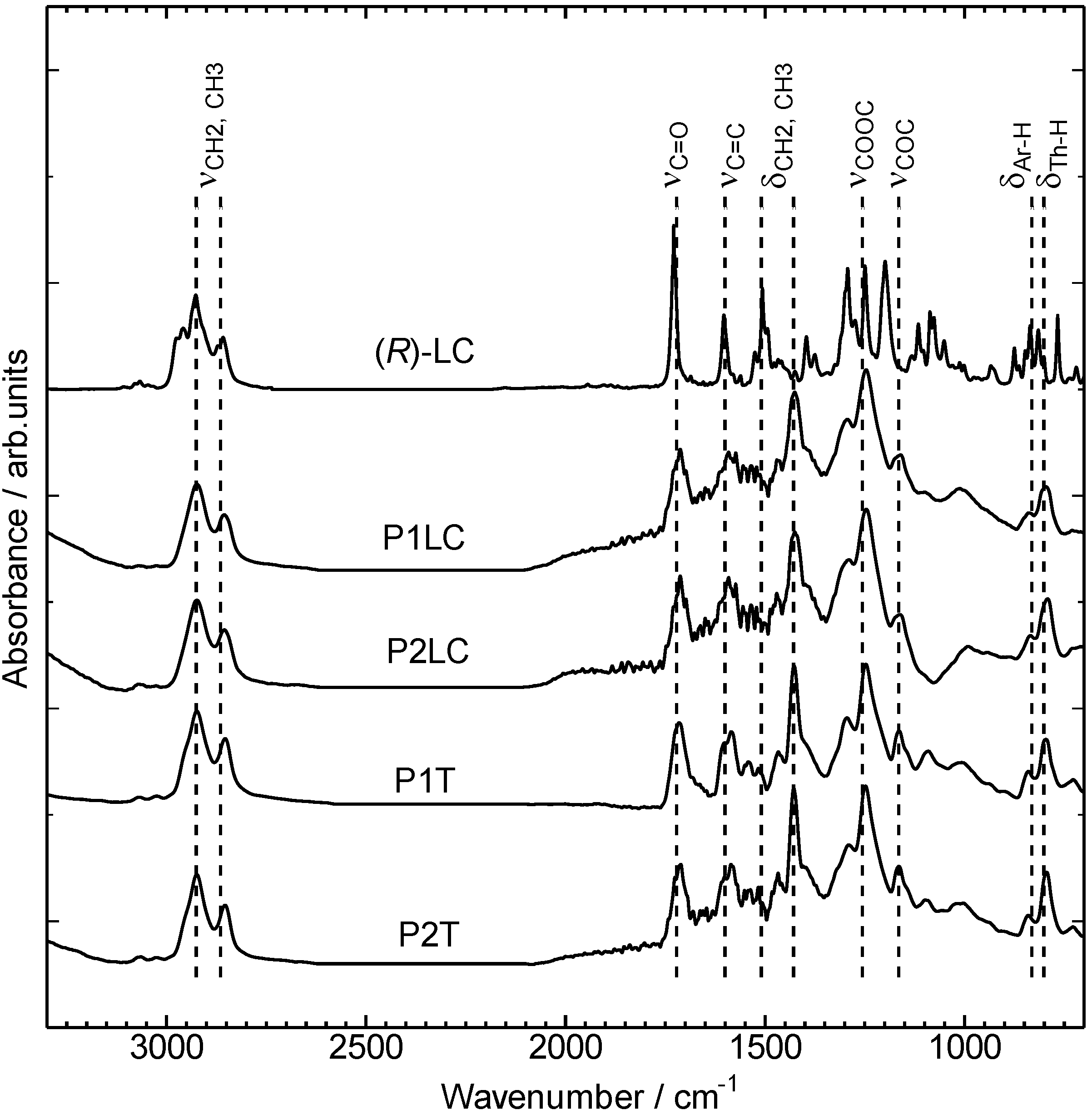
3.1. Infrared absorption spectra
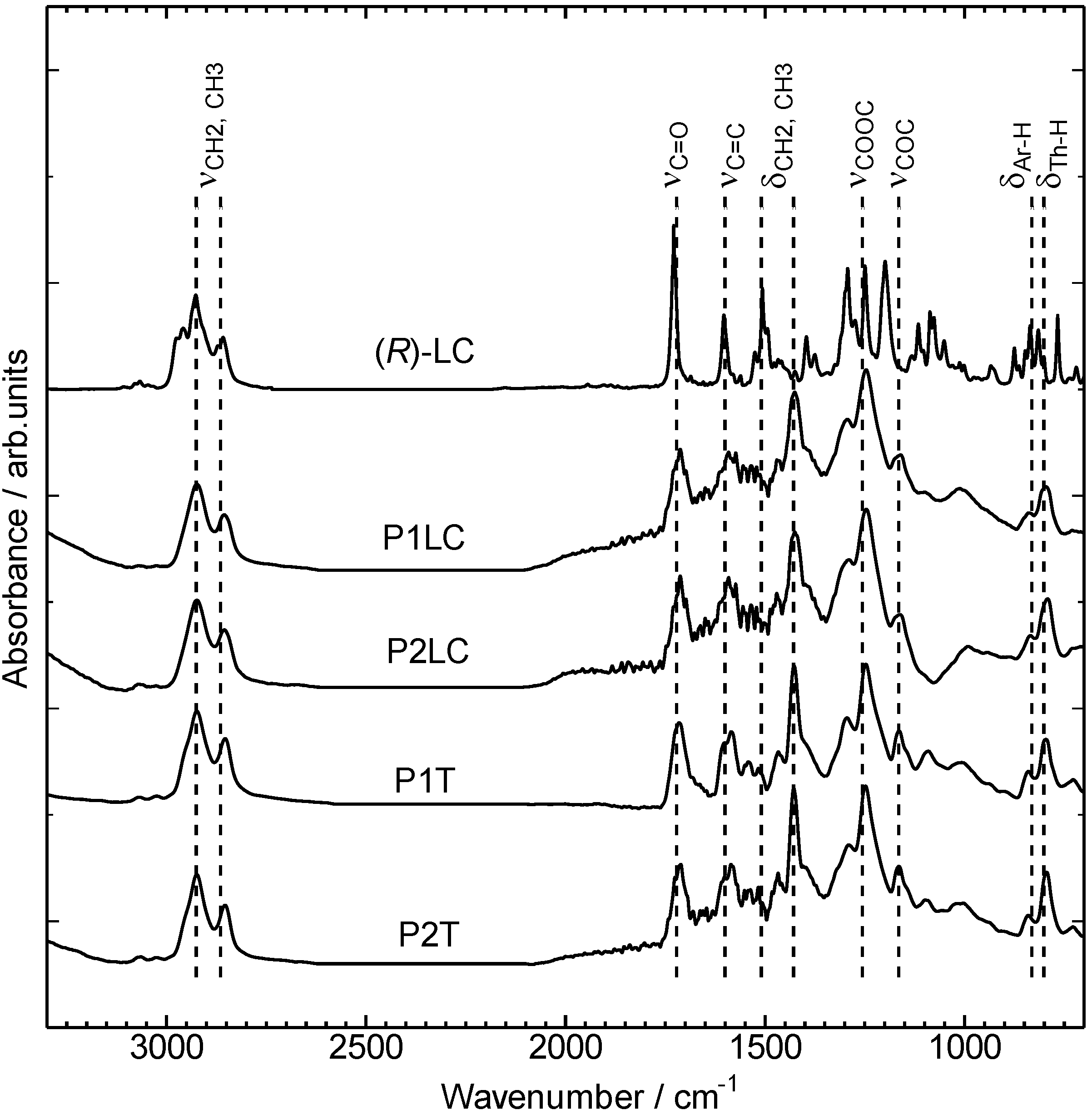
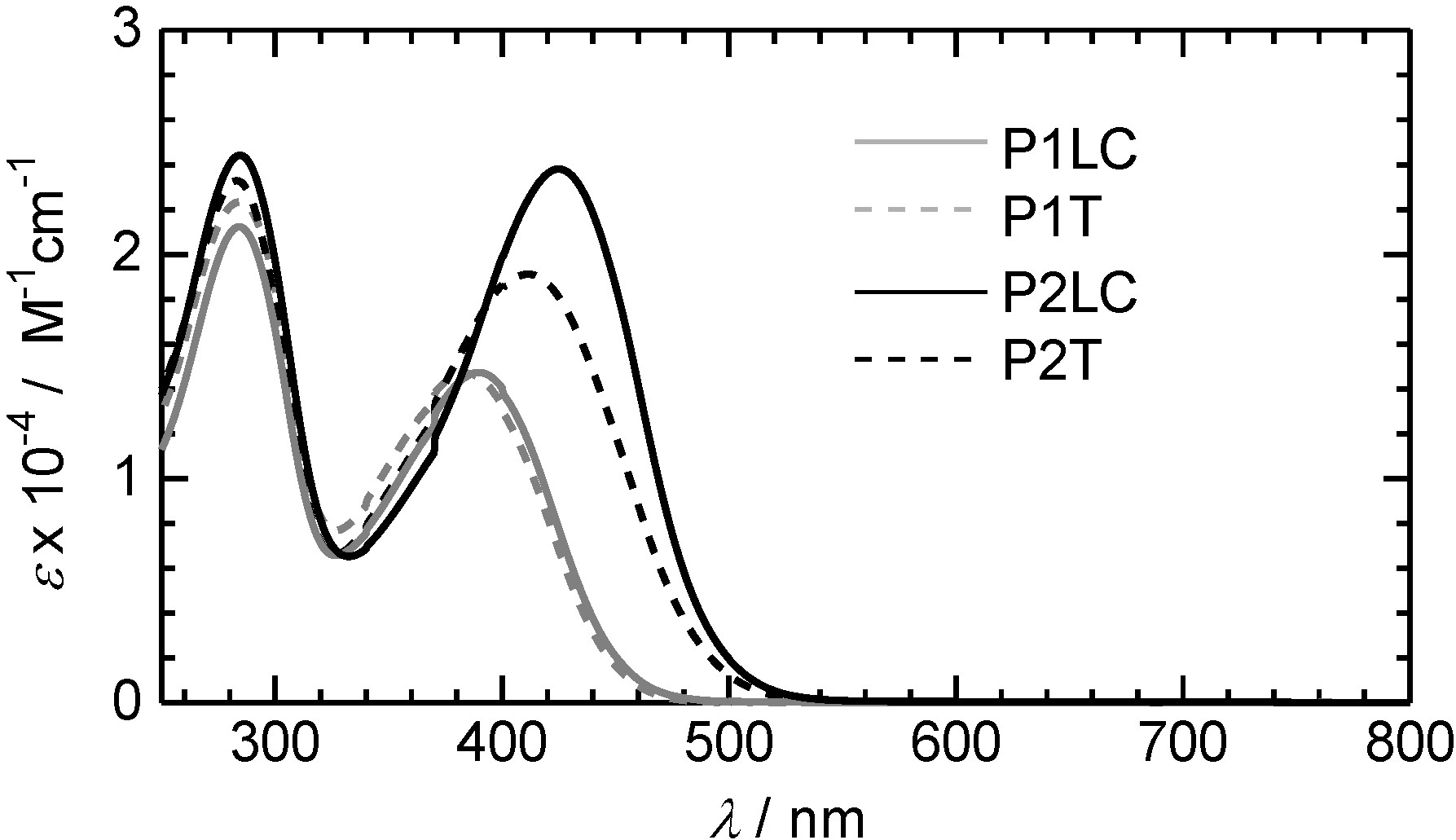
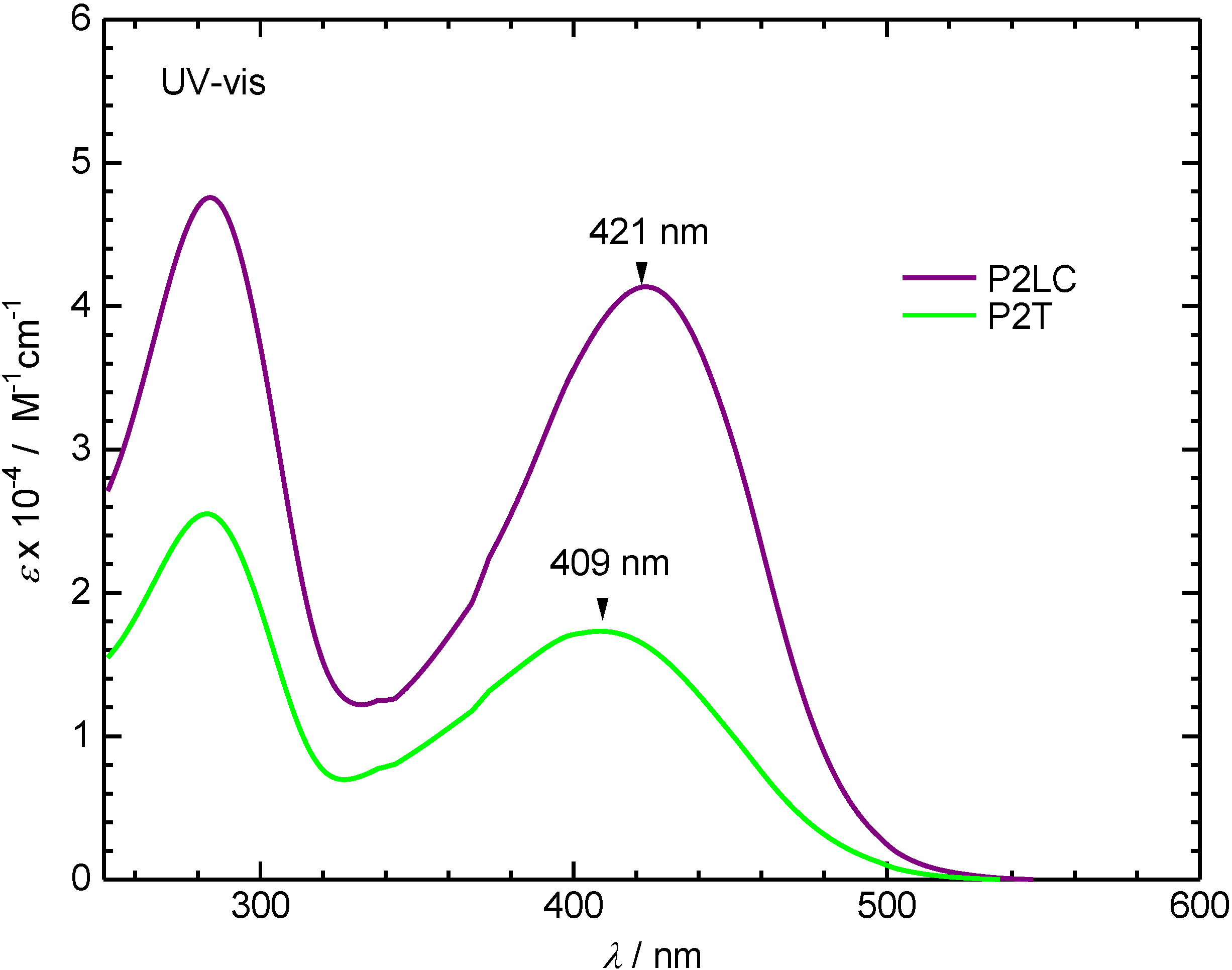
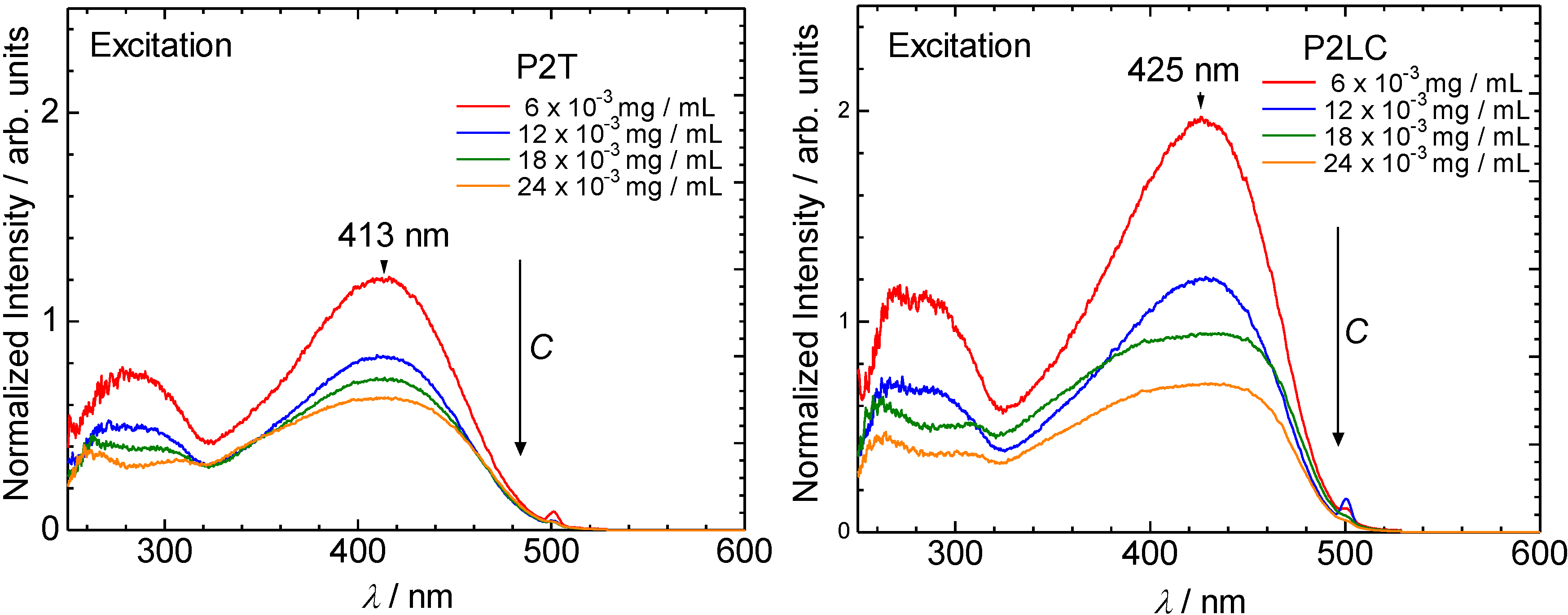
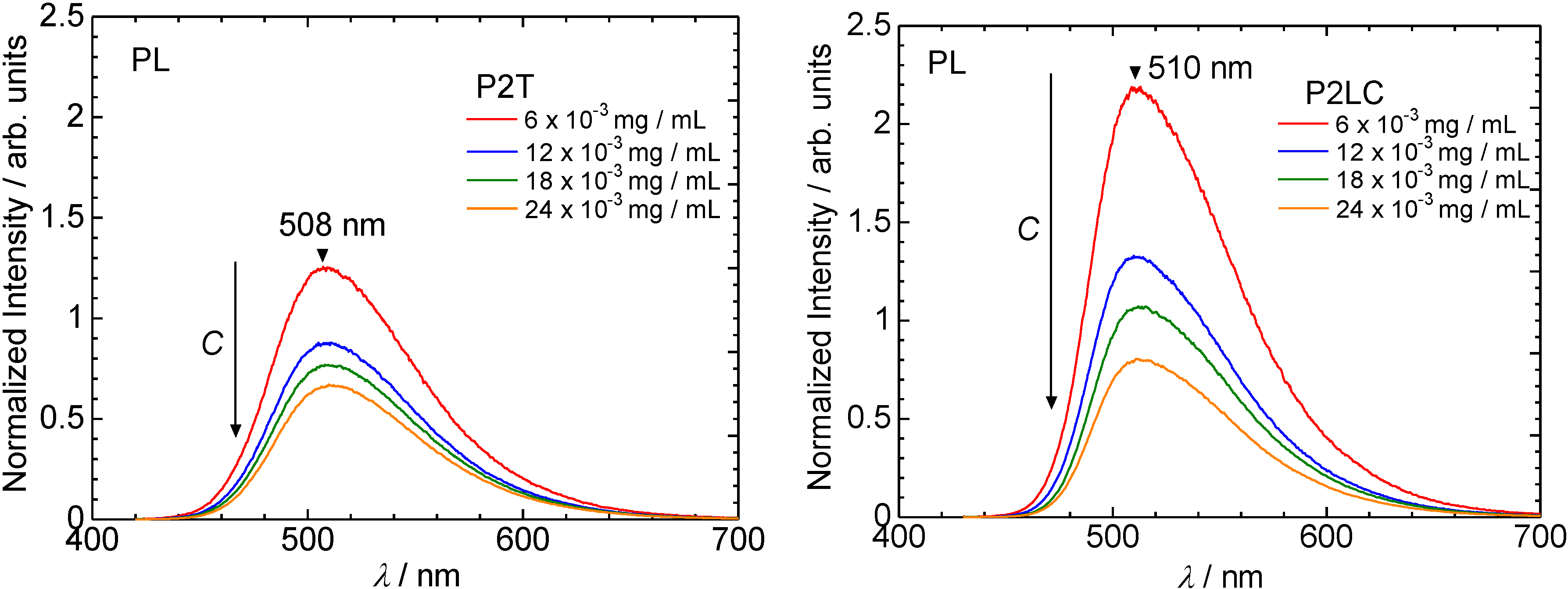
3.2. Ultraviolet-visible absorption and photoluminescence spectra
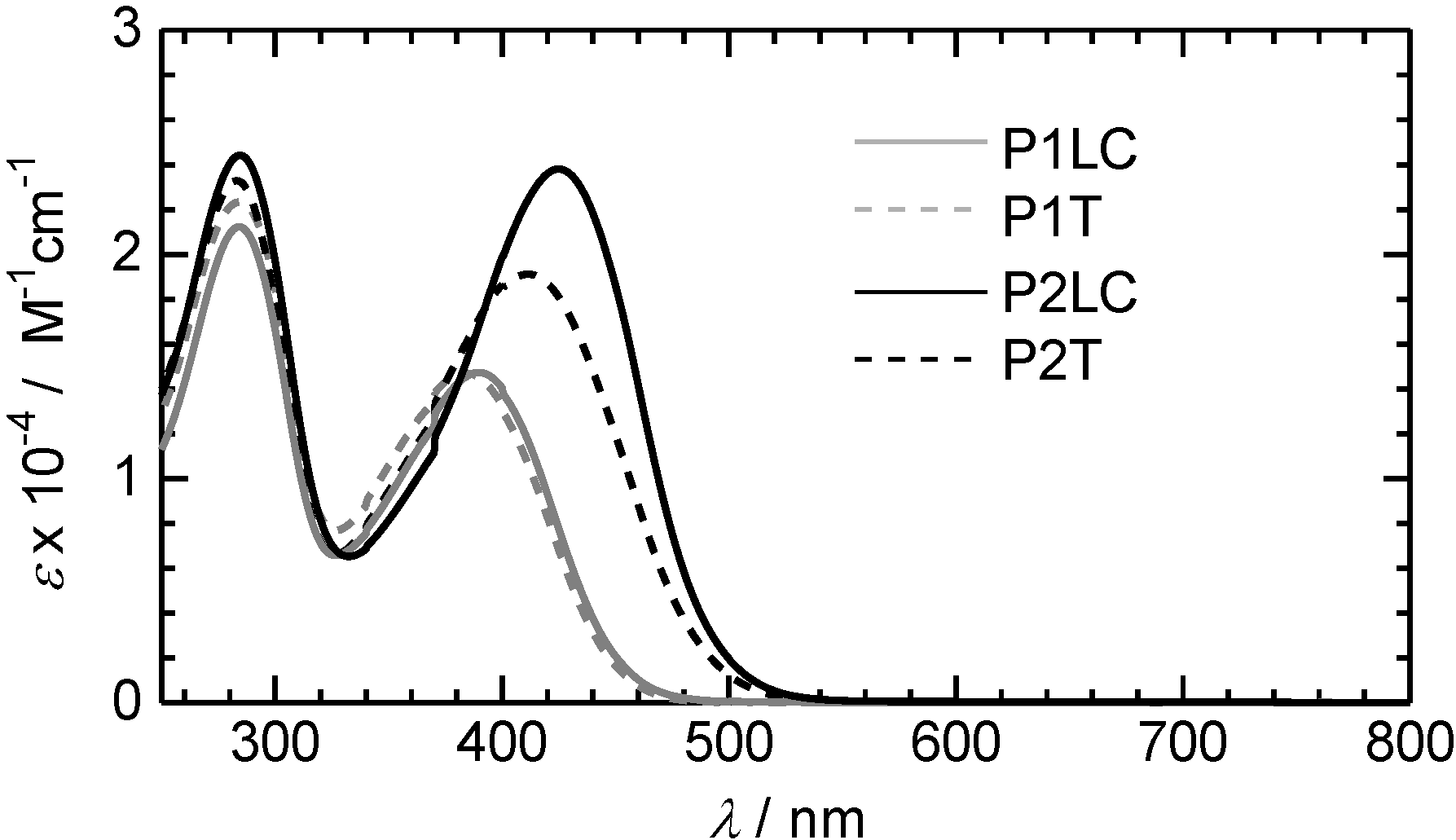
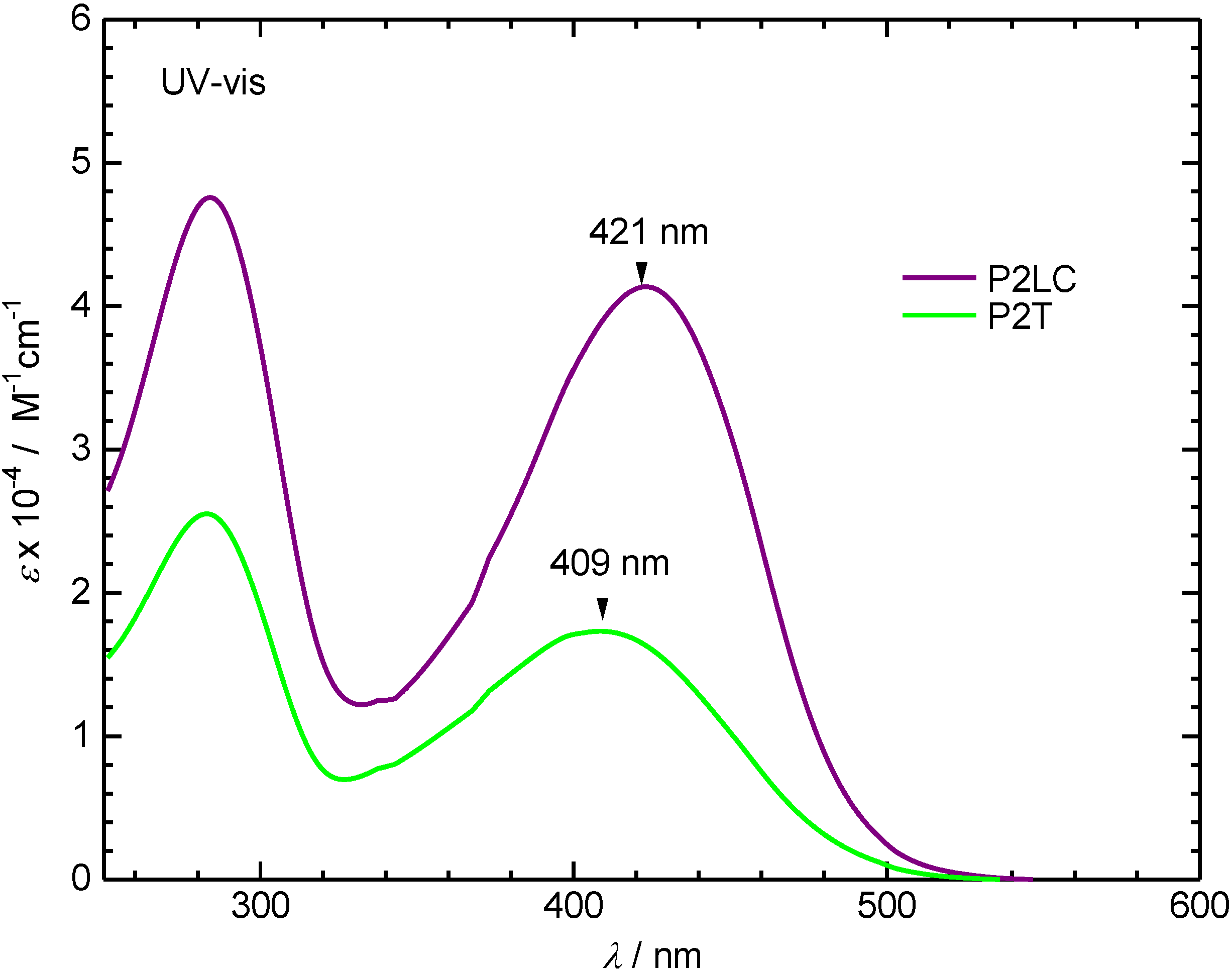
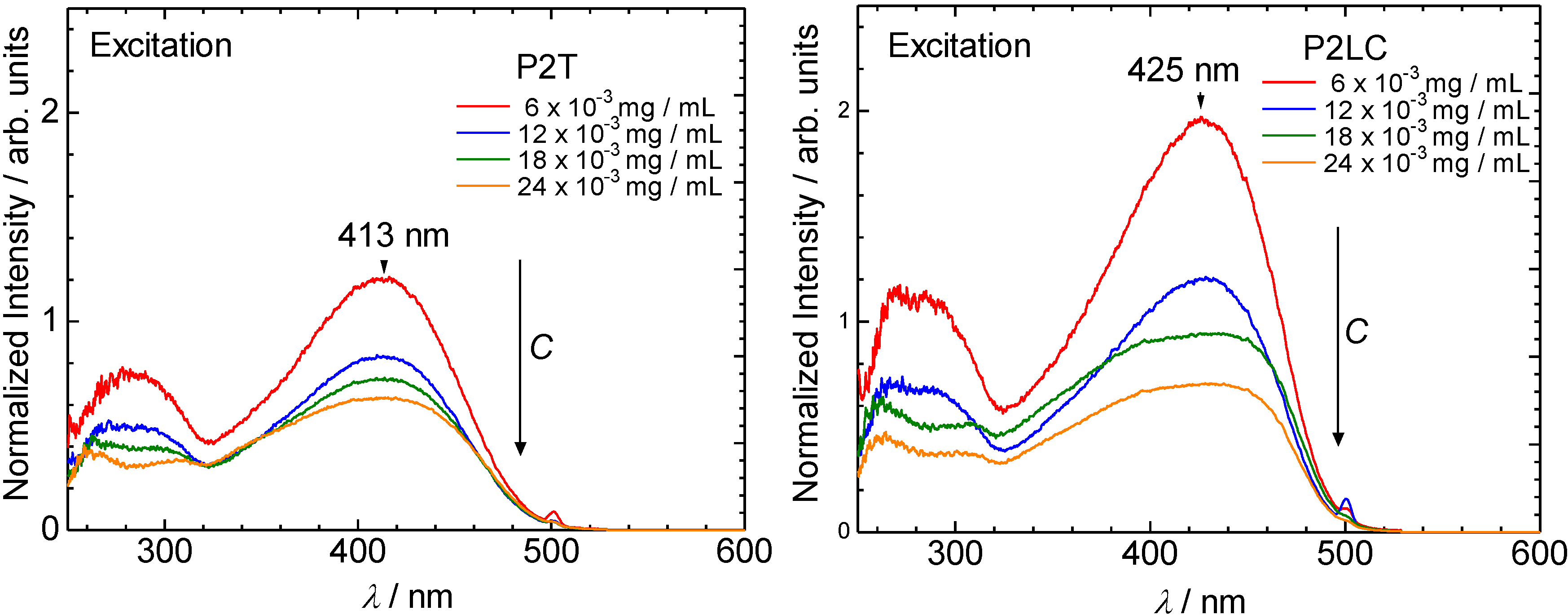

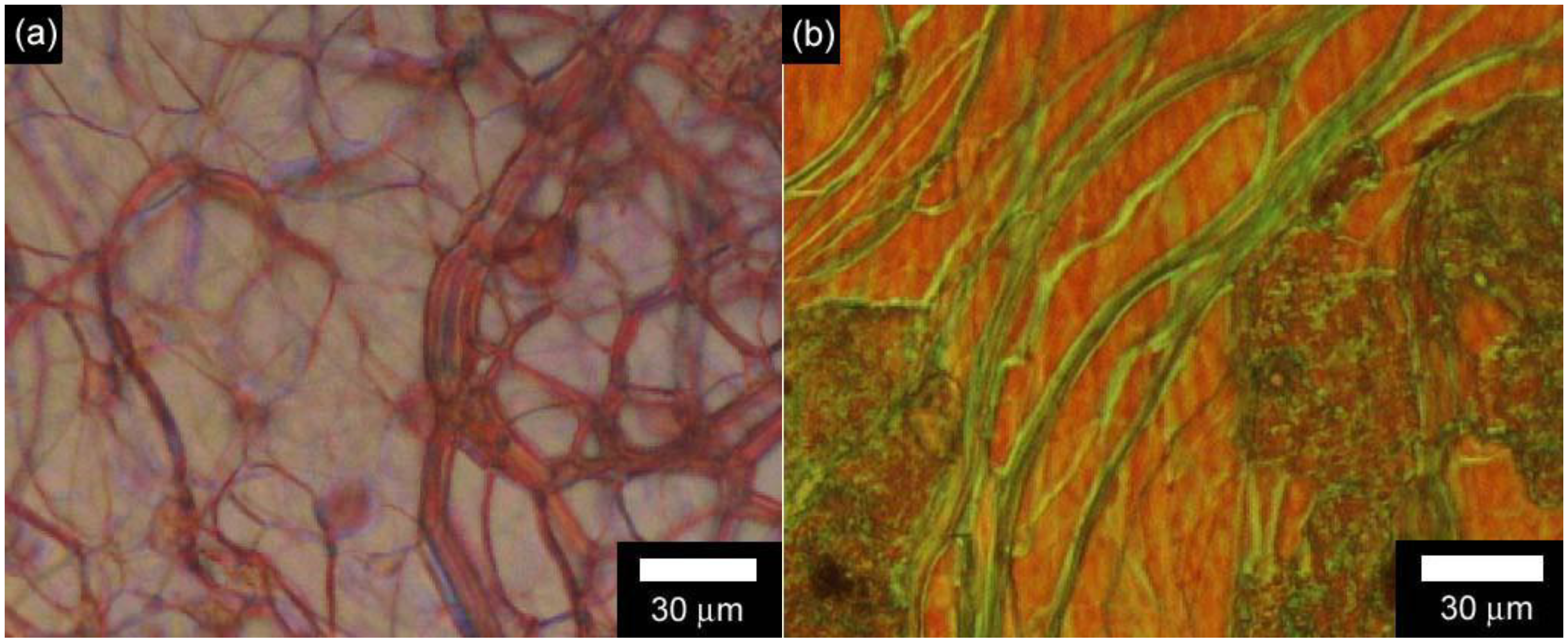
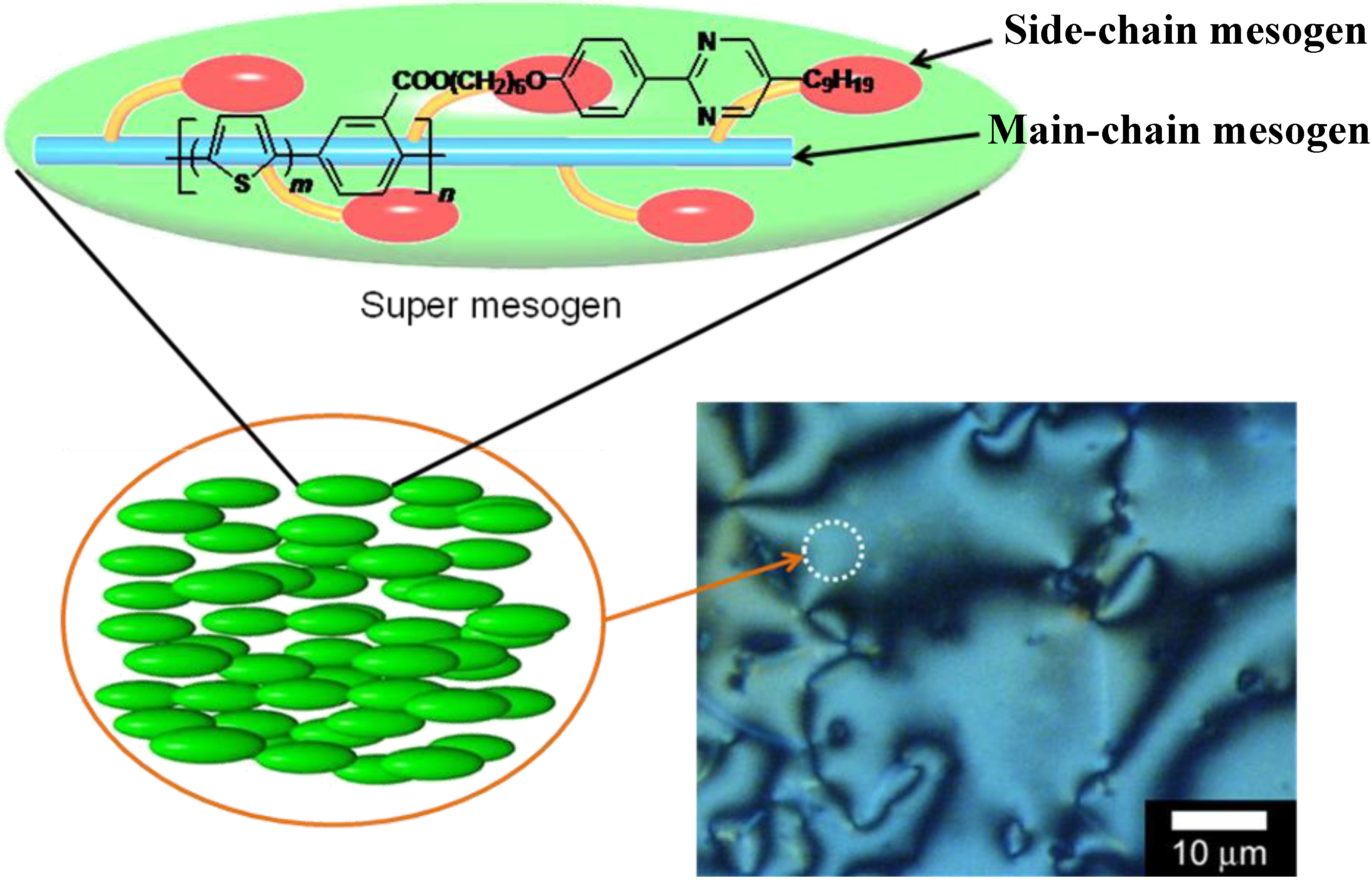
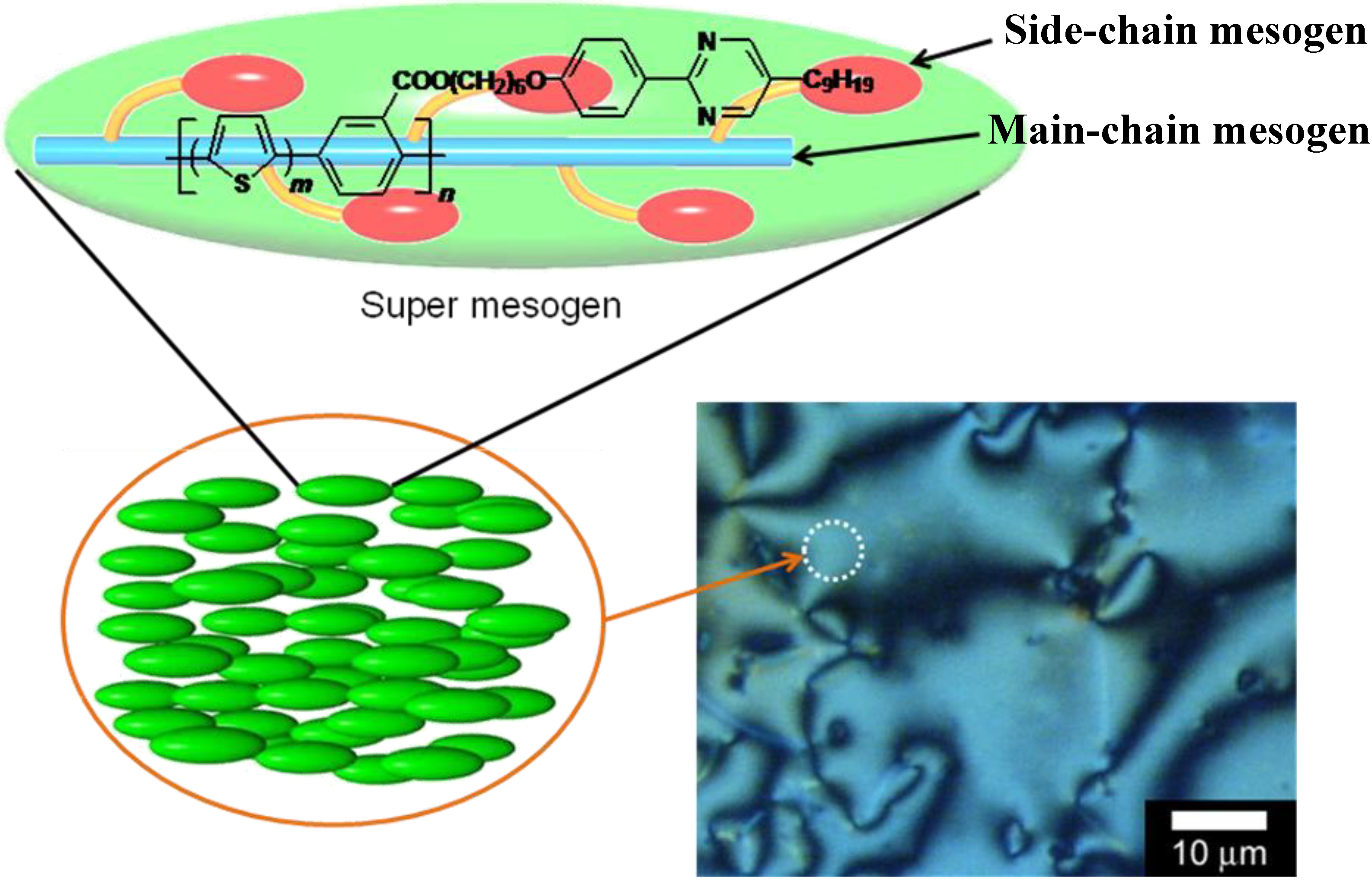
3.3. Polarizing optical microscopy observation of polymerization media


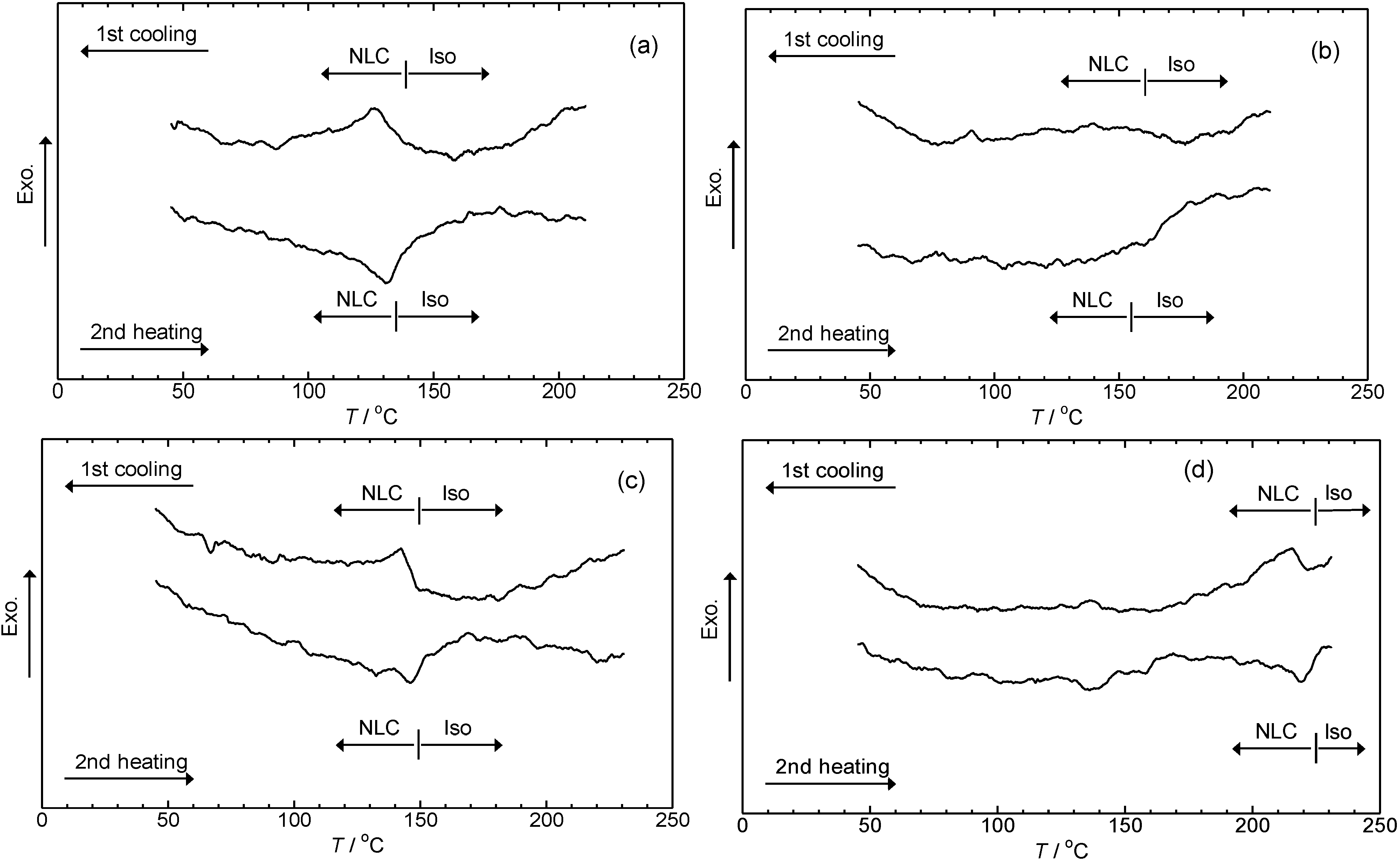
3.4. Phase transition
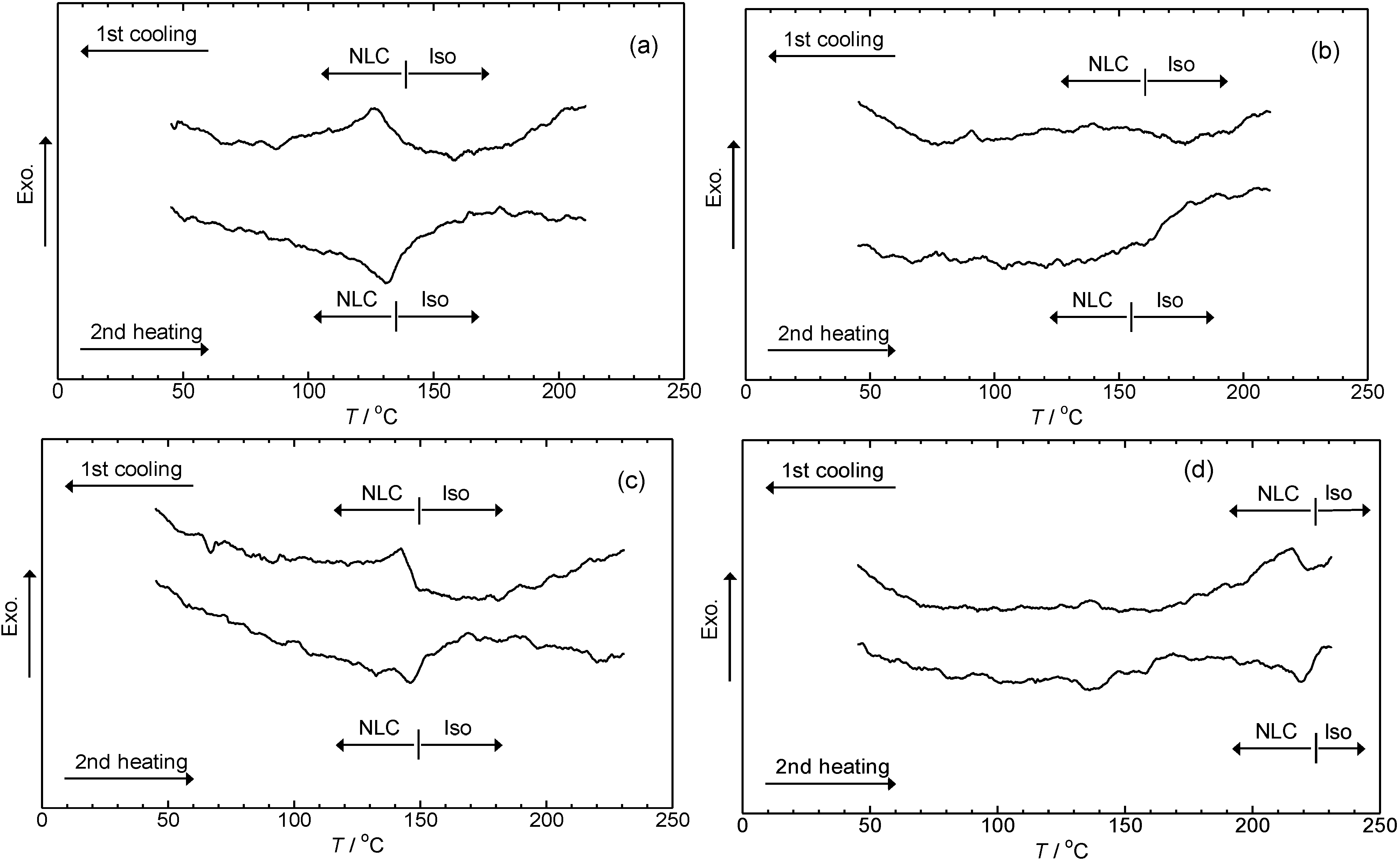


4. Conclusions
Acknowledgements
References
- Baxter, B.C.; Gin, D.L. Synthesis and Polymerization of a Chiral Liquid Crystal Diacrylate Exhibiting Smectic A* and C* Phases. Macromolecules 1998, 31, 4419–4425. [Google Scholar] [CrossRef]
- Okoshi, K.; Watanabe, J. Alternating Thick and Thin Layers Observed in the Smectic Phase of Binary Mixtures of Rigid-Rod Helical Polysilanes with Different Molecular Lengths. Macromolecules 2010, 43, 5177–5179. [Google Scholar] [CrossRef]
- Chen, S.H.; Mastrangelo, J.C.; Conger, B.M.; Kende, A.S.; Marshall, K.L. Synthesis and Characterization of Thermotropic Chiral-Nematic Polythiophenes. Macromolecules 1998, 31, 3391–3393. [Google Scholar] [CrossRef]
- Longa, L.; Pająk, G.; Wydro, T. Stability of Biaxial Nematic Phase for Systems with Variable Molecular Shape Anisotropy. Phys. Rev. E 2007, 76, 011703. [Google Scholar] [CrossRef]
- Wang, J.W.; Meng, F.B.; Li, Y.H.; Zhang, B.Y. Synthesis and Characterization of Side-Chain Cholesteric Liquid-Crystalline Polysiloxanes Containing Different Space Groups. J. Appl. Polym. Sci. 2009, 111, 2078–2084. [Google Scholar] [CrossRef]
- Yoshida, H.; Inoue, Y.; Isomura, T.; Matsuhisa, T.; Fujii, A.; Ozaki, M. Position Sensitive, Continuous Wavelength Tunable Laser Based on Photopolymerizable Cholesteric Liquid Crystals with an In-Plane Helix Alignment. Appl. Phys. Lett. 2009, 94, 093306. [Google Scholar] [CrossRef]
- Bobrovsky, A.; Boiko, N.; Shibaev, V.P.; Zavarzin, I.; Kalik, M.; Krayushkin, M. Cholesteric Mixture Containing Chiral-photochromic and Diarylethene Dopants as Novel Material with Dual Photochromism. Polym. Adv. Technol. 2002, 13, 595–600. [Google Scholar] [CrossRef]
- Liedtke, A.; Lei, C.; O’Neill, M.; Dyer, P.E.; Kitney, S.P.; Kelly, S.M. One-Step Photoembossing for Submicrometer Surface Relief Structures in Liquid Crystal Semiconductors. ACS. Nano. 2010, 4, 3248–3253. [Google Scholar] [CrossRef] [PubMed]
- Ren, W.; McMullan, P.J.; Griffin, A.C. Stress–strain Behavior in Main Chain Liquid Crystalline Elastomers: Effect of Crosslinking Density and Transverse Rod Incorporation on “Poisson’s Ratio”. Phys. Status Solid B 2009, 246, 2124–2130. [Google Scholar] [CrossRef]
- Zhu, Z.; Swager, T.M. Conjugated Polymer Liquid Crystal Solutions: Control of Conformation and Alignment. J. Am. Chem. Soc. 2002, 124, 9670–9671. [Google Scholar] [CrossRef] [PubMed]
- Nishizawa, K.; Nagano, S.; Seki, T. Novel Liquid Crystalline Organic-Inorganic Hybrid for Highly Sensitive Photoinscriptions. Chem. Mater. 2009, 21, 2624–2631. [Google Scholar] [CrossRef]
- Kim, N.; Choi, J.; Chien, L.C.; Kyu, T. Phase Equilibria of a Mixture of Side-Chain Liquid Crystalline Polymer and Low Molecular Mass Liquid Crystal. Macromolecules 2007, 40, 9582–9589. [Google Scholar] [CrossRef]
- Goto, H. Cholesteric Liquid Crystal Inductive Asymmetric Polymerization: Synthesis of Chiral Polythiophene Derivatives from Achiral Monomers in a Cholesteric Liquid Crystal. Macromolecules 2007, 40, 1377–1385. [Google Scholar] [CrossRef]
- Kawabata, K.; Goto, H. Periodic Structure in a Fluorene-based Polymer Prepared by Electrochemical Polymerization. Chem. Lett. 2009, 38, 706–707. [Google Scholar]
- Araya, K.; Mukoh, A.; Narahara, T.; Shirakawa, H. Polymerization of Acetylene in Liquid Crystal Solvent. Chem. Lett. 1984, 13, 1141–1142. [Google Scholar]
- Kilbinger, A. F. M.; Feast, W. J. Solution Processable Alternating Oligothiophene-PEO-block-copolymers: Synthesis and Evidence for Solvent Dependent Aggregation. J. Mater. Chem. 2000, 10, 1777–1784. [Google Scholar] [CrossRef]
- Mitsunobu, O.; Yamada, M. Preparation of Esters of Carboxylic and Phosphoric Acid via Quaternary Phosphonium Salts. Bull. Chem. Soc. Jpn. 1967, 40, 2380–2382. [Google Scholar] [CrossRef]
- Kawabata, K.; Goto, H. Liquid Crystalline π-Conjugated Copolymers Bearing a Pyrimidine Type Mesogenic Group. Materials 2009, 2, 22–37. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ohkawa, S.; Ohta, R.; Kawabata, K.; Goto, H. Polymerization in Liquid Crystal Medium: Preparation of Polythiophene Derivatives Bearing a Bulky Pyrimidine Substituent. Polymers 2010, 2, 393-406. https://doi.org/10.3390/polym2040393
Ohkawa S, Ohta R, Kawabata K, Goto H. Polymerization in Liquid Crystal Medium: Preparation of Polythiophene Derivatives Bearing a Bulky Pyrimidine Substituent. Polymers. 2010; 2(4):393-406. https://doi.org/10.3390/polym2040393
Chicago/Turabian StyleOhkawa, Satoshi, Reina Ohta, Kohsuke Kawabata, and Hiromasa Goto. 2010. "Polymerization in Liquid Crystal Medium: Preparation of Polythiophene Derivatives Bearing a Bulky Pyrimidine Substituent" Polymers 2, no. 4: 393-406. https://doi.org/10.3390/polym2040393
APA StyleOhkawa, S., Ohta, R., Kawabata, K., & Goto, H. (2010). Polymerization in Liquid Crystal Medium: Preparation of Polythiophene Derivatives Bearing a Bulky Pyrimidine Substituent. Polymers, 2(4), 393-406. https://doi.org/10.3390/polym2040393